

DDB2 is a multifunctional protein that participates in both nucleotide excision repair and regulation of gene transcription. In colon cancer cells, chromatin association of XRCC5/6, in the absence of DNA damage, depends on DDB2, and the DDB2–XRCC5/6 interaction promotes the transcription of the antiangiogenic gene SEMA3A.
Abstract
Damaged DNA-binding protein 2 (DDB2), a nuclear protein, participates in both nucleotide excision repair and mRNA transcription. The transcriptional regulatory function of DDB2 is significant in colon cancer, as it regulates metastasis. To characterize the mechanism by which DDB2 participates in transcription, we investigated the protein partners in colon cancer cells. Here we show that DDB2 abundantly associates with XRCC5/6, not involving CUL4 and DNA-PKcs. A DNA-damaging agent that induces DNA double-stranded breaks (DSBs) does not affect the interaction between DDB2 and XRCC5. In addition, DSB-induced nuclear enrichment or chromatin association of XRCC5 does not involve DDB2, suggesting that the DDB2/XRCC5/6 complex represents a distinct pool of XRCC5/6 that is not directly involved in DNA break repair (NHEJ). In the absence of DNA damage, on the other hand, chromatin association of XRCC5 requires DDB2. We show that DDB2 recruits XRCC5 onto the promoter of SEMA3A, a DDB2-stimulated gene. Moreover, depletion of XRCC5 inhibits SEMA3A expression without affecting expression of VEGFA, a repression target of DDB2. Together our results show that DDB2 is critical for chromatin association of XRCC5/6 in the absence of DNA damage and provide evidence that XRCC5/6 are functional partners of DDB2 in its transcriptional stimulatory activity.
INTRODUCTION
DDB2, a product of the xeroderma pigmentosum group E (XPE) gene, plays important roles in the early steps of global genomic repair through the nucleotide excision repair (NER) pathway (Tang and Chu, 2002). It associates with the CUL4-DDB1 E3 ligase (Shiyanov et al., 1999b) to ubiquitinate histones and the NER protein XPC, leading to assembly of the NER repair complex on the damaged chromatin (Wang et al., 2004, 2006; Kapetanaki et al., 2006). In addition, DDB2 has a chromatin-remodeling activity that supports NER (Luijsterburg et al., 2012). It has been suggested DDB2 cooperates with PARP1 to stimulate NER (Pines et al., 2012). DDB2 also possesses transcriptional regulatory activities. Mouse embryonic fibroblasts (MEFs) lacking DDB2 are deficient in undergoing senescence in culture, which is linked to an increase in antioxidant activities. DDB2 represses expression of the antioxidant genes MnSOD and Catalase (Minig et al., 2009; Roy et al., 2010). In DDB2−/− cells or tissues, there is derepression of MnSOD and Catalase, which inhibits accumulation of reactive oxygen species (ROS). In colon cancer cells, depletion of DDB2 leads to epithelial-to-mesenchymal transition (EMT), which is associated with aggressive tumor progression and increased liver metastasis (Roy et al., 2013). The EMT and increased metastasis was related to derepression of Zeb1, Snail, and VEGFA expression. In mesenchymal-type colon cancer cells having reduced expression of DDB2, reexpression of DDB2 causes repression of those EMT genes (Roy et al., 2013). Consistent with these observations, a colon cancer tissue microarray indicated a loss of DDB2 expression that coincided with reduced expression of the epithelial cell surface marker E-cadherin and metastatic progression of colon cancer (Roy et al., 2013). Thus the tumor suppression function of DDB2 extends far beyond its role in NER. In colon cancer, its transcriptional regulatory role is clearly important in suppression of aggressive progression and metastasis.
DDB2 also possesses transcriptional stimulatory activity, which has been implicated in tumor suppression. For example, DDB2 was shown to regulate the invasiveness of breast cancer cells by stimulating expression of the NF-κB inhibitor NFKBIA (Ennen et al., 2013). That study identified a specific sequence element, TCCCCTTA, in the NFKBIA promoter that is recognized by DDB2. The same element might also participate in DDB2-mediated repression, as a recent study showed that DDB2 represses NEDD4L, an E3 ligase for Smad2/Smad3, to enhance transforming growth factor β (TGFβ) signaling in ovarian cancer cells by binding to the same DNA element in the NEDD4L promoter (Zhao et al., 2015). This is also is related to the tumor suppression function of DDB2 because repression of NEDD4L enhances responsiveness of ovarian cancer cells to growth inhibition by the TGFβ signaling pathway (Zhao et al., 2015).
The transcriptional repressor function of DDB2 involves DDB1 and CUL4. For repression of the MnSOD and Catalase, DDB2 was shown to recruit SUV39h, a histone H3K9 methylase, onto the promoters of those genes to generate chromatin with increased trimethylation of H3K9 (Roy et al., 2010, 2013). For repression of the NEDD4L promoter, DDB2 was shown to interact with the H3K27 methylase EZH2 (Zhao et al., 2015). It is noteworthy that CUL4 proteins (A and B) have been implicated in histone methylation. Depletion of CUL4 leads to significant loss of trimethylation at both H3K9 and H3K27 (Higa et al., 2006). Therefore it is possible that the CUL4 proteins participate in the recruitment of the histone methylases onto the promoters of the genes repressed by DDB2. The role of Cul4 in the transcription stimulatory function of DDB2 is unclear.
XRCC5/6, also known as Ku86/Ku70, are essential for DNA double-stranded break repair (DSBR) through the nonhomologous end joining (NHEJ) pathway (Weaver, 1996). Moreover, Ku86/Ku70 were also reported to play a role in the transcriptional regulation of selected genes, including ERBB2 (Nolens et al., 2009), alphaMyHC (Sucharov et al., 2004), and osteocalcin (Willis et al., 2002). Of interest, XRCC5/6 interact with DDB2 and negatively regulate its E3 ubiquitin ligase activities around the damaged sites in ultraviolet (UV)-irradiated HeLa cells (Takedachi et al., 2010). Given that DDB2 does not have a direct role in NHEJ, it has been suggested that XRCC5/6-mediated inhibition of the E3 ubiquitin ligase activity is related to prevention of destabilization of DDB2 by excessive autoubiquitination (Takedachi et al., 2010). Here we show that DDB2 plays an essential role in recruiting XRCC5/6 onto chromatin in the absence of DNA DSBs and that DDB2 recruits XRCC5 onto the promoter of a DDB2-stimulated gene, SEMA3A, in colon cancer cells. Our results suggest that XRCC5/6 are important transcriptional activation partners of DDB2.
RESULTS
Identification of protein partners of DDB2
To identify proteins that could regulate the DDB2 transcription regulatory function, we used an unbiased coimmunoprecipitation (coIP) strategy. Nuclear extracts from HCT116 stably overexpressing FLAG-HA-DDB2 (FH-DDB2) or control cells (Supplemental Figure 1, A and B) were immunoprecipitated using a two-round coIP approach using an anti-FLAG antibody and an anti-hemagglutinin (HA) antibody. The coIP samples eluted with 1× Laemmli sample buffer from FH-DDB2 cells were highly enriched in FH-DDB2 as well as in known protein partners of DDB2, including DDB1 and CUL4A (Figure 1A). For tandem mass spectrometry (MS/MS) analyses, the proteins after HA IP were released using a buffer containing 0.5 M NaCl and 0.1% SDS. That condition does not elute the antigen. The samples were separated on 10% SDS–PAGE and then silver stained. The strongest bands that were specifically detected in the lane corresponding to the FH-DDB2 coIP sample were excised and subjected to MS/MS identification. This analysis led to the identification of some DDB2 binding partners that were not previously described (Figure 1B). Western blotting analyses performed on coIP samples from control or FH-DDB2–expressing cells validated the binding of JARID2, XRCC5, TBX2, and histone H3 in HCT116 cells (Figure 1C).
