

Abstract
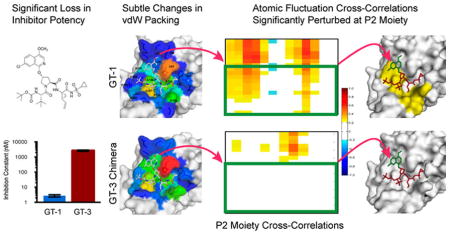
Hepatitis C virus (HCV), affecting an estimated 150 million people worldwide, is the leading cause of viral hepatitis, cirrhosis and hepatocellular carcinoma. HCV is genetically diverse with six genotypes (GTs) and multiple subtypes of different global distribution and prevalence. Recent development of direct-acting antivirals against HCV including NS3/4A protease inhibitors (PIs) has greatly improved treatment outcomes for GT-1. However, all current PIs exhibit significantly lower potency against GT-3. Lack of structural data on GT-3 protease has limited our ability to understand PI failure in GT-3. In this study the molecular basis for reduced potency of current inhibitors against GT-3 NS3/4A protease is elucidated with structure determination, molecular dynamics simulations and inhibition assays. A chimeric GT-1a3a NS3/4A protease amenable to crystallization was engineered to recapitulate decreased sensitivity of GT-3 protease to PIs. High-resolution crystal structures of this GT-1a3a bound to 3 PIs, asunaprevir, danoprevir and vaniprevir, had only subtle differences relative to GT-1 despite orders of magnitude loss in affinity. In contrast, hydrogen-bonding interactions within and with the protease active site and dynamic fluctuations of the PIs were drastically altered. The correlation between loss of intermolecular dynamics and inhibitor potency suggests a mechanism where polymorphisms between genotypes (or selected mutations) in the drug target confer resistance through altering the intermolecular dynamics of the protein–inhibitor complex.
Graphical Abstract
INTRODUCTION
Hepatitis C virus (HCV) causes chronic liver infection that affects about 3% of the global population and is the main cause of hepatitis, cirrhosis, and hepatocellular carcinoma.1,2 HCV is a highly evolved, highly diverse virus with six known genotypes (GT) and multiple subtypes.3,4 The two most prevalent viral species, GT-1 and GT-3, account for 46% and 30% of the global infections respectively,3,5 with different geographic distributions. While GT-1 is most endemic in Northern America and Europe, GT-3 is most prevalent in South Asia. The remaining GTs 4, 5, and 6 span the Middle East to Central Africa, South Africa, and Southeast Asia.3,5 Despite high genetic viral diversity, which renders HCV infections difficult to treat, enormous progress has been made toward developing effective HCV therapeutics.
Before the recent availability of direct-acting antivirals (DAAs), the standard of care for HCV infection consisted of pegylated-interferon (Peg-INF) and ribavirin (RBV); however, this treatment had moderate to low rates of cure across genotypes and was poorly tolerated.6 The development of DAAs against essential viral proteins NS5B polymerase, NS5A, and NS3/4A protease has greatly improved therapeutic options and treatment outcomes, especially when used in combination therapy.7–9 Three new combination therapies or regimens have been approved by the FDA including Harvoni (sofosbuvir and ledispasvir),10 Viekira Pak (ombitasvir, paritaprevir, ritonavir and dasabuvir),11 and most recently in January 2016, Zepatier (grazoprevir and elbasvir).12 These all-oral HCV therapies are highly effective against GT-1 with sustained virologic response (SVR) ranging 94–100%.9 However, currently approved DAA-based combination therapies and those in clinical development are significantly less effective against GT-3.9,13,14 In fact, with the exception of sofosbuvir, most DAAs have dramatically reduced potency against GT-3, and are susceptible to resistance.7,15,16 Therefore, an unmet need still exists for more robust pan-genotypic DAAs for the effective treatment of HCV.
The HCV NS3/4A protease is a major therapeutic target for HCV therapy with five FDA-approved PIs, telaprevir,17,18 boceprevir,19 simeprevir,20 paritaprevir (included in Viekira Pak),11 and grazoprevir (included in Zepatier)12 (Figure 1). In addition, a number of PIs, such as asunaprevir, vaniprevir, danoprevir, sovaprevir, GS-9451 and MK-6325 are in clinical development. The NS3/4A protein is a bifunctional enzyme containing an N-terminal serine protease domain (amino acids 1–180) with the classic catalytic triad (S139, H57, D81) of the chymotrypsin superfamily, and a C-terminal DExH/D-box helicase of superfamily II with NTPase activity.21–23 The NS3/4A protease is responsible for cleaving the viral polyprotein and host factors involved in the innate immune response, including TRIF and mitochondrial antiviral signaling (MAVS).24–26 Thus, targeting the NS3/4A protease serves as a two-pronged attack on the virus by preventing viral maturation and restoring the immune response.
Figure 1.

Chemical structure of HCV NS3/4A protease inhibitors. The three broad classes of HCV PIs: linear covalent and noncovalent (top row); P1–P3 macrocyclic (second row); and P2–P4 macrocyclic and P1–P3, P2–P4 bis-macrocyclic (third row).
Currently, NS3/4A PIs represent the largest class of FDA-approved DAAs against HCV.27–35 However, despite potent activity against wild-type GT-1, PIs suffer from a low genetic barrier to resistance.36 We have previously described the molecular basis for resistance to many of these inhibitors in terms of the substrate envelope and macrocyclization.37–41 Disturbingly, these inhibitors lack cross-genotypic activity, especially second- and third-generation PIs such as simeprevir, danoprevir, GS-9451, and grazoprevir, which are considerably less potent against GT-3 compared to GT-1 with 140- to 1500-fold loss in activity in viral subgenomic replicon models.15,16 Naturally occurring polymorphisms in GT-3 protease relative to GT-1 include drug resistance sites in GT-1 (residues 36, 123, 132, 168 and 170), several of which are outside the active site.16 Variation at position 168, a major site of drug resistance mutations in the active site, has been reported to cause decreased activity in transient replicon assays, but is not fully responsible for the loss of activity in GT-3. The role of other GT-3 polymorphisms, if any, is not clear. Moreover, no structural information exists for GT-3 protease, and the molecular mechanisms underlying differential inhibitor potency between HCV GT-1 and GT-3 remain unexplored.
