

ABSTRACT
Wilson disease (WD), a genetic disorder affecting copper transport, is characterized by hepatic and neurological manifestations with variable and often unpredictable presentation. Global DNA methylation in liver was previously modified by dietary choline in tx-j mice, a spontaneous mutant model of WD. We therefore hypothesized that the WD phenotype and hepatic gene expression of tx-j offspring could be modified by maternal methyl supplementation during pregnancy. In an initial experiment, female tx-j mice or wild type mice were fed control or choline-supplemented diets 2 weeks prior to mating through embryonic day 17. Transcriptomic analysis (RNA-seq) on embryonic livers revealed tx-j-specific differences in genes related to oxidative phosphorylation, mitochondrial dysfunction, and the neurological disorders Huntington's disease and Alzheimer disease. Maternal choline supplementation restored the transcript levels of a subset of genes to wild type levels. In a separate experiment, a group of tx-j offspring continued to receive choline-supplemented or control diets, with or without the copper chelator penicillamine (PCA) for 12 weeks until 24 weeks of age. Combined choline supplementation and PCA treatment of 24-week-old tx-j mice was associated with increased liver transcript levels of methionine metabolism and oxidative phosphorylation-related genes. Sex differences in gene expression within each treatment group were also observed. These results demonstrate that the transcriptional changes in oxidative phosphorylation and methionine metabolism genes in WD that originate during fetal life are, in part, prevented by prenatal maternal choline supplementation, a finding with potential relevance to preventive treatments of WD.
KEYWORDS: Copper, choline, mitochondria, oxidative phosphorylation, toxic-milk mouse, Wilson Disease
Abbreviations
- WD
Wilson disease
- PCA
penicillamine
- SAHH
S-adenosylhomocysteine hydrolase
- SAH
S-adenosylhomocysteine
- SAM
S-adenosylmethionine
- DNMT
DNA methyltransferase
- ALT
alanine transaminase
- Srebf1
sterol regulatory element binding transcription factor 1
- Ndufab1
NADH dehydrogenase (ubiquinone) 1 α/β subcomplex, 1
- Ndufb5
NADH dehydrogenase (ubiquinone) 1 β subcomplex, 5
- Sdha
succinate dehydrogenase complex, subunitA, flavoprotein
- Cox5a
cytochrome c oxidase subunit Va
- Atp5j
ATP synthase, H+ transporting, mitochondrial F0 complex, subunit F
- THF
tetrahydrofolate
Introduction
Wilson disease (WD) is a genetic disorder caused by mutations of the ATP7B copper transporter gene leading to copper accumulation in the liver and brain. Phenotypic presentation of WD is often highly variable and involves varying degrees of hepatic and/or neurological symptoms.1,2 Growing evidence indicates that the clinical presentation of WD is under the influence of both genetic and epigenetic factors,3-5 which may account for the high phenotypic variability. The hepatic involvement in WD ranges from mildly symptomatic to steatosis, cirrhosis, or acute liver failure. Neurological involvement is characterized by a movement disorder with Huntington's disease-like rigidity and tremors. Previous studies from our group demonstrated that tx-j mice, a model of WD, are characterized by global DNA hypomethylation and changes in hepatic gene expression, which are responsive to the provision of methyl groups through betaine or choline supplementation during both fetal and adult life.3,4
The observed DNA hypomethylation may be due, at least in part, to impaired methionine metabolism caused by hepatic copper accumulation. S-adenosylmethionine (SAM) is the main methyl donor for transmethylation reactions of DNA, RNA, histones, and proteins. S-adenosylhomocysteine (SAH) is converted into homocysteine by the bi-directional enzyme S-adenosylhomocysteine hydrolase (SAHH). If SAH is in excess, it can inhibit methylation reactions involving SAM; therefore, maintaining a balance between SAM and SAH levels is crucial for sustained gene expression and activities of DNA methyltransferases (DNMTs) and, consequently, for DNA methylation reactions. Copper accumulation has been associated with reduced protein levels6 and enzymatic activity7 of SAHH. Therefore, it is likely that changes in SAHH levels associated with copper accumulation will affect SAH levels and SAM:SAH ratios as well as DNA and histone methylation reactions, gene expression regulation, and the disease phenotype.8
Deficiencies in one-carbon metabolism or dietary methyl donors during fetal development have important pre- and post-natal consequences. Early stages of gestation and implantation are characterized by an increased demand for methyl groups, such as those found in dietary choline, betaine, or folic acid. During embryogenesis, DNA methylation confers a stable and heritable component of epigenetic markers related to cell fate and gene expression, and genome-wide DNA methylation reprogramming in the zygote can establish new methylation patterns.9,10 Environmental factors, such as oxidative stress or changes in nutrient availability during fetal development, can affect the expression of Dnmts and consequent reprogramming of DNA methylation resulting in subtle but long-lasting effects on crucial metabolic pathways.11 Importantly, the interaction of genetic susceptibility with environmental stressors affecting one-carbon metabolism during early gestational life can be carried into adult life and modify the disease penetrance.12
Given the phenotypic variability of clinical WD and the demonstrated changes in methionine metabolism in fetal and adult livers of animal models of hepatic copper accumulation, we hypothesized that maternal provision of methyl-group donor choline in a murine model of WD from pre-gestational phase, through gestation, and then continued in the offspring until 24 weeks of age (Fig. 1) would affect hepatic disease presentation and progression, as indicated by liver histology, DNA methylation, changes in select gene transcript levels, and response to the copper chelator penicillamine (PCA).
Figure 1.
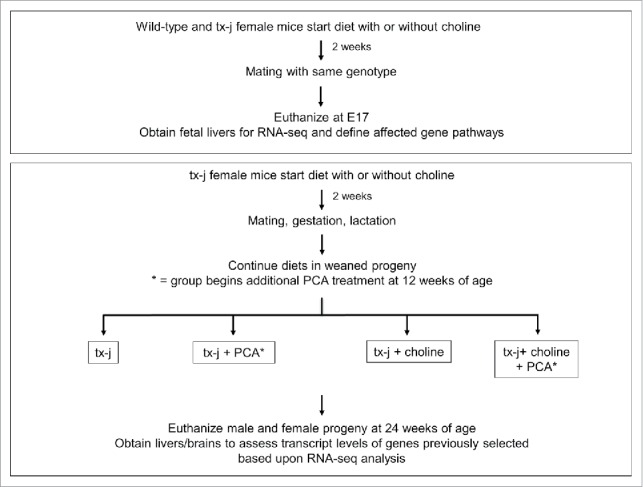
Study design. Wild type or tx-j mice were fed diets with or without choline supplementation 2 weeks before mating through embryonic day 17. Fetal livers were harvested and RNA-seq was performed. Another set of tx-j offspring were weaned and continued on diets with or without choline supplementation. Subgroups of tx-j offspring on each diet were given penicillamine (PCA) treatment.
Results
Fetal liver transcriptome analyses reveal genomic effects of tx-j mutation and choline supplementation
Given previous evidence that gene transcript levels related to methionine metabolism and DNA methylation are profoundly dysregulated in tx-j mice,3 we conducted an initial experiment in fetal liver that surveyed genome-wide RNA-seq analyses to identify differentially expressed genes in tx-j compared to wild type or tx-j with choline supplementation. Principal component analysis scores plot shows that the transcriptomes of tx-j mice with or without maternal choline supplementation are distinct based on differentially expressed fetal liver genes (Fig. 2). Gene pathway analyses of fetal liver transcriptomes revealed significant differences in gene transcript levels predominantly related to ubiquitination, protein degradation, oxidative phosphorylation, and the neurological disorders Huntington's and Alzheimer disease (Table 1). Individual genes from the top 4 pathways are shown in Table 2. We were interested in further charactering changes in oxidative phosphorylation because patients and animal models with WD often present with altered mitochondrial morphology, which may impact mitochondrial function. Oxidative phosphorylation pathway-related genes included nuclear-encoded subunits of NADH dehydrogenase (Complex I), succinate dehydrogenase complex (Complex II), ubiquinol-cytochrome c reductase (Complex III), cytochrome c oxidase (Complex IV), and nuclear-encoded subunits of mitochondria ATP synthase (Complex V). Maternal choline supplementation returned transcript levels of these genes (Table 3) in tx-j fetal livers to levels observed in fetal livers of wild type mice. A complete list of genes expressed in wild type vs. tx-j fetal livers and in tx-j vs. tx-j fetal livers after maternal choline supplementation are presented in Supplemental Table 1.
Figure 2.
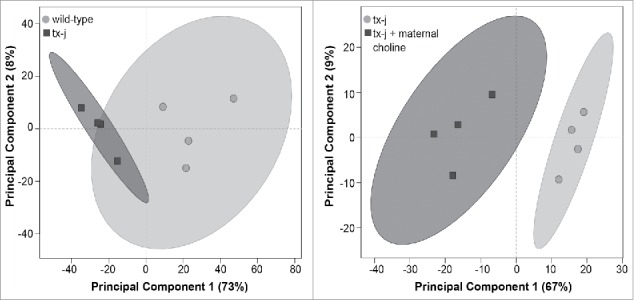
Principal component analysis scores plot of significantly differentially expressed genes in fetal liver. Each symbol represents a mouse. Symbols that are closer to one another are more similar. Ellipses represent 95% confidence intervals based on Hotelling's T2 statistic.
Table 1.
Top pathways identified by WebGestalt Pathway Analysis in fetal livers.
| Wild type vs. tx-j | ||
|---|---|---|
| Pathway | Number of genes represented in pathway | Adjusted P-value |
| Protein processing in endoplasmic reticulum | 11% (19/169) | >0.0001 |
| Ubiquitin mediated proteolysis | 12% (17/140) | >0.0001 |
| Lysosome | 12% (15/123) | >0.0001 |
| Huntington's disease | 9% (18/197) | >0.0001 |
| Purine metabolism | 10% (16/168) | >0.0001 |
| Alzheimer's disease | 9% (16/188) | >0.0001 |
| Oxidative phosphorylation | 10% (14/147) | >0.0001 |
| Vasopressin-regulated water reabsorption | 19% (8/43) | >0.0001 |
| Focal adhesion |
8% (16/200) |
>0.0001 |
| tx-j vs. tx-j + maternal choline | ||
| Pathway |
Number of genes represented in pathway |
Adjusted P-value |
| Oxidative phosphorylation | 6% (9/147) | >0.0001 |
| Alzheimer's disease | 5% (10/188) | >0.0001 |
| p53 signaling pathway | 9% (6/70) | >0.0001 |
| Proteasome | 11% (5/45) | >0.0001 |
| Huntington's disease | 4% (8/197) | 0.0006 |
| Lysosome | 5% (6/123) | 0.001 |
| Focal adhesion | 4% (7/200) | 0.0021 |
| GnRH signaling pathway | 5% (5/99) | 0.0021 |
| Phagosome | 3% (6/176) | 0.0042 |
Table 2A.
