

Abstract
The retinoblastoma tumor suppressor protein (Rb) plays a vital role in regulating mammalian cell cycle progression and inactivation of Rb is necessary for entry into S phase. Rb is inactivated by phosphorylation upon growth factor stimulation of quiescent cells, facilitating the transition from G1 phase to S phase. Although the signaling events after growth factor stimulation have been well characterized, it is not yet clear how these signals contact the cell cycle machinery. We had found previously that growth factor stimulation of quiescent cells lead to the direct binding of Raf-1 kinase to Rb, leading to its inactivation. Here we show that the Rb-Raf-1 interaction occurs prior to the activation of cyclin and/or cyclin-dependent kinases and facilitates normal cell cycle progression. Raf-1-mediated inactivation of Rb is independent of the mitogen-activated protein kinase cascade, as well as cyclin-dependent kinases. Binding of Raf-1 seemed to correlate with the dissociation of the chromatin remodeling protein Brg1 from Rb. Disruption of the Rb-Raf-1 interaction by a nine-amino-acid peptide inhibits Rb phosphorylation, cell proliferation, and vascular endothelial growth factor-mediated capillary tubule formation. Delivery of this peptide by a carrier molecule led to a 79% reduction in tumor volume and a 57% reduction in microvessel formation in nude mice. It appears that Raf-1 links mitogenic signaling to Rb and that disruption of this interaction could aid in controlling proliferative disorders.
The retinoblastoma tumor suppressor protein, Rb, plays a vital role in regulating the mammalian cell proliferation and its inactivation facilitates S-phase entry (i.e., entry into S phase) (64). Rb is inactivated during normal cell cycle progression by a cascade of phosphorylation events mediated mainly by kinases associated with D and E type cyclins (45, 55). Previous studies have shown that inhibition of Rb phosphorylation can lead to G1 arrest and that phosphorylation site mutants of Rb have enhanced growth suppressive properties (2, 17, 31). The growth-inhibitory properties of Rb are primarily mediated by its interaction with the E2F family of transcription factors (10, 18); Rb binds to E2Fs 1, 2, and 3 and suppresses their transcriptional activity (4, 33). Inactivation of Rb by phosphorylation leads to the dissociation and activation of E2F, allowing the expression of many genes required for cell cycle progression and S-phase entry (5, 7, 48).
In addition to its role in regulating cell proliferation, Rb affects chromatin structure and function as well (14, 25, 49). It has been shown recently that Rb induces heterochromatin formation and inhibition of E2F-regulated genes during cellular senescence (46). Further, Rb has been shown to localize to the chromatin and suppress abnormal endoreduplication that might occur after DNA damage (3). Rb has also been shown to possess antiapoptotic activity by repression of E2F1-regulated proapoptotic genes such as p73, Apaf-1, and caspase-3 (43, 51). These observations indicate that Rb can respond to a wide range of extracellular stimuli and execute functions that are appropriate for the signal. However, the exact pathways linking the diverse extracellular stimuli to Rb remain unclear.
Several lines of evidence indicate that receptor-mediated mitogenic signaling pathways converge on the Rb-dependent G1/S checkpoint. Growth stimulation through membrane tyrosine kinase receptors, estrogen receptors, and certain G-protein-coupled receptors requires Rb inactivation (36, 39). Moreover, members of the Ras/Raf/MEK/mitogen-activated protein (MAP) kinase signaling pathway have been implicated in the upregulation of cyclin D1 and Rb phosphorylation (39), and Rb inactivation is one of the end points of the mitogenic RAS/PI3K/AKT pathway (20). Furthermore, Ras-mediated transformation and stimulation of cell cycle progression has been found to require inhibition of the growth arrest activity of Rb mediated via cyclin D (34, 52). The importance of these observations is supported by the fact that most sporadic cancers inactivate Rb by exploiting pathways that regulate Rb phosphorylation (9).
Previous studies in our laboratory had shown that the signaling kinase c-Raf (Raf-1) can physically and functionally interact with Rb and contribute to its inactivation, facilitating cell proliferation (61). This interaction between Raf-1 and Rb is probably one of the mechanisms by which mitogenic signals received at extracellular receptors contact the cell cycle machinery in the nucleus. Raf-1 could phosphorylate Rb in vitro as well, and the results described here suggest that interaction of Raf-1 with Rb facilitates its eventual inactivation. Disruption of the Rb-Raf-1 interaction by an 9-amino-acid peptide significantly inhibits Rb phosphorylation, cell proliferation, and vascular endothelial growth factor (VEGF)-mediated angiogenic capillary tubule formation. Delivery of this peptide by a carrier molecule led to inhibition of tumor growth in nude mice. These results raise the possibility that the Rb-Raf-1 interaction is a vital event facilitating cell cycle progression and disruptors of this interaction might have antiproliferative properties.