To elucidate the molecular basis for loss of inhibitor potency against GT-3 relative to GT-1 HCV, we engineered a chimeric 1a3a protease construct that recapitulates PI susceptibility of GT-3 protease, and is amenable to crystallization for high-resolution structural studies. High-resolution cocrystal structures of the chimeric 1a3a protease bound to inhibitors asunaprevir (ASV), danoprevir (DAN) and vaniprevir (VAN) were determined. These structures enabled a detailed biophysical, structural and molecular dynamics analysis of the complexes, in comparison with the GT-1 protease. The inhibitors lost substantial intermolecular hydrogen bonding interactions and dynamic cross-correlations with the protease active site, revealing the dynamic molecular mechanism underlying lower PI potency against GT-3.
RESULTS
Inhibitors Are Significantly Less Potent against GT-3 than GT-1 Protease
The enzyme inhibition constants (Ki values) against GT-139 and GT-3 HCV NS3/4A protease for a panel of seven PIs (telaprevir, boceprevir, ASV, DAN, VAN, grazoprevir and MK-6325) representing the three general classes of inhibitors were determined (Figure 2, Figure S1, Table S1). Consistent with previous reports, GT-3 NS3/4A protease is less susceptible to all PIs tested.35,42 Among the first generation PIs, the reversible covalent inhibitors telaprevir and boceprevir have a similar Ki against GT-1 (33 nM); however, their inhibitory activity is decreased in GT-3. While telaprevir’s Ki increased 8 fold (266 nM), boceprevir’s Ki changed by only 2.2 fold (71 nM). Second-generation inhibitors (ASV, DAN, and VAN) have low to subnanomolar range (Ki = 2.7, 1.0, 0.7 nM respectively) inhibitory activities in GT-1, yet low to submicromolar Ki in GT-3. Relative to GT-1, the linear ASV experienced a 975 fold change (FC) in Ki, while the macrocyclic DAN and VAN had a 878 and 533 FC respectively. The next generation PIs, grazoprevir and MK-6325, have the best inhibitory potency with 0.1 nM Ki values against GT-1 protease, but lost 2–3 orders of magnitude in affinity in GT-3. Still even with this substantial loss of affinity these PIs, of the panel, are the most efficacious inhibitors against GT-3 (Ki = 33 and 31 nM, respectively). The improvement in inhibition with next generation PIs is due to the synergistic effects of the quinoxaline moiety, which favorably stacks against the invariant catalytic residues, and the macrocycle.37 Nevertheless, all seven inhibitors suffer from loss in potency against GT-3 compared to GT-1 HCV NS3/4A protease.
Figure 2.

Effect of GT-3 active site polymorphisms on HCV NS3/4A protease inhibition. The enzyme inhibition constants (Ki) for seven PIs were determined for four enzyme variants: GT-1a,39 GT-3a, GT-1a with D168Q, and GT-1a with all three active site polymorphisms (R123T/I132L/D168Q) in GT-3a.
Design of HCV NS3/4A Protease GT-1a3a Chimera
Overall, the viral protease is highly conserved across genotypes with sequence identities and similarities ranging from 71–84% to 82–92% respectively (Figure S2). The various polymorphic sites in GT-3 relative to GT-1 are spatially distributed throughout the protease (Figure S3). The NS3/4A active site is highly conserved: of the 36 residues in direct contact with inhibitors (within 5 Å of the ligand), GT-3 differs in 3 amino acids: Thr123, Leu132 and Gln168 (Figure S3). While these changes represent a mere 8% divergence, amino acids 123 and 168 were previously reported for their contribution to an active site salt bridge formation, inhibitor binding, and resistance in the case of residue 168.37,38,40
The single D168Q mutation and triple R123T/I132L/D168Q mutation (1a3a) variants with polymorphisms from GT-3 were engineered into GT-1 protease, and their inhibition constants measured for the seven PIs (Figure 2). In the linear covalent inhibitors telaprevir and boceprevir, the D168Q mutation did not significantly affect inhibitor potency relative to GT-1 (1.2 and 0.7 FC respectively), likely due to a lack of an extended P2 moiety reliant on interactions with the S2 subpocket and the benefits of covalency. In contrast, this polymorphism caused detrimental effects on the noncovalent inhibitors. In ASV, DAN, VAN, and grazoprevir, D168Q resulted in a 72, 30, 44, and 102 fold loss in potency relative to GT-1 WT protease. However, with only a 4-fold loss in potency, MK-6325 was able to accommodate the Asp to Gln change and maintain a subnanomolar inhibition constant. The synergistic effects of MK-6325’s bis-macrocycle and cyclopentyl P4 capping may underlie this sustained potency in the presence of D168Q. In GT-1 HCV NS3/4A protease, amino acid 168 is a multidrug resistance mutation hotspot with G/A/V/E/N/Y/K substitutions observed both experimentally and clinically in infected patients.43 Understanding MK-6325’s resistance profile and binding mode against D168 variants may provide further insights into optimizing PIs for better GT-3 targeting.
Introducing the R123T/I132L/D168Q triple polymorphism to GT-1 recapitulates the GT-3 activity profile for the PIs in the panel (Figure 2). ASV, DAN, VAN, grazoprevir and MK-6325 experienced a 2–3 orders of magnitude loss in potency relative to GT-1 WT, mirroring the observed loss of potency against GT-3. Even MK-6325 lost affinity when all three mutations are present, and has a loss similar to GT-3. Thus, introducing the three GT-3 specific active site polymorphisms into GT-1 protease yielded a chimeric 1a3a NS3/4A protease with an inhibitor susceptibility profile recapitulating GT-3.
Crystal Structures and Molecular Dynamics Simulations of Inhibitor Complexes
To gain insights into the structural basis for protease inhibitor resistance in HCV GT-3, we attempted to crystallize GT-3 protease; however, problems with large-scale expression and purification prevented extensive crystallization trials and success, in accordance with the lack of any crystal structure available for GT-3 protease in the database. Thus, crystallization was attempted with the 1a3a chimeric NS3/4A protease. Three high-resolution crystal structures (1.8–2.2 Å resolution in space group P212121) of ASV (PDB ID: 5EQS), DAN (5EGR) and VAN (5ESB) in complex with 1a3a chimeric NS3/4A protease were determined (Table S2), and compared to their corresponding GT-1 structures of ASV (4WF8), DAN (3M5L) and VAN (3SU3) which we previously determined.40,44
To complement the six crystal structures and ascertain alterations in dynamics, fully solvated 100 ns molecular dynamics (MD) simulations were performed in triplicate. For each of the inhibitor complexes the simulations were started from the cocrystal structures of either GT-1 or the GT-1a3a chimera. The structures and simulations permitted an in depth analysis of these complexes.
