

Summary
In autoantibody-mediated diseases such as pemphigus serum antibodies lead to disease. Genetic analysis of B cells has allowed characterization of antibody repertoires in such diseases but would be complemented by proteomic analysis of serum autoantibodies. Here we show using proteomic analysis that the serum autoantibody repertoire in pemphigus is much more polyclonal than found by genetic studies of B cells. In addition many B cells encode pemphigus autoantibodies that are not secreted into the serum. Heavy chain variable gene usage of serum autoantibodies is not shared among patients, implying targeting of the coded proteins will not be a useful therapeutic strategy. Analysis of autoantibodies over years in individual patients indicates that many antibody clones persist but the proportion of each changes. These studies indicate a dynamic and diverse autoantibody response not revealed by genetic studies, and explain why similar overall autoantibody titers may give variable disease activity.
Introduction
Major efforts have been made recently to characterize the autoantibody (autoab) response in patients with autoimmune diseases. In doing so, investigators ultimately hope to find commonalities that could indicate possible initiators of disease, help define how disease progresses or establish targets for therapy. Such studies have largely characterized the autoimmune B-cell response through genetic or cellular approaches including antibody phage display (APD), Epstein-Barr virus (EBV) transformation of lymphocytes, or heterohybridoma formation. More recently, next generation sequencing (NGS) has been used to characterize total B-cell repertoires (not necessarily restricted to the autoimmune B cells) of autoimmune patients (e.g., in systemic lupus erythematosus (Tipton et al., 2015)).
In studies of tissue specific autoab-mediated diseases, these approaches have suggested an oligoclonal, limited antibody (ab) response with restricted variable heavy chain gene (VH) usage. For example abs with VH4-21 gene usage are required for pathogenicity in cold agglutinin disease (Pascual et al., 1992); in thrombotic thrombocytopenic purpura VH1-69, VH1-3 and a few other genes predominate (Schaller et al., 2014); in idiopathic thrombocytopenic purpura VH3-30 predominates (Siegel, 2008). These studies have suggested a limited or oligoclonal repertoire in these patients. However, such genetic studies may not necessarily reflect the actual circulating autoabs. In fact, a recent review of serum antibodies pointed out that even after a century of immunology research, circulating serum ab clonotypes have been characterized minimally if at all (Wine et al., 2015). Since it is unclear whether genetic analysis of autoabs reflect the serum autoab repertoire, and because it is the circulating serum autoantibodies that cause disease, we characterized the circulating autoab repertoire in patients with pemphigus vulgaris (PV) and foliaceus (PF), two prototypical tissue-specific, autoab-mediated, autoimmune conditions in which the antigens are well defined as the epidermal cell adhesion molecules desmoglein (Dsg) 3 and Dsg1, respectively (Stanley and Amagai, 2006).
Previous studies, including those from our group, have characterized B-cell anti-Dsg IgG-coding sequences at the genetic level using APD, EBV transformed lymphocytes and/or heterohybridoma techniques (Cho et al., 2014; Di Zenzo et al., 2012; Ishii et al., 2008; Payne et al., 2005; Qian et al., 2009; Qian et al., 2007; Yamagami et al., 2009; Yamagami et al., 2010). These studies suggested that the autoab response is oligoclonal (defined here as less than 10 IgG heavy chain clonotypes per patient), with an average of 4 clones per PV patient (range 1-9) and PF patient (range 1-7). In some cases common IgG VH gene usage among patients was reported, e.g., VH1-46 (Cho et al., 2014) or VH3-23/VH3-30 (Qian et al., 2007) in PV and VH3-23 in PF (Qian et al., 2009). However, other studies do not show VH gene preferences (Di Zenzo et al., 2012). In addition, APD studies have shown persistence of anti-Dsg B cells over time as disease remits and relapses in patients (Hammers et al., 2015). These genetic studies have been very valuable in characterizing the non-tolerant autoimmune B-cell response and its response to therapy (Hammers et al., 2015). They have also provided valuable information about pathogenicity and non-pathogenicity of coded anti-Dsg abs and cross reactivity of these abs between Dsg3 and Dsg1 (Payne et al., 2005).
Genetic studies analyze the antibody-coding sequences of circulating B-cells. Such genetic approaches are limited by sampling issues (i.e. number of phage clones or heterohybridoma/EBV-transformed clones that can practically be analyzed). Furthermore, just because B cells have anti-Dsg coding sequences does not mean that they necessarily secrete circulating ab since they may not differentiate into ab-secreting cells (i.e., plasmablasts or plasma cells).
In order to analyze serum (i.e. circulating) abs, a proteomic approach is necessary. Isolation of antigen-specific antibodies by affinity chromatography theoretically allows analysis of the repertoire without clonal sampling limitations. Recently, mass spectrometry (MS)-based techniques have been developed to characterize serum antibody repertoires (Cheung et al., 2012; Lavinder et al., 2014; Wine et al., 2013), but such studies have not been reported in tissue-specific autoab-mediated diseases. Pemphigus is a particularly attractive disease for characterizing the autoab response at the proteomic level because the antigens are well defined; therefore, specific abs that bind those antigens can be isolated by affinity chromatography from patient sera.
In this study we characterize the antigen-specific circulating autoab response in pemphigus and compare this response to data from genetic studies. Our results indicate that the serum autoab response in pemphigus correlates very poorly with the repertoire suggested by genetic studies of B cells that encode autoantibodies, and is much more polyclonal and more diverse than suggested in all previous genetic studies. In this circulating autoab repertoire, we show that, even though the autoab response among patients is to the same two closely related antigens, there is no convergence of this response either in VH gene usage or variable heavy chain (VH)-CDR3 (H-CDR3) amino acid (AA) sequences. Furthermore, even though the anti-Dsg ab response is polyclonal, a few dominant clones produce most of the circulating ab in each patient at any given time. Finally, as illustrated by three patients studied over time, although some circulating autoab clonotypes persist over years, the entire ab landscape changes with different ab clonotypes producing various relative amounts of the total autoab.