Top four pathways identified by WebGestalt Pathway Analysis in fetal livers.
| Wild type vs. tx-j | ||||
|---|---|---|---|---|
| Pathway | Wild type Mean RPKM | tx-j Mean RPKM | Percent Difference (Wild type relative to tx-j) | |
| Protein processing in endoplasmic reticulum (KEGG Pathway 04141) | ||||
| C = 169; O = 19; E = 3.36; R = 5.65; rawP = 1.52e-09; adjP = 4.71e-08 | ||||
| Tusc3 | tumor suppressor candidate 3 | 15.1 | 21.5 | 43% |
| Dnaja1 | DnaJ (Hsp40) homolog, subfamily A, member 1 | 11.6 | 16.4 | 41% |
| Hsph1 | heat shock 105kDa/110kDa protein 1 | 8.5 | 11.5 | 35% |
| Ube2e2 | ubiquitin-conjugating enzyme E2E 2 | 4.1 | 5.5 | 34% |
| Casp12 | caspase 12 | 0.2 | 0.3 | 31% |
| Map3k5 | mitogen-activated protein kinase kinase kinase 5 | 2.5 | 3.2 | 30% |
| Ube2d2a | ubiquitin-conjugating enzyme E2D 2A | 34.0 | 43.9 | 29% |
| Ube2g1 | ubiquitin-conjugating enzyme E2G 1 | 10.6 | 13.6 | 28% |
| Rbx1 | ring-box 1 | 21.0 | 25.9 | 24% |
| Dnajc10 | DnaJ (Hsp40) homolog, subfamily C, member 10 | 16.0 | 19.5 | 22% |
| Mbtps2 | membrane-bound transcription factor peptidase, site 2 | 2.3 | 2.8 | 22% |
| Bcap31 | B cell receptor associated protein 31 | 46.7 | 55.1 | 18% |
| Ube2e1 | ubiquitin-conjugating enzyme E2E 1 | 14.0 | 16.3 | 17% |
| Sar1a | SAR1 gene homolog A (S. cerevisiae) | 29.0 | 33.8 | 16% |
| Ern1 | endoplasmic reticulum (ER) to nucleus signalling 1 | 6.7 | 5.5 | −18% |
| Rnf5 | ring finger protein 5 | 27.1 | 21.5 | −20% |
| Sil1 | endoplasmic reticulum chaperone SIL1 homolog | 10.0 | 7.9 | −20% |
| Atf6b | activating transcription factor 6 β | 4.6 | 3.3 | −27% |
| Os9 | amplified in osteosarcoma | 25.7 | 18.6 | −28% |
| Ubiquitin mediated proteolysis (KEGG Pathway 04120) | ||||
| C = 140; O = 17; E = 2.78; R = 6.10; rawP = 3.35e-09; adjP = 6.92e-08 | ||||
| Ube2e2 | ubiquitin-conjugating enzyme E2E 2 | 4.1 | 5.5 | 34% |
| Klhl9 | kelch-like 9 (Drosophila) | 9.6 | 12.6 | 31% |
| Ube2d2a | ubiquitin-conjugating enzyme E2D 2A | 34.0 | 43.9 | 29% |
| Birc2 | baculoviral IAP repeat-containing 2 | 8.1 | 10.3 | 28% |
| Ube2g1 | ubiquitin-conjugating enzyme E2G 1 | 10.6 | 13.6 | 28% |
| Rchy1 | ring finger and CHY zinc finger domain containing 1 | 11.8 | 14.9 | 27% |
| Itch | itchy, E3 ubiquitin protein ligase | 13.9 | 17.5 | 25% |
| Rbx1 | ring-box 1 | 21.0 | 25.9 | 24% |
| Vhl | von Hippel-Lindau tumor suppressor | 4.9 | 5.9 | 21% |
| Xiap | X-linked inhibitor of apoptosis | 17.4 | 20.5 | 18% |
| Ube2e1 | ubiquitin-conjugating enzyme E2E 1 | 14.0 | 16.3 | 17% |
| Klhl13 | kelch-like 13 (Drosophila) | 10.9 | 12.6 | 16% |
| Wwp2 | WW domain containing E3 ubiquitin protein ligase 2 | 9.7 | 8.3 | −14% |
| Tceb2 | transcription elongation factor B (SIII), polypeptide 2 | 33.8 | 24.9 | −26% |
| Ube2o | ubiquitin-conjugating enzyme E2O | 20.3 | 14.9 | −27% |
| Cdc34 | cell division cycle 34 | 23.6 | 16.3 | −31% |
| Fzr1 | fizzy/cell division cycle 20 related 1 (Drosophila) | 15.2 | 9.5 | −38% |
| Lysosome (KEGG Pathway 04142) | ||||
| C = 123; O = 15; E = 2.45; R = 6.13; rawP = 2.62e-08; adjP = 4.06e-07 | ||||
| Ctsk | cathepsin K | 0.7 | 1.2 | 76% |
| Hexb | hexosaminidase B | 18.3 | 24.1 | 32% |
| Atp6v1h | ATPase, H+ transporting, lysosomal V1 subunit H | 12.1 | 15.9 | 31% |
| Gla | galactosidase, α | 1.8 | 2.4 | 30% |
| Ap4e1 | adaptor-related protein complex AP-4, ϵ 1 | 2.2 | 2.9 | 29% |
| Ppt1 | palmitoyl-protein thioesterase 1 | 9.6 | 11.9 | 24% |
| Lamp2 | lysosomal-associated membrane protein 2 | 43.1 | 52.2 | 21% |
| Laptm4a | lysosomal-associated protein transmembrane 4A | 56.1 | 67.6 | 21% |
| Cd164 | CD164 antigen | 66.5 | 79.5 | 20% |
| Asah1 | N-acylsphingosine amidohydrolase 1 | 22.1 | 25.8 | 17% |
| Manba | mannosidase, beta A, lysosomal | 4.5 | 5.2 | 17% |
| Cd63 | CD63 antigen | 21.0 | 24.4 | 16% |
| Gusb | glucuronidase, beta | 10.4 | 12.1 | 16% |
| Napsa | napsin A aspartic peptidase | 2.8 | 2.2 | −22% |
| Abcb9 | ATP-binding cassette, sub-family B (MDR/TAP), member 9 | 0.4 | 0.2 | −47% |
| Huntington's disease (KEGG Pathway 05016) | ||||
| C = 197; O = 18; E = 3.92; R = 4.59; rawP = 1.04e-07; adjP = 1.29e-06 | ||||
| Atp5g3 | ATP synthase, H+ transporting, mitochondrial F0 complex, subunit C3 (subunit 9) | 172.2 | 237.1 | 38% |
| Ndufb6 | NADH dehydrogenase 1 beta subcomplex, 6 | 25.9 | 34.2 | 32% |
| Cox5a | cytochrome c oxidase, subunit Va | 67.1 | 88.2 | 32% |
| Tfam | transcription factor A, mitochondrial | 9.0 | 11.8 | 31% |
| Ndufs4 | NADH dehydrogenase Fe-S protein 4 | 22.1 | 28.5 | 29% |
| Vdac3 | voltage-dependent anion channel 3 | 64.0 | 80.0 | 25% |
| Ndufab1 | NADH dehydrogenase 1, alpha/beta subcomplex, 1 | 8.7 | 10.6 | 22% |
| Ndufb2 | NADH dehydrogenase 1 beta subcomplex, 2 | 4.3 | 5.2 | 20% |
| Atp5j | ATP synthase, H+ transporting, mitochondrial F0 complex, subunit F | 86.2 | 102.1 | 18% |
| Ndufc1 | NADH dehydrogenase 1, subcomplex unknown, 1 | 15.1 | 17.7 | 17% |
| Uqcrc2 | ubiquinol cytochrome c reductase core protein 2 | 102.6 | 119.8 | 17% |
| Ndufb5 | NADH dehydrogenase 1 beta subcomplex, 5 | 20.3 | 23.6 | 16% |
| Ap2a1 | adaptor protein complex AP-2, alpha 1 subunit | 9.6 | 7.5 | −22% |
| Gpx1 | glutathione peroxidase 1 | 474.7 | 331.8 | −30% |
| Dctn1 | dynactin 1 | 5.3 | 3.5 | −33% |
| Bbc3 | BCL2 binding component 3 | 1.9 | 1.2 | −36% |
| Ndufs7 | NADH dehydrogenase Fe-S protein 7 | 9.3 | 5.7 | −39% |
| Polr2i | polymerase (RNA) II (DNA directed) polypeptide I | 4.4 | 2.6 | −42% |
| Number of reference genes in the category (C) | ||||
| Number of differentially expressed genes in reference category (O) | ||||
| Expected number in the category (E) | ||||
| Ratio of enrichment (F) | ||||
| P-value from hypergeometric test (rawP) | ||||
| P-value adjusted by the multiple test adjustment (adjP) | ||||
| RPKM: Reads per kilobase of transcript per million mapped reads | ||||
| *Percent difference = [(tx-j - wild type)/wild type]*100 | ||||
Table 2B.