FIGURE 1:
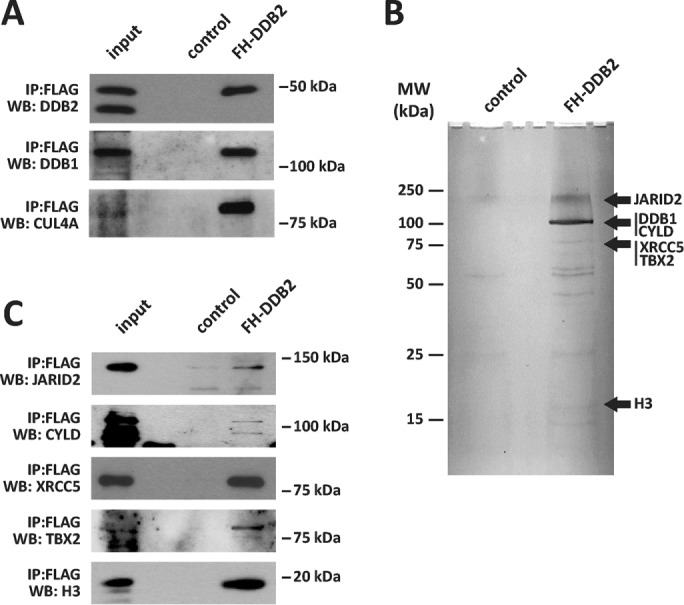
Characterization of DDB2-interacting proteins from HCT116 nuclear extracts. (A) Control and FH-DDB2 immunoprecipitates were analyzed by Western blotting using antibodies directed against the components of the DDB complex (DDB2, DDB1, and CUL4A). (B) Control and FH-DDB2 immunoprecipitates were resolved on a SDS–PAGE gel and then silver stained. (C) Proteins identified by MS/MS analysis in the DDB2 immunoprecipitates were validated by Western blotting (JARID2, CYLD, XRCC5, TBX2, H3).
Wild-type DDB2, but not XPE mutants, interacts with XRCC5/6 in HCT116 cells
One of the strong DDB2 interactors, as revealed by Western blotting analyses, is the DNA repair protein XRCC5. Of interest, the DDB2–XRCC5 interaction was already described in HeLa cells after UV irradiation (Takedachi et al., 2010). However, we were puzzled by the identification of such an interaction in the nuclei of unirradiated HCT116 cells. XRCC5 interacts with XRCC6 and DNA-PKcs for repair of DSBs by NHEJ. We further assayed the FH-DDB2 coIP samples by Western blotting and found that XRCC6, but not DNA-PKcs, coimmunoprecipitated with DDB2 (Figure 2A) in unirradiated HCT116 cells. The specificity of the DDB2–XRCC5/6 interaction was confirmed by performing coIP experiments using DDB2 mutants. Point mutation variants of DDB2 isolated from XPE patients (Nichols et al., 1996) were expressed in HCT116 cells and then coimmunoprecipitated using an anti-FLAG antibody (Figure 2B). Of interest, the 2RO and 82TO DDB2 variants were reported to have an impaired capacity of importing DDB1 to the nucleus (Shiyanov et al., 1999a). The mutant DDB2 proteins were defective in their ability to bind the XRCC5/6 proteins (Figure 2B). We also performed a coIP using anti XRCC5 antibody and nuclear extracts of control and HCT116 cells stably expressing a short hairpin RNA (shRNA) targeting DDB2 (shDDB2; Figure 2C). DDB2 was detected in coimmunoprecipitates obtained with the XRCC5 antibody. Of note, DDB1 was detected in the XRCC5 coIP samples from both control and shDDB2 cells. On the contrary, CUL4A was never found in the XRCC5 coIP samples. This suggests that the XRCC5/6 proteins may be part of a protein complex that includes DDB2 and DDB1 but not CUL4A and DNA-PKC in HCT116 cells.
FIGURE 2:
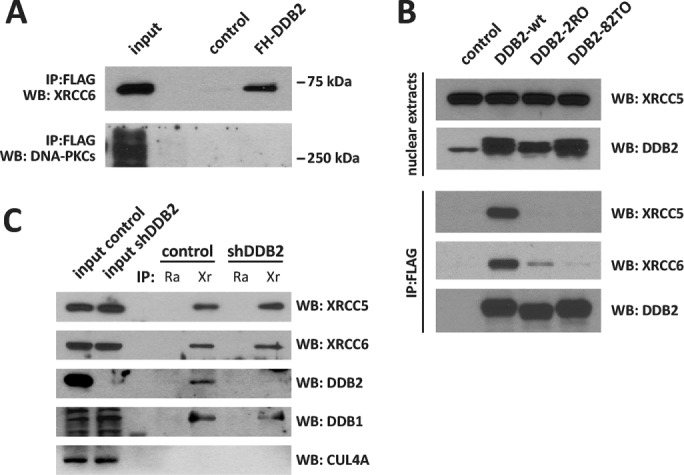
DDB2 is part of a protein complex including DDB1, XRCC5, and XRCC6. (A) DDB2 coimmunoprecipitates were analyzed by Western blotting with antibodies directed against the Ku complex subunits XRCC6 and DNA-PKcs. (B) FLAG-T7–tagged DDB2 and XPE mutants (2RO and 82TO) were overexpressed in HCT116 cells. Anti-FLAG immunoprecipitates were prepared from nuclear extracts and probed for XRCC5, XRCC6, and DDB2 by Western blotting. (C) Nuclear extracts from control and shDDB2 HCT116 cells were subjected to coIP using normal rabbit IgG (Ra) or an anti-XRCC5 antibody (Xr). Samples were probed for XRCC6 and the subunits of the DDB complex (DDB1, DDB2, and CUL4A) by Western blotting.
DDB2–XRCC5 may not be linked to the damaged-DNA response functions of XRCC5/6
We investigated whether the DDB2–XRCC5 interaction could play a role in the damaged-DNA response function of XRCC5 and XRCC6. We treated control and shDDB2 cells with the DSB-inducer phleomycin for 2 h and then replaced medium and incubated the cells for an additional 4 h. The DDB2–XRCC5 interaction remained unperturbed upon and after the phleomycin treatment, as revealed by coIP experiments followed by Western blotting analysis (Figure 3A). Of interest, control and shDDB2 cells responded to the genotoxic stress by accumulating γ-H2AX in a very similar manner (Figure 3B), unlike what we observed after depleting XRCC5 by siRNA transfection (Supplemental Figure S2). The recruitment of XRCC5 to the phleomycin-damaged chromatin was also monitored in control and shDDB2 HCT116 cells by immunofluorescence (Figure 3C). Cells were washed with a buffer containing detergent and RNase immediately before fixation in order to remove the non-chromatin-bound pool of XRCC5, as described previously (Britton et al., 2013). XRCC5 accumulation to the detergent- and RNase-resistant nuclear compartment of phleomycin-challenged cells was not prevented by DDB2 knockdown. However, the untreated control cells had a significantly higher amount of detergent- and RNase-resistant XRCC5 than did shDDB2 HCT116 cells (p < 0.001). Phleomycin did not have any significant effect on the total protein and mRNA levels of XRCC5 in the presence or absence of DDB2 (Supplemental Figure S2). Therefore we hypothesized that DDB2 expression could affect XRCC5 recruitment to the chromatin under basal conditions in HCT116 cells but not after DSB.