Results
Analysis of the anti-Dsg serum autoab repertoire by liquid chromatography—high resolution tandem MS (LC-MS/MS)
This analytic approach, based on published methods (Cheung et al., 2012; Lavinder et al., 2014; Wine et al., 2013), is summarized in Figure 1A and in detail in the Experimental Procedures. Anti-Dsg3 and anti-Dsg1 autoabs from 4 PV and 2 PF patients, respectively, were isolated by affinity chromatography and designated as “bound” fractions. IgG from the passthrough from these same columns was isolated by Protein G affinity chromatography and termed “unbound” fractions. Ab heavy chains from both fractions in each patient and time point were analyzed by LC-MS/MS. MS/MS spectra were searched against theoretical spectra inferred from a reference database comprising the AA sequences of the H-CDR3 and its immediately surrounding AAs in that patient. These sequences were obtained from peripheral blood mononuclear cells by NGS of the IgG repertoire. Stringent filtering criteria were applied to identify the peptides in the reference database whose spectra were predicted to match the results of LC-MS/MS, i.e. “peptide-spectra matches” (PSM). To confirm the specificity of our PSMs, we synthesized five stable heavy-isotope labeled peptides corresponding to LC-MS/MS identified peptides of H-CDR3 regions of bound abs. All five heavy peptides behaved the same way as their corresponding light peptides in the bound ab by being eluted in LC at the same retention time and generating matching patterns of MS/MS fragment ions (Figure S1), thus confirming that the peptide sequences assigned by database searching of MS/MS spectra were correct.
Figure 1. Proteomic platform to identify circulating pemphigus anti-Dsg abs.

(A) IgG heavy chains from Dsg-binding abs and from abs which do not bind to Dsg are analyzed by LC-MS/MS. Resultant spectra are searched against a custom database of all VH sequences from the same patient to identify ab peptides. Informative peptides match H-CDR3 AA sequences that define clonotypes of abs. (B) ELISA assays of PV or PF sera before and after affinity chromatography show that anti-Dsg ab is depleted by AP. PBMC, peripheral blood mononuclear cells. AP, affinity purification.
To be confident in the identification of only anti-Dsg abs from the proteomic analysis we required their identification in all technical replicates of the bound fraction but never in any of the technical replicates of the unbound fraction. These “strict” criteria may underestimate the number of anti-Dsg abs. For example, some low affinity pemphigus abs may be present in the unbound fraction, although enriched in the bound fraction. However, this situation seems unlikely as the affinity purification was extremely efficient and left few, if any, detectable anti-Dsg abs (Figure 1B). We may also be missing some very low abundance abs that are near the limits of detection as they may not be detected in all technical replicates of the bound fraction. Finally, there is the unlikely possibility that some clonal expansions of abs that share the same H-CDR3 clonotype may bind and some may not bind Dsg3—these we would also not include in the strict criteria as pemphigus abs. To be confident that the abs we are characterizing are pemphigus abs, in all results below we used these strict criteria unless we specifically state otherwise.
The identified anti-Dsg abs were grouped into clonotypes (also called “clones”), defined as VH AA sequences which had H-CDR3 AA sequences of the same length and at least 80% identity (Moody et al., 2011; Wine et al., 2013). Overall, this proteomic analysis of circulating antigen-specific abs in these 6 patients identified an average of 39 IgG clonotypes per patient, with a range of 18 to 63 (Tables 1, S1).
Table 1.
Circulating ab and APD B-cell clonotypes
| Patient identifier | # of IgG clonotypes identified by MS | # of B-cell clonotypes identified by APD | Overlap | % of APD clonotypes confirmed by MS |
|---|---|---|---|---|
| PV1a | 22 | 4 | 0 | 0% |
| PV8 | 37 | 12 | 3 | 25% |
| PV16 | 30 | 11 | 0 | 0% |
| PV3 | 34 | 3 | 0 | 0% |
| PV3a | 56 | 6 | 1 | 17% |
| PF1 | 18 | 4 | 3 | 75% |
| PF1a | 23 | NA | NA | NA |
| PF4 | 63 | NA | NA | NA |
| PF4a | 55 | NA | NA | NA |
| PF4b | 52 | NA | NA | NA |
MS: mass spectrometry; APD: antibody phage display
See also Tables S1, S2, S3
Serum ab repertoires identified by LC-MS/MS show greater clonal diversity and limited overlap with genetic anti-Dsg B-cell repertoires identified by APD
We previously used APD to characterize anti-Dsg3 B-cell clonotypes in the peripheral blood mononuclear cells of 2 PV (PV3a, PV1) and one PF (PF1) patients (Hammers et al., 2015; Ishii et al., 2008). Using the same techniques, we characterized two additional PV patients, PV8 and PV16 by APD. However, in these later two patients we used additional approaches to optimize APD (see Experimental Procedures). We then compared the APD B-cell clonotypes in each patient to their circulating ab clonotypes determined by LC-MS/MS. In patients in whom APD was performed at the same time point, the average anti-Dsg clonotypes per patient (+/- SEM) detected in serum was 32.8 +/-5.5, compared to 6.7 +/-1.6 detected by APD (Tables 1, S1, S2) (p=0.03 by Wilcoxon signed-rank test).
Eighteen percent (range 0-75%) of APD B cell clonotypes had corresponding clonotypes in the circulating abs as detected by LC-MS/MS (Table 1, S3). Because the strict selection criteria used to define circulating anti-Dsg abs may filter out some clonotypes identified by APD, we compared APD clones to all circulating abs in both the bound and unbound pool without applying these strict criteria. In that case 20% (range 0-75%) of the 40 APD-identified anti-Dsg B cell clones were matched to clones in this larger circulating ab pool. Only one additional APD clonotype, PV3a-2 in Table S3, identified a circulating ab that was not found by strict criteria because it was not found in all the bound technical repeats, although it was never identified in the unbound fraction. These data show that some of the memory B-cell/plasmablast clonotypes identified by APD in this and previous studies do contribute to the serologic response, however, most of the B-cell clonotypes identified by APD contribute few if any circulating abs. This is consistent with data that most memory B-cells may not contribute significantly to the serologic response (Wine et al., 2015), and is consistent with data shown below that some anti-Dsg B-cell clonotypes detected by NGS at one time point may not produce ab at that same time point.
In many cases in which APD clonotypes were found to match ab clonotypes, we detected no H-CDR3 variant AA sequences in the APD clone while in the same patient’s matching circulating ab clonotype multiple H-CDR3 variants were detected (Table S3). This finding suggests that during APD selection one variant may outcompete others by its stronger antigen binding and/or growth advantage in bacteria. In this sense, also, the circulating anti-Dsg repertoire is more varied than suggested by APD.
