

Endocytosis of VE-cadherin in response to the Kaposi sarcoma E3 ubiquitin ligase K5 is dependent on two membrane-proximal lysine residues but independent of a constitutive endocytosis motif. p120-catenin blocks endocytosis mediated by both motifs, demonstrating that p120 is a master regulator of multiple context-dependent endocytic signals.
Abstract
Vascular endothelial (VE)-cadherin undergoes constitutive internalization driven by a unique endocytic motif that also serves as a p120-catenin (p120) binding site. p120 binding masks the motif, stabilizing the cadherin at cell junctions. This mechanism allows constitutive VE-cadherin endocytosis and recycling to contribute to adherens junction dynamics without resulting in junction disassembly. Here we identify an additional motif that drives VE-cadherin endocytosis and pathological junction disassembly associated with the endothelial-derived tumor Kaposi sarcoma. Human herpesvirus 8, which causes Kaposi sarcoma, expresses the MARCH family ubiquitin ligase K5. We report that K5 targets two membrane-proximal VE-cadherin lysine residues for ubiquitination, driving endocytosis and down-regulation of the cadherin. K5-induced VE-cadherin endocytosis does not require the constitutive endocytic motif. However, K5-induced VE-cadherin endocytosis is associated with displacement of p120 from the cadherin, and p120 protects VE-cadherin from K5. Thus multiple context-dependent signals drive VE-cadherin endocytosis, but p120 binding to the cadherin juxtamembrane domain acts as a master regulator guarding cadherin stability.
INTRODUCTION
Proper vascular function requires a balance between stability and plasticity of endothelial cell–cell contacts. Adhesion must be tight enough to resist vascular leak yet also flexible enough to permit the cellular rearrangements necessary for new vessel formation during development and wound healing. Endothelial cell–cell adhesion is a dynamic and tightly regulated process, but the mechanisms controlling endothelial adhesion remain incompletely understood (Vincent et al., 2004; Giannotta et al., 2013). Because disruption of endothelial adhesion contributes to a wide variety of diseases, especially through excessive inflammation and facilitation of cancer metastasis, elucidating the basic cellular processes that determine the balance of endothelial adhesion is an important goal.
Endothelial cell–cell adhesion is mediated through the adherens junction complex. Vascular endothelial (VE)-cadherin, a member of the classical cadherin family and a principal adhesion molecule in the endothelium, joins adjacent cells through calcium-dependent homotypic trans interactions (Harris and Tepass, 2010; Ishiyama and Ikura, 2012; Dejana and Orsenigo, 2013). As with other classical cadherins, the cytoplasmic domain of VE-cadherin binds to armadillo family proteins called catenins, which perform important structural and regulatory functions. β-Catenin binds to the C-terminal catenin-binding domain of VE-cadherin and, along with α-catenin and other proteins, links the cadherin to the actin cytoskeleton, mechanically coupling adjacent cells (Yamada et al., 2005; Taguchi et al., 2011; Desai et al., 2013). The juxtamembrane domain of VE-cadherin binds to p120-catenin (p120), which stabilizes cadherins at the adherens junction. In the absence of p120 binding, cadherins are rapidly endocytosed and degraded (Davis et al., 2003; Xiao et al., 2003a, b; Miyashita and Ozawa, 2007). Thus p120 binding to cadherins can function as a regulator of adherens junction stability (Nanes and Kowalczyk, 2012).
In the endothelium, p120 plays a particularly important role balancing the stability and flexibility of cell adhesion. The VE-cadherin juxtamembrane domain contains a dual-function motif that alternately serves as a p120-binding site or as an endocytic signal. p120 binding physically masks the endocytic signal, blocking its function and stabilizing the cadherin (Nanes et al., 2012). Endothelial-specific deletion of p120 in mice results in hemorrhaging, vascular patterning defects, and embryonic lethality, underscoring the importance of p120 for maintenance of vessel stability (Oas et al., 2010). However, p120-modulation of VE-cadherin endocytosis allows a level of constitutive internalization of the cadherin. Constitutive VE-cadherin endocytosis confers plasticity to endothelial cell–cell junctions, and expression of a VE-cadherin mutant resistant to constitutive endocytosis inhibits collective migration of endothelial cells (Nanes et al., 2012). Because endothelial cell migration is an important component of angiogenesis, the junctional plasticity derived from constitutive VE-cadherin endocytosis likely has important physiologic roles.
If p120-modulated constitutive endocytosis of VE-cadherin is responsible for balancing stability and flexibility of endothelial adhesion, what causes the disruption of this mechanism in diseases associated with inappropriate loss of endothelial adhesion? One such disease is Kaposi sarcoma, an endothelial-derived tumor characterized by aberrant angiogenesis and leaky, slit-like vessels (Kaposi, 1872; Antman and Chang, 2000; Uldrick and Whitby, 2011). Kaposi sarcoma is caused by human herpesvirus 8 (HHV-8), which is always found within the lesions (Chang et al., 1994). Vascular permeability induced by HHV-8 likely occurs through multiple mechanisms, including expression of a viral G-protein–coupled receptor that activates Rac (Dwyer et al., 2011) and entry of the viral capsid into endothelial cells (Qian et al., 2008). In addition, HHV-8 encodes two membrane-associated really interesting new gene–CH (MARCH)–family ubiquitin ligases, K3 and K5, which were originally identified to target host-cell mediators of immune function, such as MHC class I, for ubiquitination, endocytosis, and down-regulation (Coscoy and Ganem, 2000; Ishido et al., 2000). K5 also targets several other endothelial cell surface proteins for ubiquitination, including PECAM-1 and VE-cadherin (Mansouri et al., 2006, 2008). Of interest, K5 expression increases permeability of endothelial monolayers (Mansouri et al., 2008), potentially linking ubiquitination of VE-cadherin to the aberrant angiogenesis and leaky vasculature seen in Kaposi sarcoma lesions.
The results presented here demonstrate that K5 disrupts endothelial cell–cell junctions by overriding the normal cellular regulation of VE-cadherin endocytosis. K5 targets VE-cadherin for ubiquitination, displaces p120 from the VE-cadherin juxtamembrane domain, and induces VE-cadherin endocytosis. Furthermore, we identify two membrane-proximal lysines within the p120-binding site as the specific VE-cadherin residues targeted by K5 and demonstrate that p120 binding can protect VE-cadherin from K5-induced down-regulation. Of interest, a VE-cadherin mutant resistant to constitutive endocytosis is still susceptible to down-regulation by K5. Thus, even though different endocytic signals drive constitutive and K5-induced VE-cadherin internalization, p120 maintains a key role as the guardian of VE-cadherin stability through protection of the cadherin juxtamembrane domain.
RESULTS
K5 targets VE-cadherin for ubiquitination and down-regulation
Previous studies showed that expression of K5 in endothelial cells leads to VE-cadherin down-regulation (Mansouri et al., 2008). We sought to determine whether K5 targets VE-cadherin directly or down-regulation of VE-cadherin is secondary to K5-induced down-regulation of other adherens junction components. In agreement with previous reports, expression of K5 caused a sharp reduction in VE-cadherin protein levels (Figure 1A). This reduction occurred rapidly and was observed within 24 h after adenoviral transduction of endothelial cells with K5 (Supplemental Figure S1A). In contrast, total protein levels of p120 and β-catenin were either slightly decreased or unchanged (Figure 1A). Neuronal cadherin (N-cadherin), which does not typically assemble into endothelial cell–cell junctions, was also decreased but not as sharply as levels of VE-cadherin, indicating that K5 has some specificity for VE-cadherin in endothelial cells (Figure 1A). Expression of K5 in keratinocytes failed to decrease levels of epithelial cadherin (E-cadherin; Supplemental Figure S1B), also indicating that K5-mediated down-regulation of VE-cadherin is a specific process rather than the result of nonspecific targeting of cell surface proteins. K5-mediated down-regulation of VE-cadherin in endothelial cells was also evident by immunofluorescence (Figure 1B). Furthermore, even though K5 did not cause a significant reduction in total protein levels of β-catenin or p120, K5 expression did displace both catenins from cell–cell junctions (Figure 1B). We also investigated whether K5 and VE-cadherin interact, using a ligase-dead version of K5 (Means et al., 2007). This allowed us to capture K5–VE-cadherin protein complexes in the absence of VE-cad down-regulation. After transient expression in COS cells, we detected coprecipitation of the K5 mutant with VE-cadherin, indicating that these proteins form a biochemical complex (Figure 1C). These data suggest that K5 targets VE-cadherin directly.