FIGURE 3:

Phleomycin-induced DNA damage does not affect the DDB2–XRCC5 interaction. (A) Cells expressing FH-DDB2 were treated with phleomycin for 2 h and further incubated for 0 h (phleo+0 h) or 4 h (phleo+4 h) at 37°C and then subjected to FLAG immunoprecipitation. XRCC5 and DDB2 levels were probed in the nuclear extracts (left) and immunoprecipitates (right) by Western blotting. (B) Control or shDDB2 HCT116 cells were treated as described for A. Whole-cell lysates were probed for β-actin and γH2Ax by Western blotting. Band intensities from three different blots were quantified using ImageJ, and relative γH2Ax signal values were plotted (*p < 0.05, **p < 0.01, ***p < 0.001; one-way analysis of variance [ANOVA]). (C) Control or shDDB2 HCT116 cells were grown on coverslips and then treated as described. Cells were washed with CSK-IGEPAL buffer supplemented with RNase and then fixed, immunostained for XRCC5, and analyzed by confocal microscopy. XRCC5 nuclear signal was quantitated using ImageJ, and values from 50 cells per condition were averaged and plotted (***p < 0.001, ****p < 0.0001; one-way ANOVA).
DDB2 promotes the accumulation of XRCC5 in a chromatin-enriched fraction of colon cancer cells
We studied the accumulation of XRCC5 in the chromatin-enriched fraction in control versus shDDB2 HCT116 cells by performing fractionation experiments followed by Western blotting. Control or shDDB2 cells were washed with phosphate-buffered saline (PBS) or with buffers containing detergent alone or detergent supplemented with RNase, similarly to what we described earlier (Figure 3C), and then harvested in 1× Laemmli sample buffer and analyzed by Western blotting (Figure 4A). Whereas control and shDDB2 cells express XRCC5 at similar levels (Figure 4A, lanes 1 and 2), only control cells accumulate XRCC5 in the RNase-resistant fraction (Figure 4A). We further studied the intracellular distribution of XRCC5 in colon cancer cell lines by performing a sequential fractionation experiment. Cell pellets were sequentially extracted with buffers containing detergent (S1 fraction), detergent supplemented with RNase (S2 fraction), and detergent supplemented with nuclease (S3 fraction) as outlined in Figure 4B. As expected, most of the histone H3 protein was released upon nuclease digestion in the S3 fraction, indicating that S3 is a chromatin-enriched fraction (Figure 4C and Supplemental Figure S3A). Western blotting analysis revealed that DDB2 accumulates in the S1 and the S3 fractions (Figure 4C). Of interest, XRCC5 accumulation in the chromatin-enriched S3 fraction was detected in the control cells but not in the shDDB2 cells (Figure 4C). Similar experiments were performed using the SW480 and SW620 colon cancer cell lines, which were isolated from the same patient (Leibovitz et al., 1976). As reported previously (Roy et al., 2013), Western blotting analysis showed that the primary site–isolated SW480 cells express more DDB2 than the lymph node metastatic variant SW620 cells (Figure 4D, left). On the contrary, the cell types expressed comparable amounts of XRCC5 (Figure 4D, left). Cells were washed with a buffer containing detergent and RNase and then harvested in 1× Laemmli sample buffer as described earlier. DDB2 was barely detectable in the RNase-resistant fraction of SW620, unlike SW480 cells. Similarly, the XRCC5 levels in the RNase-resistant fraction of SW480 cells were higher than with SW620 cells (Figure 4D, right). A sequential fractionation experiment was also performed to compare SW480 and SW620 cells and revealed a differential DDB2 distribution in the two cell types. DDB2 is depleted in the chromatin-enriched S3 fraction of SW620 cells (Figure 4E). Consistently, XRCC5 is mainly present as detergent-soluble protein in the S1 fraction of SW620 cells compared with SW480 cells. These data strongly support that DDB2 status can regulate recruitment of XRCC5 to the chromatin in colon cancer cells. It is noteworthy that we did not observe a differential XRCC5 chromatin recruitment in HeLa cells expressing control or DDB2-shRNA (Supplemental Figure S3). It is possible that HPV oncoproteins expressed by HeLa cells affect chromatin recruitment of XRCC5.
FIGURE 4:

DDB2 is required for XRCC5 recruitment in a chromatin-enriched fraction in colon cancer cell lines. (A) Whole-cell lysates (WCLs) were prepared from control (ct) or shDDB2 (sh) HCT116 cells. Alternatively, cells were washed with CSK-T buffer (CSK-T resist) or CSK-T buffer supplemented with RNase (RNAse resist) before harvesting in Laemmli sample buffer. Samples were probed for XRCC5, DDB2, and histone H3 by Western blotting. (B) Diagram outlining the sequential fractionation protocol used in the following experiments. S1, S2, S3, and P3 fractions were prepared as described in Materials and Methods. (C) Control and shDDB2 HCT116 cells were sequentially fractionated as outlined. Fractions were probed for XRCC5, DDB2, and histone H3 by Western blotting. (D) SW480 (480) and SW620 (620) cells were used to prepare WCLs or washed with CSK-T buffer supplemented with RNase and then harvested in Laemmli sample buffer (RNase resist). Samples were probed for XRCC5, DDB2, and histone H3 by Western blotting. (E) SW480 and SW620 cells were sequentially fractionated as outlined. Fractions were probed for XRCC5, DDB2, and histone H3 by Western blotting.
DDB2 and XRCC5 promote SEMA3A expression
Because DDB2 plays a role in recruiting XRCC5 to the chromatin of unchallenged colon cancer cells, we hypothesized that the DDB2–XRCC5 interaction may play a role in transcriptional regulation of selected DDB2 target genes. Our group recently found that DDB2 transcriptionally represses VEGFA, an angiogenic gene (Roy et al., 2013). In the tumor microenvironment, VEGFA effects are antagonized by a set of other antiangiogenic factors that include the protein products of several semaphorin class III genes (Maione et al., 2009, 2012). We found that HCT116 cells lacking DDB2 (shRNA expression), besides expressing ∼2.5 times more VEGFA mRNA than control cells, also express much less (>10 times less) SEMA3A mRNA (Figure 5A). The lower SEMA3A mRNA in the shDDB2 cells correlated well with the SEMA3A protein levels detected in the conditioned medium of control and shDDB2 cells (Figure 5B). Conversely, overexpression of DDB2 (Supplemental Figure S4A) led to up-regulation of SEMA3A mRNA in both HCT116 and SW620 cells (Figure 5C). This suggests that modulation of DDB2 levels can affect the transcriptional levels of SEMA3A in colon cancer cell lines. Next, we down-regulated XRCC5 in HCT116 cells by siRNA transfection (Supplemental Figure S4B) and assessed mRNA levels of the VEGFA and SEMA3A genes. XRCC5 knockdown resulted in a significant SEMA3A mRNA down-regulation, whereas VEGFA mRNA levels were unchanged (Figure 5D). The effect of XRCC5 knockdown on SEMA3A expression was not very strong because, whereas XRCC5-siRNA efficiently knocked down the mRNA level, knockdown at the protein level was less efficient (Supplemental Figure S4B), which is likely related to the long half-life of XRCC5 (Ajmani et al., 1995). Therefore SEMA3A is a transcriptional activation target of both DDB2 and XRCC5.
