

The question of whether the Golgi complex is a stable compartment or is constantly regenerated from the endoplasmic reticulum (ER) is an important issue under debate. Using an ER trapping procedure and Golgi-specific O-linked glycosylation of a resident ER protein, this study demonstrates that Golgi enzymes do not cycle through the ER during secretion and mitosis.
Abstract
Golgi-specific sialyltransferase (ST) expressed as a chimera with the rapamycin-binding domain of mTOR, FRB, relocates to the endoplasmic reticulum (ER) in cells exposed to rapamycin that also express invariant chain (Ii)-FKBP in the ER. This result has been taken to indicate that Golgi-resident enzymes cycle to the ER constitutively. We show that ST-FRB is trapped in the ER even without Ii-FKBP upon rapamycin addition. This is because ER-Golgi–cycling FKBP proteins contain a C-terminal KDEL-like sequence, bind ST-FRB in the Golgi, and are transported together back to the ER by KDEL receptor–mediated retrograde transport. Moreover, depletion of KDEL receptor prevents trapping of ST-FRB in the ER by rapamycin. Thus ST-FRB cycles artificially by binding to FKBP domain–containing proteins. In addition, Golgi-specific O-linked glycosylation of a resident ER protein occurs only upon artificial fusion of Golgi membranes with ER. Together these findings support the consensus view that there is no appreciable mixing of Golgi-resident enzymes with ER under normal conditions.
INTRODUCTION
The Golgi apparatus comprises stacks of flattened cisternae that localize to the perinuclear region of mammalian cells. Secretory cargoes and proteins of the plasma membrane and endosome/lysosome compartments are synthesized in the endoplasmic reticulum (ER) and then transported to the Golgi apparatus, where they are then sorted and trafficked to their specific destination (Schatz and Dobberstein, 1996; Lee et al., 2004). The export of cargoes from the Golgi is mediated by a large number of vesicles, including clathrin or COPI-coated vesicles and carriers of the trans-Golgi network (TGN) to the cell surface (CARTS; Pearse, 1976; Malhotra et al., 1989; Barlowe et al., 1994; Robinson, 1994; Wakana et al., 2012). During mitosis, these membranes fragment into numerous small tubules and vesicles by reactions that involve MEK1, Myt1, Plk, and Cdc2 kinases (Acharya et al., 1998; Lowe et al., 1998; Colanzi et al., 2000, 2003; Kano et al., 2000; Sütterlin et al., 2002; Preisinger et al., 2005; Villeneuve et al., 2013). For a very short period in metaphase, Golgi membranes are distributed uniformly throughout the cell, and Lippincott-Schwartz and colleagues concluded that this distribution of Golgi represented their fusion with the ER (Zaal et al., 1999; Altan-Bonnet et al., 2006). What, however, of the vesicles and tubules seen in mitotic cells by others (Lucocq et al., 1989; Jesch and Linstedt, 1998; Jokitalo et al., 2001; Puri et al., 2004)? Golgi vesicles and tubules may have been en route to fusion with the ER or instead may have been in the process of direct reassembly to generate the Golgi stacks that are observed in the late stages of mitosis. The question we sought to address is whether Golgi membranes return to the ER and reform after mitosis or do the Golgi fragments reassemble by themselves without needing to start over in the ER. This issue has been addressed in a number of previous studies and needs to be resolved unequivocally.
In previous work, we targeted a chimera of sialyltransferase (ST) and FK506-binding protein (FKBP) to the TGN and generated a major histocompatibility complex class II transporter, invariant γ chain (Ii), attached to FKBP-rapamycin–binding protein (FRB), that would be retained within the ER (Pecot and Malhotra, 2004). As FKBP forms a complex with FRB in the presence of rapamycin, the recycling of ST-FKBP to the ER would trap it by binding to Ii-FRB. The ST-FKBP and Ii-FRB constructs were expressed and found to localize to their expected cellular compartments. Addition of rapamycin did not result in the trapping of ST-FKBP to the ER. However, treatment of cells with rapamycin and brefeldin A (BFA), a fungal metabolite that fuses Golgi membranes to the ER, arrested ST-FKBP in the ER of cells expressing Ii-FRB. Moreover, an ERGIC53-FKBP fusion protein that is known to cycle through the ER (Tang et al., 1995) was arrested in the ER of cells expressing Ii-FRB upon incubation with rapamycin. The fate of ERGIC53 and our findings with the BFA treatment confirmed the use of our approach with respect to its ability to arrest proteins if and when they cycle through the ER (Pecot and Malhotra, 2004, 2006). Our approach revealed that Golgi enzymes do not cycle through the ER in general or cycle extremely slowly (t1/2 = 18 h). Furthermore, during mitosis, we found no evidence for the recycling of ST-FKBP through the ER. Based on these findings, we proposed that Golgi membranes retain their independence from the ER. Subsequently Seemann and colleagues reported that separation of Golgi from the ER during mitosis protects unregulated activation of SREBP and ATF6 (Bartz and Seemann, 2008; Bartz et al., 2008).
Recently Lippincott-Schwartz and colleagues presented data that contradict previous conclusions of numerous labs regarding Golgi and ER separation during protein secretion and mitosis (Sengupta et al., 2015). They used rapamycin-mediated trapping of FRB to FKBP but exchanged the trapping domains. They reasoned that endogenous FKBP13 (Nigam et al., 1993) competed for the binding of ST-FKBP to Ii-FRB in ER by rapamycin. They therefore targeted ST tagged with FRB to the Golgi and Ii-FKBP to the ER. Based on the behavior of these constructs, they concluded that Golgi enzymes recycle through the ER continuously and in mitosis. If correct, this would represent an important finding, as it challenges the foundation of the definition of Golgi complex as a stable compartment that exists independently of the ER.
Because of the discrepancy in their conclusions, it was important to revisit whether FKBP13 is the reason for the discrepancy in the interpretation of the data based on rapamycin-mediated binding of FKBP to FRB-appended Golgi and ER proteins used in the respective studies. We show here that the ST-FRB used by Lippincott-Schwartz and colleagues in fact redistributes to the ER even in the absence of Ii-FKBP. Moreover, ST-FRB does not cycle to the ER on its own but binds the cycling protein FKBP13, which recycles to the ER by a KDEL receptor–mediated process. Our findings, based on trapping of Golgi-specific enzymes and a new assay that monitors posttranslational modification of an ER-retained protein by Golgi-specific enzymes, further support the conclusion that Golgi enzymes do not cycle through the ER.
RESULTS
The following experiments indicate that ST-FRB, used by Lippincott-Schwartz and colleagues as a marker protein of the late Golgi cisternae (Sengupta et al., 2015), is not actually a marker of that compartment. This is because C-terminal KDEL-like sequence present in FKBP13 and likely other FKBPs localized in the ER (Nakamura et al., 1998; Shadidy et al., 1999; Boudko et al., 2014) traffic to the Golgi, bind ST-FRB in the presence of rapamycin, and are then transported by the KDEL-receptor pathway to the ER.
Knockout of FKBP13 does not trap ST-FKBP in the ER expressing Ii-FRB
Lippincott-Schwartz and colleagues argued that FKBP13 in the ER binds to Ii-FRB but did not test experimentally whether it effectively displaces ST-FKBP present in the ER in the presence of rapamycin. We performed clustered regularly interspaced short palindromic repeats (CRISPR) knockout of FKBP13 in HeLa cells. The analysis by Western blot shows a complete loss of FKBP13 (Figure 1A). Clonal cell lines were analyzed, and we chose one (HeLa B1) for the subsequent experiments. Next we transfected parental and B1 HeLa cells with ST-FKBP–green fluorescent protein (GFP) and Ii-FRB–hemagglutinin (HA) and found the proteins encoded by these constructs to be expressed in the Golgi and ER, respectively. We then treated these cells with cycloheximide (CHX) and rapamycin (or CHX only as a control) for 4 h and visualized them by microscopy. Our data reveal the segregation of Golgi-specific ST-FKBP-GFP from the ER containing Ii-FRB-HA even in the presence of rapamycin during 4 h (Figure 1B). Of importance, with or without rapamycin, ST-FKBP and Ii-FRB remain segregated in HeLa B1 cells (depleted of FKBP13). Therefore endogenous FKBP13 does not compete with ST-FKBP-GFP for binding to Ii-FRB, and the lack of trapping of the former construct cannot be explained by the presence of FKBP13 in ER lumen. Quantification of our immunofluorescence experiments shows that only in a small portion of the cells does ST-FKBP-GFP relocate completely to the ER upon rapamycin treatment (<5%), even in cells lacking FKBP13. Conversely, B1 HeLa cells cotransfected with ST-FRB-GFP and Ii-FKBP–monomeric red fluorescent protein (mRFP) show a change in ST-FRB distribution upon rapamycin treatment: in ∼90% of cells, ST-FRB-GFP is found exclusively in the ER (Figure 1C and Supplemental Figure S1).