Differences in GT-3 Active Site Alter Inhibitor Packing
The overall binding modes of the three PIs were conserved when bound to 1a3a protease relative to GT-1. To analyze the details of inhibitor packing at the active site, van der Waals (vdW) contact potentials were calculated between the inhibitor and protease residues in the crystal structures. In agreement with the decrease in inhibitor potency (Figure 2), vdW contacts were moderately lost in the 1a3a complexes relative to GT-1 (ASV: −44.4 vs −45.3 kcal/mol; DAN: −41.7 vs −43.7 kcal/mol; and VAN: −41.1 vs −41.5 kcal/mol). To capture the dynamic changes, these contacts were also calculated from the MD trajectories (Figure 3), and more extensive changes were observed as in the case of ASV where the P2 isoquinoline moiety flips onto His 57. While these changes in inhibitor packing likely contribute to decrease in affinity against GT-3 protease, they are moderate in comparison to the extent of potency loss against GT-3.
Figure 3.

Packing of inhibitors at the NS3/4A protease active site. The van der Waals (vdW) contact potentials, averaged from the MD simulations, of protease active site residues with the inhibitor: (A) GT-1 and (B) chimeric 1a3a construct complexes. The protease residues are colored blue to red for increasing contacts with the inhibitor mapped onto the protease surface of the cocrystal structures (1a/1a3a): ASV (4WF8/5EQS), DAN (3M5L/5EGR) and VAN (3SU3/5ESB). DAN’s isoindoline group is flipped between the two cocrystal structures.
Disruption of the Active Site Electrostatic Network Correlates with Loss of Inhibitor Potency
In HCV protease, high-affinity ligand binding is characterized by formation of an electrostatic network spanning residues D81, R155, D168, and R123.37,44 Within this network, R155–D168 interaction is particularly key for maintaining an electrostatic surface that accommodates high-affinity ligand binding.40 This interaction is present in all GT-1 complex crystal structures (Figure 4), with both carbonyl oxygen atoms of D168 oriented toward AR155’s Nε and Nη to form two hydrogen bonds (H-bonds) and R123’s Nε oriented toward D168’s Oδ for one H-bond formation. However, in the 1a3a complexes, the D168Q substitution resulted in simultaneous loss of the R123-Nε and R155-Nη interactions (Figure 4B).
Figure 4.

Active site hydrogen bonds. Surface representation of the active site with the bound inhibitor in (A) GT-1 and (B) chimeric 1a3a protease structures. The insets show the hydrogen-bonding network between protease active site residues R155, D/Q168 and R/T123, which is critical for ligand binding. Catalytic triad is in yellow and active site polymorphisms in GT-3 are highlighted in red.
Disruption and rearrangements of this electrostatic interaction underlie the resistance mechanism of many PIs, including ASV, VAN and DAN in GT-1, to the R155K multidrug resistance mutation,37,40 where D168 side chain shifts toward R123 for electrostatic interaction. In 1a3a chimeric structures, with D168 only engaging in one H-bond with R155, the possibility of a rotation of D168 toward R123 for electrostatic interaction is feasible. Thus, to assess the stability of the active site electrostatic network, the average time these intramolecular interactions were observed during MD simulations was computed (Table S3). The simulations revealed that, in all three GT-1 complexes, both of D168’s carbonyl oxygens were oriented toward R155’s Nε and Nη over 93% of the simulation duration. In agreement with recent reports,38 R123–D168 interaction was observed in the ASV and VAN complex only 30% of the time, suggesting that R123 might not be important for the formation of the inhibitor binding surface. Importantly, D168–R155 interaction was stable in all PI complexes of GT-1 protease.
In contrast, with the loss of a terminal carboxyl group at amino acid 168, all three 1a3a complexes had weaker Q168–R155 electrostatic interaction relative to their GT-1 counterparts. In ASV, this interaction existed during only 47% of the simulation. This reduction by half relative to GT-1 is the most severe of the three complex structures and correlates with ASV’s weaker antiviral activity in GT-3. With DAN and VAN, Q168–R155 interaction decreased by about 20% relative to their GT-1 counterparts, suggesting that the added rigidity of the macrocyclic inhibitors may help in maintaining the side chain interactions and the electrostatics of the surface. In addition, the location of the macrocycle may also affect the stability of the electrostatic surface: Comparing the three 1a3a complexes, Q168–R155 interaction is the most persistent in DAN (78%) where the macrocycle is at the P1–P3 position versus VAN (64%) and ASV (47%) representing respectively the P2–P4 macrocyclic and linear inhibitors.
In addition to changes within the protease active site, the polymorphisms in GT-3 protease relative to GT-1 resulted in the loss of at least one intermolecular H-bond at the P1′ moiety for all three PIs (Table S4). Overall, the molecular rearrangements in ASV lead to the most extensive changes in H-bonds ranging from −10 to −61%, while both DAN and VAN were better able to preserve their H-bond interactions. Thus, the active site polymorphisms severely disrupted the active site electrostatic network and consequently affected inhibitor binding and intermolecular H-bonding network.
Inhibitor Fluctuations Increase When Bound to GT-1a3a
To assess the overall consequences of the active site polymorphisms on protease–inhibitor dynamics, the root-mean-square fluctuations (RMSF) of the inhibitor atoms and protease Cα atoms during the MD simulations were calculated. The changes in 1a3a had little influence on the backbone Cα RMSF values relative to the GT-1, suggesting that the overall protein dynamics were conserved (Figure S4). However, the inhibitor’s atomic fluctuations changed depending on both the inhibitor bound and in the chimeric protein relative to GT-1. While the inhibitor backbone is more rigid, the relatively large and divergent P2 moieties sample different degrees of flexibility (Figure 5).
Figure 5.

Flexibility of inhibitor moieties when bound to HCV NS3/4A protease. Root mean square fluctuations (RMSF values) of inhibitor atoms were calculated over MD simulations when bound to GT-1 or chimeric 1a3a protease.
In ASV, the P2 isoquinoline moiety had the highest RMSF fluctuations of all the PIs (up to 4 Å RMSF) regardless of the genotype of the protease bound. DAN’s isoindoline was more rigid compared to the P2 in ASV (around 1 Å RMSF when bound to GT-1) but became more dynamic in the 1a3a complex (~2 Å RMSF). VAN, with its P2–P4 linked isoindoline, had the most rigid P2 of the three inhibitors and had no considerable change in fluctuations due to the 1a3a polymorphisms. Intriguingly, the flexibility of inhibitor P2 moiety in the simulations inversely correlates with inhibitor packing (intermolecular vdW contacts) and binding potency (Figures 2 and 4A). ASV’s isoquinoline is highly dynamic in GT-1, and among the three inhibitors ASV has the least contacts with the protease S2 residues resulting in the weakest-binding inhibitor. The isoindoline in both DAN and VAN display very low fluctuations, and make stronger contacts with the S2 residues, resulting in tighter-binding inhibitors. These results suggest a relationship between inhibitor P2 flexibility, protease-inhibitor contacts, and potency.