FIGURE 5:

DDB2 and XRCC5 control SEMA3A expression in colon cancer cells. (A) DDB2, SEMA3A, and VEGFA mRNA expression levels were assessed in control and shDDB2 HCT116 cells by reverse transcription (RT)-qPCR. Averages of three experiments are shown in the bar plots (**p < 0.01, ***p < 0.001, t test). (B) Concentrated conditioned medium from control and shDDB2 HCT116 cells was resolved by SDS–PAGE and then Coomassie stained (left) or probed for SEMA3A by Western blotting (right). (C) SEMA3A mRNA levels were assessed in HCT116 or SW620 cells transfected with an empty vector (pcDNA3) or a construct coding for FLAG-T7-DDB2 by RT-qPCR (**p < 0.01, t test). (D) XRCC5, SEMA3A, and VEGFA mRNA levels were assessed by RT-qPCR in HCT116 cells transfected with a nontargeting siRNA (siCTRL) or a siRNA against XRCC5 (siXRCC5) (*p < 0.05, ***p < 0.001, t test).
DDB2-dependent binding of XRCC5 to the SEMA3A promoter
We assessed the binding of DDB2 to the 6-kb SEMA3A proximal promoter by chromatin immunoprecipitation (ChIP), using a specific anti-DDB2 antibody or normal rabbit immunoglobulin G (IgG). We used 11 sets of primers covering the 6-kb upstream region of the SEMA3A promoter (Supplemental Figure S4C) in PCR assays of ChIP DNAs. Within the 6-kb region, there is one DDB2-cognate element at position 289 base pairs upstream of the transcription start site (Figure 6A), which corresponded to our PCR primer set 1. Consistent with this, primer set 1 showed interaction of DDB2 with the SEMA3A promoter (Figure 6B). Of interest, our ChIP PCR also detected interactions at other sites that apparently do not possess a strong DDB2-cognate element. We suspect that those PCR results are related to interactions of the DDB2 complex with other proteins binding to the SEMA3A promoter. We also carried out ChIP experiments with XRCC5 antibody in cells expressing control or DDB2 shRNA. Clearly, XRCC5 binds to the SEMA3A promoter, and that binding depends on DDB2, as there were significant losses of the fold enrichment in DDB2 shRNA–expressing cells (Figure 6C). Primer set 1, which covers the DDB2-cognate element at −284, also showed a DDB2-dependent interaction of XRCC5 with the SEMA3A promoter. XRCC5 ChIP also showed binding at other nonoverlapping sites in the promoter, which likely reflects interactions with a different set of transcription factors binding to the SEMA3A promoter. Our model is that after recruitment of the DDB2–XRCC5 onto the promoter of SEMA3A, they interact with other factors bound to the promoter to stimulate transcription. It is also possible that, in addition to the DDB2-cognate element, other DNA-bound transcription factors or a specifically modified histone H3 recruit the DDB2–XRCC5/6 complex onto the promoter of SEMA3A to activate transcription.
FIGURE 6:
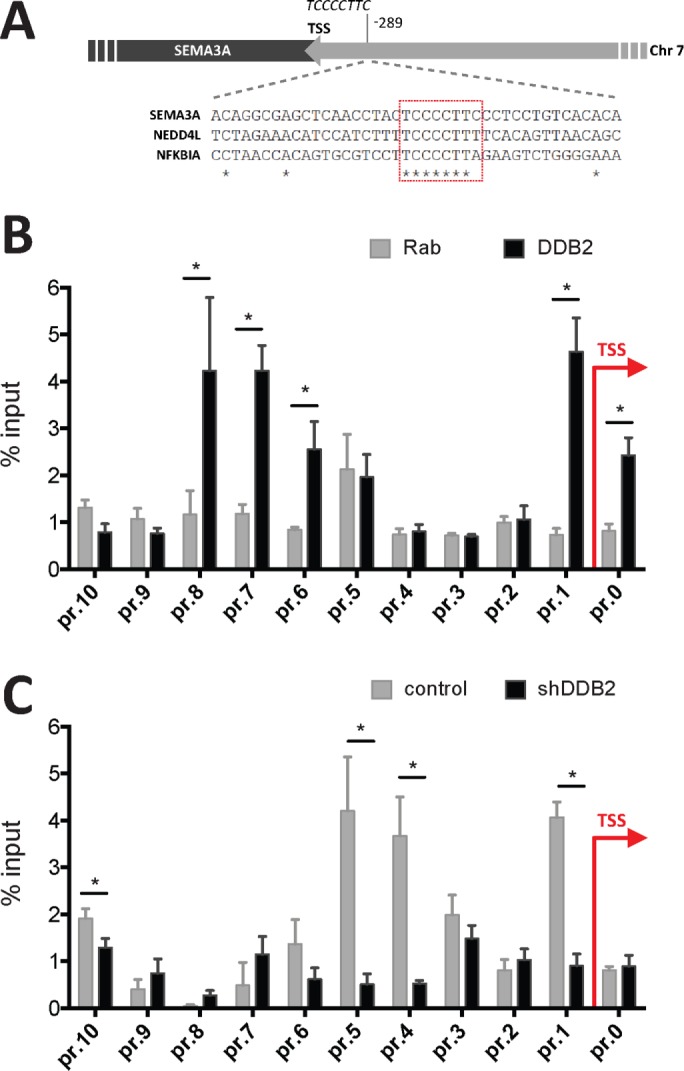
DDB2 and DDB2-dependent XRCC5 binding on the SEMA3A promoter. (A) Diagram outlining the human SEMA3A promoter, which includes a DDB2-cognate element at position 289 base pairs upstream of the transcription start site (TSS). Forty-one nucleotide sequences spanning the DDB2-cognate elements in the SEMA3A, NEDD4L, and NFKBIA promoters were aligned using Clustal Omega. (B) Chromatin from HCT116 cells was immunoprecipitated using normal rabbit IgG (Rab) or an anti-DDB2 antibody. The levels of immunoprecipitated DNA at 11 sites (pr.0 to pr.10) along the proximal SEMA3A promoter are expressed as percentage of input DNA (*p < 0.05, t test). (C) Chromatin from control and shDDB2 HCT116 cells was immunoprecipitated using an anti-XRCC5 antibody. The levels of immunoprecipitated DNA at 11 sites along the proximal SEMA3A promoter from the two cell lines are plotted as percentage of input DNA (*p < 0.05, t test).