FIGURE 1:
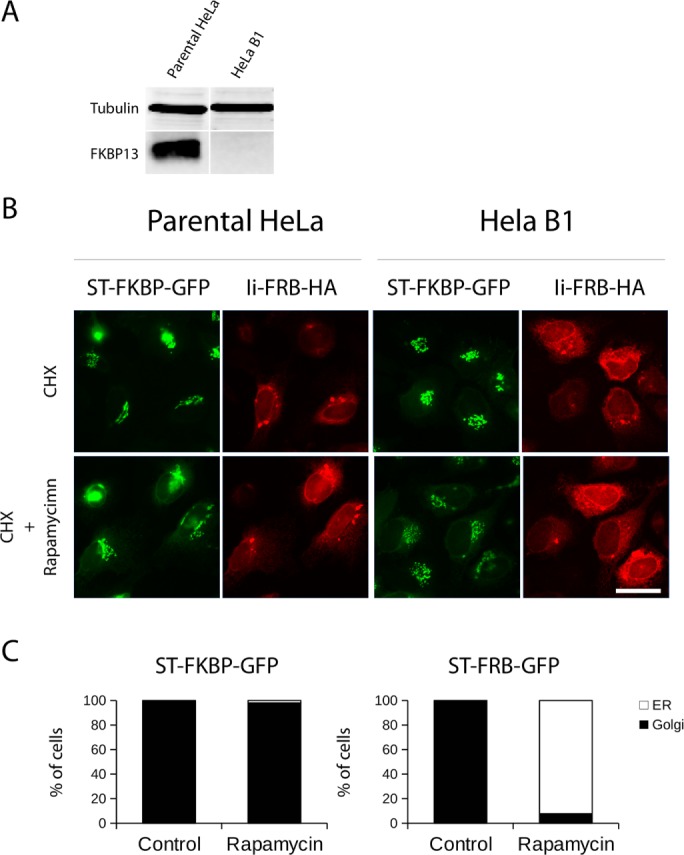
FKBP13 knockout does not induce trapping of ST-FBKP-GFP at the ER. (A) Lysates of parental HeLa and B1 HeLa cell lines were analyzed for FKBP13 levels using tubulin as a loading control. The B1 HeLa cell line has no detectable level of FKBP13. (B) Parental or B1 HeLa cells were cotransfected with ST-FKBP-GFP and Ii-FRB-HA and treated with CHX only (control) or CHX and rapamycin. After 4 h, cells were fixed, and an anti HA antibody was used to visualize Ii-FRB. There is no trapping of ST-FKBP-GFP by Ii-FRB-HA in parental or B1 HeLa cells (scale bar, 25 μm). (C) B1 HeLa cells were cotransfected either with ST-FKBP-GFP and Ii-FRB-HA or ST-FRB-GFP and Ii-FKBP-mRFP. After expression, cells were treated with CHX (Control) only or with a combination of CHX and rapamycin (Rapamycin). After 4 h, cells were fixed, and ST-FKBP or ST-FRB location was scored by immunofluorescence as cells showing total redistribution of ST to the ER or total or partial presence on the Golgi complex. Results are the average counting 100 cells in three separate experiments.
ST-FRB is trapped in the ER by rapamycin even in the absence of Ii-FKBP
HeLa cells express FKBP13 in the ER and in principle can bind Ii-FRB, suggesting that FKBP13 could also bind ST-FRB upon rapamycin treatment. In other words, rapamycin treatment should arrest ST-FRB in the ER even in cells lacking the exogenously expressed Ii-FKBP. To test this hypothesis, we cultured HeLa cells expressing ST-FRB-GFP or ST-FKBP-GFP without the corresponding binding partner (Ii-FKBP or Ii-FRB, respectively) with CHX only or with the addition of rapamycin. We fixed and visualized cells by microscopy 4 h later. ST-FKBP-GFP was localized to the Golgi regardless of the absence or presence of rapamycin, as we showed previously. Surprisingly, ST-FRB-GFP was found relocated to the ER in rapamycin-treated cells even in the absence of Ii-FKBP expression (Figure 2A). Quantification confirms that ∼90% of cells have ST-FRB partially or totally relocated to the ER (Figure 2B). These results show that ST-FRB can be trapped in the ER, most likely by the pool of endogenous FKBP expressed in the ER.
FIGURE 2:

ST-FRB-GFP redistributes to ER even in cells lacking Ii-FKBP. (A) HeLa cells growing on coverslips were transfected for 16 h with ST-FRB-GFP or ST-FKBP-GFP only and then treated with rapamycin and CHX or with CHX only as a control for 4 h. Cells were then fixed, permeabilized, and stained with DAPI. Only ST-FRB-GFP shows redistribution to the ER. (B) HeLa cells transfected as before were scored for three different patterns of distribution: Golgi only, intermediate, and fully ER redistributed. Results are the average of counting 100 cells in three separate experiments. (C) HeLa cells growing on coverslips were cotransfected with ST-FRB-GFP together with constructs expressing mCherry-tagged forms of FKBP13. Signal sequence-mCherry was used as a control. After 16 h of expression, cells were fixed, and GFP and mCherry fluorescence was visualized. Coexpression of ssmCherry-FKBP13wt and ssmCherry-FKBP13∆RTEL abrogates redistribution of ST-FRB-GFP to the ER.
ST-FRB-GFP relocation to ER is mediated by KDEL receptors
FKBP13 is an ER lumenal protein that contains a C-terminal KDEL-like sequence (RTEL) and likely traffics to the Golgi followed by retrieval due to binding to KDEL receptors. This could therefore affect the dynamics of ST-FRB in the presence of rapamycin. FKBP13 (or other FKBPs), in the presence of rapamycin, will bind ST-FRB at the Golgi and then be collected by KDEL-receptors and returned to the ER. We tested this possibility by expressing a mutant version of FKBP13 in which the KDEL-like sequence was removed (FKBP13∆RTEL). This mutant should be secreted, as it cannot be retrieved to the ER when it escapes from this compartment. In the presence of rapamycin, it should bind the FRB domain of ST-FRB-GFP and prevent the binding of endogenous FKBPs, inhibiting its redistribution. For this purpose, we overexpressed mCherry-FKBP13wt (wild-type FKBP13 with an N-terminal mCherry tag) and mCherry-FKBP13∆RTEL (lacking the C-terminal KDEL-like sequence). We used signal-sequence mCherry as an expression control. We first tested whether mCherry-FKBP13 is effectively retained in the cells. Conversely, mCherry-FKBP13∆RTEL leaks constantly into the medium (Supplemental Figure S2). When these constructs were coexpressed with ST-FRB-GFP, as expected, treatment with rapamycin induced the redistribution of ST-FRB-GFP to the ER in cells expressing the fluorescent protein mCherry (Figure 2C). However, when ST-FRB-GFP was coexpressed with mCherry-FKBP13∆RTEL, the former was retained in the Golgi, suggesting interference caused by the binding of FKBP13∆RTEL to the FRB domain of the ST-FRB (Figure 2C). Furthermore, mCherry-FKBP13∆RTEL partially colocalizes with ST-FRB-GFP in cells treated with rapamycin. Surprisingly, expression of wild-type mCherry-FKBP13wt also blocks redistribution of ST-FRB-GFP to the ER (Figure 2C). We hypothesize that overexpression of FKBP13 would saturate KDEL receptors and this would affect the redistribution of ST-FRB in the presence of rapamycin. Then the overexpression of FKBP13 (containing a KDEL-like retrieval sequence) could alter the normal functioning of the endogenous KDEL receptors. It is known that chronic stimulation of the KDEL receptors by overexpression of its ligands (like FKBP13) induces receptor self-activation and relocation to the ER (Pulvirenti et al., 2008). Together these data suggest indirect trafficking of ST-FRB by binding to FKBP13 and binding of the latter to KDEL receptors, followed by transport to the ER.