Dynamic Correlations in Protease–Inhibitor Atomic Fluctuations Are Lost in GT-1a3a
In principle, tight binding inhibitors are characterized by strong intermolecular interactions with the protein, which persist over the dynamics of the enzyme.38 To further investigate the interdependency between the protease active site surface and the inhibitor P2 isoquinoline and isoindoline fluctuations, cross-correlation coefficients between atomic fluctuations of protease active site residues and inhibitor atoms were calculated. During the MD simulations, atom pairs either fluctuate in the same direction (positive correlation, COi,j = 1), opposite direction (negative correlation, COi,j = −1) or have random movements with respect to one other (no correlation, COi,j = 0).
In the GT-1 complexes, ASV and VAN displayed strong positive (COi,j > 0.6) correlations with the protease active site, specifically with protease residues 132–157 and 168 (Figure 6), whereas DAN had both positive and negative correlations with residues 132–168 and 41–43 respectively (Figure S5). In addition to the conserved R155–D168 interaction revealed by H-bond interactions, the cross correlation analysis showed high cooperativity between the P2 moieties of ASV and DAN with R155–D168 residues in GT-1 protease complexes. VAN’s isoindoline had positive cross-correlations with R155 but not with D168. D168 possibly aids in stabilizing the R155’s conformation for stacking with the heterocyclic rings.
Figure 6.

Effect of GT-3 polymorphisms on protease-inhibitor dynamic coupling mapped onto the crystal structures of ASV. Cross-correlations between atomic fluctuations of protease active site residues and ASV atoms in (A) GT-1 and (B) chimeric 1a3a constructs. Warm and cool colors in the matrices indicate positive and negative correlations, respectively. Inhibitor P4 and P3 (magenta) and P2 isoquinoline (green) moieties are highlighted as boxes on the correlation matrix, and colored on the structures where protease residues with positive correlations with the corresponding moieties are in yellow, and those with no considerable dynamic correlation in gray.
In the 1a3a complexes, the cross-correlations were significantly altered ranging from partial losses to complete disruption compared to GT-1. In ASV, the P4–P3 moieties completely lost correlations with 8 residues, 132 through 154 (Figure 6), constituting the oxyanion hole, S1 pocket and the 310-helix located between β strands C2 and D2. This disruption in correlations with the S1 subpocket residues is in agreement with the observed loss in P1 intermolecular interactions in this inhibitor. Similar to ASV, DAN suffered from complete disruptions in correlations with respect to residues 132 through 138 (Figure S5). While protease–P4–P3 moiety correlations were relatively maintained in the 1a3a complexes, the intermolecular cooperativity of the inhibitor P2 moieties was partially lost in VAN (Figure S6) and completely abolished in ASV and DAN (Figures 6, S5). Thus, active site polymorphisms in GT-3 caused substantial losses in cross-correlations between protease–inhibitor atomic fluctuations relative to GT-1 protease, corresponding closely to the losses in binding affinities observed.
DISCUSSION
HCV therapeutic efforts have greatly benefited from structure based drug design (SBDD), specifically in developing viral protease inhibitors. The elucidation of the structural mechanism for substrate binding, coupled with rational medicinal chemistry efforts, has spawned the current arsenal of PIs with subnanomolar potency and remarkable antiviral activity. Sadly, this activity is genotype dependent and current PIs fail against GT-3 protease. In this study, we reveal the major contributors to decreased affinity of inhibitors to GT-3 protease, and demonstrate that three active site polymorphisms, R123T-I132L-D168Q, are responsible for loss of potency observed for current PIs against GT-3 HCV. In the context of GT-3, the R123T and D168Q mutations disrupt the R123-D168-R155 electrostatic network to a mere Q168–R155 interaction. Thus, our crystal structures and dynamic analysis indicate that active site electrostatics, which we previously reported to be critical for tight ligand binding,37,38,40 are disrupted by GT-3 active site polymorphisms. Notably, we discovered a correlation between increased P2 moiety fluctuations, disruption of the protease active site electrostatic network and intermolecular dynamic correlations, and binding affinity.
In viruses like HCV, due to the high replication rate (1012 new virions per day) and high mutation frequency during replication, every possible nonsense mutation is likely introduced in the viral genome on a daily basis.45 Consequently, a heterogeneous viral population exists in every patient. However, outside of the native GT-3 context, neither the single D168Q nor the simultaneous presence of the triple R123T/I132L/D168Q mutations has been reported. The absence of these mutations in GT-1 suggests other secondary GT-3 polymorphisms outside the active site may be required for maintaining protease fitness. One such candidate is I170V, which was frequent in GT-3 viral isolates but did not affect PI potency in replicon assays.16 Nevertheless, we found that these three active site polymorphisms are required and sufficient to recapitulate the loss of potency for most PIs against GT-3 protease.
The molecular mechanisms underlying loss of PI potency against GT-3 relative to GT-1 is somewhat reminiscent of drug resistance due to active site drug resistance mutations in GT-1 protease.37,38,40,41,46 The ability of single active site mutations to confer multidrug resistance has been extensively studied, specifically in the context of R155 and D168 resistance associated variants in GT-1 NS3/4A protease. Such mutations unfavorably alter or completely disrupt the protease active site electrostatic network required for tight ligand binding. Thus, inhibitors that retain potency against D168 variants of GT-1 protease may be better inhibitors against GT-3 protease as well. The chimeric 1a3a protease that we have engineered can be used as a surrogate in structural studies, enzymatic assays and potentially replicon assays to test inhibitors designed to target GT-3.
A thorough understanding of ligand–receptor interactions is key for rational drug design and further improvements in PIs to achieve broader potency. We have previously established the substrate envelope hypothesis to efficiently explain the molecular basis for drug resistance due to major active site mutations in both HCV and HIV-1 protease.38,46–49 Accordingly, active site residues are prone to resistance associated mutations where an inhibitor protrudes outside of the consensus substrate volume.50 When added as a constraint in drug design, staying within the substrate envelope leads to more robust inhibitors with enhanced resistance barriers and improved antiviral activity.48 This principle was successfully implemented to design more robust HIV-1 PIs,48 and was fundamental in our understanding of the structural basis for HCV drug resistance.44,46 In addition to staying within the substrate envelope, inhibitors need to balance the rigidity required for stable target interactions and flexibility to adapt to changes due to resistance mutations and binding site dynamics.38,49 The network hypothesis explains how changes in conformational dynamics due to mutations, located even away from the active site, can be propagated to the active site to affect inhibitor binding.51 Thus, target protein and inhibitor dynamics are essential components of molecular mechanisms leading to the emergence of drug resistance in HCV NS3/4A protease.