Top four pathways identified by WebGestalt Pathway Analysis.
| tx-j vs tx-j + maternal choline supplementation | ||||
|---|---|---|---|---|
| Pathway | tx-j Mean RPKM | tx-j + maternal choline Mean RPKM | Percent Difference (tx-j relative to tx-j + maternal choline) | |
| Oxidative phosphorylation (KEGG Pathway 00190) | ||||
| C = 147; O = 9; E = 1.15; R = 7.85; rawP = 2.61e-06; adjP = 1.48e-05 | ||||
| Atp6v1a | ATPase, H+ transporting, lysosomal V1 subunit A | 18.5 | 16.1 | −13% |
| Uqcrfs1 | ubiquinol-cytochrome c reductase, Rieske iron-sulfur polypeptide 1 | 77.4 | 66.3 | −14% |
| Ndufb6 | NADH dehydrogenase (ubiquinone) 1 β subcomplex, 6 | 34.2 | 28.6 | −16% |
| Atp6v1b2 | ATPase, H+ transporting, lysosomal V1 subunit B2 | 35.2 | 29.4 | −17% |
| Cox17 | cytochrome c oxidase, subunit XVII assembly protein homolog (yeast) | 5.0 | 4.2 | −17% |
| Atp6v1h | ATPase, H+ transporting, lysosomal V1 subunit H | 15.9 | 13.1 | −18% |
| Atp5g3 | ATP synthase, H+ transporting, mitochondrial F0 complex, subunit C3 (subunit 9) | 237.1 | 189.9 | −20% |
| Cox5a | cytochrome c oxidase, subunit Va | 88.2 | 69.8 | −21% |
| Cox11 | COX11 homolog, cytochrome c oxidase assembly protein (yeast) | 5.5 | 3.8 | −30% |
| Alzheimer's disease (KEGG Pathway 05010) | ||||
| C = 188; O = 10; E = 1.47; R = 6.82; rawP = 2.60e-06; adjP = 1.48e-05 | ||||
| Ern1 | endoplasmic reticulum (ER) to nucleus signalling 1 | 5.5 | 6.8 | 24% |
| Uqcrfs1 | ubiquinol-cytochrome c reductase, Rieske iron-sulfur polypeptide 1 | 77.4 | 66.3 | −14% |
| Casp9 | caspase 9 | 6.4 | 5.4 | −15% |
| Ndufb6 | NADH dehydrogenase (ubiquinone) 1 beta subcomplex, 6 | 34.2 | 28.6 | −16% |
| Itpr1 | inositol 1,4,5-trisphosphate receptor 1 | 3.7 | 3.1 | −17% |
| Aph1a | anterior pharynx defective 1a homolog (C. elegans) | 13.6 | 11.0 | −19% |
| Bace2 | beta-site APP-cleaving enzyme 2 | 1.2 | 1.0 | −19% |
| Atp5g3 | ATP synthase, H+ transporting, mitochondrial F0 complex, subunit C3 (subunit 9) | 237.1 | 189.9 | −20% |
| Casp7 | caspase 7 | 6.6 | 5.2 | −21% |
| Cox5a | cytochrome c oxidase, subunit Va | 88.2 | 69.8 | −21% |
| p53 signaling pathway ID:04115 | ||||
| C = 70; O = 6; E = 0.55; R = 11.00; rawP = 1.87e-05; adjP = 7.95e-05 | ||||
| Steap3 | STEAP family member 3 | 13.4 | 11.5 | −14% |
| Casp9 | caspase 9 | 6.4 | 5.4 | −15% |
| Rchy1 | ring finger and CHY zinc finger domain containing 1 | 14.9 | 12.0 | −20% |
| Perp | PERP, TP53 apoptosis effector | 15.8 | 12.5 | −21% |
| Igf1 | insulin-like growth factor 1 | 3.0 | 2.3 | −24% |
| Sesn3 | sestrin 3 | 3.2 | 2.4 | −25% |
| Proteasome (KEGG Pathway 03050) | ||||
| C = 45; O = 5; E = 0.35; R = 14.25; rawP = 2.66e-05; adjP = 9.04e-05 | ||||
| Pomp | proteasome maturation protein | 74.6 | 63.6 | −15% |
| Psmf1 | proteasome inhibitor subunit 1 | 7.2 | 6.1 | −15% |
| Psmc5 | protease 26S subunit, ATPase 5 | 33.3 | 26.7 | −20% |
| Psmd13 | proteasome 26S subunit, non-ATPase, 13 | 19.6 | 15.5 | −21% |
| Psmd2 | proteasome 26S subunit, non-ATPase, 2 | 49.3 | 38.5 | −22% |
| Number of reference genes in the category (C) | ||||
| Number of differentially expressed genes in reference category (O) | ||||
| Expected number in the category (E) | ||||
| Ratio of enrichment (F) | ||||
| P-value from hypergeometric test (rawP) | ||||
| P-value adjusted by the multiple test adjustment (adjP) | ||||
| *Percent difference = [(tx-j - tx-j+maternal choline)/tx-j]*100 | ||||
RPKM: Reads per kilobase of transcript per million mapped reads
Table 3.
Fetal RNA-seq transcript levels for select oxidative phosphorylation genes.
| Gene | Wild type | Wild type + maternal choline | tx-j | tx-j + maternal choline |
|---|---|---|---|---|
| Nuclear encoded subunit of NADH dehydrogenase (Complex I) | ||||
| Ndufa4 | 205.34 | 205.27 | 232.51 | 224.94 |
| Ndufa9 | 54.45 | 59.13 | 60.06 | 54.44 |
| Ndufab1 | 8.68a | 8.89a,b | 10.60b | 9.31a,b |
| Ndufb2 | 4.35a | 4.28a | 5.20b | 4.66a,b |
| Ndufb5 | 20.28a | 21.10a | 23.59b | 21.58a,b |
| Ndufb6 | 25.90a | 25.59a | 34.22b | 28.58a |
| Ndufc1 | 15.08 | 15.59 | 17.68 | 16.87 |
| Ndufc2 | 31.24 | 32.61 | 35.81 | 31.72 |
| Ndufs4 | 22.05a | 23.55a | 28.46b | 26.15b |
| Ndufs7 | 9.31 | 9.67 | 5.68 | 7.30 |
| Nuclear encoded subunit of succinate dehydrogenase (Complex II) | ||||
| Sdha | 74.36a,b | 76.68a | 77.06a | 67.80b |
| Nuclear encoded subunits of ubiquinol-cytochrome c reductase (Complex III) | ||||
| Uqcrc2 | 102.64 | 109.76 | 119.80 | 112.96 |
| Uqcrfs1 | 68.13a,b | 65.85a | 77.45b | 66.28a |
| Nuclear encoded subunits of cytochrome c oxidase (Complex IV) | ||||
| Cox17 | 3.80a | 4.50a,b | 5.02b | 4.17a |
| Cox5a | 67.06a | 64.76a | 88.23b | 69.81a |
| Nuclear encoded subunits of mitochondrial ATP synthase (Complex V) | ||||
| Atp5f1 | 139.19 | 140.28 | 152.81 | 146.87 |
| Atp5g3 | 172.22a | 187.34a | 237.14b | 189.94a |
| Atp5j | 86.24a | 84.33b | 102.15b | 89.90a,b |
| Atp5l | 135.50 | 131.52 | 149.37 | 135.43 |
| Atp6v1a | 15.31a | 17.00a,b | 18.52b | 16.07a,b |
| Atp6v1h | 12.12a | 13.85a,b | 15.93b | 13.07a,b |
Values are expressed as average reads per kilobase of exon per million reads mapped (RPKM). Between group differences were analyzed by 1-way ANOVA; values with different letter symbols are significantly different (P < 0.05) from each other.
Tx-j phenotypes in adulthood and response to choline and penicillamine treatment
To determine the effects of choline and PCA treatments on phenotypes of disease pathology, 24-week old tx-j mice within the 4 treatment groups (Fig. 1) were characterized in terms of anthropometric parameters: liver histology and ALT; hepatic copper, iron, and zinc levels; hepatic SAM and SAH levels. Mild chronic hepatic involvement with mild steatosis and inflammatory infiltrate was observed in untreated tx-j mice, with significantly greater inflammation in male mice compared to females (Fig. 3). Female mice treated with PCA revealed reduced hepatic steatosis compared to all other treatment groups (Fig. 3).
Figure 3.
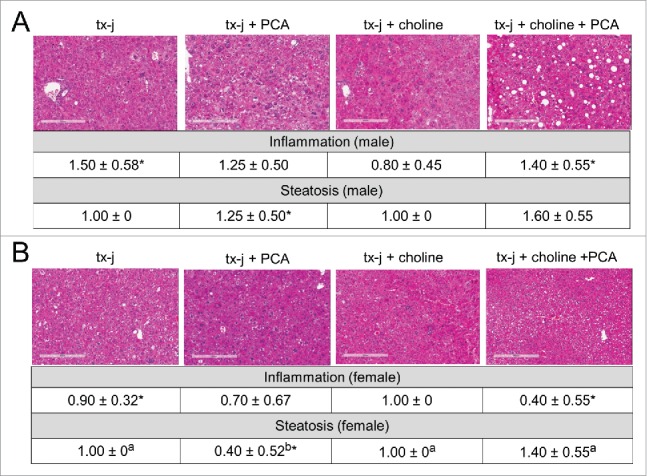
Liver histology from tx-j mice at 24 weeks of age. Hematoxylin and eosin, 100 X. Untreated tx-j mice presented mild inflammation and steatosis with more inflammation occurring in males compared to females in the same diet group. Steatosis improved in female tx-j mice after penicillamine (PCA) treatment. Whereas choline did not change histology scores, the combination of PCA and choline was associated with higher grade of steatosis compared to all other groups. Results are expressed as mean ± standard deviation. Between group differences were analyzed by 1-way ANOVA; values with different letter symbols are significantly different (P < 0.05) from each other. Differences between sexes in the same treatment group were analyzed by Student's t-test; values with an asterisk (*) are significantly different (P < 0.05) between sexes.
Hepatic copper, iron, and zinc concentrations were reduced in male mice treated with PCA alone and, surprisingly plasma ALT levels were increased in these same mice (Fig. 4 and Supplemental Tables 2–4). There was no change in copper concentration among female mice and a significant increase in iron with PCA alone or in combination with choline. Hepatic zinc concentration was increased in female mice treated with the combination of PCA and choline (Fig. 4 and Supplemental Tables 2–4).
Figure 4.