DISCUSSION
Studies in HeLa cells identified strong interactions of DDB2 with the subunits of the Cop9 signalosome (Groisman et al., 2003). Surprisingly, in our experiments with HCT116 cells, we did not detect the Cop9 signalosome as a major DDB2-interacting protein. It is likely that DDB2 possess cell type–specific partners. In colon cancer cells, we detected XRCC5 and histone H3 as strong binding partners of DDB2 (Figure 1C). The interaction with histone H3 is not surprising, given previous studies showing that DDB2 induces methylation of histone H3 to repress transcription (Roy et al., 2010, 2013; Zhao et al., 2015). It is possible that the interaction of DDB2 with histone H3 bridges the association of the XRCC5/6 proteins with chromatins. Previous studies also indicated interactions of DDB2 with histones. For example, the UV-RING1B complex, which includes DDB2, RING1B, DDB1, and Cul4b, is important for monoubiquitylation of histone H2A at the K119 residues, which is significant for nucleotide excision repair after UV irradiation (Gracheva et al., 2016). The UV-RING1B complex also recruits EZH2 and other PRC complex proteins to ubiquitylate H2A, followed by trimethylation of the histone H3 at the K27 residue, which is associated with a suppressive chromatin (Hu et al., 2012). In that regard, our observation on an association with JARID2 is interesting because JARID2 has been shown to be a functional partner of the PRC2 complex (Son et al., 2013; Kaneko et al., 2014). In addition, a recent study in ovarian cancer cells detected an interaction of DDB2 with the PRC2 complex protein EZH2 (Zhao et al., 2015). However, it is noteworthy that immunoprecipitates with the XRCC5 antibody contained DDB2 and DDB1 but not Cul4 (Figure 2). TBX2 is a transcription factor that inhibits expression of the p16Ink4a and the p14Arf tumor suppressors to block cellular senescence and promotes EMT (Jacobs et al., 2000; Wang et al., 2012). Our previous studies demonstrated exactly opposite roles of DDB2 with regard to senescence and EMT (Roy et al., 2013). Therefore it is possible that DDB2 regulates TBX2, and that regulation is important in the mechanisms by which DDB2 suppresses tumor development and progression.
XRCC5/6, like DDB2, are multifunctional proteins involved in DSBR (Milne et al., 1996), transcription (Willis et al., 2002; Sucharov et al., 2004; Kim et al., 2008; Nolens et al., 2009; Xiao et al., 2015), and telomere maintenance (Indiviglio and Bertuch, 2009). XRCC5/6 localizes to DSB sites after DNA damage (Britton et al., 2013). We provided evidence that chromatin association of XRCC5 after DSB does not involve DDB2. DDB2 was shown to be involved in the activation of ATM/ATR after UV irradiation (Ray et al., 2013). However, after DSB induced by phleomycin, we did not see evidence for a role of DDB2 in the activation of ATM, as judged by H2AX phosphorylation (Figure 3). Therefore it is likely that the checkpoint activation function of DDB2 is restricted to certain types of DNA damage—for example, UV irradiation. Moreover, our results imply that the DDB2–XRCC5 interaction may have a role in cellular processes other than DNA damage response.
The major finding of this study is that in colon cancer cells, chromatin association of XRCC5/6, in the absence of DNA damage, depends on DDB2. This was observed in colon cancer cells HCT116, SW480, and SW620. However, in HeLa cells, XRCC5/6 accumulation on the chromatin was not affected upon knockdown of DDB2 (Supplemental Figure S3). In addition, in HeLa cells, the DDB2/XRCC5 interaction was detected only after UV irradiation (Takedachi et al., 2010). Therefore it appears that in HeLa cells, other mechanisms are involved in chromatin recruitment of XRCC5/6. Immunoprecipitations with XRCC5 antibody also demonstrated coimmunoprecipitation of DDB2 and DDB1 but not CUL4. Of interest, coprecipitation of DDB1 was detected in the absence of DDB2. Therefore DDB1 plays an important role in the interaction between DDB2 and XRCC5/6. That is also consistent with the observations that naturally occurring mutants of DDB2 that fail to import DDB1 in the nucleus are impaired in binding to XRCC5/6 (Figure 2).
Our results provide evidence for a transcription function of the DDB2–XRCC5/6 interaction. DDB2 stimulates expression of the SEMA3A gene. We show that depletion of XRCC5 inhibited the expression of the SEMA3A gene, implying that SEMA3A is stimulated by XRCC5/6. In that experiment, we did not detect any effect of XRCC5 depletion on the repression function of DDB2. For example, depletion of DDB2 increased transcription of VEGFA. Depletion of XRCC5 did not increase the RNA levels of VEGFA. Therefore it is possible that the DDB2–XRCC5/6 interaction is not involved in the transcription repression function of DDB2. That would be consistent with the observation that XRCC5 does not associate with CUL4A, which is involved in the repression of VEGFA and other genes repressed by DDB2 (Roy et al., 2013). We showed that DDB2 recruits XRCC5 onto the promoter of SEMA3A, whose expression in colon cancer cells depends on both DDB2 and XRCC5. Therefore the interaction between DDB2 and XRCC5/6 is related to the transcriptional activation function of DDB2.
Previous studies on the transcriptional activation function of XRCC5/6 indicated cooperation with the transcriptional coactivator CBP (Xiao et al., 2015). In that regard, it is interesting that DDB2 also associates with CBP (Datta et al., 2001). It is noteworthy that the XRCC5/6 proteins also have been implicated in elongation steps of mRNA synthesis (Mo and Dynan, 2002). In addition, XRCC5/6 were shown to participate as corepressors in steroid hormone–regulated transcription (Jeyakumar et al., 2007). In those studies, DNA-PKC was shown to activate histone deacetylases to inhibit transcription. XRCC5/6-associated DNA-PKC also is involved in the inactivation of human GCN5 histone acetyltransferase by phosphorylation (Barlev et al., 1998). Therefore the observation that DDB2 does not associate with DNA-PKC further supports the notion that the DDB2–XRCC5/6 interaction is related to transcriptional activation and not repression. However, because chromatin association of XRCC5/6 in colon cancer cells largely depends on DDB2, we cannot rule out other, indirect functions of the DDB2-mediated localization of XRCC5/6 onto chromatin.
MATERIALS AND METHODS
Plasmids
pOZ-FH-N and pOZ-FH-N-DDB2 were a kind gift from Yoshihiro Nakatani (Dana-Farber Cancer Institute, Boston, MA). FLAG-T7-DDB2 WT, 2RO, and 82TO constructs were generated starting from T7-DDB2 expression constructs described previously (Shiyanov et al., 1999a). Briefly, the constructs were HindIII and KpnI double digested and then ligated with a double-stranded DNA fragment obtained by annealing the oligonucleotides 5′-AAG CTT ATG GAC TAC AAG GAC GAC GAT GAC AAG CTC GGT ACC-3′ and 5′-CGA GCT TGT CAT CGT CGT CCT TGT AGT CCA TA-3′.
Cell culture and transfection
HCT116 cells were grown in DMEM containing 10% fetal bovine serum (FBS) and 1% penicillin–streptomycin at 37°C and 5% CO2. SW480 and SW620 were grown in RPMI containing 10% FBS and 1% penicillin–streptomycin at 37°C and 5% CO2. HCT116 cells stably expressing FLAG-HA-tagged DDB2 were established using a retroviral vector (Groisman et al., 2003). HCT116 cells stably expressing shRNA targeting DDB2 were described previously (Roy et al., 2013). Cell transfection experiments were conducted using Lipofectamine2000 (Thermo Fisher Scientific, Waltham, MA), following the manufacturer’s recommendations. Transient knockdown of XRCC5 was carried out by transfecting ON-TARGET Plus XRCC5 siRNA SMARTpool (GE Dharmacon, Lafayette, CO) or nontargeting siRNA SMARTpool (Dharmacon) into HCT116 at a final concentration of 150 nM. Cells were harvested 72 h after siRNA transfection.