FIGURE 1:
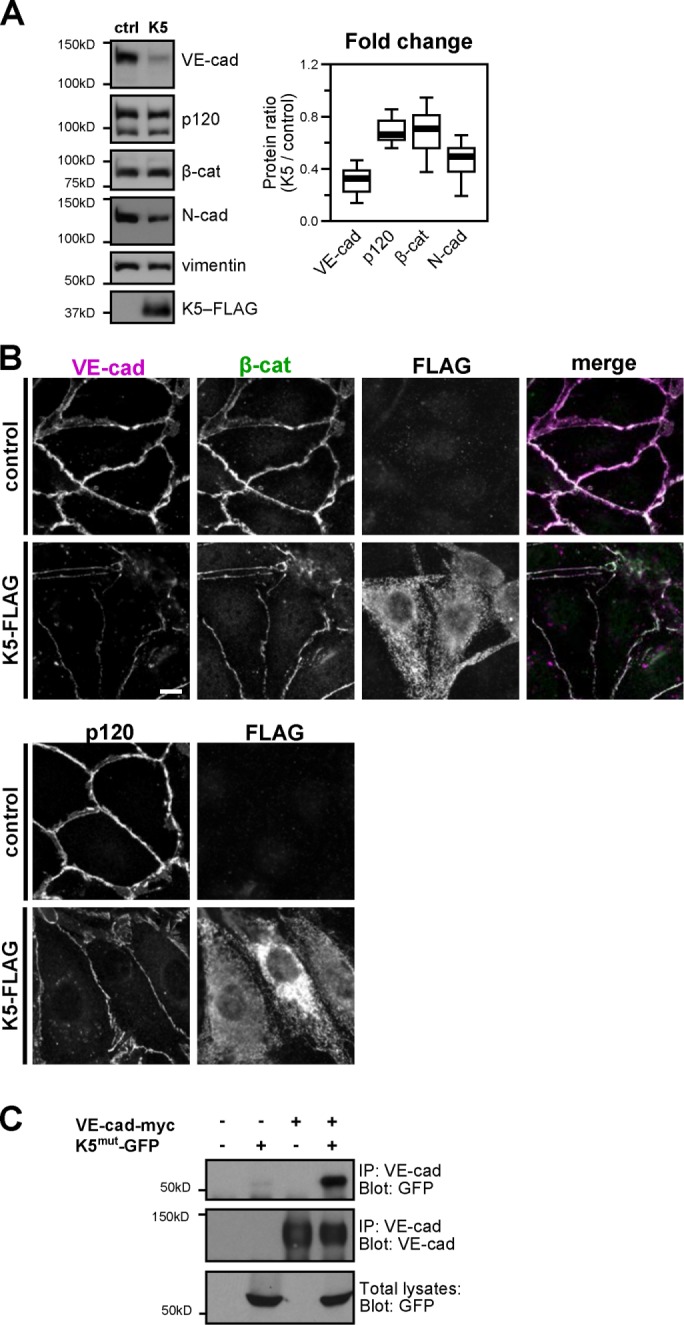
K5 down-regulates VE-cadherin. (A) K5-FLAG was expressed in cultures of the endothelial cell line HMEC-1 using adenoviral transduction. Control cells were uninfected. After 48 h, cells were harvested and the lysates analyzed by Western blot. Thick line, median band intensity; boxes, interquartile range; whiskers, 90% range (n = 7 sample pairs per protein); p < 0.01, VE-cadherin compared with p120; p < 0.05, VE-cadherin compared with β-catenin. (B) FLAG-tagged K5 was expressed in primary cultures of dermal microvascular endothelial cells. After 48 h, cells were fixed and stained for VE-cadherin, β-catenin, or FLAG (top) or p120 and FLAG (bottom). Bars, 10 μm. (C) VE-cadherin forms a biochemical complex with K5 RING mutant. VE-cadherin-myc and a ligase-dead RING mutant of K5-GFP were expressed in COS-7 cells as indicated. After 24 h, total cell lysates were immunoprecipitated with anti-VE-cadherin antibody, and the coprecipitation of mutant K5-GFP was analyzed by Western blot.
In addition, we found that K5-mediated down-regulation of VE-cadherin is associated with ubiquitination of the cadherin. Prolonged treatment of endothelial cells with MG-132 to broadly disrupt the ubiquitin–proteasome system blocked the ability of K5 to remove VE-cadherin and p120 from cell–cell junctions (Supplemental Figure S2A). Furthermore, the K5 mutant lacking ubiquitin ligase activity failed to down-regulate VE-cadherin stably expressed in a CHO cell line (Supplemental Figure S2B). We also used immunoprecipitation and Western blot to detect VE-cadherin ubiquitination directly. Expression of K5 in endothelial cells significantly increased the amount of ubiquitination detected in VE-cadherin complexes captured by immunoprecipitation (Figure 2A). However, standard immunoprecipitation conditions with nonionic detergents isolate cadherin-binding proteins along with the cadherin. Therefore this result has two possible explanations. Either K5 targets VE-cadherin directly or K5-mediated ubiquitination of another adherens junction component, such as p120, leads to the subsequent down-regulation of VE-cadherin. To determine whether K5 targets VE-cadherin for ubiquitination, we added ionic detergents to disrupt noncovalent interactions. Increased ubiquitination of VE-cadherin was still detected with the addition of ionic detergents (Figure 2B), and no K5-induced ubiquitination was detected in p120 captured by immunoprecipitation (Figure 2C), indicating that ubiquitin is ligated directly to VE-cadherin. Thus K5 targets VE-cadherin for ubiquitination and down-regulation, leading to disassembly of the endothelial adherens junction.
FIGURE 2:

K5 targets VE-cadherin for ubiquitination. K5-FLAG was expressed in HMEC-1 cultures using adenoviral transduction. After 24 h, cells were pretreated with 10 μM MG-132 for 2 h to preserve protein ubiquitination and then lysed either in nonionic detergents to preserve protein–protein interactions (A) or 0.1% SDS to disrupt noncovalent interactions (B, C). VE-cadherin (A, B) or p120 (C) was isolated by immunoprecipitation and the products analyzed by Western blot.
K5 induces VE-cadherin endocytosis
Because K5 expression caused adherens junction disassembly in cultured endothelial cells, we also asked whether biopsies of Kaposi sarcoma lesions showed evidence of junctional alterations. Kaposi sarcoma lesions are characterized by fascicles of endothelial-derived spindle cells, abnormal slit-like vascular spaces, and extravasated erythrocytes (Radu and Pantanowitz, 2013). We used immunohistochemistry to stain biopsies of Kaposi sarcoma lesions and assess the organization of endothelial cell–cell junctions. Consistent with previous reports (Dwyer et al., 2011), we found lower levels of VE-cadherin staining in endothelial cells lining the vascular spaces in Kaposi sarcoma lesions than in similarly located cells in hemangiomas (Figure 3A). Of interest, we also found substantially decreased p120 staining (Figure 3B). Because K5 expression in endothelial cells did not induce a substantial decrease in p120 protein levels (Figure 1A), decreased p120 staining in Kaposi sarcoma lesions may reflect an HHV-8 pathomechanism unrelated to K5. Given their thin profile, we were unable to determine the subcellular localization of VE-cadherin or p120 in cells lining the vascular spaces. However, the endothelial-derived spindle cells expressed both VE-cadherin and p120 and were large enough to distinguish junctional from cytoplasmic staining patterns (Figure 3, C and D). Although limited cell border localization was observed in some spindle cells (Figure 3, C and D, arrowheads), both VE-cadherin and p120 stains were predominantly diffuse and cytoplasmic. The cytoplasmic staining pattern of p120 in spindle cells was particularly striking compared with the junctional localization of p120 in keratinocytes adjacent to the lesions (Figure 3D, d′).
FIGURE 3:

Adherens junction proteins are disrupted in Kaposi sarcoma. Formalin-fixed, paraffin-embedded tissue sections from Kaposi sarcoma lesions and hemangiomas were stained for VE-cadherin or p120 by immunohistochemistry. (A, B) A linear unmixing algorithm was used to estimate diaminobenzidine absorbance, which was then quantified in the endothelial cells lining vascular spaces in each section. Thick line, median; boxes, interquartile range; whiskers, 90% range (n = 116 vessels from four Kaposi sarcoma lesions and 89 vessels from two hemangiomas). (C, D) Kaposi sarcoma spindle cells stained diffusely positive for both VE-cadherin and p120, with only occasional junctional localization (arrowheads). In epidermal keratinocytes overlying the lesion (D, asterisk), p120 maintained junctional localization. Bars, 20 μm (A, B), 100 μm (C, D, main images), 400 μm (insets).
Because VE-cadherin undergoes rapid endocytosis and degradation in the absence of p120 binding (Xiao et al., 2003a), disruption of p120 in Kaposi sarcoma lesions suggests that down-regulation of VE-cadherin in the lesions may result from increased internalization of the cadherin. In fact, we found that expression of K5 in endothelial cells significantly increased VE-cadherin endocytosis (Figure 4A). Although we cannot rule out the possibility that K5 might affect VE-cadherin synthesis or trafficking in other ways, increased endocytosis of the cadherin is consistent with the disruption of endothelial cell junction proteins seen both in cell culture and in Kaposi sarcoma lesions. Furthermore, this increased VE-cadherin internalization was associated with an increase in endothelial cell permeability as assessed by electrical cell-substrate impedance sensing (ECIS), reminiscent of the increased vascular permeability in Kaposi sarcoma lesions (Figure 4B).