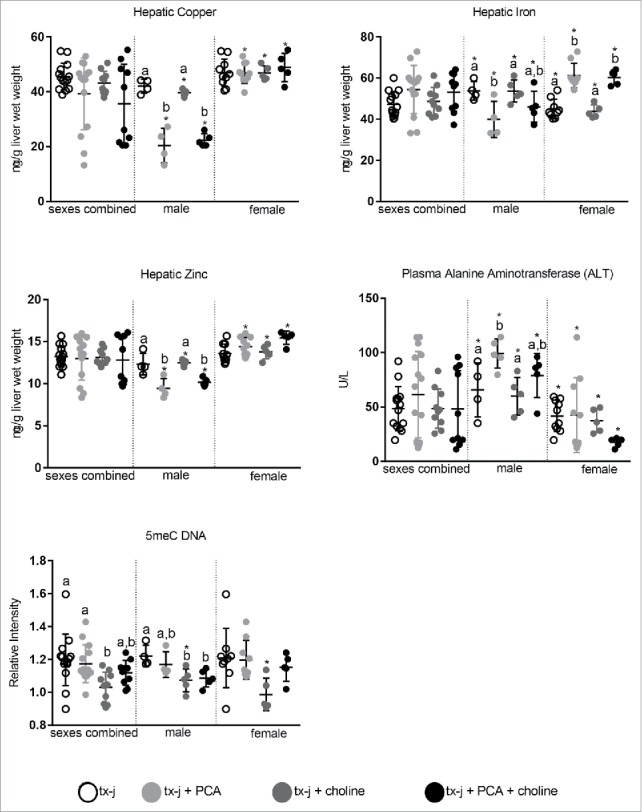
Hepatic copper, iron zinc concentrations, plasma triglycerides and ALT, and global hepatic DNA methylation. Results are expressed as mean ± standard deviation. Between group differences were analyzed by 1-way ANOVA; values with different letter symbols are significantly different (P < 0.05) from each other. Differences between sexes in the same treatment group were analyzed by Student's t-test; values with an asterisk (*) are significantly different (P < 0.05) between sexes.
PCA treatment was not associated with either changes in SAM, SAH, or SAM/SAH ratio, or global hepatic DNA methylation (Supplemental Table 2–4). Choline supplementation alone was characterized by a non-significant increase in hepatic SAM levels in both males and females. Even though choline supplementation did not affect hepatic histology in mice of either sex compared to untreated mice, a significant reduction of the nuclear to cell diameter ratio was observed when both sexes were combined (Supplemental Table 2). This is particularly relevant since tx-j mice present enlarged hepatocyte and nuclear diameter, which may be attributed to impaired cell cycle machinery.4,13 Global hepatic DNA methylation was reduced in male mice treated with choline alone and in combination with PCA compared to untreated male mice; no difference among females was observed (Fig. 4 and Supplemental Tables 2–4). There was a significant correlation between global DNA methylation and SAM levels when results from each sex were combined (r = −0.50; P = 0.0003).
To summarize, 24-week-old tx-j mice presented an overall mild phenotype with minimal hepatic histology involvement and marked enlargement of hepatocytes and nuclear diameter that was corrected by choline supplementation. Male and female mice responded differently to PCA and choline supplementation in terms of hepatic trace elements and global DNA methylation.
Changes in transcript levels of genes related to oxidative phosphorylation and neurological disease pathways persist in adult livers
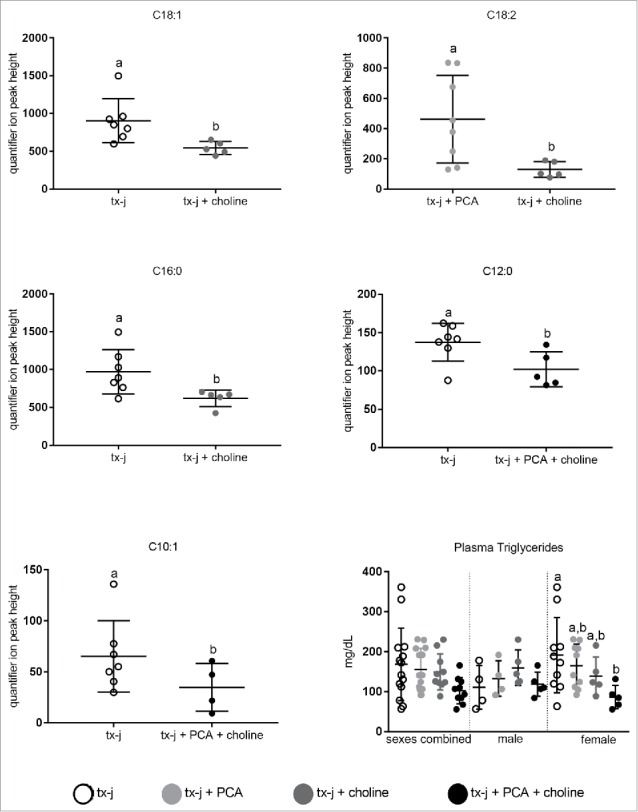
A select number of nuclear-encoded genes related to oxidative phosphorylation and overlapping neurological disease pathways that had shown a response to choline supplementation in fetal livers (Table 3) were examined in adult livers (Fig. 5). The most relevant effect on gene transcript levels studied in this pathway was achieved by the combination of PCA and choline but, remarkably, only in female mice. The combination of PCA and choline in female mice significantly increased transcript levels compared to untreated tx-j mice, whereas the provision of PCA and choline separately was not associated with a significant change. These results show for the first time the potential for a sex-specific synergistic effect of copper chelation and provision of methyl groups. Western blot analysis revealed that hepatic protein levels of SDHA were lowest in untreated tx-j mice and significantly greater in mice treated with PCA (Fig. 5). To explore the functional significance of the changes in oxidative phosphorylation genes we assessed plasma acylcarnitines. Altered levels of acylcarnitines indicate impaired β-oxidation and are considered a marker of mitochondrial function. Elevated levels of plasma acylcarnitines were observed in untreated tx-j mice compared to other treatment groups (Fig. 6). Plasma triglycerides were significantly greater in untreated female tx-j mice compared to female tx-j mice treated with the combination of PCA and choline (Fig. 6). No difference in plasma triglycerides was observed in male mice. In summary, female mice present changes in transcript levels of genes related to oxidative phosphorylation after PCA and choline treatment combined. Plasma acylcarnitines and triglycerides levels both improved in female mice after treatment.
Figure 5.
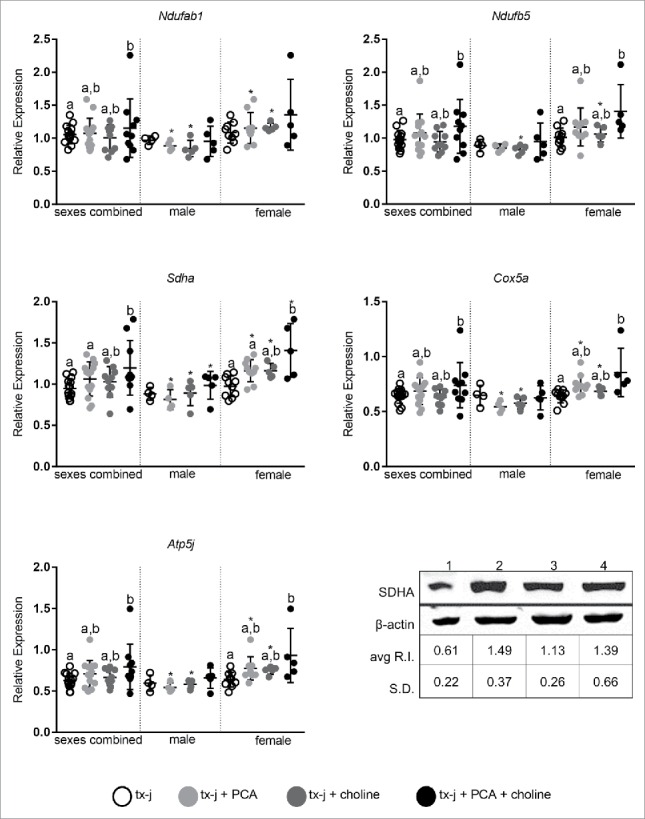
Transcript levels of genes representative of oxidative phosphorylation (Ndufab1, Ndufb5, Sdha, Cox5a, Atp5j). Transcript expression data are normalized to Gapdh (10–22 mice/group). Protein relative expression (R.I.) according to Western blots analyses for SDHA are reported; lane 1: tx-j; lane 2: tx-j + PCA; lane 3: tx-j + choline; lane 4: tx-j + choline + PCA. Results are expressed as mean ± standard deviation. Between group differences were analyzed by 1-way ANOVA; values with different letter symbols are significantly different (P< 0.05) from each other. Differences between sexes in the same treatment group were analyzed by Student's t-test; values with an asterisk (*) are significantly different (P < 0.05) between sexes.
Figure 6.
Plasma acylcarnitines and plasma triglycerides. Results are expressed as mean ± standard deviation. Plasma Acylcarnitines: Significant differences between 2 groups were assessed by Student's t-test. Values with different letter symbols are significantly different (P< 0.05) from each other. Plasma Triglycerides: Between group differences were analyzed by 1-way ANOVA; values with different letter symbols are significantly different (P < 0.05) from each other.
Transcript levels of genes related to DNA methylation and lipogenesis are affected by sex and diet
We first focused on genes related to DNA methylation and lipogenesis, since both can be affected by maternal diet and the availability of methyl groups.14,15 Our group and others have previously demonstrated downregulation of Sahh transcript and protein levels in animal models of hepatic copper accumulation compared to healthy controls.3,6 In the present study, Sahh transcript levels were increased almost 3-fold by choline supplementation, both in male and female mice (Fig. 7). Female mice responded differently to choline and PCA than male mice. Dnmt transcript levels did not change with treatment in male mice, whereas PCA supplementation or choline supplementation alone in females was associated with downregulation of Dnmt1 hepatic transcript levels without modifying hepatic copper concentration (Fig. 4). In females, Dnmt3a transcript levels increased more from combined PCA and choline treatment, whereas Dnmt3b increased in response to choline alone. Transcript levels of Srebf1 increased almost 2-fold after PCA supplementation in female mice, whereas the combination of PCA and choline was more effective in male mice in increasing Srebf1 levels. Western blot analysis revealed increased hepatic protein levels of SAHH in mice treated with PCA alone or in combination with choline and there was no difference in SREBF1 among treatment groups. To summarize, transcript levels of key genes related to DNA methylation change in female mice in response to PCA and/or choline.