HeLa cells express two isoforms of KDEL receptors: KDELR1 and KDELR2 (Raykhel et al., 2007). We transfected HeLa cells with specific small interfering RNA (siRNA) duplexes against these two isoforms. We could not assess directly protein levels of KDEL receptors with Western blotting because of the poor signal given by the antibodies available. Therefore we tested the efficiency of the specific siRNA duplexes against KDELR1 and KDELR2 at the mRNA level. siRNA-mediated knockdown reduced KDELR1 and KDELR2 expression by 75 and 80%, respectively, compared with control cells (Figure 3A). We also tested the intracellular levels of FKBP13, which, in principle, should be altered by KDEL receptor functional impairment. As expected, knockdown of KDELR1 and KDELR2 decreased the intracellular levels of FKBP13 and strongly increased FKBP13 secreted levels in the medium compared with cells transfected with control siRNA. We conclude that KDEL receptor knockdown reduces intracellular levels of FKBP13 and triggers its release in the medium because of the inability of the cells to retrieve KDEL motif–containing proteins from the Golgi (Figure 3B). These cells were transfected with ST-FRB-GFP, which localized to the Golgi apparatus, and then treated with rapamycin. Visualization by fluorescence microscopy revealed that ST-FRB was retained in the Golgi. In contrast, in HeLa cells transfected with the control siRNA, ST-FRB relocated to the ER upon rapamycin treatment (Figure 3C). Quantification shows that at 4 h of rapamycin treatment, ∼50% of the cells transfected with siRNAs for KDEL receptors retained ST-FRB-GFP construct at the Golgi, in contrast to <10% in cells transfected with the control siRNAs (Figure 3D). This can only be because KDEL-containing FKBP13 and any other FKBPs that cycle between the ER and the Golgi through a KDEL receptor–mediated pathway are not retrieved from the Golgi to the ER after KDEL receptor knockdown. It is important to note that we probably do not have a total knockdown of KDEL receptors, and this probably explains the incomplete retention of ST-FRB in the Golgi by rapamycin treatment.
FIGURE 3:

ST-FRB-GFP redistributes to the ER via KDEL receptor. (A, B) HeLa cells were transfected with a mix of siRNAs for KDEL receptor 1 and 2 or scrambled oligos. After 72 h of knockdown, cells were collected and lysed for analysis of KDELR1 and KDELR2 mRNA level by reverse transcription PCR (A) and for analysis of protein levels by Western blotting in total cell lysate and in TCA-precipitated medium with antibody against tubulin and FKBP13 (B). (C) Cells were transfected with siRNAs as in A and B, and after 48 h of knockdown, cells were transfected with ST-FRB-GFP and incubated for an additional 20 h. After 68 h of knockdown, CHX and rapamycin were added to the medium. Cells were fixed at indicated time points and stained with DAPI. DAPI and GFP fluorescence were visualized with an epifluorescence microscope (scale bar, 10 μm). (D) Cells shown in C were scored for ST-FRB-GFP localization at the Golgi only (black bars), Golgi and the ER (gray bars), and ER only (white bars) and expressed as percentage of cells. Results shown are the average of three experiments and counting 50 cells/condition.
These findings reveal that the reason for the trapping of ST-FRB-GFP in the ER even in cells that do not express Ii-FKBP-RFP is because FKBPs bind the former in the Golgi, and the bound complex is retrieved from Golgi to the ER by KDEL receptors.
The ER-resident Ii protein is O-glycosylated only when Golgi membranes fuse with the ER
We tested the fate of Golgi enzymes by testing posttranslational modification of an ER-retained, HA-tagged Ii-FRB by Golgi-specific enzymes. HeLa cells stably expressing ManII-GFP were transfected with an Ii-FRB-HA plasmid and incubated with BFA to artificially fuse Golgi membranes with the ER. When cells were visualized by immunofluorescence microscopy, after 30 min, ManII-GFP was relocated with the ER (Figure 4A). At the indicated times, cells were lysed, and total lysates were Western blotted with an anti-HA antibody. Strikingly, the Ii protein in cells incubated with BFA migrated as a higher-mass polypeptide with a peak after 2 h of incubation with BFA compared with control cells (T = 0, Figure 4B). This result demonstrated that the Ii protein is modified posttranslationally upon relocation of Golgi enzymes to the ER. The incubation of cells with CHX demonstrated that the observed posttranslational modification of the Ii protein in the presence of BFA is independent of newly synthesized proteins (Figure 4B).
FIGURE 4:
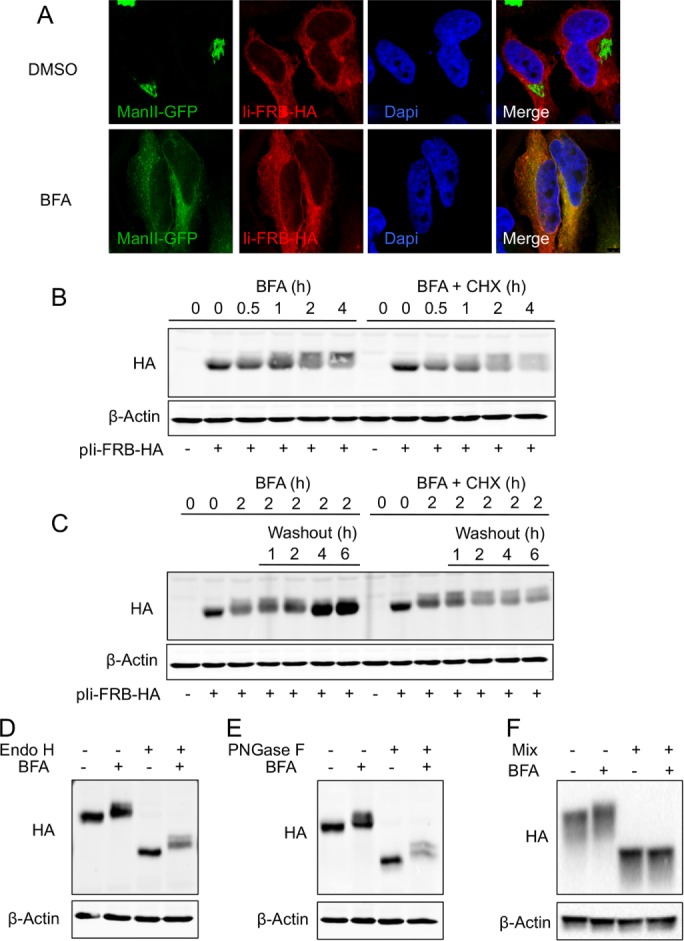
The Ii protein is O-glycosylated when Golgi membranes fuse with the ER. (A) HeLa cells expressing ManII-GFP grown on coverslip were transfected with Ii-FRB-HA plasmid. At 24 h after transfection, cells were incubated for 2 h with dimethyl sulfoxide (DMSO) or BFA, fixed, and processed for immunofluorescence microscopy with an anti-HA antibody and DAPI (scale bar, 5 μm). (B) HeLa cells were transfected or not with Ii-FRB-HA plasmid. At 24 h after transfection, cells were incubated with BFA in the presence or not of CHX. At the indicated times, cells were lysed, and total cell lysates were analyzed by Western blotting with an anti-HA antibody. Western blotting with an anti–β-actin antibody was used as a loading control. (C) HeLa cells were transfected or not with Ii-FRB-HA plasmid. At 24 h after transfection cells, were incubated for 2 h with BFA in the presence or not of CHX. Then cells were washed and incubated in normal BFA-free medium in the presence or absence of CHX. At the indicated times, cells were lysed, and total cell lysates were analyzed by Western blotting with an anti-HA antibody. Western blotting with an anti–β-actin antibody was used as a loading control. (D, F) HeLa cells were transfected with Ii-FRB-HA plasmid and incubated after 24 h with or without BFA during 2 h. Then total cell lysates were treated with or without Endo H (D), PNGase F (E), and protein deglycosylation mix supplemented with additional exoglycosidases (F) and analyzed by Western blotting with an anti-HA and an anti–β-actin antibody, respectively.
To determine whether this posttranslational modification of the Ii protein was transiently caused by Golgi enzymes, we transfected HeLa cells with Ii-FRB-HA plasmid, incubated them for 2 h with BFA, and then washed them with phosphate-buffered saline (PBS) and incubated them in normal BFA-free medium. At the indicated time points, cells were lysed and total cell lysates analyzed by Western blot with an anti-HA antibody. After BFA washout, the newly synthesized Ii protein is not subject to posttranslational modification, indicating that the enzymes involved in this process are not located in the ER after BFA washout. Moreover, when protein synthesis was inhibited with CHX, the higher-mass polypeptide remains detectable even after 6 h of incubation in normal BFA-free medium (Figure 4C). Together these results suggested that Golgi enzymes could modify the Ii protein in the ER when the latter membranes fused to the ER in presence of BFA. Moreover, the Ii protein remains modified after reorganization of the Golgi membranes in the perinuclear area, demonstrating that this posttranslational modification is an irreversible process until the Ii protein is degraded.