FIGURE 4:
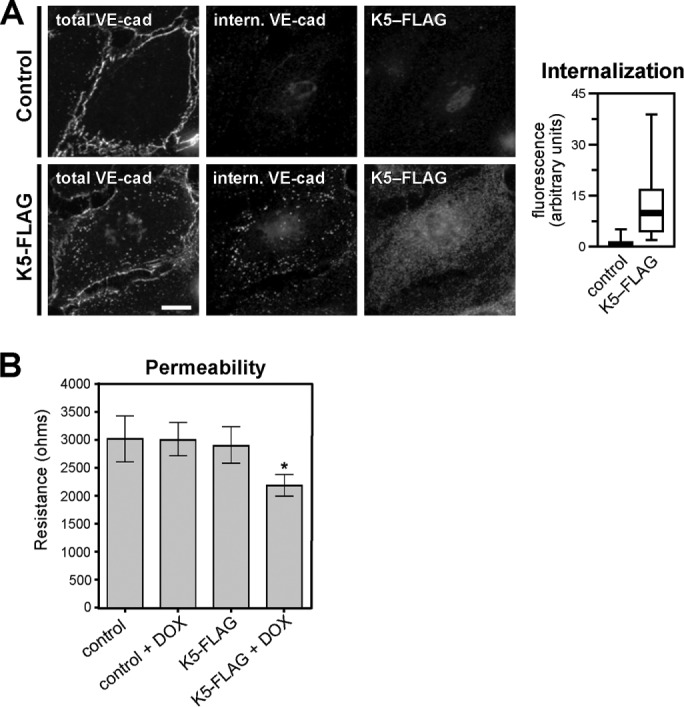
K5 induces VE-cadherin endocytosis and increased endothelial permeability. (A) K5-FLAG was expressed in primary dermal microvascular endothelial cells by adenoviral transduction. Cells were treated with 1 μg/ml doxycycline to suppress K5 expression until 6 h before beginning the assay. At 24 h after transduction, VE-cadherin endocytosis was measured using a fluorescence-based internalization assay. Internalized VE-cadherin (center column) was identified by antibody-labeling cell-surface VE-cadherin, incubating cells for 10 min to allow endocytosis, and then washing cells with a low-pH buffer to remove any antibody remaining on the cell surface. A second antibody was used to label the total VE-cadherin pool for comparison (left column). Thick line, median; boxes, interquartile range; whiskers, 90% range (n = 55–58 cells/group); p < 0.001. Bar, 20 μm. (B) Barrier function HDMECs expressing doxycycline-inducible K5-FLAG or control HDMECs was measured using an ECIS assay 48 h after K5-FLAG induction. Error bars represent SD (n = 3 control HDMEC and 21 K5-FLAG expressing HDMEC replicates); *p < 0.05 for K5-FLAG plus doxycycline compared with K5-FLAG control
Distinct endocytic motifs drive constitutive and K5-induced VE-cadherin endocytosis
VE-cadherin undergoes constitutive endocytosis and recycling driven by an internalization signal in the cadherin juxtamembrane domain. This constitutive endocytic motif is anchored by three acidic amino acids within the core p120-binding region (DEE646-648; Figure 5A), and mutation of these residues results in a cadherin variant that is resistant to constitutive endocytosis. We therefore asked whether K5-induced VE-cadherin down-regulation requires the constitutive endocytic signal. Surprisingly, the constitutive endocytosis–defective VE-cadherin mutant was not resistant to down-regulation by K5 (Figure 5B), indicating that K5-mediated ubiquitination of the cadherin is sufficient to drive cadherin endocytosis, even in the absence of the constitutive endocytic signal. A control VE-cadherin mutant that does not bind p120 but undergoes constitutive endocytosis (GGG649-651; Nanes et al., 2012) was also susceptible to K5-induced down-regulation (Figure 5B). Because the constitutive endocytic signal is not required for K5-mediated down-regulation of VE-cadherin, we reasoned that other motifs in the cadherin cytoplasmic tail might be important. In particular, two membrane-proximal lysines, K626 and K633, are potential target residues for K5-mediated ubiquitination (Figure 5A). To test whether K5 targets these particular residues, we mutated them to arginines, which are not suitable targets for ubiquitination. Unlike the constitutive endocytosis–resistant VE-cadherin mutant, the K626R, K633R mutant was resistant to down-regulation by K5 (Figures 5C and S3). The K626R, K633R mutant also failed to undergo K5-induced internalization (Figure 5D). Together these data suggest that distinct motifs in the cadherin juxtamembrane domain drive constitutive and K5-induced VE-cadherin internalization.
FIGURE 5:
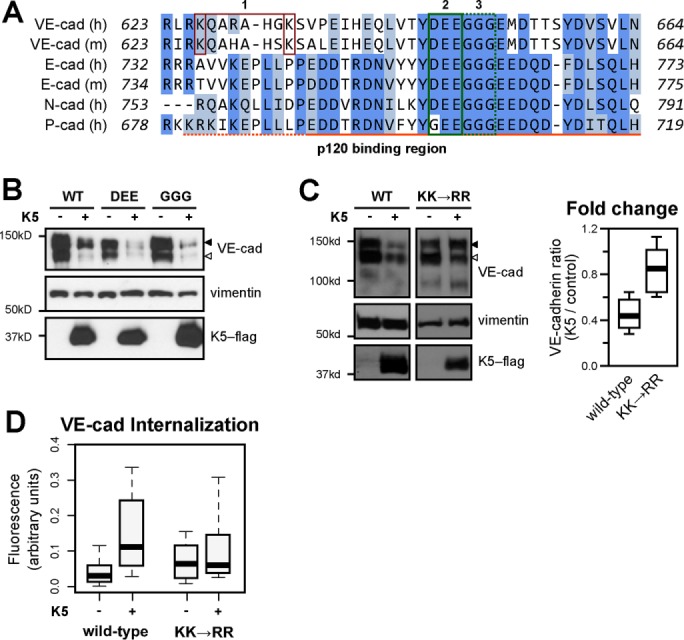
K5 induces VE-cadherin down-regulation through an alternate endocytic motif. (A) Multiple sequence alignment of the juxtamembrane domains of classical cadherins. 1, K626 and K633 mutated in C; 2, DEE646–648 mutated in B; 3, GGG649-651 mutated in B. The p120-binding region is marked with an orange line below the alignment: solid line, static binding region; dotted line, dynamic binding region. (B) Wild-type or mutant VE-cadherin–red fluorescent protein (RFP) and K5-FLAG were expressed in primary dermal microvascular endothelial cells by adenoviral transduction. VE-cadherin with a DEE646-648AAA mutation (DEE) does not bind p120 but is resistant to constitutive endocytosis (Nanes et al., 2012). VE-cadherin with a GGG649-651AAA mutation (GGG) does not bind p120 and undergoes constitutive endocytosis normally. At 48 h after transduction, cells were harvested and the lysates analyzed by Western blot. Empty arrowhead, endogenous VE-cadherin; filled arrowhead, VE-cadherin–RFP. (C) Wild-type (WT) or mutant (K626R, K633R; KK→RR) VE-cadherin–RFP was stably expressed in HMEC-1 cells using lentiviral transduction. K5-FLAG was expressed by adenoviral transduction 48 h before cells were harvested, and the lysates were analyzed by Western blot. Empty arrowhead, endogenous VE-cadherin; filled arrowhead, VE-cadherin–RFP. Thick line, median; boxes, interquartile range; whiskers, 90% range (six or seven sample pairs/group); P < 0.05. (D) Wild-type or KK mutant VE-cadherin–RFP was expressed in COS-7 cells by transient transfection. K5-FLAG was expressed by adenoviral transduction 24 h before measurement of VE-cadherin endocytosis over a 30-min period using a fluorescence-based internalization assay. KK, VE-cadherin K626R K633R, the K5-resistant mutant; Thick lines, median; boxes, interquartile range; whiskers, 90% range (25–30 cells/group). WT plus K5 compared with WT, p = 0.006.