Not only are many APD clonotypes not detected as serum abs but, conversely, most of the serum abs are undetectable by APD even when comparing APD to all ab clones detected by LC-MS/MS (Figure 2; triangles show ab clones identified by the ‘strict” criteria, that is only in bound (pemphigus) or only in unbound fraction; circles show all other antibodies identified by LC-MS/MS). Serum abs may not be detectable by APD because it has been shown that APD is biased as certain sequences may be toxic to bacteria and others may predominate because of faster growth of their phage (Derda et al., 2011). We did observe in PV8 that the sequences of about 1/3 of the serum anti-Dsg abs were present in the VH PCR amplicon used for APD, but most became undetectable after 2-4 rounds of panning (each having >12,000 unique VH sequences) when analyzed by NGS (data not shown).
Figure 2. Most of the abs in circulation were not identified by APD.
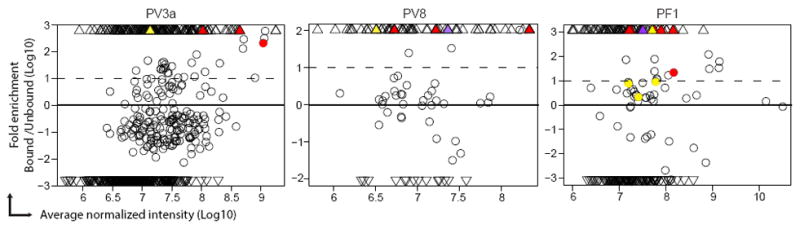
Each MS-identified ab is shown as a circle (peptide was detected in both bound and unbound fractions) or triangle (peptide was detected only in either bound or unbound fraction). Colored circle or triangle indicates that particular ab is clonally related with one B-cell clonotype identified by APD. In each patient, different APD clonoypes are indicated by different colors. APD clonotypes did not match with any abs identified by MS in PV1a, PV3 or PV16 samples.
To further address why circulating ab clones may not be identified by APD and to confirm that the VH chains detected by LC-MS/MS but not APD are valid, we synthesized nucleotide sequences of three such chains from PV3a, then combined each with the PCR-amplified VL repertoire of PV3a to generate VH-specific APD libraries. The three VH-specific libraries were selected on Dsg3-ELISA plates by at least three rounds of panning to identify single chain variable fragment abs (scFv) that bind Dsg3. Two of the 3 VH identified by LC-MS/MS (AA sequences shown in Table S4) paired with at least one VL to form scFv that bound Dsg3 by ELISA and the cell surface of keratinocytes by indirect immunofluorescence (Figure 3A, B). The VH of GB4 paired with two VLs (from VL genes IGKV3-20 and IGKV3-15) and GB8 paired with 3 VLs (all from gene IGLV2-14) to form scFvs that bound Dsg3. The relevance of these recombinant abs to those in PV3a sera was confirmed by showing that serum from PV3a blocked their binding to Dsg3 (Figure 3C). These data illustrate that VH identified by LC-MS/MS but not originally identified by APD are capable of producing anti-Dsg3 abs that bind the same or similar epitopes as the patient’s serum; and that these abs are not toxic in APD but are likely outcompeted by other, faster growing clones in a full APD library from this patient. The third VH chain could not be analyzed because its VH-specific APD library did not permit growth of transfected E.coli, presumably because that particular VH was toxic.
Figure 3. Recombinant scFv with VH AA sequences identified by MS from PV3a serum bind Dsg3.
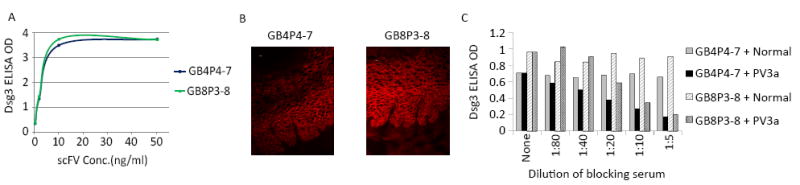
(A) Two scFv (GB4P4-7 and GB8P3-8) with different VH AA sequences as identified in MS, synthesized and isolated as in Experimental Procedures, bind Dsg3 by ELISA. (B) Same two scFv bind cell surface of keratinocytes in monkey esophagus where Dsg3 locates. Scale bar, 10 μm (C) PV3a serum but not normal human serum inhibits binding of these scFv to Dsg3 on ELISA. See also Table S4.
Taken together, the above data demonstrate that the serum ab repertoire revealed by a proteomic approach and the B-cell repertoire as analyzed by APD have limited overlap, that the serological response in pemphigus is more polyclonal than previously reported, and that B cells encoding anti-Dsg abs may produce little if any circulating ab.
Circulating anti-Dsg abs in pemphigus are divergent among patients
A variety of VH genes are used in the coding sequences of anti-Dsg abs in each patient, coming from the IGHV1, IGHV2, IGHV3, IGHV4 and IGHV5 gene families (Figure 4A, B). Among them, IGHV1-69 and IGHV5-51 were used by all PV and PF patients. The VH gene usage frequency is variable among patients. Some but not all patients showed high frequency usage of certain VH genes, but the predominant VH gene usage in various patients was different, e.g., VH1-69 and VH4-4 in PV8, VH3-30 in PV1a, and VH1-69 and VH4-34 in PF4.
Figure 4. Characterization of circulating ab clonotypes in PV and PF patients.
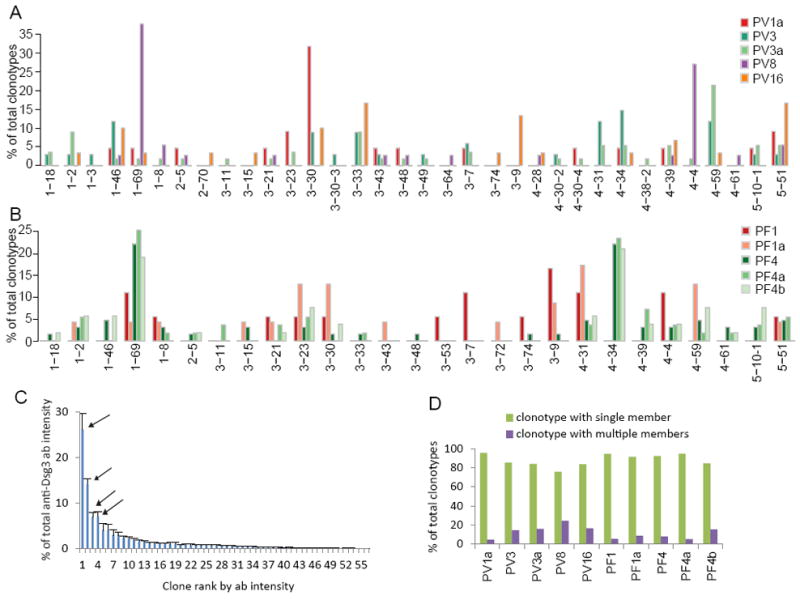
VH gene usage of clonotypes in PV patients (A) and PF patients (B). (C) Intensity (as a measure of amount of ab) of each of the 56 clonotypes found in PV3a. Arrows indicate the 4 “dominant” clones which together produce more than 50% of the total ab. Data were from 4 bound replicates. Error bar, SEM. (D) Most clonotypes do not have variable H-CDR3 AA sequences.