In addition to substrate envelope constraints, we propose here that the conservation of inhibitor–protease dynamic cross-correlations should be incorporated into the rational design and computational evaluation of PIs in structure-based drug design (SBDD). The significance of dynamic cross-correlations with the target active site for ligand binding likely extends well beyond HCV NS3/4A protease to other systems as well. In such cases, inhibitors that are able to maintain dynamic correlations with the target active site despite mutations will be less susceptible to resistance. These correlations can be probed by MD simulations of the inhibitor-target complex structure. In the absence of GT-3 protease structures, the 1a3a chimeric protease structures can serve as an ideal platform to test the designed PIs for the preservation of cross-correlations with the protease active site in GT-3. For instance, inhibitor moieties could be first modeled in 1a3a and tested for their ability to maintain or increase cross-correlations relative to GT-1. As both the P4–P3 and P2 moieties of ASV, DAN, and VAN were shown to lose correlations in the chimeric complex, SAR studies to identify moieties with stronger cross correlations could yield more robust inhibitors. This hypothesis is supported by a recent report detailing a P4 capping SAR in a faldaprevir derivative.52
In conclusion, we demonstrated that the molecular mechanism underlying loss of PI potency against HCV GT-3 is associated with alterations in the active site electrostatics. We also demonstrated that the chimeric 1a3a protease is a useful model system for structural studies and future design and assessment of more robust GT-3 NS3/4A inhibitors. By incorporating the major polymorphisms responsible for the loss of PI activity against GT-3 into GT-1 protease, the chimera enables understanding PI failure against HCV GT-3, and testing potential novel inhibitors by a direct comparison of ligand binding modes against the extensive repertoire of GT-1 inhibitor complexes. The commonality between mechanisms underlying drug resistance due to D168 active site mutations in GT-1 and loss of potency against GT-3 promises the possibility of designing inhibitors robust against both GT-1 resistance mutations and GT-3 polymorphisms.
METHODS
Protein Constructs
The HCV genotype 1a NS3/4A protease domain gene37 was synthesized by GenScript and cloned into the pET28a expression vector (Novagen). The highly soluble single-chain construct consists of NS3/4A protease domain (residues 4–181) fused to a fragment of the cofactor NS4A (residues 12–23) via an SD linkage. A similar protease construct exhibited catalytic activity comparable to that of the authentic full-length protein.53 All protease variants were generated using the QuikChange Site-Directed Muta-genesis Kit from Stratagene.
The HCV genotype 3a NS3/4A protease domain gene was constructed and synthesized similarly to that of genotype 1a.
Protein Expression and Purification
Protein expression and purification were carried out as previously described.37 Briefly, transformed BL21(DE3) E. coli cells were grown at 37 °C and induced at an optical density of 0.6 by adding 1 mM IPTG. Cells were harvested after 5 h of expression, pelleted, and frozen at −80 °C for storage. Cell pellets were thawed, resuspended in 5 mL/g of resuspension buffer (50 mM phosphate buffer, 500 mM NaCl, 10% glycerol, 2 mM β-ME, pH 7.5) and lysed with a cell disruptor. The soluble fraction was retained, applied to a nickel column (Qiagen), washed with resuspension buffer, and eluted with resuspension buffer supplemented with 200 mM imidazole. The eluent was dialyzed overnight (MWCO 10 kDa) to remove the imidazole, and the His-tag was simultaneously removed with thrombin treatment. The nickel-purified protein was then flash frozen and stored at −80 °C.
Crystallization
The above-mentioned protein solution was thawed, concentrated to 3 mg/mL and loaded on a HiLoad Superdex75 16/60 column equilibrated with gel filtration buffer, 25 mM 2-(N-morpholino)ethanesulfonic acid (MES), 500 mM NaCl, 10% glycerol, 30 mM zinc chloride, and 2 mM 1,4-dithiothreitol, pH 6.5. The protease fractions were pooled and concentrated to 20–25 mg/mL with an Amicon Ultra-15 10 kDa device (Millipore). The concentrated samples were incubated for 1 h with 1–3 molar excess of inhibitor. Diffraction-quality crystals were obtained overnight by mixing equal volume of concentrated protein solution with precipitant solution (20–26% PEG-3350, 0.1 M sodium MES buffer, 4% ammonium sulfate, pH 6.5) in 24-well VDX hanging drop trays.
Data Collection and Structure Solution
X-ray diffraction data were collected on an in-house Rigaku X-ray system with a Saturn 944 CCD detector. Diffraction intensities were indexed, integrated and scaled using the program HKL2000.54 All structure solutions were generated using molecular replacement with PHASER.55 The B chain model of viral substrate product 4A-4B (PDB ID: 3M5M)46 was used as the starting model for all structure solutions. Initial refinement was carried out in the absence of modeled ligand, which was subsequently built in during later stages of refinement. Subsequent crystallographic refinement was carried out within the Phenix program suite, with iterative rounds of TLS or restrained refinement until convergence was achieved.56 1a3a–VAN complex was refined using the CCP4 program suite.57 The final structures were evaluated with MolProbity58 prior to deposition in the Protein Data Bank. To limit the possibility of model bias throughout the refinement process, 5% of the data were reserved for the free R-value calculation.59 Interactive model building and electron density viewing were carried out using the program COOT.60
Enzyme Inhibition Assays
All enzyme inhibition assays were performed in nonbinding surface 96-well black half-area plates (Corning) in a reaction volume of 60 μL. The NS3/4A protease (2 nM) was preincubated with increasing concentration of inhibitors in 50 mM Tris, 2.5% glycerol, 0.1% OβG, 5 mM Tris(2-carboxyethyl)-phosphine, 1% dimethyl sulfoxide, pH 7.5 for an hour. The reaction was initiated by the rapid injection of 5 μL of HCV NS3/4A protease substrate, Ac-DE-Dap(QXL-520)-EE-Abu-ψ-[COO]AS-C(5-FAMsp)-NH2 (Anaspec), to a final concentration of 200 nM and kinetically monitored using a PerkinElmer EnVision plate reader (excitation at 485 nm, emission at 530 nm). At least four independent data sets were collected for each inhibitor with each protease construct. Each inhibitor titration included at least 12 inhibitor concentration points, which were globally fit to the Morrison equation to obtain the Ki value.39
Structural Analysis
Superpositions were performed in PyMOL61 using Cα atoms of the active site protease residues 137–139 and 154–160. The A chain of WT–asunaprevir complex was used as the reference structure for each alignment. Hydrogen bonding analysis was carried out in Maestro, Schrodinger suite, with bond distance and donor–acceptor angles cutoff of 3.5 Å and 120°, respectively. van der Waals contact energies between protease–inhibitor residues were computed using a simplified Lennard–Jones potential, as described previously.47
Molecular Dynamics Simulations
Molecular dynamics simulations were carried out in triplicate, following previously published protocols38 using Desmond61 with the OPLS2005 force field.62,63 After equilibration, each trajectory was run for 100 ns at 300 K and the coordinates recorded every 5 ps. The percentage of time a hydrogen bond existed between the protease and an inhibitor was calculated using VMD.64 A hydrogen bond was defined by a distance between the donor and acceptor of less than 3.5 Å and a hydrogen–donor–acceptor angle of less than 30°. Only the hydrogen bonds that existed more than 20% of the time were considered in the analyses. Salt bridges were defined as an interaction between a side-chain oxygen atom of Asp or Glu within 4.0 Å of a nitrogen atom of Arg or Lys.65
Atomic Fluctuation Dynamics
The normalized cross-correlations of residue pairs were defined as
where ΔRi is the fluctuation in the position vector R of site i and ΔRj is the fluctuation in the position vector R of site j. The brackets represent time averages over recorded snapshots. The cross-correlations vary in the range [−1, 1], with the lower and upper limits indicating fully anticorrelated and correlated atomic fluctuations, respectively. COi,j = 0 reflects uncorrelated atomic fluctuations.