K5 displaces p120 from VE-cadherin
Binding of p120 to the VE-cadherin juxtamembrane domain modulates cadherin endocytosis by masking the constitutive endocytic signal. Therefore we asked whether p120 might similarly regulate K5-induced VE-cadherin endocytosis. In particular, we tested whether K5-induced VE-cadherin endocytosis was associated with separation of p120 from the cadherin cytoplasmic tail and, conversely, whether p120 could protect VE-cadherin from down-regulation by K5. Expressing K5 in endothelial cells did not measurably decrease the amount of p120 immunoprecipitated with VE-cadherin (Figure 2A). However, VE-cadherin that is not bound to p120 is rapidly internalized and degraded, so the size of any p120-unbound cadherin pool is likely quite small. To overcome this challenge, we treated endothelial cells with chloroquine, an inhibitor of lysosomal acidification, which causes proteins targeted for degradation in the lysosome to instead accumulate in intracellular vesicles. Chloroquine treatment of K5-expressing endothelial cells resulted in the loss of VE-cadherin from cell–cell junctions and accumulation of the cadherin intracellularly, consistent with K5-induced VE-cadherin endocytosis (Figure 6, A and B). Of interest, most of the VE-cadherin–containing vesicles did not contain p120, indicating that in the process of K5-induced VE-cadherin endocytosis, p120 is displaced from the cadherin (Figure 6C). Because K5-induced VE-cadherin endocytosis is associated with displacement of p120 from the cadherin, we also tested whether p120 could protect VE-cadherin from down-regulation by K5. Indeed, increased expression of p120 in endothelial cells limited the reduction of VE-cadherin protein levels induced by K5 expression (Figure 7, A and B). To confirm that this protection from down-regulation involved direct binding between p120 and VE-cadherin, we examined the VE-cadherin DEE mutant, which fails to bind to p120. In contrast to wild-type VE-cadherin, p120 failed to prevent down-regulation of the DEE mutant in response to K5 expression (Figure 7C). Together these results suggest that p120 binding to VE-cadherin directly controls K5-induced down-regulation of VE-cadherin. Thus, even though constitutive- and K5-induced VE-cadherin endocytosis are driven by distinct internalization signals, p120 functions as a common modulator of both mechanisms (Figure 8).
FIGURE 6:

K5 displaces p120 from VE-cadherin. K5-FLAG was expressed in primary dermal microvascular endothelial cells by adenoviral transduction. Cells were treated with vehicle (bottom) or 100 μM chloroquine (top). (A) After 24 h, cells were fixed and immunostained for VE-cadherin, p120, and FLAG as indicated. (B) The density of VE-cadherin–containing vesicles in individual cells was quantified. Thick line, median; boxes, interquartile range; whiskers, 90% range (20–22 cells/group); p < 0.001. (C) Individual vesicles were selected from K5-expressing chloroquine-treated cells, and the amount of VE-cadherin and p120 fluorescence signals was quantified. Most vesicles contain high levels of VE-cadherin fluorescence or high levels of p120 fluorescence but not both. Bars, 20 μm (main image), 80 μm (inset).
FIGURE 7:
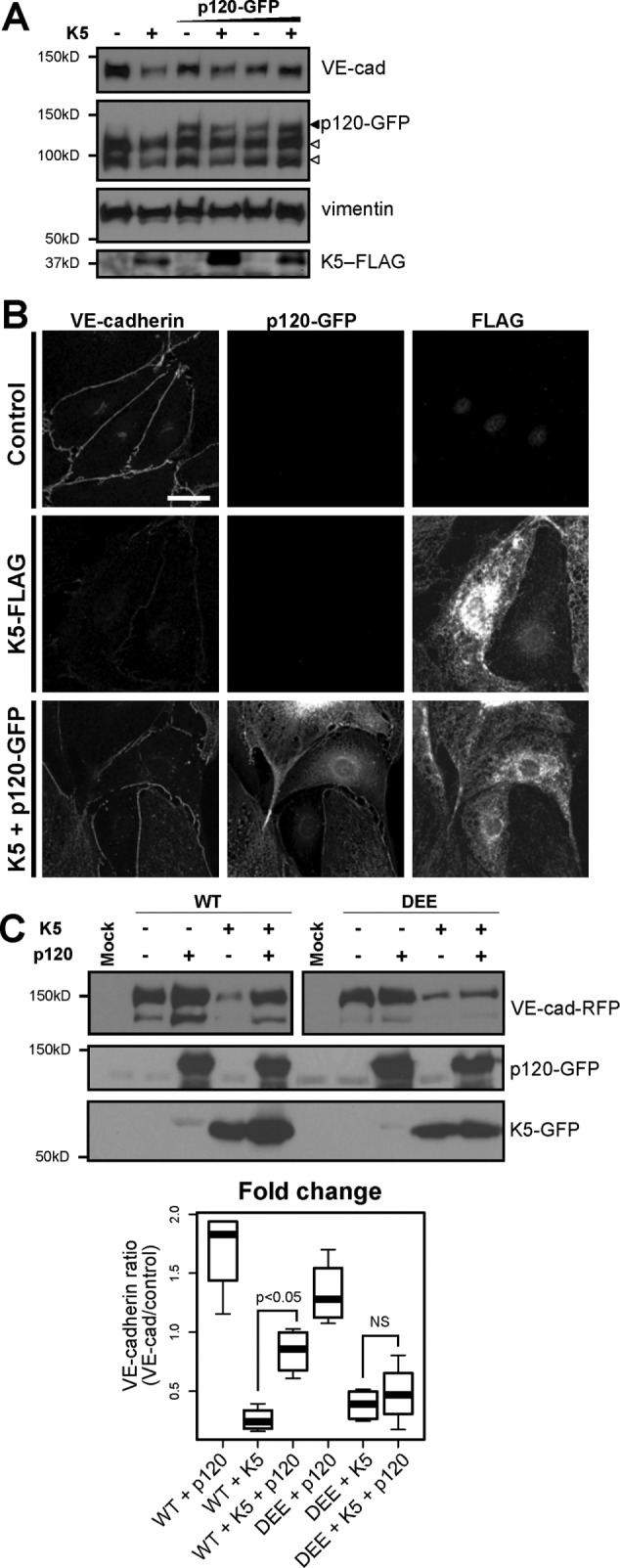
p120 binding protects VE-cadherin from down-regulation by K5. (A) K5-FLAG was expressed in primary dermal microvascular endothelial cells by adenoviral transduction. After 24 h, cells were additionally transduced to express varying levels of p120-GFP or transduced with an empty adenovirus as a control. After another 24 h, cells were lysed and analyzed by Western blot. Empty arrowheads, endogenous p120 isoforms; filled arrowhead, exogenous p120-GFP. (B) K5-FLAG or K5-FLAG and p120-GFP were expressed in primary dermal microvascular endothelial cells by adenoviral transduction. After 24 h, cells were fixed and processed for immunofluorescence. Bar, 20 μm. (C) p120 fails to protect the DEE mutant from K5-mediated down-regulation. Wild-type or DEE mutant VE-cad-RFP was expressed with K5-GFP in COS-7 cells by transient transfection as indicated. p120-GFP was expressed by adenoviral transduction. After 24 h, cells were lysed and analyzed by Western blot. For VE-cadherin (top), the right sample (DEE) was exposed longer than the left one (WT) because of decreased expression of the DEE mutant compared with wild type in COS-7 cells. Thick line, median; boxes, interquartile range; whiskers, 90% range (four sample pairs/group). Cadherin levels for cells expressing K5, p120, or K5 plus p120 were normalized to control cells expressing only WT cadherin or the DEE mutant, respectively. WT plus K5 plus p120 vs. WT plus K5: p < 0.05. DEE plus K5 plus p120 vs. DEE plus K5: not significant.
FIGURE 8:

p120 guards against multiple internalization signals. (A) Displacement of p120 from VE-cadherin by an unknown mechanism unmasks an endocytic motif in the cadherin juxtamembrane domain. This motif drives constitutive VE-cadherin endocytosis, which confers plasticity to the endothelial adherens junction. (B) K5 targets VE-cadherin for ubiquitination, displaces p120 from the cadherin, and induces VE-cadherin endocytosis and down-regulation. K5-induced VE-cadherin endocytosis does not require the constitutive endocytic motif.
DISCUSSION
The aberrant angiogenesis and leaky vasculature observed in Kaposi sarcoma lesions indicate that normal cellular control of VE-cadherin stability has been overridden. We identified one potentially responsible mechanism. The HHV-8 ubiquitin ligase K5 targets two membrane-proximal lysine residues on VE-cadherin (Figure 5), displaces p120 from the cadherin (Figure 6), and induces removal of VE-cadherin from the membrane by endocytosis (Figures 4A and 5D). Of interest, K5-induced internalization of VE-cadherin does not depend on the same motif that drives constitutive endocytosis of VE-cadherin required for collective migration of endothelial cells (Figure 5B; Nanes et al., 2012). However, p120 maintains a key role modulating VE-cadherin endocytosis by protecting against both the constitutive and K5-mediated endocytic signals (Figure 7). The p120 binding site in the cadherin juxtamembrane domain can be divided into a region mediating tight binding and a region mediating more dynamic interactions (Ishiyama et al., 2010). Because the VE-cadherin lysines targeted by K5 lie within the dynamic p120-binding region, our results support a model in which p120 and K5 compete for access to the VE-cadherin juxtamembrane domain. Thus p120 protects VE-cadherin from down-regulation by K5, and K5-induced ubiquitination of VE-cadherin displaces p120 (Figure 8). These findings indicate that a variety of signals may trigger VE-cadherin endocytosis both in the context of normal physiology and in disease states, but p120 binding to the cadherin juxtamembrane domain serves as a common control point guarding cadherin stability.