Closer examination of the H-CDR3 sequences revealed no shared ab clonotypes among patients.
These data show that there is no convergence of the immune response among various pemphigus patients, even though they all form abs to two closely related Dsgs.
Although the anti-Dsg ab response is polyclonal a few dominant clones produce most of the circulating ab
By using peptide intensity in LC-MS/MS as an indicator for the ab protein level, we found that >50% of the detectable serum ab response is produced by only 5-20% (mean 11%) of the total ab clonotypes in each patient (Table 2). We defined “dominant clones” as the highest producing clones that together produce greater than 50% of the total circulating anti-Dsg ab. The average ab production per clone of the dominant clones in each patient ranged between 7-52% of the total circulating ab (mean 17%); and the non-dominant clones produced only 1-3% of the total ab per clone (Table 2). For example, of 56 ab clones in patient PV3a, most produce little detectable ab, but 4 clones produce over 50% of the total ab (Figure 4C).
Table 2.
A few “dominant” clonotypes produce most of the circulating abs in pemphigus patients
| Patient identifier | # of total clones | # of dominanta clones | % of dominant clones | % of total ab production per clone | |
|---|---|---|---|---|---|
| dominant clones | non-dominant clones | ||||
| PV1a | 22 | 3 | 14 | 19 | 2 |
| PV8 | 37 | 3 | 8 | 17 | 1 |
| PV16 | 30 | 6 | 20 | 9 | 2 |
| PV3 | 34 | 5 | 15 | 10 | 2 |
| PV3a | 56 | 4 | 7 | 13 | 1 |
| PF1 | 18 | 1 | 6 | 52 | 3 |
| PF1a | 23 | 3 | 13 | 18 | 2 |
| PF4 | 63 | 7 | 11 | 7 | 1 |
| PF4a | 55 | 3 | 5 | 19 | 1 |
| PF4b | 52 | 7 | 13 | 7 | 1 |
| Mean ± SEM | 11 ± 1b | 17 ± 4 | 2 ± 0c | ||
Dominant clones are defined as the highest producing circulating ab clonotypes that together produce more than 50% of the total abs in any patient
p<0.0005 compared to 50% using one-sample test of proportion
p=0.005 for % of total ab per clone dominant compared to non-dominant by Wilcoxon signed-rank test
H-CDR3 somatic mutations causing AA changes are unusual in anti-Dsg abs
For all patients, most of the anti-Dsg clonotypes (74% -95%) contain only one H-CDR3 AA variant (Figure 4D), suggesting that the epitopes on Dsg have stringent requirements for ab binding.
Individual circulating ab clonotypes may persist over time although the overall circulating ab landscape changes in pemphigus patients
Previously, we have shown by APD that the same genetic B-cell ab coding sequences persist over time in 2 PV patients (Hammers et al., 2015). Here, we asked whether serum ab clonotypes persist over time in one PV and 2 PF patients (labeled “Ab repertoire” in Figure 5A). In patient PV3 13 circulating ab clonotypes persisted over 6 years. Three of 38 circulating ab clonotypes were detected over 6.6 years in patient PF1. Patient PF4 abs were characterized at time 0 (PF4), 0.7 year later (PF4a) and then another 1.8 years later (PF4b). This analysis is particularly interesting in that we can determine that of 20 abs found at the PF4 and PF4b time points, 18 (or 90%) were also detected at the intermediate PF4a time point, showing the robustness of this technique to detect ab clones even at different time points (Figures 5A, labeled “ab repertoire”, and S4). We also analyzed the presence of B-cell clonotypes at each time point as defined by NGS sequences that matched anti-Dsg serum abs detected at any time point or inferred from serum abs that matched NGS from a different time point (labeled “B cell/ab” repertoire in Figure 5A). In this analysis of B-cell and/or ab clonotypes over time, the persistence of clones is even more impressive, and is consistent with what we have previously reported on a much smaller dataset from APD combined with clone-specific PCR in two PV patients (Hammers et al., 2015). However, it is also clear from these data that some B-cell or ab clones are not detected at all time points. Whether this is due to sampling, disappearance of some clones with time or appearance of new clones cannot be determined by our data.
Figure 5. Anti-Dsg Ab and B-cell genetic clonotypes over time in 3 patients.
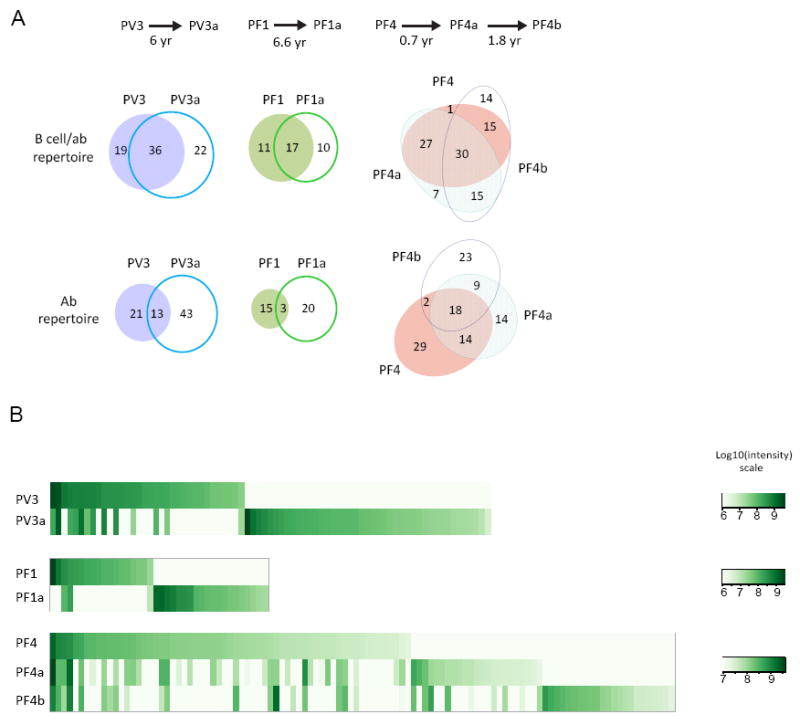
(A) Circulating ab clonotypes at each time point. “Ab repertoire”: these are MS-identified abs at each time point and correspond to the # of IgG clonotypes in Table 1; “B cell/ab repertoire”: refers to both the ab detected at a particular time point by MS plus genetic ab sequences found at that time point identified by an pemphigus ab found at another time point by MS (i.e. NGS genetic sequences identified as pemphigus abs by matches to MS sequences of the affinity-purified pemphigus ab from a different time point). Many anti-Dsg B cell and circulating ab clones persist over many years. (B) Intensity of circulating ab clones at different time points. Each column is one clonotype. Data indicates that the repertoire of circulating ab clonotypic expression is variable over time and that although some ab clonotypes may persist their ab production varies over time. See also Figures S2, S3, S4 and Tables S5, S6
Although these data show that some individual circulating ab clonotypes may persist up to 6.6 years in PF and PV patients, the overall circulating ab landscape changes over time (Figures 5B, S2, S3, S4). Not only are sets of circulating clonotypes expressed at one time point but not others, but also those that are expressed at different time points are expressed at different levels. Statistical analysis of technical repeats shows good reproducibility (i.e. small standard error of the normalized mean intensities) allowing discrimination of statistically significant changes of most antibody clones over time. Detailed analysis in these patients further showed that anti-Dsg sequences could be present by NGS at one time but the circulating abs corresponding to those sequences could not be detected at that time point but only at a different time point (Table S5). These data show that anti-Dsg B-cell clones often persist in pemphigus but they may produce little if any circulating ab at certain times, which is consistent with the findings above that many anti-Dsg B-cell clones detected by APD do not produce detectable circulating ab.