Antibodies
We used the following commercial antibodies: JARID2 (NB100-2214; Novus Biologicals, Littleton, CO), CYLD (sc-28211; Santa Cruz Biotechnology, Santa Cruz, CA), XRCC5 (sc-9034; Santa Cruz Biotechnology), XRCC6 (ALS14031; Abgent, San Diego, CA), TBX2 (H00006909-M01; Novus Biologicals), H3 (4499; Cell Signaling, Danvers, MA), DDB2 (Western blotting: 5416; Cell Signaling; ChIP: sc-25368; Santa Cruz Biotechnology), DNA-PKcs (NBP2-22128; Novus Biologicals), SEMA3A (AF1250; Novus Biologicals), actin (A4700; Sigma-Aldrich, St. Louis, MO), and y-H2AX (9718; Cell Signaling). DDB1 and CUL4A antibodies were described previously (Nag et al., 2004). Horseradish peroxidase–conjugated secondary antibodies were purchased from Bio-Rad.
Nuclei extraction and coimmunoprecipitation
Nuclear extracts for coIP were prepared starting from 100 million cells that were harvested by scraping. Cell pellets were resuspended in hypotonic buffer containing 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), pH 7.4, 10 mM KCl, 1.5 mM MgCl2, and 0.5 mM ethylene glycol tetraacetic acid (EGTA) supplemented with 1 mM phenylmethylsulfonyl fluoride (PMSF) and 1× protease inhibitor cocktail (Roche, Basel, Switzerland) and then lysed using a 7-ml Glass Tenbroek homogenizer (Wheaton, Millville, NJ). Cell lysates were spun through a sucrose cushion (15 mM Tris-HCl, pH 7.5, 15 mM NaCl, 60 mM KCl, 5 mM MgCl2, 0.1 mM EGTA, and 1.2 M sucrose supplemented with 1 mM PMSF) at 10,000 × g for 20 min at 4°C. Nuclei pellets were extracted in extraction buffer containing 50 mM Tris-HCl, pH 7.4, 320 mM NaCl, 1.5 mM MgCl2, 1 mM EGTA, 10% glycerol, and 0.25% Triton X-100, supplemented with 1× protease inhibitor cocktail, 1 mM NaF, and 1 mM sodium orthovanadate. Samples were spun at 16,000 × g for 20 min at 4°C, and supernatants were collected, diluted with 1.2 volumes of dilution buffer (50 mM Tris-HCl, pH 7.4, 8.34 mM NaCl, 1.5 mM MgCl2, 1 mM EGTA, and 0.15% Triton X-100 supplemented with 1 mM PMSF, 1 mM NaF, and 1 mM sodium orthovanadate), and saved as nuclear extracts. CoIP was performed immediately by rotating 5-mg nuclear extracts and 100 μL of anti-FLAG M2 agarose bead slurry (Sigma-Aldrich) for 12 h, followed by four washes in IP buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.5 mM EGTA, and 0.05% Triton X-100 supplemented with 1 mM PMSF). Beads were eluted for 30 min with 250 μl of 0.15 mg/ml 3× FLAG peptide (Sigma-Aldrich) in IP buffer. For dual-round coIPs, supernatants were further rotated for 5 h at 4°C with 15 μg of anti-HA monoclonal antibody (Roche). Protein G–Sepharose beads were added, and samples were further rocked for 1 h. After three washes with IP buffer, beads were eluted with 1× Laemmli sample buffer (Western blotting) or in 50 mM Tris-HCl, 500 mM NaCl, 0.1% SDS, and 1 mM PMSF (MS/MS identification). Coimmunoprecipitates were separated on a 10% SDS–PAGE, and then gels were silver stained, and selected bands were excised from the gel for MS/MS identification.
Conditioned medium preparation
Cells were grown for 48 h at 37°C in DMEM, high glucose, HEPES, and no Phenol Red (Thermo Fisher Scientific). Cell supernatant was filtered through a Millex-GP Syringe Filter Unit with 0.22-μm cutoff (EMD Millipore), spun down at 8000 × g to remove insoluble material, and then 10-fold concentrated using an Amicon Ultra-4 10-kDa filter unit (EMD Millipore, Billerica, MA). Concentrated conditioned medium was diluted in 1 volume of 4× Laemmli sample buffer (120 mM Tris-HCl, pH 6.8, 40% glycerol, 8% SDS, 60 mM dithiothreitol) and stored at −20°C.
Immunofluorescence
Immunofluorescence experiments were performed on cells grown on 12-mm coverslips for 48 h after seeding. Cells were treated with 200 μg/ml phleomycin (Sigma-Aldrich) for 2 h, medium was replaced, and cells were further incubated at 37°C for the indicated times. Cells were washed with PBS or CSK buffer (10 mM 1,4-piperazinediethanesulfonic acid, pH 6.8, 100 mM NaCl, 3 mM MgCl2, 300 mM glycerol) supplemented with 0.7% IGEPAL CA630 and 0.1 mg/ml RNase A (Thermo Fisher Scientific) for 3 min at room temperature (Britton et al., 2013). After two washes with ice-cold PBS, cells were cross-linked for 15 min with 4% paraformaldehyde at room temperature. Coverslips were blocked for 1 h at room temperature in blocking buffer (5% BSA in PBS). XRCC5 antibody was diluted 1:500 in blocking buffer and incubated for 2 h at room temperature. Anti-rabbit secondary antibody Alexa Fluor 488 (Thermo Fisher Scientific) was then added for 1 h at room temperature. Coverslips were counterstained with Hoechst 33342 (Thermo Fisher Scientific) and then mounted onto glass slides using ProLong Gold Antifade reagent (Thermo Fisher Scientific). Images were acquired using a Zeiss LSM 510 microscope.
Protein extraction experiments and Western blotting
For preextraction experiments, cells were seeded in six-well multiwall plates at a density of 400,000 cells/well. At 48 h later, cells were washed twice with PBS and then incubated for 5 min at room temperature with PBS, CSK-T (CSK buffer supplemented with 0.7% Triton X-100 and 0.5 mM PMSF), or CSK-T RNase (CSK-T supplemented with 0.1 mg/ml RNase A). Wells were further washed with 1 ml of ice-cold PBS, and 1× Laemmli sample buffer was then added; samples were boiled at 95°C for 5 min and loaded on gels. For sequential fractionation experiments, 2.0 M cells were seeded in a 10-cm plate. At 48 h later, cells were washed with PBS and harvested by scraping. Samples were spun at 800 × g for 5 min at 4°C. Pellets were resuspended in 500 μl of CSK-T buffer and incubated on ice for 5 min. Samples were centrifuged at 3000 × g for 6 min at 4°C, supernatants were saved as S1 fractions, and pellets were washed twice with ice-cold PBS and then resuspended in 500 μl of CSK-T RNase buffer. Samples were incubated at room temperature for 5 min and then centrifuged at 6000 × g for 6 min at 4°C. Supernatants were collected as S2 fractions, and pellets were washed twice with PBS and then resuspended in 500 μl of nuclease-containing CSK buffer (CSK-T supplemented with Benzonase [EMD Millipore] and 0.1% SDS). Samples were rocked at room temperature for 10 min and then centrifuged at 16,000 × g for 10 min at 4°C. Supernatants were collected as S3 fractions, and pellets were washed once with PBS, boiled for 5 min at 95°C in 1× Laemmli sample buffer, and saved as P3 fractions. Whole-cell lysates were prepared by boiling cell pellets in 1× Laemmli sample buffer for 5 min at 95°C. Samples were separated on 12% SDS–PAGE (unless otherwise stated), and Western blotting was performed using standard procedures and the antibodies described diluted in 5% nonfat dry milk in Tris-buffered saline supplemented with 0.1% Tween 20 (Sigma-Aldrich).