Because p120 binding to classical cadherins both in the endothelium and in other tissues is an important regulator of adherens junction dynamics, understanding other processes that may displace p120 from cadherins remains an important area for research. Many studies of VE-cadherin focused on the potential of inflammatory mediators such as histamine and vascular endothelial growth factor (VEGF) to induce phosphorylation of the cadherin (Esser et al., 1998; Andriopoulou et al., 1999). However, there is considerable disagreement over which sites may be phosphorylated under different conditions and whether p120 binding is disrupted (Wallez et al., 2007; Hatanaka et al., 2011). Furthermore, it is unclear whether phosphorylation alone is sufficient to drive cadherin down-regulation (Adam et al., 2010). Nonetheless, VEGF signaling has been linked to phosphorylation and β-arrestin–dependent endocytosis of VE-cadherin (Gavard and Gutkind, 2006; Hebda et al., 2013). Even if the precise mechanisms activated in different physiological contexts remain unclear, VE-cadherin phosphorylation, disruption of p120 binding, and cadherin endocytosis are emerging hallmarks of the endothelial response to inflammatory signals.
Most studies of cadherin ubiquitination have focused on epithelial (E)-cadherin rather than VE-cadherin. The c-Cbl–like ligase Hakai targets E-cadherin for ubiquitination in a manner dependent on Src-mediated phosphorylation of two juxtamembrane domain tyrosine residues that are not conserved in VE-cadherin (Fujita et al., 2002). Hakai-mediated ubiquitination of E-cadherin is associated with increased endocytosis and down-regulation of the cadherin. Although the specific residues ubiquitinated by Hakai are unknown, the phosphotyrosines required for targeting of E-cadherin are within the p120-binding region. This raises the possibility that p120 binding may compete with phosphorylation and Hakai-mediated ubiquitination of the cadherin. Although this hypothesis has not been tested, it would, if correct, represent another instance of p120 modulation of a cadherin endocytic mechanism. One study supporting this possibility used mitochondria-targeting assays to demonstrate that E-cadherin ubiquitination and p120 recruitment were mutually exclusive (Hartsock and Nelson, 2012). However, there is also evidence that Hakai does not trigger E-cadherin endocytosis directly. Instead, Hakai-mediated ubiquitination may target E-cadherin that has already entered the endocytic pathway for lysosomal degradation rather than recycling back to the cell surface, a mechanism dependent on Hrs and Src (Palacios et al., 2005). Of interest, depletion of Hrs causes up-regulation of E-cadherin, suggesting that this mechanism may play a role in balancing cellular cadherin levels (Toyoshima et al., 2007). The role of p120 in this pathway is unclear. A second ubiquitin ligase, MDM2, has also been reported to target E-cadherin, and in human breast carcinomas, high levels of MDM2 expression correlated with decreased E-cadherin protein levels (Yang et al., 2006). However, as with cadherin down-regulation by Hakai, the relevance of p120 to E-cadherin down-regulation by MDM2 is unknown.
In addition to the disease-associated K5-mediated ubiquitination and down-regulation of VE-cadherin reported here, VE-cadherin ubiquitination may also play a role in normal cellular processes. Proteasome inhibitors can block constitutive VE-cadherin endocytosis (Xiao et al., 2003b), as well as K5-induced VE-cadherin down-regulation (Supplemental Figure S2A). Furthermore, treatment with the inflammatory signal bradykinin induces VE-cadherin ubiquitination in vitro and in vivo and drives VE-cadherin endocytosis in vitro (Orsenigo et al., 2012). These findings suggest that cellular ubiquitin ligases may participate in the regulation of cadherin stability. One intriguing possibility is that cellular homologues of K5, MARCH-family ubiquitin ligases (Bartee et al., 2004; Nicholas et al., 1997), might serve such a function. As with many viral pathomechanisms, K5-induced down-regulation of VE-cadherin may result from the hijacking of existing cellular machinery. Indeed, we observed that several MARCH-family ubiquitin ligases are expressed endogenously in vascular endothelial cells (Supplemental Figure S4A). Of interest, both MARCH 2 and MARCH 4 disrupt VE-cadherin localization at cell–cell junctions when expressed exogenously, whereas only MARCH 4 disrupts both VE-cadherin and PECAM-1 distribution (Supplemental Figure S4, B and D). These data suggest that the MARCH family of ligases may play critical roles in regulating endothelial barrier function and migration in the context of development or inflammatory conditions characterized by vascular leak. Because these MARCH proteins display varying abilities to target other endothelial proteins in addition to VE-cadherin, individual members may function to regulate specific subsets of endothelial proteins in particular biological contexts. The role of endogenous MARCH proteins in endothelial function will be an interesting area for future studies.
Somewhat surprisingly, the junctional remodeling induced by K5 in cultured endothelial cells was not accompanied by the retraction of cells to form large gaps in the monolayer (Figure 1). Such changes sometimes accompany endothelial cell–cell junction disassembly in response to inflammatory mediators (e.g., Chen et al., 2012; Gong et al., 2014). This raises the possibility that inflammatory mediators may induce changes in endothelial cells beyond the removal of VE-cadherin from junctions, such as activation of myosin (Vandenbroucke et al., 2008; Wallez and Huber, 2008). It is also likely that HHV-8 expresses factors in addition to K5 that disrupt endothelial cell–cell adhesion, potentially with more inflammatory-like effects. Additional HHV-8 virulence factors could also explain why expression of K5 in cultured endothelial cells did not decrease p120 protein levels (Figure 1), whereas p120 staining in Kaposi sarcoma lesion biopsies was substantially decreased (Figure 3B). Because disruption of cell–cell junctions can change cell morphology, and changing cell morphology, such as through cytoskeletal remodeling, can disrupt cell–cell junctions, disentangling the causal mechanisms in cases where both phenotypes occur may be difficult. Of interest, p120 can influence both cell–cell junctions and cell morphology. In addition to regulating cadherin stability, p120 also affects cytoskeletal dynamics through regulation of Rho GTPases (Anastasiadis, 2007; Beckers et al., 2010). This function is separable from p120 modulation of cadherin endocytosis (Chiasson et al., 2009), but recruitment of p120 to cell borders can influence the spreading of individual cells on an adhesive substrate (Oas et al., 2013). Further work is needed to fully understand the connections between adherens junction disassembly and cytoskeletal remodeling in inflammatory conditions and the roles of p120 in both processes.
Our results indicate that p120 binding to the cadherin juxtamembrane domain modulates VE-cadherin endocytosis driven by two very different signals. p120 regulates both the constitutive endocytosis and recycling of VE-cadherin, which establishes junctional plasticity necessary for collective migration of endothelial cells, and the K5-induced ubiquitination, endocytosis, and down-regulation of VE-cadherin associated with the endothelial-derived tumor Kaposi sarcoma. Constitutive and K5-induced VE-cadherin endocytosis are driven by different internalization signals, but both signals are located within the p120-binding region of the cadherin. Furthermore, sequence analysis and a review of the literature suggest that a number of classical cadherins contain functional endocytic motifs within the p120-binding domain (Cadwell et al., 2016). Thus the cadherin juxtamembrane domain serves as the integration site for multiple mechanisms regulating cadherin stability. These studies suggest that a variety of different endocytic signals drive cadherin internalization and promote junctional plasticity in different contexts, whereas p120 functions as the master regulator stabilizing the cadherin and guarding junctional stability. It will be important to better understand the hierarchy of this regulation. For example, p120 is displaced from the cadherin by K5-mediated ubiquitination (Figure 6), whereas overexpression of p120 protects the cadherin from K5-mediated down-regulation (Figure 7). These two observations suggest that p120 and K5 compete for cadherin binding or that p120 modulates the ligase activity of K5, among other possibilities. Future experiments are needed to address this issue and determine whether common regulatory mechanisms extend from the viral K5 ligase to the mammalian MARCH proteins.