Figure 7.
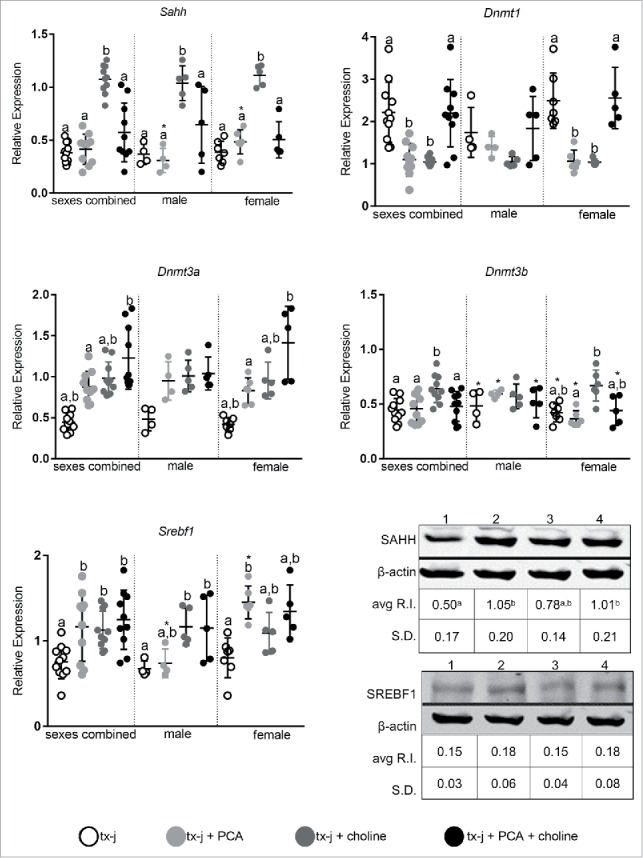
Transcript levels of genes representative of methionine metabolism, DNA methylation (Sahh, Dnmt1, Dnmt3a, Dnmt3b), and lipid metabolism (Srebf1). Transcript expression data are normalized to Gapdh (10–14 mice/group). Protein relative expression (R.I.) according to Western blot analyses for SAHH and SREBP1 are reported; lane 1: tx-j; lane 2: tx-j + PCA; lane 3: tx-j + choline; lane 4: tx-j + choline + PCA. Results are expressed as mean ± standard deviation. Between group differences were analyzed by 1-way ANOVA; values with different letter symbols are significantly different (P < 0.05) from each other. Differences between sexes in the same treatment group were analyzed by Student's t-test; values with an asterisk (*) are significantly different (P < 0.05) between sexes. PCA, penicillamine.
Discussion
Clinical observations and experimental evidence indicate that factors in addition to ATP7B gene mutations, including epigenetic mechanisms, are involved in the pathogenesis of WD and its various phenotypic presentations.4,16,17 Our study supplemented the methyl donor choline in the diets of tx-j mice, an animal model of WD, and found several novel changes in offspring liver phenotype and gene expression patterns. First, fetal liver gene expression of oxidative phosphorylation pathway genes were significantly upregulated in tx-j mice and were modified by choline supplementation. Second, dietary choline supplementation in adult tx-j mice modified transcript levels of genes related to DNA methylation (Dnmt1 and Srebf1) similarly to PCA but without having any effect on copper concentration. Third, there were sex differences in response to PCA and choline supplementation in adult tx-j mice validating the use of this model to explore mechanisms behind the sex phenotypic differences observed in patients with WD.18
In addition to epigenetic regulation changes, there is evidence for mitochondrial dysfunction in WD. Patients with WD19 and animal models of copper accumulation20 exhibit aberrant hepatocyte mitochondrial morphologies (e.g., pleomorphism, dilated and elongated cristae, and changes in matrix density), which can precede hepatic inflammation or copper accumulation. Interestingly, pathway analyses of fetal livers identified genes related to neurological diseases Huntington's and Alzheimer, which were differentially regulated between wild type and tx-j mice and in tx-j mice with or without maternal choline supplementation. These diseases are known to involve mitochondrial dysfunction21,22 and may provide insight into the neurological manifestations of WD. Decreased expressions and activities of Complex II, a component of the respiratory transport chain in the mitochondria, are well-described and are considered major contributors to the pathogenesis of Huntington's disease,23 which, like WD, has the characteristic features of resting tremors, rigidity and gait disturbances, and presents alterations in mitochondria morphology and function.
Oxidative phosphorylation gene transcript levels (Ndufb5, Sdha, Cox5a, Atp5j) in 24-week-old tx-j offspring livers were, for the most part, unaffected in males and were upregulated in female mice treated with PCA and choline. In our experiment, transcript levels of Sdha, a component of Complex II, were upregulated by choline treatment with an additional upregulation by the combination of PCA and choline. Others found that knockdown of Sdha in a mouse model of ovarian cancer was associated with upregulation of gene transcript levels related to histone methylation and mitochondrial dysfunction, which resulted in greater sensitivity to oxidative stress.24 Human mutations of SDHA have been associated with Leigh syndrome, which is characterized by progressive neurodegenerative disease and developmental delay.25
We observed upregulation of oxidative phosphorylation-related gene transcript levels in fetal livers, but downregulation of most of the same gene transcripts in adult livers. This finding may be due to differences in hepatic copper accumulation between fetal and adult tx-j mice, suggesting copper accumulation may affect the mitochondrial capability to respond to insults. We previously reported low copper concentrations in tx-j fetal liver, which is most likely due to low maternal blood ceruloplasmin levels with resultant reduction in available copper for transport across the placenta.4,26 Previous studies showed ATP7B is expressed by human placenta throughout gestation. Therefore, it is also possible that mutated ATP7B in WD patients or in tx-j dams reduces hepatic copper transport to the fetus.26
Choline supplementation was associated with increased SAM levels and Sahh transcript levels in 24-week-old mice. SAHH is the central enzyme in methionine metabolism affected by both relative copper deficiency in fetal liver and copper accumulation in adult liver, and is regulated by the availability of methyl groups, as provided by choline.3,4 However, choline supplementation was not associated with significant histological improvement in the liver, though it should be noted significant improvements would be difficult to assess in our tx-j model because overall histological disease severity is mild until at least 6 months of age.27 More importantly, choline was associated with a reduced hepatic nuclear-to-cell diameter ratio indicating changes in cell cycle and mitosis. Atp7b knockout mice have greater expression of cell cycle related genes compared to wild type mice.28 These findings indicate that the provision of methyl groups may counter-act cell cycle and mitosis abnormalities related to copper accumulation.
Results from our study indicate males and females responded differently to PCA and choline alone or in combination. In particular, Ndufab1, Ndufb5, Sdha, Cox5a, and Atp5j transcript levels were 1.2–1.4 times higher in female mice treated with PCA or choline alone or in combination compared to their male counterparts, indicating PCA and choline supplementation exert a sex-dependent effect. There are several possible clinical correlates to these results. It is well known that women with WD tend to present more frequently with acute liver failure than men18 and that men present with neurological involvement more frequently than women.18,29 While these findings have been attributed to hormonal factors and differences in hepatic iron accumulation over time,30 our study indicates epigenetic factors may contribute to the different presentations between sexes and opens the possibility for personalized medical treatment of WD according to sex. In addition, others have shown sex may have a significant role in affecting mechanisms of DNA methylation.31
Lastly, we explored differences in adult hepatic gene expression of Srebf1, a transcription factor responsible for regulating hepatocyte cholesterol homeostasis.32 Previous studies in Atp7b knockout mice have reported a downregulation of cholesterol biosynthesis genes and reduced levels of liver and blood cholesterol.28 We found PCA treatment in females and choline as well as PCA in combination with choline in males to increase hepatic gene expression of Srebf1, indicating this gene is responsive to treatment. Of note, reductions in transcript and protein levels of nuclear receptors including farnesoid X receptor (FXR), liver X receptor (LXR), retinoid X receptor (RXR), and glucocorticoid receptor (GR) have been reported in Atp7b knockout mice.33-35 In our study, untreated tx-j fetal mice liver exhibited a non-significant reduction of 19–35% for Fxr, Lxra, Lxrb, Rxra, and Rxrb. These reductions did not appear to be corrected by maternal choline supplementation.
A potential limitation of this study was it did not include a group of 24-week-old wild type mice with normal copper metabolism and treated with choline and/or penicillamine. However, our previous data indicate essentially no effect in gene expression from methyl group supplementation in adult wild type mice.3 In addition, PCA can cause copper deficiency in wild type mice with related changes in methionine and lipid metabolism.36-40 Also, this study did not explore the role of choline beyond methyl donation. Choline contributes to mitochondrial membrane integrity41 and can reduce lipid peroxidation,42 which may have contributed to the observed phenotypic effects. RNAseq was performed to explore pathways that may be impacted in fetal tx-j liver and normalized by maternal choline supplementation. The majority of fetal liver gene expression differences were between 1.2- and 1.5-fold. We consider that a 20–50% difference in gene expression may be biologically relevant and, therefore, we included genes with a fold change greater than 1.2 and P-value ≤ 0.05 in pathway analysis.
In conclusion, our study found that (1) mitochondrial dysregulation in a mouse-model of WD originates during fetal life; (2) maternal choline supplementation prevented this aberrant fetal hepatic gene expression; and (3) the tx-j mouse model may be a valid model to explore the sexual dimorphism that occurs in WD. This study has important transgenerational implications of disease progression and raises a possible preventative treatment for pregnant women with a heterozygous ATP7B mutation via methyl donor supplementation. Transgenerational studies are very challenging to perform in humans, whereas animal models provide a means to explore maternal and fetal nutritional factors that could modify WD phenotype. Also, there is a growing level of interest to understand how mitochondrial and epigenetic mechanisms work alone and in combination to impact disease phenotype.43 The model used in this study could become a model to explore the epigenetic-mitochondrial relationship for other more common conditions exhibiting variable and unexplained clinical course, such as neurological disorders.