MATERIALS AND METHODS
Plasmids.
The constructs pDCE2F1, pE2CAT, pCDNA3-cdk2wt, pCDNA3-cdk2dn, pCDNA3-Raf-1, pCDNA3-Raf-1Δ28, and pSVRb have been described before (61). The Raf-MEK inhibitor RKIP, A-Raf, and B-Raf plasmids were kind gifts from J. Sedivy, D. Anderson, and Ann Vojtek. pCDNA3-MEK1 and pGEX-4TK-MEK1 were obtained from Ron Prywes, Columbia University. The adenovirus (Ad) constructs Ad-green fluorescent protein (GFP) and Ad-E2F1 were obtained from W. D. Cress. Ad-cyclin D was kindly provided by I. Cozar-Castellano.
Cell culture and transfection.
The human promyelocytic leukemia cell line U937 was cultured in RPMI (Mediatech) containing 10% fetal bovine serum (FBS; Mediatech). HSF-8, U2OS, and Saos2 were cultured in Dulbecco modified Eagle medium (DMEM; Mediatech Cellgro) containing 10% FBS. A549 cells were maintained in Ham F-12K supplemented with 10% FBS. Human aortic endothelial cells (HAECs) were obtained from Clonetics and cultured in endothelial growth medium, supplemented with 5% FBS, according to the manufacturer's instructions. Saos-2 osteosarcoma cells were transfected by calcium phosphate precipitation method according to standard protocols. Generally, 2 μg of plasmids was used unless noted otherwise, and a pSVβ-Gal vector was included in all transfections. Assays for chloramphenicol acetyltransferase (CAT) and β-galactosidase were performed by using standard protocols. Colony formation assays were done by transfecting A549 cells with Lipofectamine 2000 (Invitrogen Corp.) according to the manufacturer's instructions. Infection with adenoviral constructs was done as described elsewhere (37).
In vitro binding assays.
Glutathione S-transferase (GST) fusion of Rb, MEK1, and Raf-1 have been previously described (23, 61). 35S-labeled proteins were generated by in vitro transcription translation in rabbit reticulocyte lysate according to the manufacturer's instructions (Promega, Madison, Wis.). First, 8 μl of the lysates was incubated with glutathione beads carrying an equal amount of the GST fusion proteins in 200 μl of protein binding buffer (20 mM Tris [pH 7.5], 50 mM KCl, 0.5 mM EDTA, 1 mM dithiothreitol, 0.5% NP-40, 3 mg of bovine serum albumin/ml) at 4°C for 2 h as described earlier (61). The protein amounts in the input lanes were approximately one-fourth of that used in the binding assay.
Whole-cell extracts, nuclear extracts, lysate preparation, immunoprecipitation, and Western blotting.
Whole-cell extracts were prepared by hypotonic shock, followed by salt extraction, as described previously (24). Lysates from cells treated with different agents were prepared by NP-40 lysis as described earlier (61). Nuclear and cytosolic fractions were made as described in reference 23. The purity of the nuclear fraction was ascertained by performing a Western blot for PARP (Cell Signaling). Physical interaction between proteins in vivo was analyzed by immunoprecipitation-Western blot analyses with 200 μg of lysate with 1 μg of the appropriate antibody as previously described (61). Polyclonal Raf-1, B-Raf, monoclonal E2F1, cyclin D, HP1, Brg1, and HDAC1 antibodies were obtained from Santa Cruz Biotechnology. Monoclonal Rb antibody was supplied by Oncogene Research Products, Cambridge, Mass. Monoclonal antibody to Raf-1 was obtained from Transduction Laboratories.
In vitro kinase assay.