RNA extraction and quantitative PCR
Total RNA was extracted using TRIzol (Thermo Fisher Scientific) and following manufacturer’s instructions. cDNA was synthesized using an iScript cDNA Synthesis Kit (Bio-Rad), and quantitative PCR (qPCR) was performed using iTaq Universal SYBR Green Supermix (Bio-Rad) and a CFX96 system (Bio-Rad). Oligonucleotides used as primers are described in Supplemental Table S1.
Chromatin immunoprecipitation
Chromatin immunoprecipitation experiments were performed as previously described (Roy et al., 2013). De-cross-linked chromatin was analyzed by qPCR, using the primers described in Supplemental Table S2.
Supplementary Material
Acknowledgments
We thank Vania Vidimar for insightful discussions and editorial assistance in the writing of the manuscript. The work was supported by National Institutes of Health/National Cancer Institute Grants CA156164 and CA77637 to P.R. and S.B. P.R. is also supported by VA Merit Grant BX000131.
Abbreviations used:
- CUL4
Cullin 4
- DDB1
Damaged-DNA binding protein 1
- DDB2
Damaged-DNA binding protein 2
- DNA-PKcs
DNA-dependent protein kinase catalytic subunit
- NHEJ
nonhomologous end joining
- SEMA3A
semaphorin 3A
- VEGFA
vascular endothelial growth factor A
- XRCC5/6
x-ray repair cross complementing 5 and 6
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E16-08-0573) on November 9, 2016.
REFERENCES
- Ajmani AK, Satoh M, Reap E, Cohen PL, Reeves WH. Absence of autoantigen ku in mature human neutrophils and human promyelocytic leukemia line (Hl-60) cells and lymphocytes undergoing apoptosis. J Exp Med. 1995;181:2049–2058. doi: 10.1084/jem.181.6.2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlev NA, Poltoratsky V, Owen-Hughes T, Ying C, Liu L, Workman JL, Berger SL. Repression of GCN5 histone acetyltransferase activity via bromodomain-mediated binding and phosphorylation by the Ku-DNA-dependent protein kinase complex. Mol Cell Biol. 1998;18:1349–1358. doi: 10.1128/mcb.18.3.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britton S, Coates J, Jackson SP. A new method for high-resolution imaging of Ku foci to decipher mechanisms of DNA double-strand break repair. J Cell Biol. 2013;202:579–595. doi: 10.1083/jcb.201303073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta A, Bagchi S, Nag A, Shiyanov P, Adami GR, Yoon T, Raychaudhuri P. The p48 subunit of the damaged-DNA binding protein DDB associates with the CBP/p300 family of histone acetyltransferase. Mutat Res. 2001;486:89–97. doi: 10.1016/s0921-8777(01)00082-9. [DOI] [PubMed] [Google Scholar]
- Ennen M, Klotz R, Touche N, Pinel S, Barbieux C, Besancenot V, Brunner E, Thiebaut D, Jung AC, Ledrappier S, et al. DDB2: a novel regulator of NF-kappa B and breast tumor invasion. Cancer Res. 2013;73:5040–5052. doi: 10.1158/0008-5472.CAN-12-3655. [DOI] [PubMed] [Google Scholar]
- Gracheva E, Chitale S, Wilhelm T, Rapp A, Byrne J, Stadler J, Medina R, Cardoso MC, Richly H. ZRF1 mediates remodeling of E3 ligases at DNA lesion sites during nucleotide excision repair. J Cell Biol. 2016;213:185–200. doi: 10.1083/jcb.201506099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groisman R, Polanowska J, Kuraoka I, Sawada J, Saijo M, Drapkin R, Kisselev AF, Tanaka K, Nakatani Y. The ubiquitin ligase activity in the DDB2 and CSA complexes is differentially regulated by the COP9 signalosome in response to DNA damage. Cell. 2003;113:357–367. doi: 10.1016/s0092-8674(03)00316-7. [DOI] [PubMed] [Google Scholar]
- Higa LA, Wu M, Ye T, Kobayashi R, Sun H, Zhang H. CUL4-DDB1 ubiquitin ligase interacts with multiple WD40-repeat proteins and regulates histone methylation. Nat Cell Biol. 2006;8:1277-U1248. doi: 10.1038/ncb1490. [DOI] [PubMed] [Google Scholar]
- Hu HL, Yang Y, Ji QH, Zhao W, Jiang BC, Liu RQ, Yuan JP, Liu Q, Li X, Zou YX, et al. CRL4B catalyzes H2AK119 monoubiquitination and coordinates with PRC2 to promote yumorigenesis. Cancer Cell. 2012;22:781–795. doi: 10.1016/j.ccr.2012.10.024. [DOI] [PubMed] [Google Scholar]
- Indiviglio SM, Bertuch AA. Ku’s essential role in keeping telomeres intact. Proc Natl Acad Sci USA. 2009;106:12217–12218. doi: 10.1073/pnas.0906427106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs JJL, Keblusek P, Robanus-Maandag E, Kristel P, Lingbeek M, Nederlof PM, van Welsem T, van de Vijver MJ, Koh EY, Daley GQ, van Lohuizen M. Senescence bypass screen identifies TBX2, which represses Cdkn2a (p19(ARF)) and is amplified in a subset of human breast cancers. Nat Genet. 2000;26:291–299. doi: 10.1038/81583. [DOI] [PubMed] [Google Scholar]
- Jeyakumar M, Liu XF, Erdjument-Bromage H, Tempst P, Bagchi MK. Phosphorylation of thyroid hormone receptor-associated nuclear receptor corepressor holocomplex by the DNA-dependent protein kinase enhances its histone deacetylase activity. J Biol Chem. 2007;282:9312–9322. doi: 10.1074/jbc.M609009200. [DOI] [PubMed] [Google Scholar]
- Kaneko S, Bonasio R, Saldana-Meyer R, Yoshida T, Son J, Nishino K, Umezawa A, Reinberg D. Interactions between JARID2 and noncoding RNAs regulate PRC2 recruitment to chromatin. Mol Cell. 2014;53:290–300. doi: 10.1016/j.molcel.2013.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapetanaki MG, Guerrero-Santoro J, Bisi DC, Hsieh CL, Rapic-Otrin V, Levine AS. The DDB1-CUL4ADDB2 ubiquitin ligase is deficient in xeroderma pigmentosum group E and targets histone H2A at UV-damaged DNA sites. Proc Natl Acad Sci USA. 2006;103:2588–2593. doi: 10.1073/pnas.0511160103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Li K, Lieu C, Tong S, Kawai S, Fukutomi T, Zhou Y, Wands J, Li J. Expression of apolipoprotein C-IV is regulated by Ku antigen/peroxisome proliferator-activated receptor gamma complex and correlates with liver steatosis. J Hepatol. 2008;49:787–798. doi: 10.1016/j.jhep.2008.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leibovitz A, Stinson JC, Mccombs WB, Mccoy CE, Mazur KC, Mabry ND. Classification of human colorectal adenocarcinoma cell lines. Cancer Res. 1976;36:4562–4569. [PubMed] [Google Scholar]