Materials and methods
Wild type and tx-j mice and diets
Wild type (C3HeB/FeJ) and tx-j (C3HeB/FeJ-Atp7btx-J/J) mouse colonies were maintained at 20–23°C, 45–65% relative humidity, and a light cycle of 14 h light/10 h dark. Mice were maintained on LabDiet chow (Purina Lab, catalog # 5001). Mice used for this study were then switched to a common purified AIN-76A diet (Dyets, Inc., catalog # D100000) with (36 mmol/kg diet) or without (8 mmol/kg diet) choline supplementation 2 weeks before mating (Fig. 1). Food and deionized water were provided ad libitum. In the initial experiment, diets were continued throughout gestation and at embryonic day 17 fetal livers were collected for RNA-sequencing and identification of affected gene pathways.
In the second experiment, tx-j female mice received AIN-76A diets with or without choline supplementation 2 weeks before mating, which were continued through gestation and lactation. Since the concentration of copper in tx-j mouse breast milk is insufficient to maintain neonatal growth and development beyond day 10 on average, all tx-j pups were fostered to a lactating wild type dam by day 7 post-partum. Offspring were weaned at approximately 3 weeks of age and were fed AIN-76A diets with the same amount of choline that had been provided to their mothers and a subset of progeny received additional treatment at 12 weeks of age with oral PCA for a total of 4 treatment groups: 1) tx-j (control); 2) tx-j + PCA; 3) tx-j + choline; and 4) tx-j choline + PCA. Diet group 1 was used as a relative control group. Offspring were subdivided into treatment groups 2 and 4, which received oral PCA treatment beginning at 12 weeks of age. D-penicillamine (Sigma, catalog # P4575) was dissolved in deionized water and was administered at 100 mg/kg b.w./day, as previously described.3
Mice were anesthetized via isoflurane at 24 weeks of age. Approximately 1 ml of blood was collected from the retro-orbital sinus, and then the mice were euthanized by cervical dislocation. Livers were harvested and sections from each liver were either placed in formalin for subsequent blocking in paraffin or flash-frozen in liquid nitrogen and stored at −80°C for further analysis. Brain cortex and striatum were dissected, flash-frozen in liquid nitrogen, and stored at −80°C for further analysis.
All mouse protocols followed the guidelines of the American Association for Accreditation of Laboratory Animal Care and were reviewed and approved annually by the UC Davis Institutional Animal Care and Use Committee. All animals received humane care according to the criteria outlined in the “Guide for the Care and Use of Laboratory Animals” prepared by the National Academy of Sciences and published by the National Institutes of Health (NIH publication 86–23 revised 1985).
DNA and RNA isolation
Genomic DNA and total RNA were isolated from frozen fetal and adult liver using the DNeasy Blood and Tissue Kit (QIAGEN, catalog # 69504) and RNeasy Mini Kit (QIAGEN, catalog # 74104). E17 fetal livers from a single dam were pooled together at harvest as a single sample. Due to the limited amount of tissue available, total RNA was extracted from striatum samples via TRIzol reagent (Life Technologies, catalog # 15996) in order to adequately concentrate the final RNA resuspension based on pellet size. The concentration and purity of samples was determined by measuring the absorbency at A230, A260, and A280. RNA integrity was further evaluated by agarose gel electrophoresis. Genomic DNA was stored at −20°C and total RNA was stored at −80°C until further use.
cDNA synthesis and quantitative real-time PCR
Reverse transcription was carried out using 5 µg of total RNA following the protocol provided for the SuperScript III First-Strand cDNA Synthesis kit (Invitrogen, catalog # 18080051). Primers for mouse cDNA sequences were designed using Primer Express 3.0 (Applied Biosystems, Foster City, CA) or NCBI Primer-BLAST (http://www.ncbi.nlm.nih.gov/tools/primer-blast/) and blasted against the mouse genome using NCBI blastn to check primer specificity (http://blast.ncbi.nlm.nih.gov/Blast.cgi). The amplification efficiency (E) of all assays was calculated from the slope of a standard curve generated via 10-fold serial dilution of pooled control cDNA using the formula E = 10(−1/slope) −1. Primer sequences used are listed in Supplemental Table 5.
Transcriptome analysis by RNA-sequencing
Sixteen samples were chosen from wild type and tx-j dams receiving AIN-76A and AIN-76A + choline diets (n = 4 per group). RNA Integrity Number (RIN) value was determined on the Agilent Bioanalyzer 2100 (Agilent Technologies). The RIN values ranged from 9.6 to 10.0, indicating high quality. Sequencing libraries were constructed using the TrueSeq Stranded mRNA Sample Preparation kit (Illumina, Inc., catalog # RS-122). Briefly, mRNA was purified, fragmented, and converted to double-stranded cDNA. Adapters were ligated to the ends of double-stranded cDNA and PCR-amplified to create libraries. Sequencing was performed in 2 lanes multiplexing 8 samples per lane on a HiSeq 2000 sequencer analyzer (Illumina, Inc.) at the Vincent J. Coates Genomics Sequencing Laboratory at UC Berkeley.
Quality control (QC) analysis was performed using the application NGS quality control tool of CLC Genomics workbench software (CLC Bio, http://www.clcbio.com). This tool assesses sequence quality indicators based on the FastQC-project (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Quality was measured taking into account sequence-read lengths and base-coverage, nucleotide contributions and base ambiguities, quality scores as emitted by the base caller, and over-represented sequences. Samples passed all the QC parameters having the same length (100 bp); 100% coverage in all bases; 25% of A, T, G, and C nucleotide contributions; 50% GC on base content; and less than 0.1% over-represented sequences, indicating a very good quality.
Sequence paired-end reads (100 bp) were assembled against the annotated mouse reference genome (release 76) (ftp://ftp.ensembl.org/pub/release-76/genbank/mus_musculus/). Data was normalized by calculating the ‘reads per kilo base per million mapped reads’ (RPKM) for each gene.44 To select expressed genes, a threshold of RPKM ≥ 0.2 in at least one treatment group was used.45
Normalization and transformation of data were performed to transform the expression data from negative binomial distribution to normal distribution. Differential expression analysis was performed using t-test (P ≤ 0.05 and fold change ≥ 1.2) on transformed data to identify genes with significant change in expression between the 2 genotypes (wild type and tx-j) and between the 2 treatments (with or without maternal choline supplementation). Pathway analysis was performed using WebGestalt.46,47 A KEGG pathway must have had a minimum of 5 differentially expressed genes to be considered significantly impacted.
Light microscopy and histology
Liver sections were stained with hematoxylin and eosin for histology and with Masson Trichrome stain for fibrosis. Slides were scanned using an Aperio Scanscope XT (Leica Biosystems, Inc.) to produce digital whole slide images. Images were analyzed using Aperio Imagescope (Leica Biosystems, Inc.), and hepatocyte and nuclei diameters were measured using the ruler tool with a resolution of 0.504 µm per pixel. Fuji software was implemented to verify reproducibility of diameter measurements. For each slide, an average of 46 ± 2 hepatocytes in 4–6 mice/treatment or genotype were measured. To calculate mean nuclei area in each treatment group, hepatocyte nuclei were located and their area measured using the Fuji measure command. The Fuji software was considered reliable to automatically calculate nuclei area but not cell area due to high variability in hepatocyte dimensions in tx-j mice.48 Lobular and portal inflammation were graded on a 4-point scale based on inflammatory foci within the liver section: grade 0 (none); 1 with 1–2 foci; 2 with 3–4 foci; 3 with >4 foci. Steatosis was graded 0 (none), 1 (1%–25%), 2 (26%–50%), 3 (51%–75%), and 4 (76%–100%). Fibrosis was graded 0 (none), 1 (expansion of portal fibrous tissue), 2 (early bridging, no nodules), 3 (bridging fibrosis, early nodule formation), and 4 (cirrhosis).
Hepatic copper and iron quantification
Sections of liver (˜180 mg) were digested with concentrated nitric acid and wet-ashed.49 Samples were analyzed at 10X dilution using an Agilent 7500CE ICP-MS (Agilent Technologies) and values expressed as µg/g liver. Hepatic copper and iron quantifications were performed by the Interdisciplinary Center for Plasma Mass Spectrometry at UC Davis.
HPLC with fluorescence detection of methionine metabolites
Fifty mg of frozen liver was homogenized in 0.4 ml of cold 0.5 N perchloric acid and then centrifuged for 10 min at 4°C and 14000 rpm. The supernatant was passed through a 0.2 µm syringe filter and frozen at −80°C until HPLC analysis of SAH and SAM. Perchloric acid sample preparation was performed within 4 weeks from tissue harvest to ensure sample stability.50
Plasma biochemical measurements
Plasma alanine transaminase (ALT) levels were assessed using an enzymatic colorimetric assay from Bioo Scientific (catalog # 3460–01). ALT measurement was performed by the MMPC at UC Davis.
Plasma triglycerides were assessed using and enzymatic colorimetric assay from Thermo-Scientific (catalog # TR22203). Plasma triglyceride measurement was performed by the MMPC at UC Davis.
Plasma acylcarnitines
Plasma acylcarnitines were assessed as previously described51 at the West Coast Metabolomics Center at UC Davis.
Western blot
Protein (40–50 ug) was subjected to 4–20% precast polyacrylamide gel (Bio-Rad, catalog # 4561096). The samples were separated and transferred onto nitrocellulose membranes for 130 min. at a constant current of 100 mA. The membranes were blocked in Odyssey Blocking Buffer (PBS) (LI-COR, catalog # 927) containing 0.2% Tween (BioRad, catalog # 170–6531) for 1 h. Anti-SDHA and anti-SAHH (Santa Cruz Biotechnology, Inc., catalog # sc-27992 and sc-55759), anti-SREBP1 (Abcam, catalog # ab3259), and anti-β-Actin (Sigma-Aldrich, catalog # A5441) were incubated with the membranes in blocking buffer overnight at 4°C. IRDye 800CW Donkey anti-Mouse IgG (LI-COR, catalog #925–32212) and IRDye 680RD Donkey anti-Goat IgG (LI-COR, catalog # 925–68074) were used as secondary antibodies for imaging on the infrared Odyssey Imager (LI-COR). Protein quantification was performed using the Odyssey infrared imaging system according to manufacturer's instructions.