U937 cells were serum starved for 48 h and subsequently serum stimulated for various times in the presence or absence of 1 μM penetratin-Raf-1 peptide conjugate. Subsequently, lysates were made as described previously (40). Immunoprecipitations with the monoclonal cyclin D antibody were performed, and the cyclin-dependent kinase 4 (cdk4) assay was carried out according to a protocol described previously (40). The kinase reaction was carried out on protein G beads in a total volume of 10 μl containing 1 μg of full-length Rb protein (QED Biosciences) as the substrate, 10 μM ATP, and 10 μCi of [γ-32P]ATP in kinase assay buffer at 30°C for 30 min as previously described (40, 47, 63). Rb phosphorylation was assessed by autoradiography. For Raf-1 kinase assays, immunoprecipitations were done with Raf-1 monoclonal antibody as described previously (61). The kinase reaction was carried out at 30°C for 30 min by using 1 μg of bacterially produced human Rb as the substrate.
Double immunofluorescence assay.
U2OS osteosarcoma cells were plated on poly-d-lysine treated chamber slides and rendered quiescent by serum starvation for 48 h. Thereafter, the cells were restimulated with serum for 1 h in the presence of 1 μM concentrations of the penetratin-Raf-1 peptide conjugate or the scrambled peptide conjugate. Cells were fixed with 4% paraformaldehyde and permeabilized with phosphate-buffered saline containing 0.2% Triton X-100. Monoclonal anti-Rb antibody (1:50) and polyclonal Raf-1 antibody (1:200) were added in blocking buffer, and the cells were incubated overnight at 4°C. Secondary antibodies, goat anti-mouse immunoglobulin G (IgG)-Alexa Fluor 488 (green fluorochrome), and goat anti-rabbit IgG-Alexa Fluor 568 (red fluorochrome) (Molecular Probes) were used as described previously (23). Nuclear staining was performed by using Hoechst 33258. Immunostained Rb and Raf-1 were visualized by confocal microscopy using a Zeiss laser scanning microscope model 510 system equipped with argon (458/488 nm) and helium neon (543 nm) laser systems.
Chromatin immunoprecipitation (ChIP) assay.
U937 cells were rendered quiescent by serum starvation and subsequently restimulated with serum for 2 h or 16 h in the presence or absence of 1 μM penetratin-Raf-1 peptide conjugate or a 1 μM concentration of the scrambled peptide. Cells were cross-linked with 1% formaldehyde for 10 min at room temperature. Subsequently, the cells were harvested and lysates were prepared. Immunoprecipitations were analyzed for the presence of E2F1, Rb, Raf-1, Brg1, HP1, and HDAC1 by PCR as described previously (23, 60). Rabbit anti-mouse secondary antibody was used as the control for all reactions. The sequences of the PCR primers used in the PCRs were as follows: Cdc6 promoter (forward primer), 5′-GGCCTCACAGCGACTCTAAGA-3′; and Cdc6 promoter (reverse primer), 5′-CTCGGACTCACCACAAGC-3′. The cdc25A and c-fos primers are described in references 23 and 60.
Colony formation assay.
A549 cells were stably transfected with 4 μg of indicated plasmids in 35-mm six-well plates in duplicate by using Lipofectamine 2000 reagent according to the manufacturer's instructions. Cells were allowed to recover for 48 h in DMEM supplemented with 20% FBS prior to being placed in G418-containing DMEM. After 48 h, cells were treated with 400 μg of G418/ml for 6 days, 200 μg of G418/ml for 6 days, 100 μg of G418/ml for 6 days, and finally with 40 μg of G418/ml for 6 days to allow for selection of colonies expressing the transfected plasmids. Colonies were fixed and stained with crystal violet as described previously (30). The number of colonies containing more than 20 cells was scored.
Proliferation assays and angiogenesis.
Bromodeoxyuridine (BrdU) labeling kits were obtained from Roche Biochemicals. Cells were plated in poly-d-lysine coated chamber slides at a density of 10,000 cells per well and rendered quiescent by serum starvation for 48 h. Cells were then restimulated with serum or 100 ng of EGF/ml for 16 h in the presence or absence of 1 μM concentrations of either penetratin alone, Raf-1 peptide, penetratin-Raf-1 peptide conjugate, or scrambled peptide conjugate. S-phase cells were visualized by microscopy and quantitated. HAECs were rendered quiescent by incubating the cells in endothelial growth medium lacking VEGF, supplemented with 0.5% FBS for 24 h. Thereafter, endothelial cells were restimulated with 100 ng of VEGF/ml for 16 h, and the BrdU staining was performed to determine the number of cells in S phase.
BrdU-GFP assay for S-phase entry.