Discussion
Proteomic analysis of circulating antigen-specific autoabs in our cohort of PV and PF patients indicates poor correlation with genetic analysis of antigen-specific B-cell clones in this and previous studies. We found a much more polyclonal and diverse circulating autoab pool than previously suggested by genetic methods that characterize autoab-coding sequences in B cells, such as APD, heterohybridoma and cell immortalization approaches. We used “strict” criteria to define the anti-Dsg serum clones here to minimize false positive identification. Therefore, it is likely the number of serum ab clones we identify is a minimum. A major conclusion here is that even identifying what is probably a lower limit of circulating pemphigus ab clones, the numbers are significantly greater than those identified using standard B-cell based genetic techniques as typically reported in the literature.
Furthermore, in patients whose anti-Dsg B-cell clones have been characterized by APD, most do not contribute to detectable levels of circulating abs. However, interestingly, we are able to detect circulating abs matching APD clones in three patients, including two in whom the matching APD clones identified pathogenic ab sequences (i.e., scFv expressed from those sequences caused pemphigus pathology in organ culture; specifically pathogenic APD PF1-8-15 in reference (Yamagami et al., 2010) matches ab clonotypes PF1-1 in Table S3; and V(D3) in the (Hammers et al., 2015) reference matches ab clonotypes PV3a-1 in Table S3, pathogenicity data unpublished). These findings demonstrate that APD does recognize ab sequences that are relevant to the circulating ab response in PF and PV. Further underscoring the relevance of APD clones to circulating abs is the observation that circulating abs recognized by APD were always enriched by binding to Dsg (i.e., above the 0 line in Figure 2).
There are several possible explanations of why there is little overlap between the APD-defined anti-Dsg B-cell repertoire and the proteomic circulating ab repertoire. First, some APD clones may be artifactual due to random heavy and light chain pairing that does not occur in the patients but that leads to abs that bind Dsg. This explanation seems unlikely as in individuals without pemphigus as well as in pemphigus patients in long-term remission, APD does not identify any abs that bind mature cell-surface expressed Dsg (Hammers et al., 2015; Yamagami et al., 2009), indicating that random pairing does not result in artifactual Dsg-binding abs. Second, we show here that in cases in which we analyzed anti-Dsg abs over time, there were many cases in which the genetic sequences for the abs were present by NGS at one time point, but the circulating abs corresponding to those sequences were only present at another (Table S5). These results indicate that B cells that encode anti-Dsg abs may be present but not secreting detectable abs at any particular time point. Third, it is known that APD can be biased because some phage outgrow others (Derda et al., 2011). Finally, some anti-Dsg ab sequences may be toxic to bacteria in APD cloning. We did not exhaustively analyze these potential explanations, but we did study three VH chains, identified by LC-MS/MS but not APD, by constructing VH chain-specific APD libraries. One VH chain was toxic to bacteria so that an APD library could not be made with this chain. The two other VH chains were able to produce Dsg-specific abs as identified by APD selection on Dsg. This result suggests that in full libraries from this patient the APD clones representing these MS-identified abs were outcompeted by better growing or binding clones, but in VH-specific libraries there is not such competition so they can be found. In short we think it likely that many B cells that have coding sequences for anti-Dsg abs may not differentiate to produce circulating abs and/or those ab coding sequences are outcompeted by others in phage libraries.
Proteomic analysis of anti-Dsg autoabs in these pemphigus patients indicates a wide use of VH genes, indicating a non-convergent ab response. Although certain VH genes contributed up to 20% or more of the autoab response in some individual patients, the predominant VH gene was variable among patients. Even in patients with a predominant VH gene usage, many other abs were found in the circulation using other VH genes. Looking at VH gene usage in anti-Dsg abs across patients we found that VH1-69 and 5-51 were used for some abs in 4 of 4 PV and 2 of 2 PF patients. VH1-46 was detected in 4 of the 4 PV patients, consistent with its previously reported finding in many PV patients by APD (Cho et al., 2014). This gene has been thought to be possibly important in initiating the immune response in PV. These data indicate that there is some common usage of various VH genes among different pemphigus patients but that the predominant VH gene usage (if any) varies. These data differ from studies in some other autoab-mediated diseases such as cold agglutinin disease, thrombotic thrombocytopenic purpura, and idiopathic thrombocytopenic purpura (Pascual et al., 1992; Schaller et al., 2014; Siegel, 2008), or even a non-tissue specific disease like systemic lupus erythematosus in which VH4-34 can predominate (Tipton et al., 2015). However, study of these autoimmune diseases mostly used genetic techniques, as opposed to proteomic approaches (with the exception of 9G4-idiotype-reactive abs in lupus for which VH4-34 predominates (Tipton et al., 2015)), with the limitations discussed above. Not only is VH gene usage among patients variable, but also analysis of the AA sequences of the VH-CDR3s indicates no identical, or even 80% similar, sequences among the anti-Dsg3 or anti-Dsg1 abs in different patients. These data show that even though the pemphigus patients all produce antibodies against only two closely related Dsgs, there is no obvious convergence of the autoab response at a gene or AA sequence level. This finding contrasts with recent analysis of B-cell repertoires in patients with dengue in which H-CDR3 sequences are convergent among patients (Parameswaran et al., 2013). There have also been a few convergent or public B-cell IgG repertoires (based on shared H-CDR3 sequences) for presumed tetanus toxoid and influenza reactivity (Galson et al., 2015). These are a small part of the total repertoires and are not found among many different people. The lack of public repertoires of circulating abs in pemphigus may be because autoimmunity is less likely to cause public antibody repertories than is the immune response against foreign antigens in infectious disease or vaccination, because Dsg antigens are not prone to producing shared H-CDR3 sequences in different individuals (e.g., because they do not have “hot spots” for ab binding), because viruses like dengue may have fewer epitopes capable of eliciting an ab response and do have “hot spots” of ab binding (Dejnirattisai et al., 2015) or because the public ab clusters are so minor they are difficult to detect in our patients.