- Luijsterburg MS, Lindh M, Acs K, Vrouwe MG, Pines A, van Attikum H, Mullenders LH, Dantuma NP. DDB2 promotes chromatin decondensation at UV-induced DNA damage. J Cell Biol. 2012;197:267–281. doi: 10.1083/jcb.201106074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maione F, Capano S, Regano D, Zentilin L, Giacca M, Casanovas O, Bussolino F, Serini G, Giraudo E. Semaphorin 3A overcomes cancer hypoxia and metastatic dissemination induced by antiangiogenic treatment in mice. J Clin Invest. 2012;122:1832–1848. doi: 10.1172/JCI58976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maione F, Molla F, Meda C, Latini R, Zentilin L, Giacca M, Seano G, Serini G, Bussolino F, Giraudo E. Semaphorin 3A is an endogenous angiogenesis inhibitor that blocks tumor growth and normalizes tumor vasculature in transgenic mouse models. J Clin Invest. 2009;119:3356–3372. doi: 10.1172/JCI36308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne GT, Jin SF, Shannon KB, Weaver DT. Mutations in two Ku homologs define a DNA end-joining repair pathway in Saccharomyces cerevisiae. Mol Cell Biol. 1996;16:4189–4198. doi: 10.1128/mcb.16.8.4189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minig V, Kattan Z, van Beeumen J, Brunner E, Becuwe P. Identification of DDB2 protein as a transcriptional regulator of constitutive SOD2 gene expression in human breast cancer cells. J Biol Chem. 2009;284:14165–14176. doi: 10.1074/jbc.M808208200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo XM, Dynan WS. Subnuclear localization of Ku protein: functional association with RNA polymerase II elongation sites. Mol Cell Biol. 2002;22:8088–8099. doi: 10.1128/MCB.22.22.8088-8099.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nag A, Bagchi S, Raychaudhuri P. Cul4A physically associates with MDM2 and participates in the proteolysis of p53. Cancer Res. 2004;64:8152–8155. doi: 10.1158/0008-5472.CAN-04-2598. [DOI] [PubMed] [Google Scholar]
- Nichols AF, Ong P, Linn S. Mutations specific to the xeroderma pigmentosum group E Ddb- phenotype. J Biol Chem. 1996;271:24317–24320. doi: 10.1074/jbc.271.40.24317. [DOI] [PubMed] [Google Scholar]
- Nolens G, Pignon JC, Koopmansch B, Elmoualij B, Zorzi W, De Pauw E, Winkler R. Ku proteins interact with activator protein-2 transcription factors and contribute to ERBB2 overexpression in breast cancer cell lines. Breast Cancer Res. 2009;11:R83–00. doi: 10.1186/bcr2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pines A, Vrouwe MG, Marteijn JA, Typas D, Luijsterburg MS, Cansoy M, Hensbergen P, Deelder A, de Groot A, Matsumoto S, et al. PARP1 promotes nucleotide excision repair through DDB2 stabilization and recruitment of ALC1. J Cell Biol. 2012;199:235–249. doi: 10.1083/jcb.201112132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray A, Milum K, Battu A, Wani G, Wani AA. NER initiation factors, DDB2 and XPC, regulate UV radiation response by recruiting ATR and ATM kinases to DNA damage sites. DNA Repair. 2013;12:273–283. doi: 10.1016/j.dnarep.2013.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy N, Bommi PV, Bhat UG, Bhattacharjee S, Elangovan I, Li J, Patra KC, Kopanja D, Blunier A, Benya R, et al. DDB2 suppresses epithelial-to-mesenchymal transition in colon cancer. Cancer Res. 2013;73:3771–3782. doi: 10.1158/0008-5472.CAN-12-4069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy N, Stoyanova T, Dominguez-Brauer C, Park HJ, Bagchi S, Raychaudhuri P. DDB2, an essential mediator of premature senescence. Mol Cell Biol. 2010;30:2681–2692. doi: 10.1128/MCB.01480-09. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Shiyanov P, Hayes SA, Donepudi M, Nichols AF, Linn S, Slagle BL, Raychaudhuri P. The naturally occurring mutants of DDB are impaired in stimulating nuclear import of the p125 subunit and E2F1-activated transcription. Mol Cell Biol. 1999a;19:4935–4943. doi: 10.1128/mcb.19.7.4935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiyanov P, Nag A, Raychaudhuri P. Cullin 4A associates with the UV-damaged DNA-binding protein DDB. J Biol Chem. 1999b;274:35309–35312. doi: 10.1074/jbc.274.50.35309. [DOI] [PubMed] [Google Scholar]
- Son J, Shen SS, Margueron R, Reinberg D. Nucleosome-binding activities within JARID2 and EZH1 regulate the function of PRC2 on chromatin. Genes Dev. 2013;27:2663–2677. doi: 10.1101/gad.225888.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sucharov CC, Helmke SM, Langer SJ, Perryman MB, Bristow M, Leinwand L. The Ku protein complex interacts with YY1, is up-regulated in human heart failure, and represses alpha myosin heavy-chain gene expression. Mol Cell Biol. 2004;24:8705–8715. doi: 10.1128/MCB.24.19.8705-8715.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takedachi A, Saijo M, Tanaka K. DDB2 complex-mediated ubiquitylation around DNA damage is oppositely regulated by XPC and Ku and contributes to the recruitment of XPA. Mol Cell Biol. 2010;30:2708–2723. doi: 10.1128/MCB.01460-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J, Chu G. Xeroderma pigmentosum complementation group E and UV-damaged DNA-binding protein. DNA Repair (Amst) 2002;1:601–616. doi: 10.1016/s1568-7864(02)00052-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Lindley LE, Fernandez-Vega V, Rieger ME, Sims AH, Briegel KJ. The T box transcription factor TBX2 promotes epithelial-mesenchymal transition and invasion of normal and malignant breast epithelial cells. PLoS One. 2012;7:e41355. doi: 10.1371/journal.pone.0041355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Zhai L, Xu J, Joo HY, Jackson S, Erdjument-Bromage H, Tempst P, Xiong Y, Zhang Y. Histone H3 and H4 ubiquitylation by the CUL4-DDB-ROC1 ubiquitin ligase facilitates cellular response to DNA damage. Mol Cell. 2006;22:383–394. doi: 10.1016/j.molcel.2006.03.035. [DOI] [PubMed] [Google Scholar]
- Wang QE, Zhu Q, Wani G, Chen J, Wani AA. UV radiation-induced XPC translocation within chromatin is mediated by damaged-DNA binding protein, DDB2. Carcinogenesis. 2004;25:1033–1043. doi: 10.1093/carcin/bgh085. [DOI] [PubMed] [Google Scholar]
- Weaver DT. Regulation and repair of double-strand DNA breaks. Crit Rev Eukaryot Gene Expr. 1996;6:345–375. doi: 10.1615/critreveukargeneexpr.v6.i4.20. [DOI] [PubMed] [Google Scholar]
- Willis DM, Loewy AP, Charlton-Kachigian N, Shao JS, Ornitz DM, Towler DA. Regulation of osteocalcin gene expression by a novel Ku antigen transcription factor complex. J Biol Chem. 2002;277:37280–37291. doi: 10.1074/jbc.M206482200. [DOI] [PubMed] [Google Scholar]
- Xiao Y, Wang JS, Qin Y, Xuan Y, Jia YL, Hu WX, Yu WD, Dai M, Li ZL, Yi CH, et al. Ku80 cooperates with CBP to promote COX-2 expression and tumor growth. Oncotarget. 2015;6:8046–8061. doi: 10.18632/oncotarget.3508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao R, Cui TT, Han CH, Zhang XL, He JS, Srivastava AK, Yu JH, Wani AA, Wang QE. DDB2 modulates TGF-beta signal transduction in human ovarian cancer cells by downregulating NEDD4L. Nucleic Acids Res. 2015;43:7838–7849. doi: 10.1093/nar/gkv667. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.