What is the nature of the posttranslational modification affecting the Ii protein when Golgi membranes fuse with the ER? Ii protein is a chaperone that assists in processing and transport of the MHC class II antigen along the secretory pathway. Without its binding partners, the Ii protein is arrested in the ER, but upon binding the β chain of the MHC class II antigen, it is transported to the Golgi apparatus and then to the endosomal/lysosomal system, where it can undergo proteolysis or reach the cell surface. Along its route of transport, the Ii protein is posttranslationally modified by the addition of N- and O-linked glycosylations on several amino acid residues. We tested first whether BFA treatment induced specific N-linked glycosylation on the Ii protein. HeLa cells transfected with Ii-FRB-HA plasmid were cultured with or without BFA during 2 h and lysed. Total cell lysates were then incubated in the presence or absence of endoglycosidases (Endo H or PNGase F) for 1 h at 37°C and analyzed by Western blot with an anti-HA antibody. Endo H and PNGase F specifically remove N-linked glycans from glycosylated proteins. Compared to control conditions, when cells were cultured with BFA, the Ii protein migrated at a higher mass (Figure 4, D and E, lanes 1 and 2). When total cells lysates were incubated with Endo H or PNGase F, the Ii protein migrated at a smaller mass, but the shift detected in the presence of BFA was still detectable (Figure 4, D and E, lanes 3 and 4). These results indicate that 1) the Ii protein is N-glycosylated when it localizes in the ER and 2) the posttranslational modification affecting the Ii protein in BFA is not due to N-linked glycosylation.
Next we tested whether BFA treatment induced specific O-linked glycosylation on the Ii protein. HeLa cells transfected with Ii-FRB-HA plasmid were cultured with or without BFA for 2 h and lysed. Total cell lysates were then incubated in the presence or absence of protein deglycosylation mix supplemented with additional exoglycosidases during 8 h at 37°C and analyzed by Western blot with an anti-HA antibody. This protein deglycosylation mix supplemented with additional exoglycosidases is used to remove both N- and O-linked glycosylation (Magnelli et al., 2012). Compared to control conditions, when cells were cultured with BFA, the Ii protein migrated at a higher mass (Figure 4F, lanes 1 and 2). When total cells lysates were incubated with the protein deglycosylation mix, the Ii protein migrated at a smaller mass, and the shift seen in the presence of BFA was not detectable (Figure 4F, lanes 3 and 4). These results indicated that when Golgi membranes fuse with the ER by BFA treatment, the Ii protein is O-glycosylated.
The Ii protein is known to be O-glycosylated on the Thr-203 and Ser-281 by the addition of an O-linked N-acetylgalactosamine (O-GalNAc; Halim et al., 2013). O-GalNAc glycosylation modifications are found in >50% of the proteins that enter in the secretory pathway, and this modification requires the cooperative action of GalNAc transferases (GalNac-Ts) that are localized in the Golgi (Bennett et al., 2012). It was reported that Golgi enzymes under special circumstances can be relocated from the Golgi apparatus to the ER (Lee and Linstedt, 1999; Gill et al., 2010; Tu et al., 2012; Eckert et al., 2014). In addition, the relocation of GalNac-Ts from the Golgi apparatus to the ER in a Src kinase– and Erk8-dependent manner increases O-GalNAc addition to endogenous O-glycosylated proteins and on ER-trapped reporter molecules (Gill et al., 2010; Chia et al., 2014). However, these observations are not related to a complete fusion of Golgi membranes with the ER. Can O-GalNAc addition to ER-trapped reporter be used to investigate whether Golgi membranes remain independent or fuse with the ER during mitosis? Farmaki et al. (1999) showed that CD8, which during the interphase is transported to the cell surface, is retained at the ER during mitosis. They monitored O-linked glycosylation of the newly synthesized and ER-localized CD8 by pulse labeling. With this sensitive approach, they demonstrated that newly synthesized, ER-localized CD8 in mitotic cells was modified by O-GalNAc addition, but this was due to newly synthesized GalNac-Ts. In our study, we used Ii protein as an ER-retained reporter at every step of the cell cycle. Moreover, our findings clearly demonstrate that when Golgi enzymes are relocated in the ER by BFA treatment, most of the pool of Ii protein is modified by O-GalNAc addition. Our simple but efficient assay establishes that modification by O-linked oligosaccharides can be used to monitor Golgi enzyme relocation when Golgi membranes fuse with the ER. We took advantage of this observation to reinvestigate with this approach whether Golgi membranes remain independent or fuse with the ER during the mitosis.
The Ii protein is not O-glycosylated during mitosis
We tested whether Golgi membranes remain segregated from the ER or instead fuse with the ER during mitosis by monitoring GalNac-Ts–mediated O-linked glycosylation of the Ii protein. First, we monitored whether the pattern of endogenous O-glycosylated proteins by GalNac-Ts was affected during mitosis. To this end, we used Helix pomatia lectin (HPL), which recognizes mostly O-GalNAc–containing Ser/Thr residues, a posttranslational modification that occurs in the Golgi. Indeed, HPL staining colocalized exclusively with a Golgi marker when cells were cultured in normal complete medium (Supplemental Figure S3A). Next we assessed whether the GalNac-T enzymes were active during the whole cell cycle. HeLa cells were arrested in S phase with a double thymidine block and in prometaphase with a double thymidine block followed by 18 h of incubation in thymidine-free medium containing nocodazole. HeLa cells were also arrested in metaphase with a double thymidine block followed by an incubation of 18 h in thymidine-free medium containing nocodazole and then by 2 h of incubation in nocodazole-free medium containing MG123. MG132 is a proteasome inhibitor used to arrest cells in metaphase (Lioutas and Vernos, 2013). Cells were lysed and total cells lysates analyzed by Western blot with horseradish peroxidase (HRP)–conjugated HPL. In each condition, synchronized cells in S phase, prometaphase, and metaphase exhibited the same general pattern of O-glycosylated proteins, suggesting that GalNac-Ts remain active and that glycans are available during mitosis progression (Supplemental Figure S3B).
We tested whether Ii protein was susceptible to O-glycosylation during mitosis. HeLa cells were arrested in S phase with a double thymidine block; the cells were then washed to remove thymidine and incubated in normal thymidine-free medium. At the indicated times, cells were stained with the DNA-binding dye 4′,6-diamidino-2-phenylindole (DAPI) to identify mitotic cells and with an anti–phospho-histone H3 antibody to identify cells in late G2 phase and in mitosis (Colanzi et al., 2007). A mitotic index (cells in mitosis/total number of cells) of 25% was observed at 10 h after removal of the cells from S-phase block (Figure 5, A and B). Based on the same experimental approach, at the indicated times, cells were lysed and total cell lysates analyzed by Western blot with an anti-HA and an anti–phospho-histone H3 antibody. The phosphorylation of histone H3 is maximal at 10 h after release from thymidine block; however, we could not detect a higher-mass polypeptide form of the Ii protein at any time during the release from the thymidine arrest (Figure 5C). This result suggests that during the progression of cells into mitosis, the Ii protein is not O-glycosylated.
FIGURE 5:

The Ii protein is not O-glycosylated in cells synchronized in prometaphase and metaphase. (A) HeLa cells grown on coverslips were transfected with Ii-FRB-HA plasmid and synchronized in S phase after a double thymidine block. Cells were washed to remove thymidine, and at the indicated times, cells were fixed and stained with an anti–phospho-histone H3 antibody and DAPI and visualized by fluorescence microscopy (scale bar, 100 μm). (B) The percentage of cells in mitosis (mitotic index) was determined by staining DNA with DAPI and an anti–phospho-histone H3 antibody at the indicated times after a double thymidine block and release. We counted 400 cells for each time point (mean ± SD, n = 3). (C) HeLa cells were transfected or not with Ii-FRB-HA plasmid and synchronized in S phase after a double thymidine block. Cells were washed to remove thymidine and lysed at the indicated times cells. Total cell lysates were analyzed by Western blotting with an anti-HA antibody and an anti–phospho-histone H3 antibody. Western blotting with an anti–β-actin antibody was used as a loading control. (D) HeLa cells expressing ManII-GFP grown on coverslip were transfected with Ii-FRB-HA plasmid and synchronized in S phase (double thymidine block), prometaphase (double thymidine block plus nocodazole), and metaphase (double thymidine block plus nocodazole plus MG132 during 2 h). Then cells were fixed and analyzed by immunofluorescence with an anti–phospho-histone H3 antibody and DAPI. Top, low magnification (scale bar, 100 μm). Bottom, high magnification (scale bar, 10 μm). (E) HeLa cells expressing ManII-GFP were transfected with Ii-FRB-HA plasmid and synchronized in S phase (double thymidine block), prometaphase (double thymidine block plus nocodazole or STLC), and metaphase (double thymidine block plus nocodazole or STLC plus MG132). At the indicated time, cells were lysed and total cell lysates analyzed by Western blotting with an anti-HA antibody and an anti–phospho-histone H3 antibody. Western blotting with an anti–β-actin antibody was used as a loading control. (F) HeLa cells expressing ManII-GFP grown on coverslips were transfected with Ii-FRB-HA plasmid and synchronized in metaphase (double thymidine block plus nocodazole plus MG132 during 2 h). BFA was added to the medium at the same time as MG132. Then cells were fixed and analyzed by immunofluorescence with an anti–phospho-histone H3 antibody and DAPI (scale bar, 10 μm). (G) HeLa cells expressing ManII-GFP were transfected with Ii-FRB-HA plasmid and synchronized in S phase (double thymidine block), prometaphase (double thymidine block plus nocodazole), and metaphase (double thymidine block plus nocodazole plus MG132 with or without BFA). At the indicated time, cells were lysed and total cell lysates analyzed by Western blotting with an anti-Ha antibody and an anti–phospho-histone H3 antibody. Western blotting with an anti–β-actin antibody was used as a loading control.