MATERIALS AND METHODS
Cell culture
Primary human dermal microvascular endothelial cells and keratinocytes were isolated from neonatal foreskin. Primary endothelial cells and HMEC-1 immortalized endothelial cells (Ades et al., 1992) were cultured in Endothelial Growth Medium 2 Microvascular (Lonza, Walkersville, MD) and grown on tissue culture–treated plastic or glass coverslips coated with 0.1% gelatin. Keratinocytes were cultured in Keratinocyte Growth Medium Gold (Lonza). African green monkey kidney fibroblast-like cell line COS-7 (American Type Culture Collection, Manassas, VA), human embryonic kidney cell line HEK-293T (American Type Culture Collection), and, for adenovirus production, human embryonic kidney cell line QBI-293A (MP Biomedicals, Santa Ana, CA) were cultured in DMEM with 4.5 g/l glucose, l-glutamine, and sodium pyruvate (Corning, Oneonta, NY) supplemented with 10% fetal bovine serum (Thermo Fisher Scientific) and 1% antibiotic/antimycotic solution (Corning). Cells were transfected using Lipofectamine 2000 (Life Technologies, Carlsbad, CA). Stable lentiviral transduction of HMEC-1 cells was achieved through selection in 20 μg/ml blasticidin (Valeant Pharmaceuticals, Laval, Canada), followed by further enrichment for cells expressing fluorescently tagged constructs by fluorescence-activated cell sorting (FACS Aria II; BD Biosciences, Columbus, NE). After selection, cells were maintained in 1–5 μg/ml blasticidin.
cDNA constructs
The K5–green fluorescent protein (GFP) constructs, containing HHV-8 K5 ligated between EcoRI and BamHI in pEGFP-N1 (Clontech, Mountain View, CA) in-frame with GFP, were provided by R. Means (Yale University, New Haven, CT) and J. Jung (University of Southern California, Los Angeles, CA). The ligase-dead K5 mutant (C30A, C32A, H40A, C43A), described previously, was created by site-directed mutagenesis (Means et al., 2007). The construct encoding human VE-cadherin, ligated between EcoRI sites in pECE, was provided by E. Dejana (Italian Foundation for Cancer Research, Institute of Molecular Oncology, Milan, Italy). The pECE-VE-cadherin-myc construct was generated by PCR from the C-terminus of the IL-2R-VE-cadcyto-myc construct (Venkiteswaran et al., 2002). The PCR product was then inserted into the C-terminus of pECE-VE-cadherin using BstEII/XbaI cloning sites. The K5-resistant mutant (K626R, K633R), constitutive endocytosis–resistant mutant (DEE646–648AAA; Nanes et al., 2012), and p120-binding control mutant (GGG649–651AAA) were created by site-directed mutagenesis (primers described in Supplemental Table S1). For virus production, VE-cadherin constructs were subcloned between BamHI and AgeI restriction sites in Gateway TagRFP-AS-N (Evrogen, Farmingdale, NY), in-frame with monomeric C-terminal TagRFP, then shuttled into pAd/Cmv/V5-DEST for transient transfection or for adenovirus production or pLenti6/V5-DEST for lentivirus production using LR Clonase recombination (Life Technologies). The pSV2-neo plasmid was used to confer neomycin/G-418 resistance (Southern and Berg, 1982). cDNA constructs encoding MARCH-family ligases were constructed as previously described (Bartee et al., 2004). The MARCH-2 mutant contains C64S and C67S point mutations.
Virus production
To create replication-deficient human adenovirus type 5 packaged with a gene of interest, the gene was cloned into pAd/CMV/V5-DEST and then digested with PacI to expose the viral inverted terminal repeats. Linearized DNA was transfected into virus-producing QBI-293A cells, which were harvested, concentrated, and lysed after 48–72 h to recover adenovirus. To create replication-deficient second-generation lentivirus packaged with a gene of interest, the gene was cloned into pLenti6/V5-DEST and transfected into HEK-293T cells using a kit combining transfection reagent with the necessary lentiviral regulatory genes (LENTI-Smart; InvivoGen, San Diego, CA). Lentivirus was collected from culture supernatants 48–72 h after transfection.
Immunoprecipitation and Western blot analysis
For immunoprecipitation experiments, cells were harvested either in 0.5% Triton X-100 (Roche) to preserve noncovalent interactions or in 1% Triton X-100 with 0.1% SDS (Fisher, Hampton, NH). Both buffers also contained protease inhibitor cocktails (Complete Mini tablets, ethylenediaminetetraacetic acid [EDTA] free; Roche, Switzerland), 5 mg/ml N-ethylmaleimide to inhibit deubiquitinase enzymes, 10 μM MG-132 to inhibit the proteasome, 150 mM sodium chloride, 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, 1 mM ethylene glycol tetraacetic acid, and 0.1 mM magnesium chloride. In addition, pretreatment of cells with 10 μM MG-132 for 2 h before harvesting was used to increase the amount of ubiquitinated protein recovered. After a 30-min incubation at 4°C, cell lysates were centrifuged at 16,100 × g for 10 min, and the soluble fraction was diluted to a final protein concentration of 1 mg/ml. Cell lysates were then incubated with 2 μg of antibody against VE-cadherin or p120 (Supplemental Table S2) conjugated to ferromagnetic beads (Dynabeads, Life Technologies) for 1 h at 4°C. The beads were then washed with 0.1% Triton X-100 and eluted into Laemmli sample buffer (Bio-Rad Laboratories, Hercules, CA) with 5% β-mercaptoethanol. For other Western blot experiments, cells were harvested directly into sample buffer. Samples were heated at 95°C for 5 min and then separated by SDS–PAGE and analyzed by immunoblotting on nitrocellulose membranes (Whatman, GE Healthcare Life Sciences, Pittsburgh, PA). To increase detection of ubiquitin, membranes were covered with deionized water and autoclaved for 30 min. Primary antibodies are listed in Supplemental Table S2. Horseradish peroxidase–conjugated secondary antibodies (Bio-Rad Laboratories), a luminol-based detection system (ECL, GE Healthcare), and autoradiography film (Denville Scientific, Holliston, MA) were used for detection.
Immunofluorescence
Cells cultured on glass coverslips were fixed either in methanol for 2 min at 4°C or in 4% paraformaldehyde for 10 min, followed by 0.1% Triton X-100 for 8 min at room temperature, depending on the performance of the primary antibodies used (Supplemental Table S2). Secondary antibodies conjugated to fluorescent dyes (Alexa Fluor 488, 555, or 647 nm; Life Technologies) were used to identify target molecules. Microscopy was performed using an epifluorescence microscope (DMRXA2; Leica, Wetzlar, Germany) equipped with 63×/1.32 numerical aperture (NA) and 100×/1.40 NA oil immersion objectives with apochromatic aberration and flat-field corrections, narrow-bandpass filters, and a digital camera (ORCA-ER C4742-80; Hamamatsu Photonics, Naka-ku, Hamamatsu, Japan). Images were captured using Simple PCI software (Hamamatsu Photonics).
Immunohistochemistry
Formalin-fixed, paraffin-embedded tissue blocks were cut to 5-μm sections, affixed to glass slides, deparaffinized in Xylene, and processed for heat-induced antigen retrieval in either 10 mM sodium citrate, pH 6.0, for VE-cadherin staining or Tris/EDTA, pH 9.0, for p120 staining. Primary antibodies are described in Supplemental Table S2. Horseradish peroxidase–conjugated secondary antibodies and diaminobenzidine substrate were used to detect antibody labeling. Hematoxylin was used as a counterstain. Digital images were captured using whole-slide scanning (Nanozoomer 2.0HT; Hamamatsu Photonics). For quantification, each image channel was log-transformed, and then a linear unmixing algorithm was used to separate the resulting red, green, and blue absorbances into diaminobenzidine and hematoxylin absorbance components. Vascular spaces were outlined, and, for each vessel, average diaminobenzidine absorbance was calculated within 1.1 μm of the border.
Internalization assay and vesicle analysis
To measure K5-induced or constitutive internalization of VE-cadherin, cells were incubated in antibody against the VE-cadherin extracellular domain dissolved in culture medium for 30 min at 4°C. For K5-induced internalization, cells expressing VE-cadherin proteins were infected with K5 virus 24 h before assay. Unbound antibody was removed by washing with cold phosphate-buffered saline (PBS). Cells were then incubated in culture medium for various time periods at 37°C to allow internalization to occur. At the end of the internalization period, cells were returned to 4°C and washed with PBS. Any antibody remaining at the cell surface was removed by washing cells with a low pH buffer (PBS with 100 mM glycine, 20 mM magnesium acetate, and 50 mM potassium chloride, pH 2.2). Cells were then fixed and processed for immunofluorescence, with a second antibody against VE-cadherin, distinguished based on isotype, used to label the total cadherin pool or the total cadherin pool was detected by a c-terminus red fluorescent protein tag. Internalization was quantified as the ratio of fluorescence signals corresponding to the internalized and total cadherin pools. For vesicle analysis experiments, cells were treated with 100 μM chloroquine (Sigma-Aldrich, St. Louis, MO) dissolved in culture medium for 24 h, refreshed after 12 h, and then fixed and processed for immunofluorescence. Vesicles were identified by automated selection of four-connected regions of pixels above background thresholds with areas of 2.56 × 10−2 to 16.0 × 10−2 μm2.
Measurement of transendothelial electric resistance
Human dermal microvascular endothelial cells (HDMECs) grown in EGM-2 MV (Lonza) were infected with K5 3XF pINDUCER20 lentivirus (pINDUCER20; a generous gift of Thomas F. Westbrook, Baylor College of Medicine, Houston, TX) and allowed to grow to confluence. Cells were then suspended and seeded along with control HDMECs at 4.8 × 104 cells/well of an ECIS 8W10E PET plate (Applied Biophysics, Troy, NY). After 24 h, cells were treated with 1 μg/ml doxycycline hyclate (Enzo Life Sciences, Farmingdale, NY) in EGM-2 MV. Transendothelial resistance was then measured for the following 48 h after treatment with doxycycline .