Interestingly, although proteomic analysis of circulating ab clones in pemphigus indicates a polyclonal response, we found that only a few clones in each patient account for most of the circulating ab. This finding is similar to what has been noted in the circulating ab clonotypes after tetanus toxoid vaccination in which a single clonotype can contribute as much as 10-15% of the antigen specific ab response and most clonotypes contribute very little (Lavinder et al., 2014; Wine et al., 2015). However, we also found that the highly producing B-cell clones vary at different time points (Figure 5B). What causes these changes in ab clonal expression is unclear but it has been speculated that variations in B-cell clonotypes over time may result from subclinical immune activation (Galson et al., 2015). For example, in pemphigus this might result from variations in Dsg fragments released by the epidermis depending on the extent or type of inflammation at various times.
Previously, using APD, we have shown that anti-Dsg B-cell clonotypes persist over years in PV patients as their disease remits and relapses (Hammers et al., 2015). Here we show that some serum ab clones, as well as B cell clones, can persist over years in PV and PF patients, including one previously shown by APD (V(D3) in ref. (Hammers et al., 2015) corresponds to ab PV3a-1 in Table S2). Even though some serum abs persist over time, the overall serum ab repertoire changes over time in these patients, with some abs expressed in the circulation at one time and not another. Contributing to this dynamic ab repertoire is the observation that the actual B cell encoding an ab might be present at a particular time point but not producing abs at that time point but only at another time point. Therefore, genetic methods, such as APD, may be more appropriate than proteomics to ask the question as to whether non-tolerant B-cells persist over time.
In summary, this proteomic approach to the analysis of circulating pemphigus abs provides a much more comprehensive spectrum of the actual ab response than previously described, and indicates a very diverse immune response both in individual patients and among different patients. These findings demonstrate that the autoab response in pemphigus does not result from an expansion of a few oligoclonal non-tolerant B cells but is a more generalized immune response against Dsgs. Furthermore, we show that, although some clonotypes of circulating abs may persist over years in both PV and PF patients, there is a changing autoab profile of the repertoire, which is consistent with the clinical observation that ELISA anti-Dsg titers do not always correlate with disease activity (Harman et al., 2001; Kwon et al., 2008), presumably because of differential expression of pathogenic and non-pathogenic abs. This was the case in our patients PF1 and PF4 in which at certain time points there was no disease activity but positive anti-Dsg ab (Table S6). Finally, these data suggest that approaches to therapy that target B cells expressing abs with specific B-cell VH genes will not be adequate for eliminating the autoimmune response and that either targeting all B cells (through B cell-depleting such as anti-CD20 ab therapy) or all B cells that express anti-Dsg B-cell receptors will be necessary (Ellebrecht et al., 2016).
Experimental Procedures
Patients
Four patients with PV (PV1, PV3, PV8, PV16) and two with PF (PF1, PF4) were studied. These patients had typical clinical presentations, histology, and anti-Dsg ELISA positivity (Stanley and Amagai, 2006). The anti-Dsg genetic repertoires of two of the PV patients were previously characterized at various time points by APD (Cho et al., 2014; Hammers et al., 2015; Payne et al., 2005); and that of the one PF patient was characterized at one (the initial) time point by APD (Ishii et al., 2008; Yamagami et al., 2009; Yamagami et al., 2010).
APD characterization of anti-Dsg3 B-cells/plasmablasts from PV8 and PV16
APD was performed as described previously (Barbas et al., 2001; Ishii et al., 2008; Payne et al., 2005) with modifications as detailed in Supplemental Experimental Procedures.
NGS sequencing of IgVH repertoire
Two NGS platforms were used in this study. Pan IgG VH repertoires (PV1a, PV3, PV3a, PF1, PF1a, PF4, PF4a, PF4b), IgG1 or IgG4 VH repertoires (PV8, PV16), and the VH regions of PV8 panned phage libraries were sequenced using Illumina Miseq 300bp paired-end sequencing (Institute for Genome Science, University of Maryland; Next-Generation Sequencing Core, University of Pennsylvania). The VH regions of PV3a panned phage libraries were sequenced using Pacific Biosciences (PacBio) sequencing (Yale Center for Genome Analysis).
All sequences from various sequencing platforms (Illumina, PacBio, Sanger for APD clones) from each patient were combined to construct the reference databases used to analyze LC-MS/MS spectra for subsequent analysis. See Supplemental Experimental Procedures.
Construction of reference databases for identification of LC-MS/MS spectra
We implemented a customized pipeline to build the antibody reference databases for LC-MS/MS from raw FASTQ reads called by the corresponding NGS instruments. See Supplemental Experimental Procedures.
LC-MS/MS of serum antibodies
The pemphigus serum was affinity purified on Dsg. The passthrough was analyzed as the “unbound fraction” and the eluate from the Dsg was analyzed as the “bound” fraction, which represents the anti-Dsg autoabs.
LC-MS/MS analysis was performed by the Wistar Proteomics Facility (Philadelphia, PA) using a Q Exactive Plus mass spectrometer (Thermo Scientific) coupled with a Nano-ACQUITY UPLC system (Waters). All LC-MS/MS samples were prepared in 3-4 technical replicates both for bound and unbound samples. Technical replicates included at least one repeat of the affinity chromatography and at least one repeat in LC-MS/MS of the abs from the same affinity chromatography run. See Supplemental Experimental Procedures.