To confirm these results, we synchronized cells in different stages of the cell cycle. HeLa cells expressing ManII-GFP were arrested in S phase with a double thymidine block and in prometaphase with a double thymidine block followed by an incubation of 18 h in thymidine-free medium containing nocodazole or S-trityl-l-cysteine (STLC). STLC is a specific inhibitor of the kinesin Eg5 that prevents bipolar spindle formation (Skoufias et al., 2006). HeLa cells were arrested in metaphase with a double thymidine block followed by an incubation of 18 h in thymidine-free medium containing nocodazole or STLC and then by an incubation of 2 h in nocodazole- or STLC-free medium containing MG123. As visualized by immunofluorescence, with this procedure, ∼70–90% of the cells are synchronized in metaphase (identified by DAPI and phospho-histone H3 staining) and have their Golgi membranes completely fragmented (Figure 5D). At the indicated times, mitotic cells were easily detached and harvested by a “shake-off” procedure and lysed, and total cell lysates was analyzed by Western blotting with anti-HA and anti–phospho-histone H3 antibodies. In cells synchronized in prometaphase or metaphase, we could not detect a higher-mass Ii protein (Figure 5E). As demonstrated earlier, the O-glycosylation of Ii requires a delay of 2 h to be easily detected by Western blotting in the presence of BFA. Cells were indeed maintained in metaphase from 2 to 4 h. However, no posttranslational modification of the Ii protein was detected (Figure 5E). To test whether under these experimental conditions GalNac-Ts were able to O-glycosylate the Ii protein, we incubated cells synchronized in metaphase with BFA to force the relocation of the enzyme to the ER. BFA did not affect cell synchronization in metaphase (Figure 5F). The analysis of cells maintained in metaphase from 2 to 4 h in the presence of BFA revealed a higher-mass form of the Ii protein and demonstrated the addition of O-GalNac glycans on the Ii protein when Golgi membranes are artificially forced to fuse with the ER (Figure 5G and Supplemental Figure S4).
DISCUSSION
The Golgi complex is easily identified by its characteristic shape and by the presence of enzymes that modify cargoes en route to endosomes, the cell surface, or for secretion. When cells enter mitosis, the Golgi complex loses its shape and starts to fragment, which makes them difficult to identify based on their enzymatic composition (Sütterlin et al., 2002). There is extensive high-quality data demonstrating that Golgi membranes fragment during mitosis and then rebuild from preexisting fragments (Jokitalo et al., 2001; Seemann et al., 2002; Axelsson and Warren, 2004; Feinstein and Linstedt, 2008). Sengupta et al. (2015) recently concluded that Golgi membranes relocate to the ER and are rebuilt by export of the specific Golgi components from the ER in late mitosis. The experiments presented here identify why the conclusions of those investigators did not mesh with other results—the constructs used were artificially interacting with proteins that normally are retrieved from the Golgi by KDEL receptors. This background phenomenon was missed in the previous studies, which resolves an important disagreement.
We previously used a procedure to test whether the ultimate fate of the Golgi in mitosis, before reassembly late in mitosis, was relocation to the ER. The approach was to express a Golgi enzyme tagged with FKBP at the Golgi and a protein that cannot leave the ER (Ii) tagged with FRB at the ER. If FKBP-containing Golgi enzymes cycles through the ER, then, in the presence of rapamycin, they will be trapped by binding the corresponding partner containing FRB. In previous work, we presented data that revealed no appreciable mixing of Golgi enzymes to the ER during protein secretion and any time during mitosis (Pecot and Malhotra, 2004, 2006).
More recently, Sengupta et al. (2015) used the same trapping procedure, but the FRB-bearing component was placed in the Golgi and the FKBP bearer at the ER. They reasoned that the ER contains soluble FKBP-containing protein (FKBP13), which competes with binding of ST-FKBP to Ii-FRB in the ER in the presence of rapamycin. However, the following observations make their hypothesis unlikely:
When Golgi membranes are fused to the ER by BFA treatment, the ST-FKBP construct is trapped in the ER by Ii-FRB. In other words, if these constructs are proximal to each other in the ER, then rapamycin treatment traps the Golgi-specific protein in the ER, regardless of other endogenous FKBP-containing proteins.
ERGIC53, which is known to recycle through the ER, is in fact trapped (ERGIC53-FKBP) at the ER (by Ii-FRB) in the presence of rapamycin. Again, this points to the fact that if two proteins are in close proximity, then rapamycin-based procedures trap them regardless of the presence of FKBP-containing proteins in the ER.
The original authors did not test whether depletion of FKBP13 affected relocation of ST-FKBP to the ER in cells expressing Ii-FRB. We tested their hypothesis and found that ST-FKBP does not relocate to the ER irrespective of whether the ER contained FKBP13 or not (Pecot and Malhotra, 2004, 2006).
Sengupta et al. (2015) did not test the possibility that FKBP13 or other ER-located FKBP-containing proteins retrieve their ST-FRB construct by binding it in the Golgi in the presence of rapamycin and then returning to the ER by a KDEL receptor–mediated pathway. It is known that the KDEL receptor can reach even distal Golgi compartments, such as the trans-Golgi (Miesenböck and Rothman, 1995), where ST-FRB is located. Our new data show transport of ST-FRB to the ER by binding FKBP13 (or other FKBPs) and transport of the latter by KDEL receptor (Figure 6).
Our new approach shows that Ii-FRB undergoes O-linked glycosylation when Golgi membranes are artificially fused to the ER. The results also reveal that this modification does not occur ordinarily during interphase or in mitosis.
FIGURE 6:
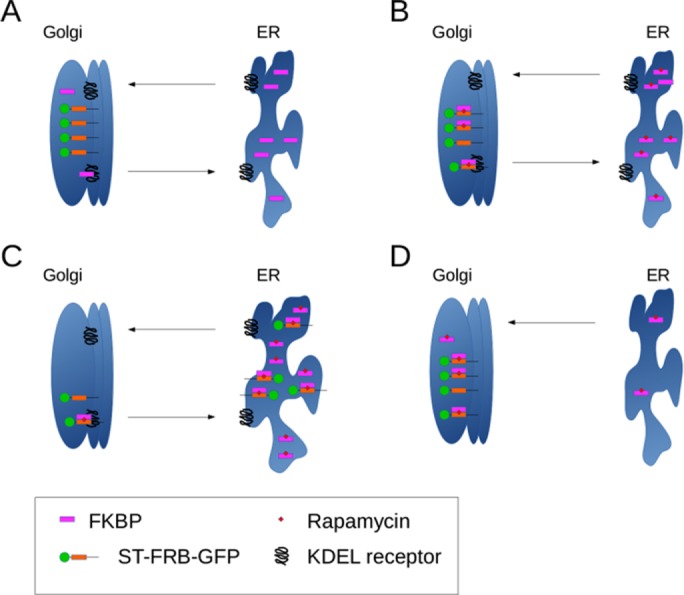
Model of ST-FRB relocalization mediated by FKBP and KDELR. (A) ST-FRB-GFP localizes to the Golgi apparatus and FKBPs are located in the ER but cycle through the Golgi, being retrieved by the KDEL receptor. (B) When rapamycin is added, FKBPs passing through the Golgi bind to the FRB domain of ST-FRB-GFP and to the KDEL receptor due to their C-terminal KDEL-like sequences. (C) Retrieval of FKBP by KDEL receptor redistributes bound ST-FRB-GFP to the ER over time. (D) When rapamycin is added in the absence of KDEL receptor, ST-FRB-GFP stays in the Golgi apparatus. FKBPs are not retained in the ER and bind to the FRB domain as they pass through the Golgi.
Together these findings demonstrate clearly that the Golgi and the ER remain segregated during protein secretion and mitosis. Still not understood are the precise molecular events underlying Golgi fragmentation and how these fragments fuse and reassemble to generate a polarized stack of Golgi cisternae. There is also the issue of why blocking Golgi fragmentation affects or delays entry of cells into mitosis. Together these issues should ultimately reveal why Golgi membranes are kept in the pericentriolar region of the cells, why and how they are stacked, and how they are dispersed and rebuilt during repeated cycles of cell division.