A549 cells were plated in poly-d-lysine-coated chamber slides at a density of 10,000 cells/well. Subsequently, the cells were transfected with the indicated plasmids, along with GFP, by using Lipofectamine 2000 (Invitrogen Corp.) according to the manufacturer's protocols. At 48 h after transfection, the cells were incubated with BrdU solution (BrdU Staining Kit; Roche Diagnostics). Subsequently, the cells were fixed with 2% paraformaldehyde and permeabilized with 70% ethanol. The rest of the BrdU staining procedure was done according to the manufacturer's instructions. Each sample was done in duplicate. Two independent fields of 50 cells were counted per well by using a Leica DMLB fluorescence microscope (Welzar) by using IPLab (version 3.0) software. Of these fields, the number of cells that were double positive for both GFP and BrdU were used for calculating the percentage of transfected cells entering S phase.
Matrigel assay.
Matrigel (Collaborative Biomedical Products) was used to promote the differentiation of HAECs into capillary tube-like structures (15). A total of 100 μl of thawed Matrigel was added to 96-well tissue culture plates, followed by incubation for 60 min at 37°C to allow polymerization. Then, 1.5 × 104 quiescent HAECs were seeded on the gels in EGM medium supplemented with 0.5% FBS containing 100 ng of VEGF/ml in the presence or absence of 1 μM concentrations of penetratin, Raf-1 peptide, penetratin-Raf-1 peptide conjugate, or scrambled peptide conjugate, followed by incubation for 36 h at 37°C. The peptides were added every 12 h during the 36-h incubation. Capillary tube formation was assessed by using a Leica DMIL phase-contrast microscope (Weltzar).
Antitumor studies in nude mice.
A total of 107 A549 cells were injected subcutaneously into the left and right flanks of 8-week-old female nude mice. When the tumors reached ∼100 mm3, the animals were randomized and dosed intratumorally with 50 μl of H2O as the vehicle or with 50 mg of Raf-1 peptide or penetratin-Raf-1-peptide conjugate/kg/day. Tumor volumes were determined as described previously (6). Microvessel formation in tissue sections was assessed by immunohistochemistry by using an anti-CD31 antibody. For micro-blood vessel counting, five areas were microscopically examined, and the averages and standard deviations were recorded. Brown-staining endothelial cells or endothelial cell clusters were considered as a single, countable microvessel.
RESULTS
Raf-1 peptide inhibits Rb-Raf-1 binding in vitro.
Previous studies by Wang et al. (61) had shown that the Rb-Raf-1 interaction is mediated through the amino-terminal 28 amino acids of Raf-1, which contacts the pocket domain of Rb. Attempts were made to further delineate the Rb-binding region of Raf-1 by using an in vitro GST-binding assay. As shown in Fig. 1A, wild-type (full-length) Raf-1 could efficiently bind to GST-Rb. Deletion of 10 amino acids from the N terminus of Raf-1 (Raf-1Δ10) had no effect on the binding, but deletion of the N-terminal 18 amino acids abolished the binding, suggesting that residues 10 to 18 were probably involved in binding to Rb. To examine whether this region of Raf-1 was indeed involved in binding to Rb, a peptide was synthesized that corresponding to amino acids 10 to 18 of Raf-1 (ISNGFGFK; a C was added at the carboxy-terminal end for coupling to carrier molecules). This peptide (referred to as the Raf-1 peptide) could compete for the binding of 35S-labeled Raf-1 (35S-Raf-1) to GST-Rb in a dose-dependent manner (Fig. 1B). The binding of 35S-Raf-1 to GST-Rb was abolished by a 1 μM concentration of the Raf-1 peptide, whereas a 1 μM concentration of an unrelated peptide, penetratin, had no effect. The specificity of the Raf-1 peptide was examined by using a scrambled peptide sequence containing the same amino acid content as the Raf-1 peptide. As shown in Fig. 1C, 1 μM concentrations of the scrambled peptide or a peptide derived from the C terminus of Raf-1 did not inhibit the Rb-Raf-1 binding. Furthermore, peptides derived from the Rb-binding region of proteins such as E2F1 and HPV E7 had no effect on the binding of Raf-1 to Rb, showing that the disruption of Rb-Raf-1 interaction required the specific Raf-1 peptide sequence. These findings were further confirmed in an enzyme-linked immunosorbent assay-based binding assay, wherein 10 μg of the aforementioned peptides was coated on the plate. It was found that the Raf-1 peptide bound efficiently with GST-Rb, whereas the scrambled peptide or the other control peptides manifested no binding whatsoever to GST-Rb (data not shown).