Supplementary Material
Acknowledgments
The National Institute of Allergy and Infectious Disease (R01-AI085051) supported this work. DIS was also supported by National Institute of General Medical Sciences of the NIH (F31-GM103259) as well as the HOPE scholarship sponsored by the Biomedical Science Career Program (BSCP). We thank W. Royer, F. Massi and D. Lambright for helpful discussions.
Footnotes
The authors declare the following competing financial interest(s): The PI has received sponsored research support from Merck for the study of HCV inhibitors that however are not the major focus of this study.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b06454.
Crystallographic statistics, tables of intra- and intermolecular hydrogen bonds, table of enzyme inhibition constants, the percent identity and similarity matrix for the protease sequences of the 6 HCV genotypes, comparison of GT-1 and GT-3 protease sequences, root-mean-square fluctuations of protease Cα atoms in MD simulations, figures of protease-danoprevir and vaniprevir dynamic cross-correlations, titration curves for determination of inhibition constants. (PDF)
References
- 1.Hajarizadeh B, Grebely J, Dore GJ. Nat Rev Gastroenterol Hepatol. 2013;10:553. doi: 10.1038/nrgastro.2013.107. [DOI] [PubMed] [Google Scholar]
- 2.Webster DP, Klenerman P, Dusheiko GM. Lancet. 2015;385:1124. doi: 10.1016/S0140-6736(14)62401-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gower E, Estes C, Blach S, Razavi-Shearer K, Razavi H. J Hepatol. 2014;61:S45. doi: 10.1016/j.jhep.2014.07.027. [DOI] [PubMed] [Google Scholar]
- 4.Simmonds P, Bukh J, Combet C, Deleage G, Enomoto N, Feinstone S, Halfon P, Inchauspe G, Kuiken C, Maertens G, Mizokami M, Murphy DG, Okamoto H, Pawlotsky JM, Penin F, Sablon E, Shin IT, Stuyver LJ, Thiel HJ, Viazov S, Weiner AJ, Widell A. Hepatology. 2005;42:962. doi: 10.1002/hep.20819. [DOI] [PubMed] [Google Scholar]
- 5.Messina JP, Humphreys I, Flaxman A, Brown A, Cooke GS, Pybus OG, Barnes E. Hepatology. 2015;61:77. doi: 10.1002/hep.27259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fried MW, Shiffman ML, Reddy KR, Smith C, Marinos G, Goncales FL, Jr, Haussinger D, Diago M, Carosi G, Dhumeaux D, Craxi A, Lin A, Hoffman J, Yu J. N Engl J Med. 2002;347:975. doi: 10.1056/NEJMoa020047. [DOI] [PubMed] [Google Scholar]
- 7.Bartenschlager R, Lohmann V, Penin F. Nat Rev Microbiol. 2013;11:482. doi: 10.1038/nrmicro3046. [DOI] [PubMed] [Google Scholar]
- 8.Kohli A, Shaffer A, Sherman A, Kottilil S. JAMA. 2014;312:631. doi: 10.1001/jama.2014.7085. [DOI] [PubMed] [Google Scholar]
- 9.Asselah T, Boyer N, Saadoun D, Martinot-Peignoux M, Marcellin P. Liver Int. 2016;36:47. doi: 10.1111/liv.13027. [DOI] [PubMed] [Google Scholar]
- 10.Afdhal N, Reddy KR, Nelson DR, Lawitz E, Gordon SC, Schiff E, Nahass R, Ghalib R, Gitlin N, Herring R, Lalezari J, Younes ZH, Pockros PJ, Di Bisceglie AM, Arora S, Subramanian GM, Zhu Y, Dvory-Sobol H, Yang JC, Pang PS, Symonds WT, McHutchison JG, Muir AJ, Sulkowski M, Kwo P, Investigators ION. N Engl J Med. 2014;370:1483. doi: 10.1056/NEJMoa1316366. [DOI] [PubMed] [Google Scholar]
- 11.Andreone P, Colombo MG, Enejosa JV, Koksal I, Ferenci P, Maieron A, Mullhaupt B, Horsmans Y, Weiland O, Reesink HW, Rodrigues L, Jr, Hu YB, Podsadecki T, Bernstein B. Gastroenterology. 2014;147:359. doi: 10.1053/j.gastro.2014.04.045. [DOI] [PubMed] [Google Scholar]
- 12.Lawitz E, Gane E, Pearlman B, Tam E, Ghesquiere W, Guyader D, Alric L, Bronowicki JP, Lester L, Sievert W, Ghalib R, Balart L, Sund F, Lagging M, Dutko F, Shaughnessy M, Hwang P, Howe AYM, Wahl J, Robertson M, Barr E, Haber B. Lancet. 2015;385:1075. doi: 10.1016/S0140-6736(14)61795-5. [DOI] [PubMed] [Google Scholar]
- 13.WHO. WHO Hepatitis C Fact Sheet. 2016 http://www.who.int/mediacentre/factsheets/fs164/en/
- 14.Panel AIHG. Hepatology. 2015;62:932. [Google Scholar]
- 15.Yu M, Corsa AC, Xu S, Peng B, Gong R, Lee YJ, Chan K, Mo H, Delaney IW, Cheng G. Antiviral Res. 2013;100:439. doi: 10.1016/j.antiviral.2013.08.018. [DOI] [PubMed] [Google Scholar]
- 16.Chan K, Yu M, Peng B, Corsa A, Worth A, Gong R, Xu S, Chen X, Appleby TC, Taylor J, Delaney WE, Cheng G. International Workshop on HIV & Hepatitis Virus Drug Resistance and Curative Strategies. Toronto, ON, Canada: 2013. [Google Scholar]
- 17.Kwong AD, Kauffman RS, Hurter P, Mueller P. Nat Biotechnol. 2011;29:993. doi: 10.1038/nbt.2020. [DOI] [PubMed] [Google Scholar]