MATERIALS AND METHODS
Reagents, antibodies, and constructs
Reagents used in this study were obtained as follows: BFA, CHX, MG132 (Z-Leu-Leu-Leu-al), nocodazole, STLC, and thymidine were from Sigma-Aldrich (St. Louis, MO). Rapamycin was from Calbiochem (San Diego, CA). The endoglycosidases Endo H and PNGase F, the protein deglycosylation mix, and the exoglycosidases α-N-acetylgalactosaminidase, α1-2 fucosidase, β1-3 galactosidase and α1-3,6 galactosidase were from New England Biolabs (Ipswich, MA). Antibodies used in this study were purchased from the following sources: mouse monoclonal antibodies against β-actin and α-tubulin were from Sigma-Aldrich. Anti-HA (clone HA.11) was from Biolegend (San Diego, CA). The rabbit polyclonal antibody against phospho-histone H3 was from Upstate Biotechnology (Lake Placid, NY). Anti–red fluorescent protein (Cherry) antibody was from Abcam (Cambridge, MA). Anti-FKBP13 antibody was from Santa Cruz Biotechnology (sc-390753; Dallas, TX)). Alexa 647–conjugated HPL was from Life Technologies (Camarillo, CA), and HRP-conjugated HPL was from EY Laboratories (San Mateo, CA). Secondary antibodies for immunofluorescence microscopy and Western blotting were from Invitrogen (Carlsbad, CA). ST-FKBP-GFP and Ii-FRB-HA plasmids were previously described (Pecot and Malhotra, 2004). Secreted mCherry (ssCherry) was created by ligating mCherry cDNA containing interleukin 2 signal sequence for its import into the ER between the XhoI and BamHI sites of pCDNA3.1 (−) A myc/his plasmid (Invitrogen). FKBP cDNA was amplified from human liver cDNA and cloned into ssCherry digested with BamHI and HindIII to yield the plasmids ssCherry-FKBP13wt (using primers forward, TTTAGATCTACGGGGGCCGAGGGCAAAA; and reverse, TTTAAGCTTTTACAGCTCAGTTCGTCGCTC) and ssCherry-FKBP13∆RTEL (using primers forward, AGTCGGATCCACGGGGGCCGAGGGCAAAA; and reverse, GCATAAGCTTTTATCGCTCTATTTTGAGCAGC). Note that tagged versions of FKBP13 lack their endogenous signal sequence.
CRISPR of FKBP13
FKBP13 expression was knocked out from HeLa cells using a CRISPR/Cas9 knockout plasmid from Santa Cruz Biotechnology (sc-409613) following manufacturer’s recommendations. Briefly, HeLa cells were transfected with FKBP13 CRISPR/Cas9 plasmid containing a GFP cassette, and, after 24 h of expression, single GFP-positive cells were sorted with fluorescence-activated cell sorting and placed in 96-well plates. After the isolation of individual clones, lack of FKBP13 expression was assessed by Western blotting.
Cell culture and cell synchronization
HeLa cells and HeLa cells stably expressing ManII-GFP were grown in complete medium consisting of DMEM (PAA Laboratories) containing 10% (vol/vol) fetal calf serum (FCS), 100 U/ml penicillin, and 100 μg/ml streptomycin at 37°C with 7% CO2. OptiMEM was from Invitrogen.
For the transfection of DNA plasmids, cells were transfected with X-tremeGENE 9 DNA transfection reagent (Roche, Indianapolis, IN), following the manufacturer’s recommendations. For the knockdown of KDEL receptors, pooled siRNA duplexes for KDELR1 and KDELR2 were transfected using Hiperfect (Qiagen, Germantown, MD), following manufacturer’s recommendations. The same amounts of a control siRNA duplexes were used as a control. Control siRNAs consisted of a pool of ON-TARGETplus Non-Targeting siRNAs (D-001810-10-05; Thermo Scientific, Waltham, MA). The siRNA target sequence for KDELR1 was GGUGUUCACUGCCCGAUAU and for KDELR2 was CCUUCAGGGUGCUCGGACA (Ruggiero et al., 2015). Experiments were started 68 h after siRNA transfection.
To study cell progression into the mitosis, we first synchronized HeLa cells in S phase with double thymidine block. Briefly, cells were plated at 60% confluence in 24-well plates on 12-mm coverslips, maintained in growth medium for 6 h, and incubated for 18 h with 2 mM thymidine. Then cells were washed three times with PBS and maintained in growth medium for 10 h. Cells were then incubated in 2 mM thymidine for an additional 18 h before final release. After release, at the indicated times, cells were stained with DAPI and an anti–phospho-histone H3 antibody and analyzed by immunofluorescence microscopy to calculate mitotic index. Mitotic cells are positive for the phospho-histone H3 staining and exhibit condensed DNA. To synchronize cells in prometaphase, first, cells were synchronized in S phase with a double thymidine block as previously described and then washed three times with PBS and maintained in growth medium in the presence of 1.5 μM nocodazole or 5 μM STLC for 12 h. Cells were synchronized in metaphase with double thymidine block and then washed three times with PBS and maintained in growth medium in the presence of 1.5 μM nocodazole or 5 μM STLC for 12 h. The cells were washed three times with PBS and incubated for an additional 2 h in the presence of 10 μM MG123. For Western blot analysis, the mitotic cells were easily detached from the Petri dishes by a “shake-off” procedure.
Immunofluorescence microscopy
Cells were fixed with 3.7% paraformaldehyde (PFA) in PBS at room temperature for 20 min, permeabilized with 0.2% Triton X-100 for 20 min, and then blocked with blocking buffer (2.5% FCS, 0.1% Triton X-100 in PBS) for 30 min. Cells were then incubated with the indicated primary antibody for 45 min, followed by a PBS wash and an incubation with fluorescently labeled secondary antibodies for 45 min (donkey anti-mouse or anti-rabbit Alexa Fluor 488 or 594). DAPI was used to stain the DNA. Samples were analyzed with a Leica DMI6000 B inverted epifluorescence microscope or a Leica SPE confocal microscope using a 63× ACS Apo numerical aperture 1.3 objective and LASAF software. When necessary, cells were scored for three different patterns of sialyltransferase distribution: Golgi (only Golgi-like pattern of sialyltransferase), intermediate (Golgi and ER reticular patterns in the same cell), and fully ER redistributed (only reticular pattern, with no Golgi-like staining).
Reverse transcription PCR
RNAs were prepared using the RNAeasy Mini kit (Qiagen) following the manufacturer’s instructions. cDNAs were synthesized with the cloned AMV First-Strand cDNA Synthesis Kit (Invitrogen). RPL0, KDELR1, and KDELR2 were amplified with specific primers, and PCR products were analyzed on a 1.5% agarose gel.
ER trapping procedure
HeLa cells were transfected with ST-FKBP-GFP and Ii-FRB-HA plasmids or ST-FRB-GFP and Ii-FKBP-mRFP plasmids. After 24 h, cells were incubated with 10 μg/ml BFA with or without 250 nM of rapamycin for 1 h at 37°C. Cells were then washed three times with PBS and incubated with normal BFA-free medium for 90 min at 37°C. Cells were fixed and analyzed by immunofluorescence microscopy. ST-FKBP and ST-FRB were visualized by GFP fluorescence. Ii-FRB was observed with the monoclonal mouse antibody against the HA epitope and Ii-FKBP-mRFP using RFP fluorescence.
Immunoblotting
Cells were lysed in lysis buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 0,1% SDS, 1% NP-40, and 0.5% sodium deoxycholate) supplemented with protease inhibitors, 1 mM Na3VO4, and 25 mM sodium fluoride and centrifuged at 16,000 × g for 15 min. Total protein was resuspended in 1× SDS sample buffer containing dithiothreitol (DTT) and incubated at 95°C for 5 min. For medium samples concentrated by trichloroacetic acid (TCA) protein precipitation, 30 μg of bovine serum albumin and 150 μl of 55% TCA were added to 1 ml of medium and incubated for 30 min on ice. Medium was then centrifuged at 16,000 × g for 30 min at 4°C and supernatant removed. After another centrifugation at 16,000 × g for 1 min at 4°C, pellet was dried and resuspended in 1× SDS sample buffer containing DTT and incubated at 95°C for 5 min. The samples were then separated on 10% SDS–PAGE, transferred to nitrocellulose, and immunoblotted with the primary antibodies mentioned. Proteins were detected using the specific secondary antibody, and fluorescent bands were visualized using the Odyssey Infrared Imaging System (LI-COR Biosciences, San Diego, CA).