Analysis of global DNA methylation levels by dot blot analyses
Relative methylation dot blots were performed as described in Woods et al.52 Briefly, 50 ng of genomic DNA was alkaline denatured and spotted in triplicate on a nitrocellulose membrane, followed by UV crosslinking. The membrane was blocked in Odyssey Blocking Buffer (PBS) (LI-COR Biosciences, catalog # 927), then incubated in Blocking Buffer + 0.1% Tween-20 with anti-5-methylcitidine primary antibody (Eurogentec, catalog # BI-MECY-0100) overnight at 4°C. The membrane was washed in 1x PBS + 0.2% Tween-20, then incubated in Blocking Buffer + 0.1% Tween-20 with 680-IR secondary antibody (LI-COR Biosciences, catalog # 926–68072). The blot was imaged using the infrared Odyssey Imager (LI-COR Biosciences). The blot was then washed in 1x PBS + 0.1% Tween-20 followed by equilibration in PerfectHyb Plus Hybridization Buffer (Sigma, catalog # H7033) at 42°C. Biotin-labeled gDNA was hybridized overnight at 42°C as a loading control. The blot was incubated with Streptavidin 800-IR secondary antibody (LI-COR Biosciences, catalog # 926–32230) in Blocking Buffer + 0.1% Tween-20 for one hour at room temperature. The blot was imaged on the Odyssey Imager and integrated intensities were quantified using Odyssey software (LI-COR Biosciences, https://www.licor.com). Methylation signal was normalized to total DNA signal.
Statistical analyses
Statistical analyses were performed using SPSS Statistics, version 23 (IBM, http://www-01.ibm.com/software/analytics/spss/products/statistics/) and GraphPad Prism 6 (GraphPad Software). One-way ANOVA with Tukey's post hoc test were utilized to determine statistically significant differences among treatment groups. Both sexes were combined, but included sex as a covariate to adjust for potential sex differences. Each sex was also analyzed separately. Differences between sexes were assessed by 2-tailed Student's t-test. Data are presented as means ± SD. Pearson correlation coefficients and their P-values were calculated to assess the magnitude and direction of an association between 2 given measures. A P-value < 0.05 was considered significant. Principal component analysis was performed on significantly differentially expressed fetal liver genes. Principal component analysis is an unsupervised multivariate analysis, meaning that the model is generated without information regarding treatment groups.
Supplementary Material
Disclosure of potential conflicts of interest
The content is the sole responsibility of the authors and does not necessarily represent the official views of the National Institute of Health.
Funding
This research was supported by grant number K08DK084111, R03DK099427, and R01DK104770 from the National Institute of Diabetes and Digestive and Kidney Diseases, by Department and Divisional funds (to V.M.), by NIH R01ES021707, NIH P01ES011269, EPA 83543201 (to J.M.L.), and by a Veterans Health Administration Biomedical Laboratory Research and Development National Merit Review grant (to K.K.K.). V.M. is a full member of the University of California San Francisco Liver Center (Liver Center grant number P30 DK026743). This work used the Vincent J. Coates Genomics Sequencing Laboratory at UC Berkeley, supported by NIH S10 Instrumentation Grants S10RR029668 and S10RR027303.
References
- 1.Riordan SM, Williams R. The Wilson's disease gene and phenotypic diversity. J Hepatol 2001; 34:165-71; PMID:11211896; http://dx.doi.org/ 10.1016/S0168-8278(00)00028-3 [DOI] [PubMed] [Google Scholar]
- 2.Lutsenko S. Modifying factors and phenotypic diversity in Wilson's disease. Ann N Y Acad Sci 2014; 1315:56-63; PMID:24702697; http://dx.doi.org/ 10.1111/nyas.12420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Medici V, Shibata NM, Kharbanda KK, LaSalle JM, Woods R, Liu S, Engelberg JA, Devaraj S, Torok NJ, Jiang JX, et al.. Wilson's disease: changes in methionine metabolism and inflammation affect global DNA methylation in early liver disease. Hepatology 2013; 57:555-65; PMID:22945834; http://dx.doi.org/ 10.1002/hep.26047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Medici V, Shibata NM, Kharbanda KK, Islam MS, Keen CL, Kim K, Tillman B, French SW, Halsted CH, LaSalle JM. Maternal choline modifies fetal liver copper, gene expression, DNA methylation, and neonatal growth in the tx-j mouse model of Wilson disease. Epigenetics 2014; 9:286-96; PMID:24220304; http://dx.doi.org/ 10.4161/epi.27110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huster D, Kuhne A, Bhattacharjee A, Raines L, Jantsch V, Noe J, Schirrmeister W, Sommerer I, Sabri O, Berr F, et al.. Diverse functional properties of Wilson disease ATP7B variants. Gastroenterology 2012; 142:947-56.e5; PMID:22240481; http://dx.doi.org/ 10.1053/j.gastro.2011.12.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bethin KE, Petrovic N, Ettinger MJ. Identification of a major hepatic copper binding protein as S-adenosylhomocysteine hydrolase. J Biol Chem 1995; 270:20698-702; PMID:7657650; http://dx.doi.org/ 10.1074/jbc.270.35.20698 [DOI] [PubMed] [Google Scholar]
- 7.Li M, Li Y, Chen J, Wei W, Pan X, Liu J, Liu Q, Leu W, Zhang L, Yang X, et al.. Copper ions inhibit S-adenosylhomocysteine hydrolase by causing dissociation of NAD+ cofactor. Biochemistry 2007; 46:11451-8; PMID:17892301; http://dx.doi.org/ 10.1021/bi700395d [DOI] [PubMed] [Google Scholar]
- 8.Halsted CH, Medici V. Vitamin-dependent methionine metabolism and alcoholic liver disease. Adv Nutr 2011; 2:421-7; PMID:22332083; http://dx.doi.org/ 10.3945/an.111.000661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reik W, Dean W, Walter J. Epigenetic reprogramming in mammalian development. Science 2001; 293:1089-93; PMID:11498579; http://dx.doi.org/ 10.1126/science.1063443 [DOI] [PubMed] [Google Scholar]
- 10.Morgan HD, Santos F, Green K, Dean W, Reik W. Epigenetic reprogramming in mammals. Hum Mol Genet 2005; 14(Spec No 1):R47-58; PMID:15809273; http://dx.doi.org/ 10.1093/hmg/ddi114 [DOI] [PubMed] [Google Scholar]
- 11.Heijmans BT, Tobi EW, Stein AD, Putter H, Blauw GJ, Susser ES, Slagboom PE, Lumey LH. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc Natl Acad Sci U S A 2008; 105:17046-9; PMID:18955703; http://dx.doi.org/ 10.1073/pnas.0806560105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barker DJ, Osmond C. Infant mortality, childhood nutrition, and ischaemic heart disease in England and Wales. Lancet (London, England) 1986; 1:1077-81; PMID:2871345; http://dx.doi.org/ 10.1016/S0140-6736(86)91340-1 [DOI] [PubMed] [Google Scholar]
- 13.Huster D, Purnat TD, Burkhead JL, Ralle M, Fiehn O, Stuckert F, Olson NE, Teupser D, Lutsenko S. High copper selectively alters lipid metabolism and cell cycle machinery in the mouse model of Wilson disease. J Biol Chem 2007; 282:8343-55; PMID:17205981; http://dx.doi.org/ 10.1074/jbc.M607496200 [DOI] [PubMed] [Google Scholar]
- 14.Cordero P, Gomez-Uriz AM, Campion J, Milagro FI, Martinez JA. Dietary supplementation with methyl donors reduces fatty liver and modifies the fatty acid synthase DNA methylation profile in rats fed an obesogenic diet. Genes Nutr 2013; 8:105-13; PMID:22648174; http://dx.doi.org/ 10.1007/s12263-012-0300-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cooney CA, Dave AA, Wolff GL. Maternal methyl supplements in mice affect epigenetic variation and DNA methylation of offspring. J Nutr 2002; 132:2393S-400S; PMID:12163699 [DOI] [PubMed] [Google Scholar]
- 16.Ala A, Schilsky M. Genetic modifiers of liver injury in hereditary liver disease. Semin Liver Dis 2011; 31:208-14; PMID:21538285; http://dx.doi.org/ 10.1055/s-0031-1276648 [DOI] [PubMed] [Google Scholar]
- 17.Steindl P, Ferenci P, Dienes HP, Grimm G, Pabinger I, Madl C, Maier-Dobersberger T, Herneth A, Dragosics B, Meryn S, et al.. Wilson's disease in patients presenting with liver disease: a diagnostic challenge. Gastroenterology 1997; 113:212-8; PMID:9207280; http://dx.doi.org/ 10.1016/S0016-5085(97)70097-0 [DOI] [PubMed] [Google Scholar]
- 18.Weiss KH, Gotthardt DN, Klemm D, Merle U, Ferenci-Foerster D, Schaefer M, Ferenci P, Stremmel W. Zinc monotherapy is not as effective as chelating agents in treatment of Wilson disease. Gastroenterology 2011; 140:1189-98.ne1; PMID:21185835; http://dx.doi.org/ 10.1053/j.gastro.2010.12.034 [DOI] [PubMed] [Google Scholar]
- 19.Sternlieb I. Mitochondrial and fatty changes in hepatocytes of patients with Wilson's disease. Gastroenterology 1968; 55:354-67; PMID:5675366 [PubMed] [Google Scholar]
- 20.Sternlieb I, Quintana N, Volenberg I, Schilsky ML. An array of mitochondrial alterations in the hepatocytes of Long-Evans Cinnamon rats. Hepatology (Baltimore, Md) 1995; 22:1782-7; PMID:7489989; http://dx.doi.org/ 10.1002/hep.1840220626 [DOI] [PubMed] [Google Scholar]