- 18.Perni RB, Almquist SJ, Byrn RA, Chandorkar G, Chaturvedi PR, Courtney LF, Decker CJ, Dinehart K, Gates CA, Harbeson SL, Heiser A, Kalkeri G, Kolaczkowski E, Lin K, Luong YP, Rao BG, Taylor WP, Thomson JA, Tung RD, Wei Y, Kwong AD, Lin C. Antimicrob Agents Chemother. 2006;50:899. doi: 10.1128/AAC.50.3.899-909.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Malcolm BA, Liu R, Lahser F, Agrawal S, Belanger B, Butkiewicz N, Chase R, Gheyas F, Hart A, Hesk D, Ingravallo P, Jiang C, Kong R, Lu J, Pichardo J, Prongay A, Skelton A, Tong X, Venkatraman S, Xia E, Girijavallabhan V, Njoroge FG. Antimicrob Agents Chemother. 2006;50:1013. doi: 10.1128/AAC.50.3.1013-1020.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rosenquist A, Samuelsson B, Johansson PO, Cummings MD, Lenz O, Raboisson P, Simmen K, Vendeville S, de Kock H, Nilsson M, Horvath A, Kalmeijer R, de la Rosa G, Beumont-Mauviel M. J Med Chem. 2014;57:1673. doi: 10.1021/jm401507s. [DOI] [PubMed] [Google Scholar]
- 21.Appleby TC, Anderson R, Fedorova O, Pyle AM, Wang R, Liu X, Brendza KM, Somoza JR. J Mol Biol. 2011;405:1139. doi: 10.1016/j.jmb.2010.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Love RA, Parge HE, Wickersham JA, Hostomsky Z, Habuka N, Moomaw EW, Adachi T, Hostomska Z. Cell. 1996;87:331. doi: 10.1016/s0092-8674(00)81350-1. [DOI] [PubMed] [Google Scholar]
- 23.Yao N, Reichert P, Taremi SS, Prosise WW, Weber PC. Structure. 1999;7:1353. doi: 10.1016/s0969-2126(00)80025-8. [DOI] [PubMed] [Google Scholar]
- 24.Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, Ishii KJ, Takeuchi O, Akira S. Nat Immunol. 2005;6:981. doi: 10.1038/ni1243. [DOI] [PubMed] [Google Scholar]
- 25.Li K, Foy E, Ferreon JC, Nakamura M, Ferreon AC, Ikeda M, Ray SC, Gale M, Jr, Lemon SM. Proc Natl Acad Sci U S A. 2005;102:2992. doi: 10.1073/pnas.0408824102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Seth RB, Sun L, Ea CK, Chen ZJ. Cell. 2005;122:669. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 27.Arasappan A, Bennett F, Bogen SL, Venkatraman S, Blackman M, Chen KX, Hendrata S, Huang Y, Huelgas RM, Nair L, Padilla AI, Pan W, Pike R, Pinto P, Ruan S, Sannigrahi M, Velazquez F, Vibulbhan B, Wu W, Yang W, Saksena AK, Girijavallabhan V, Shih NY, Kong J, Meng T, Jin Y, Wong J, McNamara P, Prongay A, Madison V, Piwinski JJ, Cheng KC, Morrison R, Malcolm B, Tong X, Ralston R, Njoroge FG. ACS Med Chem Lett. 2010;1:64. doi: 10.1021/ml9000276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bartolini B, Giombini E, Zaccaro P, Selleri M, Rozera G, Abbate I, Comandini UV, Ippolito G, Solmone M, Capobianchi MR. Virus Res. 2013;177:205. doi: 10.1016/j.virusres.2013.08.001. [DOI] [PubMed] [Google Scholar]
- 29.Bogen SL, Arasappan A, Bennett F, Chen K, Jao E, Liu YT, Lovey RG, Venkatraman S, Pan W, Parekh T, Pike RE, Ruan S, Liu R, Baroudy B, Agrawal S, Chase R, Ingravallo P, Pichardo J, Prongay A, Brisson JM, Hsieh TY, Cheng KC, Kemp SJ, Levy OE, Lim-Wilby M, Tamura SY, Saksena AK, Girijavallabhan V, Njoroge FG. J Med Chem. 2006;49:2750. doi: 10.1021/jm060077j. [DOI] [PubMed] [Google Scholar]
- 30.Clark VC, Peter JA, Nelson DR. Liver Int. 2013;33(Suppl 1):80. doi: 10.1111/liv.12061. [DOI] [PubMed] [Google Scholar]
- 31.Harper S, McCauley JA, Rudd MT, Ferrara M, DiFilippo M, Crescenzi B, Koch U, Petrocchi A, Holloway MK, Butcher JW, Romano JJ, Bush KJ, Gilbert KF, McIntyre CJ, Nguyen KT, Nizi E, Carroll SS, Ludmerer SW, Burlein C, DiMuzio JM, Graham DJ, McHale CM, Stahlhut MW, Olsen DB, Monteagudo E, Cianetti S, Giuliano C, Pucci V, Trainor N, Fandozzi CM, Rowley M, Coleman PJ, Vacca JP, Summa V, Liverton NJ. ACS Med Chem Lett. 2012;3:332. doi: 10.1021/ml300017p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Niu D, Hagel M, Qiao L, Martin T, StSheets M, Chaturvedi P, Labenski M, Nacht M, Westlin W, Petter RC, Singh J. J Hepatol. 2010;52:S297. [Google Scholar]
- 33.Pompei M, Di Francesco ME, Pesci S, Koch U, Vignetti SE, Veneziano M, Pace P, Summa V. Bioorg Med Chem Lett. 2010;20:168. doi: 10.1016/j.bmcl.2009.11.005. [DOI] [PubMed] [Google Scholar]
- 34.Prongay AJ, Guo Z, Yao N, Pichardo J, Fischmann T, Strickland C, Myers J, Jr, Weber PC, Beyer BM, Ingram R, Hong Z, Prosise WW, Ramanathan L, Taremi SS, Yarosh-Tomaine T, Zhang R, Senior M, Yang RS, Malcolm B, Arasappan A, Bennett F, Bogen SL, Chen K, Jao E, Liu YT, Lovey RG, Saksena AK, Venkatraman S, Girijavallabhan V, Njoroge FG, Madison V. J Med Chem. 2007;50:2310. doi: 10.1021/jm060173k. [DOI] [PubMed] [Google Scholar]