LC-MS/MS spectrum data analysis to identify H-CDR3s of anti-Dsg abs
The LC-MS/MS data for each patient were analyzed using MaxQuant 1.5.2.8 software (Cox and Mann, 2008) against the patient-specific reference database, described above, appended with a contaminants protein list. All bound and unbound LC-MS/MS replicate experiments of a same sample were searched together using MaxQuant’s “Match Between Runs” option. After initial search parameters (see Supplemental Experimental Procedures), a set of universal MS spectrum match filters were applied to the representative PSMs for all samples, including an unambiguous one-peptide-one-CDR3 relationship and other MaxQuant specific criteria (PEP ≤0.01, Score ≥70, mass error PPM ≤1.5). At last, for most analyses, we applied a “strict” filter defined as only H-CDR3s found in all of 3-4 bound technical replicates but never in 3-4 unbound technical replicates.
Confirmation of CDR3 peptide identification
In some cases, stable isotopically labeled peptides were used to confirm peptide identification by the PSM criteria described above. See Supplemental Experimental Procedures.
Construction of ab clonotypes and analysis of their intensity of expression
Construction of recombinant scFv containing VH chains identified by LC-MS/MS
VH nucleotide sequences randomly selected from circulating PV3a anti-Dsg ab clusters that were not detected by APD were synthesized as gBlock gene fragments (Integrated DNA technologies) and randomly combined with the VL repertoire from PV3a-PBMC-RNA to screen by APD for binding to Dsg. See Supplemental Experimental Procedures.
Indirect immunofluorescence and anti-Dsg ELISA with scFv
Cloned scFv (which contained a hemagglutinin tag (HA)) were used for indirect immunofluorescence on frozen sections of monkey esophagus and ELISA on Dsg3 and Dsg1 plates (MBL/Euroimmun). Both were developed with anti-HA ab as previously described (Payne et al., 2005).
Statistics
Wilcoxon signed-rank test and the one-sample test of proportion was calculated with STATA 14.1 software. In Figures S2, S3 and S4 the expression differences between multiple time points were tested by fitting linear models and subsequent ANOVA (analysis of variance) analysis for each ab signature using the R package “lme4”. The signature intensities were log10 transformed. For all replicates used in the barplots in which no ab was detected by LC-MS/MS, instead of 0 we assigned a background intensity as the minimum intensity observed of the entire experiment, to avoid infinite values at log scale. “*”, “**”, and “***” indicate p-values les than 0.05, 0.01, and 0.001, respectively.
Supplementary Material
Acknowledgments
We thank Drs. David Margolis, Joel Gelfand and Mark Cary for statistical analysis. This work was supported by grants from the National Institutes of Arthritis, Musculoskeletal and Skin Diseases of the National Institutes of Health (JRS, R01-AR052672; ASP, R01-AR057001; JC, T32-AR007465; EMM, T32-AR007465 and F30-AR065870); a grant from the Dermatology Foundation to JC; grants from the DFG (CMH, HA6736/1-1, 2-1 and GRK1727; CTE, EL711/1-1) and support from the Section of Medicine at the University of Luebeck (J03-2015) to CMH
Footnotes
Author Contributions
JC designed and conducted experiments, acquired data, analyzed data, and helped write the manuscript; QZ analyzed data and helped write the manuscript; CMH designed and conducted experiments, acquired and analyzed data, and edited the manuscript; CTE designed and conducted experiments, acquired data, helped with data analysis and figure preparation, and edited the manuscript; EM conducted experiments, acquired data and helped with data analysis; HYT conducted MS and helped analyze MS data, and edited the manuscript; CL conducted experiments; HY conducted experiments; MP conducted some experiments; JL synthesized and contributed the Dsg matrix reagent; LK contributed the Dsg matrix reagents and edited the manuscript; DLS helped interpret and analyze data and edited the manuscript; ASP helped design experiments and analyze data, and edited the manuscript; JRS conceived the initial concept, oversaw the project, designed experiments, analyzed data and helped write the manuscript.
Conflict of Interest
JL and LK are employees of the Euroimmun AG, a company that develops, produces and manufactures immunoassays for the detection of disease-associated antibodies. The other authors have nothing to disclose.
Accession Numbers
The accession number for the NGS antibody sequencing in this paper is SRA: PRJNA309663. The mass spectrometry proteomics data have been deposited to the MassIVE data repository (http://massive.ucsd.edu) with the MassIVE accession MSV000080368 and ProteomeXchange accession PXD005474
Study approval. Written informed consent was received from participants prior to inclusion in the study. Human participation was approved by the University of Pennsylvania Institutional Review Board, Protocol #704768.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Barbas CFI, Burton DR, Scott JK, Silverman GJ. Phage Display: A Laboratory Manual. Cold Spring Harbor: Cold Spring Harbor Laboratory Press; 2001. [Google Scholar]
- Cheung WC, Beausoleil SA, Zhang X, Sato S, Schieferl SM, Wieler JS, Beaudet JG, Ramenani RK, Popova L, Comb MJ, et al. A proteomics approach for the identification and cloning of monoclonal antibodies from serum. Nat Biotechnol. 2012;30:447–452. doi: 10.1038/nbt.2167. [DOI] [PubMed] [Google Scholar]