- 21.Schapira AHV. Mitochondrial involvement in Parkinson's disease, Huntington's disease, hereditary spastic paraplegia and Friedreich's ataxia. Biochim Biophys Acta 1999; 1410:159-70; PMID:10076024; http://dx.doi.org/ 10.1016/S0005-2728(98)00164-9 [DOI] [PubMed] [Google Scholar]
- 22.Wang X, Wang W, Li L, Perry G, Lee H-g, Zhu X. Oxidative stress and mitochondrial dysfunction in Alzheimer's disease. Biochim Biophys Acta 2014; 1842:1240-7; PMID:24189435; http://dx.doi.org/ 10.1016/j.bbadis.2013.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Benchoua A, Trioulier Y, Zala D, Gaillard M-C, Lefort N, Dufour N, Saudou F, Elalouf J-M, Hirsch E, Hantraye P, et al.. Involvement of mitochondrial complex II defects in neuronal death produced by N-terminus fragment of mutated huntingtin. Mol Biol Cell 2006; 17:1652-63; PMID:16452635; http://dx.doi.org/ 10.1091/mbc.E05-07-0607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aspuria PJ, Lunt SY, Varemo L, Vergnes L, Gozo M, Beach JA, Salumbides B, Reue K, Wiedemeyer WR, Nielsen J, et al.. Succinate dehydrogenase inhibition leads to epithelial-mesenchymal transition and reprogrammed carbon metabolism. Cancer Metab 2014; 2:21; PMID:25671108; http://dx.doi.org/ 10.1186/2049-3002-2-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bourgeron T, Rustin P, Chretien D, Birch-Machin M, Bourgeois M, Viegas-Pequignot E, Munnich A, Rotig A. Mutation of a nuclear succinate dehydrogenase gene results in mitochondrial respiratory chain deficiency. Nat Genet 1995; 11:144-9; PMID:7550341; http://dx.doi.org/ 10.1038/ng1095-144 [DOI] [PubMed] [Google Scholar]
- 26.Hardman B, Michalczyk A, Greenough M, Camakaris J, Mercer J, Ackland L. Distinct Functional Roles for the Menkes and Wilson Copper Translocating P-type ATPases in Human Placental Cells. Cell Physiol Biochem 2007; 20:1073-84; PMID:17975309; http://dx.doi.org/ 10.1159/000110718 [DOI] [PubMed] [Google Scholar]
- 27.Roberts EA, Robinson BH, Yang S. Mitochondrial structure and function in the untreated Jackson toxic milk (tx-j) mouse, a model for Wilson disease. Mol Genet Metab 2008; 93:54-65; PMID:17981064; http://dx.doi.org/ 10.1016/j.ymgme.2007.08.127 [DOI] [PubMed] [Google Scholar]
- 28.Huster D, Purnat TD, Burkhead JL, Ralle M, Fiehn O, Stuckert F, Olson NE, Teupser D, Lutsenko S. High Copper Selectively Alters Lipid Metabolism and Cell Cycle Machinery in the Mouse Model of Wilson Disease. J Biol Chem 2007; 282:8343-55; PMID:17205981; http://dx.doi.org/ 10.1074/jbc.M607496200 [DOI] [PubMed] [Google Scholar]
- 29.Litwin T, Gromadzka G, Czlonkowska A. Gender differences in Wilson's disease. J Neurol Sci 2012; 312:31-5; PMID:21917273; http://dx.doi.org/ 10.1016/j.jns.2011.08.028 [DOI] [PubMed] [Google Scholar]
- 30.Litwin T, Gromadzka G, Członkowska A. Gender differences in Wilson's disease. J Neurol Sci 2012; 312:31-5; PMID:21917273; http://dx.doi.org/ 10.1016/j.jns.2011.08.028 [DOI] [PubMed] [Google Scholar]
- 31.Tobi EW, Lumey LH, Talens RP, Kremer D, Putter H, Stein AD, Slagboom PE, Heijmans BT. DNA methylation differences after exposure to prenatal famine are common and timing- and sex-specific. Hum Mol Genet 2009; 18:4046-53; PMID:19656776; http://dx.doi.org/ 10.1093/hmg/ddp353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang X, Sato R, Brown MS, Hua X, Goldstein JL. SREBP-1, a membrane-bound transcription factor released by sterol-regulated proteolysis. Cell 1994; 77:53-62; PMID:8156598; http://dx.doi.org/ 10.1016/0092-8674(94)90234-8 [DOI] [PubMed] [Google Scholar]
- 33.Wilmarth P, Short K, Fiehn O, Lutsenko S, David L, Burkhead JL. A systems approach implicates nuclear receptor targeting in the Atp7b(−/−) mouse model of Wilson's Disease. Metallomics 2012; 4:660-8; PMID:22565294; http://dx.doi.org/ 10.1039/c2mt20017a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wooton-Kee CR, Jain AK, Wagner M, Grusak MA, Finegold MJ, Lutsenko S, Moore DD. Elevated copper impairs hepatic nuclear receptor function in Wilson's disease. J Clin Invest 2015; 125:3449-60; PMID:26241054; http://dx.doi.org/ 10.1172/JCI78991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hamilton JP, Koganti L, Muchenditsi A, Pendyala VS, Huso D, Hankin J, Murphy RC, Huster D, Merle U, Mangels C, et al.. Activation of liver X receptor/retinoid X receptor pathway ameliorates liver disease in Atp7B−/− (Wilson disease) mice. Hepatology 2016; 63:1828-41; PMID:26679751; http://dx.doi.org/ 10.1002/hep.28406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lei KY. Alterations in plasma lipid, lipoprotein and apolipoprotein concentrations in copper-deficient rats. J Nutr 1983; 113:2178-83; PMID:6631537 [DOI] [PubMed] [Google Scholar]
- 37.Kaya A, Altıner A, ÖZpınar A. Effect of copper deficiency on blood lipid profile and haematological parameters in broilers. J Vet Med Series A 2006; 53:399-404; PMID:16970628; http://dx.doi.org/ 10.1111/j.1439-0442.2006.00835.x [DOI] [PubMed] [Google Scholar]
- 38.Bo S, Durazzo M, Gambino R, Berutti C, Milanesio N, Caropreso A, Gentile L, Cassader M, Cavallo-Perin P, Pagano G. Associations of Dietary and Serum Copper with Inflammation, Oxidative Stress, and Metabolic Variables in Adults. J Nutr 2008; 138:305-10; PMID:18203896 [DOI] [PubMed] [Google Scholar]
- 39.Relling DP, Esberg LB, Johnson WT, Murphy EJ, Carlson EC, Lukaski HC, Saari JT, Ren J. Dietary Interaction of High Fat and Marginal Copper Deficiency on Cardiac Contractile Function. Obesity 2007; 15:1242-57; PMID:17495201; http://dx.doi.org/ 10.1038/oby.2007.146 [DOI] [PubMed] [Google Scholar]
- 40.Koupparis AJ, Jeremy J, Angelini G, Persad RAJ, Shukla N. Penicillamine administration reverses the inhibitory effect of hyperhomocysteinaemia on endothelium-dependent relaxation in the corpus cavernosum in the rabbit. BJU Int 2006; 98:440-4; PMID:16879692; http://dx.doi.org/ 10.1111/j.1464-410X.2006.06212.x [DOI] [PubMed] [Google Scholar]
- 41.Jungalwala F, Dawson R. The origin of mitochondrial phosphatidylcholine within the liver cell. Eur J Biochem 1970; 12:399-402; PMID:5459578; http://dx.doi.org/ 10.1111/j.1432-1033.1970.tb00865.x [DOI] [PubMed] [Google Scholar]
- 42.Sachan DS, Hongu N, Johnsen M. Decreasing Oxidative Stress with Choline and Carnitine in Women. J Am Coll Nutr 2005; 24:172-6; PMID:15930482; http://dx.doi.org/ 10.1080/07315724.2005.10719462 [DOI] [PubMed] [Google Scholar]
- 43.Castegna A, Iacobazzi V, Infantino V. The mitochondrial side of epigenetics. Physiol Genomics 2015; 47:299-307; PMID:26038395; http://dx.doi.org/ 10.1152/physiolgenomics.00096.2014 [DOI] [PubMed] [Google Scholar]
- 44.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 2008; 5:621-8; PMID:18516045; http://dx.doi.org/ 10.1038/nmeth.1226 [DOI] [PubMed] [Google Scholar]
- 45.Wickramasinghe S, Rincon G, Islas-Trejo A, Medrano JF. Transcriptional profiling of bovine milk using RNA sequencing. BMC Genomics 2012; 13:1471-2164; http://dx.doi.org/ 10.1186/1471-2164-13-45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang B, Kirov S, Snoddy J. WebGestalt: an integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res 2005; 33:W741-W8; PMID:15980575; http://dx.doi.org/ 10.1093/nar/gki475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang J, Duncan D, Shi Z, Zhang B. WEB-based GEne SeT AnaLysis Toolkit (WebGestalt): update 2013. Nucleic Acids Res 2013; 41:W77-83; PMID:23703215; http://dx.doi.org/ 10.1093/nar/gkt439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cope-Yokoyama S, Finegold MJ, Sturniolo GC, Kim K, Mescoli C, Rugge M, Medici V. Wilson disease: Histopathological correlations with treatment on follow-up liver biopsies. World J Gastroenterol 2010; 16:1487-94; PMID:20333789; http://dx.doi.org/ 10.3748/wjg.v16.i12.1487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Clegg MS, Keen CL, Lönnerdal B, Hurley LS. Influence of ashing techniques on the analysis of trace elements in animal tissue. Biol Trace Elem Res 1981; 3:107-15; PMID:24271640; http://dx.doi.org/ 10.1007/BF02990451 [DOI] [PubMed] [Google Scholar]
- 50.Giulidori P, Stramentinoli G. A radioenzymatic method for S-adenosyl-l-methionine determination in biological fluids. Anal Biochem 1984; 137:217-20; PMID:6731799; http://dx.doi.org/ 10.1016/0003-2697(84)90373-7 [DOI] [PubMed] [Google Scholar]
- 51.Aguer C, Fiehn O, Seifert EL, Bézaire V, Meissen JK, Daniels A, Scott K, Renaud J-M, Padilla M, Bickel DR, et al.. Muscle uncoupling protein 3 overexpression mimics endurance training and reduces circulating biomarkers of incomplete β-oxidation. FASEB J 2013; 27:4213-25; PMID:23825224; http://dx.doi.org/ 10.1096/fj.13-234302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Woods R, Vallero RO, Golub MS, Suarez JK, Ta TA, Yasui DH, Chi LH, Kostyniak PJ, Pessah IN, Berman RF, et al.. Long-lived epigenetic interactions between perinatal PBDE exposure and Mecp2308 mutation. Hum Mol Genet 2012; 21:2399-411; PMID:22343140; http://dx.doi.org/ 10.1093/hmg/dds046 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.