Image analysis and statistics
The Fiji distribution of ImageJ (Schindelin et al., 2012) with custom plug-ins was used for all image analysis and automated quantification (Nanes, 2015). The JAMA linear algebra library (version 1.0.3; National Institute of Standards and Technology) was used to implement the linear unmixing algorithm. Statistical analyses were implemented in R (version 2.15; R Foundation for Statistical Computing). The Kruskal–Wallis rank sum test with Dunn’s method for multiple comparisons was used to evaluate nonparametric scaled data (Dunn, 1964). One-way analysis of variance followed by Dunnett’s test was used to evaluate ECIS data.
Supplementary Material
Acknowledgments
We thank D. Alexis and the Emory Pathology Core Laboratory for histology preparations, N. Ishiyama and M. Ikura for thoughtful discussions, S. Summers for help with primary cell isolations, S. N. Stahley for reviewing the manuscript, and members of the Kowalczyk laboratory for help and advice. This work was supported by grants from the National Institutes of Health (R01AR050501 and R01AR048266 to A.P.K.). B.A.N. was supported by fellowships from the National Institutes of Health (F30HL110447 and T32GM008367) and the American Heart Association.
Abbreviations used:
- E-cadherin
epithelial cadherin
- ECIS
electric cell-substrate impedance sensing
- HHV-8
human herpesvirus 8
- MARCH
membrane-associated really interesting new gene–CH
- N-cadherin
neuronal cadherin
- p120
p120-catenin
- VE-cadherin
vascular endothelial cadherin.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E16-06-0459) on October 26, 2016.
REFERENCES
- Adam AP, Sharenko AL, Pumiglia K, Vincent PA. Src-induced tyrosine phosphorylation of VE-cadherin is not sufficient to decrease barrier function of endothelial monolayers. J Biol Chem. 2010;285:7045–7055. doi: 10.1074/jbc.M109.079277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ades EW, Candal FJ, Swerlick RA, George VG, Summers S, Bosse DC, Lawley TJ. HMEC-1: establishment of an immortalized human microvascular endothelial cell line. J Invest Dermatol. 1992;99:683–690. doi: 10.1111/1523-1747.ep12613748. [DOI] [PubMed] [Google Scholar]
- Anastasiadis PZ. p120-ctn: a nexus for contextual signaling via Rho GTPases. Biochim Biophys Acta. 2007;1773:34–46. doi: 10.1016/j.bbamcr.2006.08.040. [DOI] [PubMed] [Google Scholar]
- Andriopoulou P, Navarro P, Zanetti A, Lampugnani MG, Dejana E. Histamine induces tyrosine phosphorylation of endothelial cell-to-cell adherens junctions. Arterioscler Thromb Vasc Biol. 1999;19:2286–2297. doi: 10.1161/01.atv.19.10.2286. [DOI] [PubMed] [Google Scholar]
- Antman K, Chang Y. Kaposi’s sarcoma. N Engl J Med. 2000;342:1027–1038. doi: 10.1056/NEJM200004063421407. [DOI] [PubMed] [Google Scholar]
- Bartee E, Mansouri M, Hovey Nerenberg BT, Gouveia K, Fruh K. Downregulation of major histocompatibility complex class I by human ubiquitin ligases related to viral immune evasion proteins. J Virol. 2004;78:1109–1120. doi: 10.1128/JVI.78.3.1109-1120.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckers CM, van Hinsbergh VW, van Nieuw Amerongen GP. Driving Rho GTPase activity in endothelial cells regulates barrier integrity. Thromb Haemost. 2010;103:40–55. doi: 10.1160/TH09-06-0403. [DOI] [PubMed] [Google Scholar]
- Cadwell CM, Su W, Kowalczyk AP. Cadherin tales: regulation of cadherin function by endocytic membrane trafficking. Traffic. 2016;17:1262–1271. doi: 10.1111/tra.12448. [DOI] [PubMed] [Google Scholar]
- Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, Moore PS. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science. 1994;266:1865–1869. doi: 10.1126/science.7997879. [DOI] [PubMed] [Google Scholar]
- Chen XL, Nam JO, Jean C, Lawson C, Walsh CT, Goka E, Lim ST, Tomar A, Tancioni I, Uryu S, et al. VEGF-induced vascular permeability is mediated by FAK. Dev Cell. 2012;22:146–157. doi: 10.1016/j.devcel.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiasson CM, Wittich KB, Vincent PA, Faundez V, Kowalczyk AP. p120-catenin inhibits VE-cadherin internalization through a Rho-independent mechanism. Mol Biol Cell. 2009;20:1970–1980. doi: 10.1091/mbc.E08-07-0735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coscoy L, Ganem D. Kaposi’s sarcoma-associated herpesvirus encodes two proteins that block cell surface display of MHC class I chains by enhancing their endocytosis. Proc Natl Acad Sci USA. 2000;97:8051–8056. doi: 10.1073/pnas.140129797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MA, Ireton RC, Reynolds AB. A core function for p120-catenin in cadherin turnover. J Cell Biol. 2003;163:525–534. doi: 10.1083/jcb.200307111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejana E, Orsenigo F. Endothelial adherens junctions at a glance. J Cell Sci. 2013;126:2545–2549. doi: 10.1242/jcs.124529. [DOI] [PubMed] [Google Scholar]
- Desai R, Sarpal R, Ishiyama N, Pellikka M, Ikura M, Tepass U. Monomeric alpha-catenin links cadherin to the actin cytoskeleton. Nat Cell Biol. 2013;15:261–273. doi: 10.1038/ncb2685. [DOI] [PubMed] [Google Scholar]
- Dunn OJ. Multiple comparisons using rank sums. Technometrics. 1964;6:241–252. [Google Scholar]
- Dwyer J, Le Guelte A, Galan Moya EM, Sumbal M, Carlotti A, Douguet L, Gutkind JS, Grange PA, Dupin N, Gavard J. Remodeling of VE-cadherin junctions by the human herpes virus 8 G-protein coupled receptor. Oncogene. 2011;30:190–200. doi: 10.1038/onc.2010.411. [DOI] [PubMed] [Google Scholar]
- Esser S, Lampugnani MG, Corada M, Dejana E, Risau W. Vascular endothelial growth factor induces VE-cadherin tyrosine phosphorylation in endothelial cells. J Cell Sci. 1998;111:1853–1865. doi: 10.1242/jcs.111.13.1853. [DOI] [PubMed] [Google Scholar]
- Fujita Y, Krause G, Scheffner M, Zechner D, Leddy HE, Behrens J, Sommer T, Birchmeier W. Hakai, a c-Cbl-like protein, ubiquitinates and induces endocytosis of the E-cadherin complex. Nat Cell Biol. 2002;4:222–231. doi: 10.1038/ncb758. [DOI] [PubMed] [Google Scholar]
- Gavard J, Gutkind JS. VEGF controls endothelial-cell permeability by promoting the beta-arrestin-dependent endocytosis of VE-cadherin. Nat Cell Biol. 2006;8:1223–1234. doi: 10.1038/ncb1486. [DOI] [PubMed] [Google Scholar]
- Giannotta M, Trani M, Dejana E. VE-cadherin and endothelial adherens junctions: active guardians of vascular integrity. Dev Cell. 2013;26:441–454. doi: 10.1016/j.devcel.2013.08.020. [DOI] [PubMed] [Google Scholar]
- Gong H, Gao X, Feng S, Siddiqui MR, Garcia A, Bonini MG, Komarova Y, Vogel SM, Mehta D, Malik AB. Evidence of a common mechanism of disassembly of adherens junctions through Galpha13 targeting of VE-cadherin. J Exp Med. 2014;211:579–591. doi: 10.1084/jem.20131190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris TJ, Tepass U. Adherens junctions: from molecules to morphogenesis. Nat Rev Mol Cell Biol. 2010;11:502–514. doi: 10.1038/nrm2927. [DOI] [PubMed] [Google Scholar]
- Hartsock A, Nelson WJ. Competitive regulation of E-cadherin juxtamembrane domain degradation by p120-catenin binding and Hakai-mediated ubiquitination. PLoS One. 2012;7:e37476. doi: 10.1371/journal.pone.0037476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatanaka K, Simons M, Murakami M. Phosphorylation of VE-cadherin controls endothelial phenotypes via p120-catenin coupling and Rac1 activation. Am J Physiol Heart Circ Physiol. 2011;300:H162–H172. doi: 10.1152/ajpheart.00650.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebda JK, Leclair HM, Azzi S, Roussel C, Scott MG, Bidere N, Gavard J. The C-terminus region of beta-arrestin1 modulates VE-cadherin expression and endothelial cell permeability. Cell Commun Signal. 2013;11:37. doi: 10.1186/1478-811X-11-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishido S, Wang C, Lee BS, Cohen GB, Jung JU. Downregulation of major histocompatibility complex class I molecules by Kaposi’s sarcoma-associated herpesvirus K3 and K5 proteins. J Virol. 2000;74:5300–5309. doi: 10.1128/jvi.74.11.5300-5309.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiyama N, Ikura M. The three-dimensional structure of the cadherin-catenin complex. Subcell Biochem. 2012;60:39–62. doi: 10.1007/978-94-007-4186-7_3. [DOI] [PubMed] [Google Scholar]