- 35.Summa V, Ludmerer SW, McCauley JA, Fandozzi C, Burlein C, Claudio G, Coleman PJ, Dimuzio JM, Ferrara M, Di Filippo M, Gates AT, Graham DJ, Harper S, Hazuda DJ, Huang Q, McHale C, Monteagudo E, Pucci V, Rowley M, Rudd MT, Soriano A, Stahlhut MW, Vacca JP, Olsen DB, Liverton NJ, Carroll SS. Antimicrob Agents Chemother. 2012;56:4161. doi: 10.1128/AAC.00324-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Delang L, Vliegen I, Froeyen M, Neyts J. Antimicrob Agents Chemother. 2011;55:4103. doi: 10.1128/AAC.00294-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Romano KP, Ali A, Aydin C, Soumana D, Ozen A, Deveau LM, Silver C, Cao H, Newton A, Petropoulos CJ, Huang W, Schiffer CA. PLoS Pathog. 2012;8:e1002832. doi: 10.1371/journal.ppat.1002832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ozen A, Sherman W, Schiffer CA. J Chem Theory Comput. 2013;9:5693. doi: 10.1021/ct400603p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ali A, Aydin C, Gildemeister R, Romano KP, Cao H, Ozen A, Soumana D, Newton A, Petropoulos CJ, Huang W, Schiffer CA. ACS Chem Biol. 2013;8:1469. doi: 10.1021/cb400100g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Soumana DI, Ali A, Schiffer CA. ACS Chem Biol. 2014;9:2485. doi: 10.1021/cb5006118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Soumana DI, Kurt Yilmaz N, Prachanronarong KL, Aydin C, Ali A, Schiffer CA. ACS Chem Biol. 2016;11:900. doi: 10.1021/acschembio.5b00647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jensen SB, Serre SB, Humes DG, Ramirez S, Li YP, Bukh J, Gottwein JM. Antimicrob Agents Chemother. 2015;59:7426. doi: 10.1128/AAC.01953-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sarrazin C, Zeuzem S. Gastroenterology. 2010;138:447. doi: 10.1053/j.gastro.2009.11.055. [DOI] [PubMed] [Google Scholar]
- 44.Romano KP, Laine JM, Deveau LM, Cao H, Massi F, Schiffer CA. J Virol. 2011;85:6106. doi: 10.1128/JVI.00377-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Susser S, Welsch C, Wang Y, Zettler M, Domingues FS, Karey U, Hughes E, Ralston R, Tong X, Herrmann E, Zeuzem S, Sarrazin C. Hepatology. 2009;50:1709. doi: 10.1002/hep.23192. [DOI] [PubMed] [Google Scholar]
- 46.Romano KP, Ali A, Royer WE, Schiffer CA. Proc Natl Acad Sci U S A. 2010;107:20986. doi: 10.1073/pnas.1006370107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nalam MN, Ali A, Altman MD, Reddy GS, Chellappan S, Kairys V, Ozen A, Cao H, Gilson MK, Tidor B, Rana TM, Schiffer CA. J Virol. 2010;84:5368. doi: 10.1128/JVI.02531-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nalam MN, Ali A, Reddy GS, Cao H, Anjum SG, Altman MD, Yilmaz NK, Tidor B, Rana TM, Schiffer CA. Chem Biol. 2013;20:1116. doi: 10.1016/j.chembiol.2013.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kurt Yilmaz N, Swanstrom R, Schiffer CA. Trends Microbiol. 2016;24:547. doi: 10.1016/j.tim.2016.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.King NM, Prabu-Jeyabalan M, Nalivaika EA, Schiffer CA. Chem Biol. 2004;11:1333. doi: 10.1016/j.chembiol.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 51.Ragland DA, Nalivaika EA, Nalam MN, Prachanronarong KL, Cao H, Bandaranayake RM, Cai Y, Kurt-Yilmaz N, Schiffer CA. J Am Chem Soc. 2014;136:11956. doi: 10.1021/ja504096m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.O’Meara JA, Lemke CT, Godbout C, Kukolj G, Lagace L, Moreau B, Thibeault D, White PW, Llinas-Brunet M. J Biol Chem. 2013;288:5673. doi: 10.1074/jbc.M112.439455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Taremi SS, Beyer B, Maher M, Yao N, Prosise W, Weber PC, Malcolm BA. Protein Sci. 1998;7:2143. doi: 10.1002/pro.5560071011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Otwinowski Z, Minor W. Methods Enzymol. 1997;276:307. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 55.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. J Appl Crystallogr. 2007;40:658. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. Acta Crystallogr, Sect D: Biol Crystallogr. 2010;66:213. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Collaborative Computational Project, N. Acta Crystallogr, Sect D: Biol Crystallogr. 1994;50:760. [Google Scholar]
- 58.Chen VB, Arendall WB, 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. Acta Crystallogr, Sect D: Biol Crystallogr. 2010;66:12. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brunger AT. Nature. 1992;355:472. doi: 10.1038/355472a0. [DOI] [PubMed] [Google Scholar]
- 60.Emsley P, Cowtan K. Acta Crystallogr, Sect D: Biol Crystallogr. 2004;60:2126. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 61.PyMOL. 3.9. D. E. Shaw Research; New York, NY: 2014. [Google Scholar]
- 62.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. J Chem Phys. 1983;79:926. [Google Scholar]
- 63.Shivakumar D, Williams J, Wu Y, Damm W, Shelley J, Sherman W. J Chem Theory Comput. 2010;6:1509. doi: 10.1021/ct900587b. [DOI] [PubMed] [Google Scholar]
- 64.Humphrey W, Dalke A, Schulten K. J Mol Graphics. 1996;14:33. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 65.Xu D, Tsai CJ, Nussinov R. Protein Eng, Des Sel. 1997;10:999. doi: 10.1093/protein/10.9.999. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.