- Cho MJ, Lo AS, Mao X, Nagler AR, Ellebrecht CT, Mukherjee EM, Hammers CM, Choi EJ, Sharma PM, Uduman M, et al. Shared VH1-46 gene usage by pemphigus vulgaris autoantibodies indicates common humoral immune responses among patients. Nature communications. 2014;5:4167. doi: 10.1038/ncomms5167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- Dejnirattisai W, Wongwiwat W, Supasa S, Zhang X, Dai X, Rouvinski A, Jumnainsong A, Edwards C, Quyen NT, Duangchinda T, et al. A new class of highly potent, broadly neutralizing antibodies isolated from viremic patients infected with dengue virus. Nat Immunol. 2015;16:170–177. doi: 10.1038/ni.3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derda R, Tang SK, Li SC, Ng S, Matochko W, Jafari MR. Diversity of phage-displayed libraries of peptides during panning and amplification. Molecules. 2011;16:1776–1803. doi: 10.3390/molecules16021776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Zenzo G, Di Lullo G, Corti D, Calabresi V, Sinistro A, Vanzetta F, Didona B, Cianchini G, Hertl M, Eming R, et al. Pemphigus autoantibodies generated through somatic mutations target the desmoglein-3 cis-interface. J Clin Invest. 2012;122:3781–3790. doi: 10.1172/JCI64413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellebrecht CT, Bhoj VG, Nace A, Choi EJ, Mao X, Cho MJ, Di Zenzo G, Lanzavecchia A, Seykora JT, Cotsarelis G, et al. Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease. Science. 2016;353:179–184. doi: 10.1126/science.aaf6756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galson JD, Truck J, Fowler A, Munz M, Cerundolo V, Pollard AJ, Lunter G, Kelly DF. In-Depth Assessment of Within-Individual and Inter-Individual Variation in the B Cell Receptor Repertoire. Front Immunol. 2015;6:531. doi: 10.3389/fimmu.2015.00531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammers CM, Chen J, Lin C, Kacir S, Siegel DL, Payne AS, Stanley JR. Persistence of Anti-Desmoglein 3 IgG(+) B-Cell Clones in Pemphigus Patients over Years. J Invest Dermatol. 2015;135:742–749. doi: 10.1038/jid.2014.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harman KE, Seed PT, Gratian MJ, Bhogal BS, Challacombe SJ, Black MM. The severity of cutaneous and oral pemphigus is related to desmoglein 1 and 3 antibody levels. Br J Dermatol. 2001;144:775–780. doi: 10.1046/j.1365-2133.2001.04132.x. [DOI] [PubMed] [Google Scholar]
- Ishii K, Lin C, Siegel DL, Stanley JR. Isolation of pathogenic monoclonal anti-desmoglein 1 human antibodies by phage display of pemphigus foliaceus autoantibodies. J Invest Dermatol. 2008;128:939–948. doi: 10.1038/sj.jid.5701132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon EJ, Yamagami J, Nishikawa T, Amagai M. Anti-desmoglein IgG autoantibodies in patients with pemphigus in remission. J Eur Acad Dermatol Venereol. 2008;22:1070–1075. doi: 10.1111/j.1468-3083.2008.02715.x. [DOI] [PubMed] [Google Scholar]
- Lavinder JJ, Wine Y, Giesecke C, Ippolito GC, Horton AP, Lungu OI, Hoi KH, DeKosky BJ, Murrin EM, Wirth MM, et al. Identification and characterization of the constituent human serum antibodies elicited by vaccination. Proc Natl Acad Sci U S A. 2014;111:2259–2264. doi: 10.1073/pnas.1317793111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moody MA, Zhang R, Walter EB, Woods CW, Ginsburg GS, McClain MT, Denny TN, Chen X, Munshaw S, Marshall DJ, et al. H3N2 influenza infection elicits more cross-reactive and less clonally expanded anti-hemagglutinin antibodies than influenza vaccination. PLoS One. 2011;6:e25797. doi: 10.1371/journal.pone.0025797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parameswaran P, Liu Y, Roskin KM, Jackson KK, Dixit VP, Lee JY, Artiles KL, Zompi S, Vargas MJ, Simen BB, et al. Convergent antibody signatures in human dengue. Cell host & microbe. 2013;13:691–700. doi: 10.1016/j.chom.2013.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual V, Victor K, Spellerberg M, Hamblin TJ, Stevenson FK, Capra JD. VH restriction among human cold agglutinins. The VH4-21 gene segment is required to encode anti-I and anti-i specificities. J Immunol. 1992;149:2337–2344. [PubMed] [Google Scholar]
- Payne AS, Ishii K, Kacir S, Lin C, Li H, Hanakawa Y, Tsunoda K, Amagai M, Stanley JR, Siegel DL. Genetic and functional characterization of human pemphigus vulgaris monoclonal autoantibodies isolated by phage display. J Clin Invest. 2005;115:888–899. doi: 10.1172/JCI24185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian Y, Clarke SH, Aoki V, Hans-Filhio G, Rivitti EA, Diaz LA. Antigen selection of anti-DSG1 autoantibodies during and before the onset of endemic pemphigus foliaceus. J Invest Dermatol. 2009;129:2823–2834. doi: 10.1038/jid.2009.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian Y, Diaz LA, Ye J, Clarke SH. Dissecting the anti-desmoglein autoreactive B cell repertoire in pemphigus vulgaris patients. J Immunol. 2007;178:5982–5990. doi: 10.4049/jimmunol.178.9.5982. [DOI] [PubMed] [Google Scholar]
- Schaller M, Vogel M, Kentouche K, Lammle B, Kremer Hovinga JA. The splenic autoimmune response to ADAMTS13 in thrombotic thrombocytopenic purpura contains recurrent antigen-binding CDR3 motifs. Blood. 2014;124:3469–3479. doi: 10.1182/blood-2014-04-561142. [DOI] [PubMed] [Google Scholar]
- Siegel DL. Translational applications of antibody phage display. Immunol Res. 2008;42:118–131. doi: 10.1007/s12026-008-8044-y. [DOI] [PubMed] [Google Scholar]
- Stanley JR, Amagai M. Pemphigus, bullous impetigo, and the staphylococcal scalded-skin syndrome. N Engl J Med. 2006;355:1800–1810. doi: 10.1056/NEJMra061111. [DOI] [PubMed] [Google Scholar]
- Tipton CM, Fucile CF, Darce J, Chida A, Ichikawa T, Gregoretti I, Schieferl S, Hom J, Jenks S, Feldman RJ, et al. Diversity, cellular origin and autoreactivity of antibody-secreting cell population expansions in acute systemic lupus erythematosus. Nat Immunol. 2015;16:755–765. doi: 10.1038/ni.3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wine Y, Boutz DR, Lavinder JJ, Miklos AE, Hughes RA, Hoi KH, Jung ST, Horton AP, Murrin EM, Ellington AD, et al. Molecular deconvolution of the monoclonal antibodies that comprise the polyclonal serum response. Proc Natl Acad Sci U S A. 2013;110:2993–2998. doi: 10.1073/pnas.1213737110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wine Y, Horton AP, Ippolito GC, Georgiou G. Serology in the 21st century: the molecular-level analysis of the serum antibody repertoire. Curr Opin Immunol. 2015;35:89–97. doi: 10.1016/j.coi.2015.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagami J, Kacir S, Ishii K, Payne AS, Siegel DL, Stanley JR. Antibodies to the desmoglein 1 precursor proprotein but not to the mature cell surface protein cloned from individuals without pemphigus. J Immunol. 2009;183:5615–5621. doi: 10.4049/jimmunol.0901691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagami J, Payne AS, Kacir S, Ishii K, Siegel DL, Stanley JR. Homologous regions of autoantibody heavy chain complementarity-determining region 3 (H-CDR3) in patients with pemphigus cause pathogenicity. J Clin Invest. 2010;120:4111–4117. doi: 10.1172/JCI44425. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.