- Ishiyama N, Lee SH, Liu S, Li GY, Smith MJ, Reichardt LF, Ikura M. Dynamic and static interactions between p120 catenin and E-cadherin regulate the stability of cell-cell adhesion. Cell. 2010;141:117–128. doi: 10.1016/j.cell.2010.01.017. [DOI] [PubMed] [Google Scholar]
- Kaposi M. Idiopathisches multiples Pigmentsarkom der Haut. Arch Dermatol. 1872;4:265–273. [Google Scholar]
- Mansouri M, Douglas J, Rose PP, Gouveia K, Thomas G, Means RE, Moses AV, Fruh K. Kaposi sarcoma herpesvirus K5 removes CD31/PECAM from endothelial cells. Blood. 2006;108:1932–1940. doi: 10.1182/blood-2005-11-4404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansouri M, Rose PP, Moses AV, Fruh K. Remodeling of endothelial adherens junctions by Kaposi’s sarcoma-associated herpesvirus. J Virol. 2008;82:9615–9628. doi: 10.1128/JVI.02633-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Means RE, Lang SM, Jung JU. The Kaposi’s sarcoma-associated herpesvirus K5 E3 ubiquitin ligase modulates targets by multiple molecular mechanisms. J Virol. 2007;81:6573–6583. doi: 10.1128/JVI.02751-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyashita Y, Ozawa M. Increased internalization of p120-uncoupled E-cadherin and a requirement for a dileucine motif in the cytoplasmic domain for endocytosis of the protein. J Biol Chem. 2007;282:11540–11548. doi: 10.1074/jbc.M608351200. [DOI] [PubMed] [Google Scholar]
- Nanes BA. Slide Set: reproducible image analysis and batch processing with ImageJ. Biotechniques. 2015;59:269–278. doi: 10.2144/000114351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanes BA, Chiasson-MacKenzie C, Lowery AM, Ishiyama N, Faundez V, Ikura M, Vincent PA, Kowalczyk AP. p120-catenin binding masks an endocytic signal conserved in classical cadherins. J Cell Biol. 2012;199:365–380. doi: 10.1083/jcb.201205029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanes BA, Kowalczyk AP. Adherens junction turnover: regulating adhesion through cadherin endocytosis, degradation, and recycling. Subcell Biochem. 2012;60:197–222. doi: 10.1007/978-94-007-4186-7_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholas J, Ruvolo V, Zong J, Ciufo D, Guo HG, Reitz MS, Hayward GS. A single 13-kilobase divergent locus in the Kaposi sarcoma-associated herpesvirus (human herpesvirus 8) genome contains nine open reading frames that are homologous to or related to cellular proteins. J Virol. 1997;71:1963–1974. doi: 10.1128/jvi.71.3.1963-1974.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oas RG, Nanes BA, Esimai CC, Vincent PA, Garcia AJ, Kowalczyk AP. p120-catenin and beta-catenin differentially regulate cadherin adhesive function. Mol Biol Cell. 2013;24:704–714. doi: 10.1091/mbc.E12-06-0471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oas RG, Xiao K, Summers S, Wittich KB, Chiasson CM, Martin WD, Grossniklaus HE, Vincent PA, Reynolds AB, Kowalczyk AP. p120-Catenin is required for mouse vascular development. Circ Res. 2010;106:941–951. doi: 10.1161/CIRCRESAHA.109.207753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orsenigo F, Giampietro C, Ferrari A, Corada M, Galaup A, Sigismund S, Ristagno G, Maddaluno L, Koh GY, Franco D, et al. Phosphorylation of VE-cadherin is modulated by haemodynamic forces and contributes to the regulation of vascular permeability in vivo. Nat Commun. 2012;3:1208. doi: 10.1038/ncomms2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palacios F, Tushir JS, Fujita Y, D’Souza-Schorey C. Lysosomal targeting of E-cadherin: a unique mechanism for the down-regulation of cell-cell adhesion during epithelial to mesenchymal transitions. Mol Cell Biol. 2005;25:389–402. doi: 10.1128/MCB.25.1.389-402.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian L-W, Greene W, Ye F, Gao S-J. Kaposi’s sarcoma-associated herpesvirus disrupts adherens junctions and increases endothelial permeability by inducing degradation of VE-cadherin. J Virol. 2008;82:11902–11912. doi: 10.1128/JVI.01042-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radu O, Pantanowitz L. Kaposi sarcoma. Arch Pathol Lab Med. 2013;137:289–294. doi: 10.5858/arpa.2012-0101-RS. [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southern PJ, Berg P. Transformation of mammalian cells to antibiotic resistance with a bacterial gene under control of the SV40 early region promoter. J Mol Appl Genet. 1982;1:327–341. [PubMed] [Google Scholar]
- Taguchi K, Ishiuchi T, Takeichi M. Mechanosensitive EPLIN-dependent remodeling of adherens junctions regulates epithelial reshaping. J Cell Biol. 2011;194:643–656. doi: 10.1083/jcb.201104124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyoshima M, Tanaka N, Aoki J, Tanaka Y, Murata K, Kyuuma M, Kobayashi H, Ishii N, Yaegashi N, Sugamura K. Inhibition of tumor growth and metastasis by depletion of vesicular sorting protein Hrs: its regulatory role on E-cadherin and beta-catenin. Cancer Res. 2007;67:5162–5171. doi: 10.1158/0008-5472.CAN-06-2756. [DOI] [PubMed] [Google Scholar]
- Uldrick TS, Whitby D. Update on KSHV epidemiology, Kaposi sarcoma pathogenesis, and treatment of Kaposi sarcoma. Cancer Lett. 2011;305:150–162. doi: 10.1016/j.canlet.2011.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenbroucke E, Mehta D, Minshall R, Malik AB. Regulation of endothelial junctional permeability. Ann NY Acad Sci. 2008;1123:134–145. doi: 10.1196/annals.1420.016. [DOI] [PubMed] [Google Scholar]
- Venkiteswaran K, Xiao K, Summers S, Calkins CC, Vincent PA, Pumiglia K, Kowalczyk AP. Regulation of endothelial barrier function and growth by VE-cadherin, plakoglobin, and beta-catenin. Am J Physiol Cell Physiol. 2002;283:C811–C821. doi: 10.1152/ajpcell.00417.2001. [DOI] [PubMed] [Google Scholar]
- Vincent PA, Xiao K, Buckley KM, Kowalczyk AP. VE-cadherin: adhesion at arm’s length. Am J Physiol Cell Physiol. 2004;286:C987–C997. doi: 10.1152/ajpcell.00522.2003. [DOI] [PubMed] [Google Scholar]
- Wallez Y, Cand F, Cruzalegui F, Wernstedt C, Souchelnytskyi S, Vilgrain I, Huber P. Src kinase phosphorylates vascular endothelial-cadherin in response to vascular endothelial growth factor: identification of tyrosine 685 as the unique target site. Oncogene. 2007;26:1067–1077. doi: 10.1038/sj.onc.1209855. [DOI] [PubMed] [Google Scholar]
- Wallez Y, Huber P. Endothelial adherens and tight junctions in vascular homeostasis, inflammation and angiogenesis. Biochim Biophys Acta. 2008;1778:794–809. doi: 10.1016/j.bbamem.2007.09.003. [DOI] [PubMed] [Google Scholar]
- Xiao K, Allison DF, Buckley KM, Kottke MD, Vincent PA, Faundez V, Kowalczyk AP. Cellular levels of p120 catenin function as a set point for cadherin expression levels in microvascular endothelial cells. J Cell Biol. 2003a;163:535–545. doi: 10.1083/jcb.200306001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao K, Allison DF, Kottke MD, Summers S, Sorescu GP, Faundez V, Kowalczyk AP. Mechanisms of VE-cadherin processing and degradation in microvascular endothelial cells. J Biol Chem. 2003b;278:19199–19208. doi: 10.1074/jbc.M211746200. [DOI] [PubMed] [Google Scholar]
- Yamada S, Pokutta S, Drees F, Weis WI, Nelson WJ. Deconstructing the cadherin-catenin-actin complex. Cell. 2005;123:889–901. doi: 10.1016/j.cell.2005.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JY, Zong CS, Xia W, Wei Y, Ali-Seyed M, Li Z, Broglio K, Berry DA, Hung MC. MDM2 promotes cell motility and invasiveness by regulating E-cadherin degradation. Mol Cell Biol. 2006;26:7269–7282. doi: 10.1128/MCB.00172-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.