Endoglycosidase treatment
Cells were lysed in lysis buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 0,1% SDS, 1% NP-40, and 0.5% sodium deoxycholate) supplemented with protease inhibitors, 1 mM Na3VO4, and 25 mM sodium fluoride and centrifuged at 16,000 × g for 15 min. First, 1 μl of 10× glycoprotein denaturating buffer was added to 9 μl of cell extracts and incubated for 10 min at 100°C. For PNGase F treatment, in a final volume of 20 μl, 2 μl of 10× G7 reaction buffer, 1 μl of 10% NP-40, and 2 μl of PNGase F were added to the denatured proteins and incubated 1 h at 37°C. For Endo-H treatment, in a final volume of 20 μl, 2 μl of 10× G5 reaction buffer and 5 μl of Endo-H were added to the denatured proteins and incubated 1 h at 37°C. The endoglycosidase reaction was stopped by the addition of SDS sample buffer, and the samples were analyzed by Western blotting.
Protein deglycosylation mix and exoglycosidase treatment
Cells were lysed in lysis buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 0,1% SDS, 1% NP-40 and 0.5% sodium deoxycholate) supplemented with protease inhibitors, 1 mM Na3VO4, and 25 mM sodium fluoride and centrifuged at 16,000 × g for 15 min. First, 1 μl of 10× glycoprotein denaturing buffer was added to 9 μl of cell extracts and incubated for 10 min at 100°C. In a final volume of 20 μl, 2 μl of 10× G7 reaction buffer, 2 μl of 10% NP-40, and 5 μl of protein deglycosylation mix including O-glycosidase, PGNase F, neuraminidase, β1-4 galactosidase, and β-N-acetylglucosaminidase supplemented with additional exoglycosidases (α-N-acetylgalactosaminidase, α1-2 fucosidase, β1-3 galactosidase, α1-3,6 galactosidase) were added to the denatured proteins and incubated 8 h at 37°C. The deglycosylation reaction was stopped by the addition of SDS sample buffer, and the samples were analyzed by Western blotting.
Supplementary Material
Acknowledgments
We thank all members of the Malhotra laboratory for valuable discussions and Arrate Mallabiabarrena and Raquel García for help with fluorescence microscopy. J.V. acknowledges support from a Marie Curie International Outgoing Fellowship within the 7th European Community Framework Program. V.M. is an Institució Catalana de Recerca i Estudis Avançats Professor at the Center for Genomic Regulation, and the work in his laboratory is funded by grants from Plan Nacional (BFU2008-00414), Consolider (CSD2009-00016), the Agència de Gestió d’Ajuts Universitaris i de Recerca Grups de Recerca Emergents (SGR2009-1488; AGAUR-Catalan Government), and the European Research Council (268692). The project received research funding from the European Union. We acknowledge support of the Spanish Ministry of Economy and Competitiveness, Centro de Excelencia Severo Ochoa 2013-2017. This article reflects the views of only the authors. The European Union is not liable for any use that may be made of the information contained herein.
Abbreviations used:
- BFA
brefeldin A
- CHX
cycloheximide
- FKBP
FK506-binding protein
- FRB
FKBP-rapamycin–binding domain
- GalNac-T
N-acetylgalactosamine transferase
- HPL
Helix pomatia lectin
- Ii
invariant γ chain
- ST
sialyltransferase
- STLC
S-trityl-l-cysteine.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E16-08-0560) on November 2, 2016.
REFERENCES
- Acharya U, Mallabiabarrena A, Acharya JK, Malhotra V. Signaling via mitogen-activated protein kinase kinase (MEK1) is required for Golgi fragmentation during mitosis. Cell. 1998;92:183–192. doi: 10.1016/s0092-8674(00)80913-7. [DOI] [PubMed] [Google Scholar]
- Altan-Bonnet N, Sougrat R, Liu W, Snapp EL, Ward T, Lippincott-Schwartz J. Golgi inheritance in mammalian cells is mediated through endoplasmic reticulum export activities. Mol Biol Cell. 2006;17:990–1005. doi: 10.1091/mbc.E05-02-0155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axelsson MAB, Warren G. Rapid, endoplasmic reticulum-independent diffusion of the mitotic Golgi haze. Mol Biol Cell. 2004;15:1843–1852. doi: 10.1091/mbc.E03-07-0459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlowe C, Orci L, Yeung T, Hosobuchi M, Hamamoto S, Salama N, Rexach MF, Ravazzola M, Amherdt M, Schekman R. COPII: a membrane coat formed by Sec proteins that drive vesicle budding from the endoplasmic reticulum. Cell. 1994;77:895–907. doi: 10.1016/0092-8674(94)90138-4. [DOI] [PubMed] [Google Scholar]
- Bartz R, Seemann J. Mitotic regulation of SREBP and ATF6 by separation of the Golgi and ER. Cell Cycle. 2008;7:2100–2105. doi: 10.4161/cc.7.14.6327. [DOI] [PubMed] [Google Scholar]
- Bartz R, Sun L-P, Bisel B, Wei J-H, Seemann J. Spatial separation of Golgi and ER during mitosis protects SREBP from unregulated activation. EMBO J. 2008;27:948–955. doi: 10.1038/emboj.2008.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett EP, Mandel U, Clausen H, Gerken TA, Fritz TA, Tabak LA. Control of mucin-type O-glycosylation: a classification of the polypeptide GalNAc-transferase gene family. Glycobiology. 2012;22:736–756. doi: 10.1093/glycob/cwr182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudko SP, Ishikawa Y, Nix J, Chapman MS, Bächinger HP. Structure of human peptidyl-prolyl cis-trans isomerase FKBP22 containing two EF-hand motifs. Protein Sci. 2014;23:67–75. doi: 10.1002/pro.2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chia J, Tham KM, Gill DJ, Bard-Chapeau EA, Bard FA. ERK8 is a negative regulator of O-GalNAc glycosylation and cell migration. eLife. 2014;3:e01828. doi: 10.7554/eLife.01828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colanzi A, Deerinck TJ, Ellisman MH, Malhotra V. A specific activation of the mitogen-activated protein kinase kinase 1 (Mek1) is required for Golgi fragmentation during mitosis. J Cell Biol. 2000;149:331–340. doi: 10.1083/jcb.149.2.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colanzi A, Hidalgo Carcedo C, Persico A, Cericola C, Turacchio G, Bonazzi M, Luini A, Corda D. The Golgi mitotic checkpoint is controlled by BARS-dependent fission of the Golgi ribbon into separate stacks in G2. EMBO J. 2007;26:2465–2476. doi: 10.1038/sj.emboj.7601686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colanzi A, Sutterlin C, Malhotra V. RAF1-activated MEK1 is found on the Golgi apparatus in late prophase and is required for Golgi complex fragmentation in mitosis. J Cell Biol. 2003;161:27–32. doi: 10.1083/jcb.200208099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckert ESP, Reckmann I, Hellwig A, Röhling S, El-Battari A, Wieland FT, Popoff V. Golgi phosphoprotein 3 triggers signal-mediated incorporation of glycosyltransferases into coatomer-coated (COPI) vesicles. J Biol Chem. 2014;289:31319–31329. doi: 10.1074/jbc.M114.608182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmaki T, Ponnambalam S, Prescott AR, Clausen H, Tang BL, Hong W, Lucocq JM. Forward and retrograde trafficking in mitotic animal cells. ER-Golgi transport arrest restricts protein export from the ER into COPII-coated structures. J Cell Sci. 1999;112:589–600. doi: 10.1242/jcs.112.5.589. [DOI] [PubMed] [Google Scholar]
- Feinstein TN, Linstedt AD. GRASP55 regulates Golgi ribbon formation. Mol Biol Cell. 2008;19:2696–2707. doi: 10.1091/mbc.E07-11-1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill DJ, Chia J, Senewiratne J, Bard F. Regulation of O-glycosylation through Golgi-to-ER relocation of initiation enzymes. J Cell Biol. 2010;189:843–858. doi: 10.1083/jcb.201003055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halim A, Rüetschi U, Larson G, Nilsson J. LC-MS/MS characterization of O-glycosylation sites and glycan structures of human cerebrospinal fluid glycoproteins. J Proteome Res. 2013;12:573–584. doi: 10.1021/pr300963h. [DOI] [PubMed] [Google Scholar]
- Jesch SA, Linstedt AD. The Golgi and endoplasmic reticulum remain independent during mitosis in HeLa cells. Mol Biol Cell. 1998;9:623–635. doi: 10.1091/mbc.9.3.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jokitalo E, Cabrera-Poch N, Warren G, Shima DT. Golgi clusters and vesicles mediate mitotic inheritance independently of the endoplasmic reticulum. J Cell Biol. 2001;154:317–330. doi: 10.1083/jcb.200104073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kano F, Takenaka K, Yamamoto A, Nagayama K, Nishida E, Murata M. MEK and Cdc2 kinase are sequentially required for Golgi disassembly in MDCK cells by the mitotic Xenopus extracts. J Cell Biol. 2000;149:357–368. doi: 10.1083/jcb.149.2.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MCS, Miller EA, Goldberg J, Orci L, Schekman R. Bi-directional protein transport between the ER and Golgi. Annu Rev Cell Dev Biol. 2004;20:87–123. doi: 10.1146/annurev.cellbio.20.010403.105307. [DOI] [PubMed] [Google Scholar]
- Lee TH, Linstedt AD. Osmotically induced cell volume changes alter anterograde and retrograde transport, Golgi structure, and COPI dissociation. Mol Biol Cell. 1999;10:1445–1462. doi: 10.1091/mbc.10.5.1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lioutas A, Vernos I. Aurora A kinase and its substrate TACC3 are required for central spindle assembly. EMBO Rep. 2013;14:829–836. doi: 10.1038/embor.2013.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe M, Rabouille C, Nakamura N, Watson R, Jackman M, Jämsä E, Rahman D, Pappin DJ, Warren G. Cdc2 kinase directly phosphorylates the cis-Golgi matrix protein GM130 and is required for Golgi fragmentation in mitosis. Cell. 1998;94:783–793. doi: 10.1016/s0092-8674(00)81737-7. [DOI] [PubMed] [Google Scholar]
- Lucocq JM, Berger EG, Warren G. Mitotic Golgi fragments in HeLa cells and their role in the reassembly pathway. J Cell Biol. 1989;109:463–474. doi: 10.1083/jcb.109.2.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnelli P, Bielik A, Guthrie E. Identification and characterization of protein glycosylation using specific endo- and exoglycosidases. In: Hartley JL, editor. Protein Expression in Mammalian Cells. Totowa, NJ: Humana Press; 2012. pp. 189–211. [DOI] [PubMed] [Google Scholar]
- Malhotra V, Serafini T, Orci L, Shepherd JC, Rothman JE. Purification of a novel class of coated vesicles mediating biosynthetic protein transport through the Golgi stack. Cell. 1989;58:329–336. doi: 10.1016/0092-8674(89)90847-7. [DOI] [PubMed] [Google Scholar]
- Miesenböck G, Rothman JE. The capacity to retrieve escaped ER proteins extends to the trans-most cisterna of the Golgi stack. J Cell Biol. 1995;129:309–319. doi: 10.1083/jcb.129.2.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Yabe D, Kanazawa N, Tashiro K, Sasayama S, Honjo T. Molecular cloning, characterization, and chromosomal localization of FKBP23, a novel FK506-binding protein with Ca2+-binding ability. Genomics. 1998;54:89–98. doi: 10.1006/geno.1998.5571. [DOI] [PubMed] [Google Scholar]
- Nigam SK, Jin YJ, Jin MJ, Bush KT, Bierer BE, Burakoff SJ. Localization of the FK506-binding protein, FKBP 13, to the lumen of the endoplasmic reticulum. Biochem J. 1993;294:511–515. doi: 10.1042/bj2940511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearse BM. Clathrin: a unique protein associated with intracellular transfer of membrane by coated vesicles. Proc Natl Acad Sci USA. 1976;73:1255–1259. doi: 10.1073/pnas.73.4.1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pecot MY, Malhotra V. Golgi membranes remain segregated from the endoplasmic reticulum during mitosis in mammalian cells. Cell. 2004;116:99–107. doi: 10.1016/s0092-8674(03)01068-7. [DOI] [PubMed] [Google Scholar]
- Pecot MY, Malhotra V. The Golgi apparatus maintains its organization independent of the endoplasmic reticulum. Mol Biol Cell. 2006;17:5372–5380. doi: 10.1091/mbc.E06-06-0565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preisinger C, Körner R, Wind M, Lehmann WD, Kopajtich R, Barr FA. Plk1 docking to GRASP65 phosphorylated by Cdk1 suggests a mechanism for Golgi checkpoint signalling. EMBO J. 2005;24:753–765. doi: 10.1038/sj.emboj.7600569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulvirenti T, Giannotta M, Capestrano M, Capitani M, Pisanu A, Polishchuk RS, San Pietro E, Beznoussenko GV, Mironov AA, Turacchio G, et al. A traffic-activated Golgi-based signalling circuit coordinates the secretory pathway. Nat Cell Biol. 2008;10:912–922. doi: 10.1038/ncb1751. [DOI] [PubMed] [Google Scholar]
- Puri S, Telfer H, Velliste M, Murphy RF, Linstedt AD. Dispersal of Golgi matrix proteins during mitotic Golgi disassembly. J Cell Sci. 2004;117:451–456. doi: 10.1242/jcs.00863. [DOI] [PubMed] [Google Scholar]
- Raykhel I, Alanen H, Salo K, Jurvansuu J, Nguyen VD, Latva-Ranta M, Ruddock L. A molecular specificity code for the three mammalian KDEL receptors. J Cell Biol. 2007;179:1193–1204. doi: 10.1083/jcb.200705180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MS. The role of clathrin, adaptors and dynamin in endocytosis. Curr. Opin Cell Biol. 1994;6:538–544. doi: 10.1016/0955-0674(94)90074-4. [DOI] [PubMed] [Google Scholar]
- Ruggiero C, Fragassi G, Grossi M, Picciani B, Di Martino R, Capitani M, Buccione R, Luini A, Sallese M. A Golgi-based KDELR-dependent signalling pathway controls extracellular matrix degradation. Oncotarget. 2015;6:3375–3393. doi: 10.18632/oncotarget.3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schatz G, Dobberstein B. Common principles of protein translocation across membranes. Science. 1996;271:1519–1526. doi: 10.1126/science.271.5255.1519. [DOI] [PubMed] [Google Scholar]
- Seemann J, Pypaert M, Taguchi T, Malsam J, Warren G. Partitioning of the matrix fraction of the Golgi apparatus during mitosis in animal cells. Science. 2002;295:848–851. doi: 10.1126/science.1068064. [DOI] [PubMed] [Google Scholar]
- Sengupta P, Satpute-Krishnan P, Seo AY, Burnette DT, Patterson GH, Lippincott-Schwartz J. ER trapping reveals Golgi enzymes continually revisit the ER through a recycling pathway that controls Golgi organization. Proc Natl Acad Sci USA. 2015;112:E6752–E6761. doi: 10.1073/pnas.1520957112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shadidy M, Caubit X, Olsen R, Seternes OM, Moens U, Krauss S. Biochemical analysis of mouse FKBP60, a novel member of the FKPB family. Biochim Biophys Acta. 1999;1446:295–307. doi: 10.1016/s0167-4781(99)00080-9. [DOI] [PubMed] [Google Scholar]
- Skoufias DA, DeBonis S, Saoudi Y, Lebeau L, Crevel I, Cross R, Wade RH, Hackney D, Kozielski F. S-trityl-L-cysteine is a reversible, tight binding inhibitor of the human kinesin Eg5 that specifically blocks mitotic progression. J Biol Chem. 2006;281:17559–17569. doi: 10.1074/jbc.M511735200. [DOI] [PubMed] [Google Scholar]
- Sütterlin C, Hsu P, Mallabiabarrena A, Malhotra V. Fragmentation and dispersal of the pericentriolar Golgi complex is required for entry into mitosis in mammalian cells. Cell. 2002;109:359–369. doi: 10.1016/s0092-8674(02)00720-1. [DOI] [PubMed] [Google Scholar]
- Tang BL, Low SH, Hauri HP, Hong W. Segregation of ERGIC53 and the mammalian KDEL receptor upon exit from the 15 degrees C compartment. Eur J Cell Biol. 1995;68:398–410. [PubMed] [Google Scholar]
- Tu L, Chen L, Banfield DK. A conserved N-terminal arginine-motif in GOLPH3-family proteins mediates binding to coatomer. Traffic. 2012;13:1496–1507. doi: 10.1111/j.1600-0854.2012.01403.x. [DOI] [PubMed] [Google Scholar]
- Villeneuve J, Scarpa M, Ortega-Bellido M, Malhotra V. MEK1 inactivates Myt1 to regulate Golgi membrane fragmentation and mitotic entry in mammalian cells. EMBO J. 2013;32:72–85. doi: 10.1038/emboj.2012.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakana Y, van Galen J, Meissner F, Scarpa M, Polishchuk RS, Mann M, Malhotra V. A new class of carriers that transport selective cargo from the trans Golgi network to the cell surface. EMBO J. 2012;31:3976–3990. doi: 10.1038/emboj.2012.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaal KJ, Smith CL, Polishchuk RS, Altan N, Cole NB, Ellenberg J, Hirschberg K, Presley JF, Roberts TH, Siggia E, et al. Golgi membranes are absorbed into and reemerge from the ER during mitosis. Cell. 1999;99:589–601. doi: 10.1016/s0092-8674(00)81548-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.