FIG. 1.

Binding of 35S-labeled Raf-1 deletion fragments to GST-Rb in vitro. (A) 35S-Raf-1WT and the deletion fragments 35S-Raf-1Δ10, 35S-Raf-1Δ18, and 35S-Raf-1Δ28 were tested for binding to GST-Rb or unprimed GST beads. Lysate lane has one-fourth of the protein used for binding. (B) Dose-dependent inhibition of the binding of Raf-1 to GST-Rb by the Raf-1 peptide in a GST pull-down assay. A 1 μM concentration of an unrelated peptide (penetratin) was used as a negative control. (C) The Rb-Raf-1 interaction is abolished by 1 μM of the Raf-1 peptide but is unaffected by a 1 μM concentration of a scrambled peptide, a control peptide from the C terminus of Raf-1, or peptides corresponding to the Rb-binding domain on E2F1 (E2F peptide) and HPV E7 protein (E7 peptide). (D) The Raf-1 peptide is unable to disrupt the binding of B-Raf to GST-Rb and GST-MEK1 in vitro. (E) 35S-A-Raf does not bind to GST-Rb but binds to the positive control GST-MEK1. (F) The Raf-1 peptide specifically disrupts the Rb-Raf-1 interaction but does not affect the binding of HDAC1, cyclin D, and prohibitin to GST-Rb. (G) The Raf-1 peptide does not affect the interaction of other Raf-1-binding proteins such as MEK1 and Ras to GST-Raf-1.
We next assessed whether other Raf family kinases, namely, A-Raf and B-Raf, were able to bind to Rb. As shown in Fig. 1D, 35S-B-Raf could bind to Rb, but its binding was not competed for by the Raf-1 peptide. In contrast, A-Raf did not bind to Rb at all. Both A-Raf and B-Raf were found to bind to GST-MEK1, which was used as the positive control for the assay (Fig. 1E). It is interesting that A-Raf lacks the corresponding N-terminal region of c-Raf; a similar region is present on B-Raf, but the sequence is not identical. Thus, Raf-1 and B-Raf, but not A-Raf, can bind to Rb.
The effect of the Raf-1 peptide on the association of Rb with other cellular proteins was examined. Although 1 μM concentrations of the peptide could efficiently compete for the binding of Raf-1, it had no effect on the binding of cyclin D (44), HDAC1 (8) or prohibitin (62) to Rb (Fig. 1F). Similarly, the Raf-1 peptide did not affect the binding of other proteins, such as MEK1 and Ras, to GST-Raf-1 (Fig. 1G). This series of experiments demonstrate that the eight-amino-acid Raf-1 peptide specifically disrupts the binding of Raf-1, but not other proteins, to Rb in vitro.
Raf-1 peptide specifically inhibits Rb-Raf-1 binding in vivo.
The ability of the Raf-1 peptide to inhibit Rb-Raf-1 interaction in vivo was analyzed. To do this, Raf-1 peptide was conjugated with a carrier molecule, penetratin, which can transport peptides into both the nucleus and cytosol of live cells (19). The ability of the penetratin-Raf-1 peptide conjugate to disrupt the interaction of Raf-1 with Rb in vivo was examined by double immunofluorescence experiments on U2OS cells (Fig. 2A to D). Figure 2A shows that quiescent U2OS cells contained a low amount of Raf-1, and it was predominantly located in the cytoplasm. However, upon serum stimulation for 1 h, a significant amount of Raf-1 translocated to the nucleus, where it colocalized with Rb (Fig. 2B). The presence of 1 μM concentrations of penetratin-Raf-1 peptide conjugate during serum stimulation reduced the Rb-Raf-1 interaction considerably (Fig. 2C). In contrast, penetratin-scrambled Raf-1 peptide conjugate had no effect on the colocalization of Rb and Raf-1 (Fig. 2D). This experiment suggests that the Raf-1 peptide can specifically disrupt the binding of Rb with Raf-1 in vivo. We next examined whether the disruption of the binding of Rb to Raf-1 seen in the double immunofluorescence experiments was due to reduced nuclear translocation of Raf-1 in the presence of the penetratin-Raf-1 peptide conjugate. This was verified by subcellular fractionation of cells. Quiescent U2OS cells were serum stimulated for 1 h in the presence or absence of 1 μM penetratin-Raf-1 peptide conjugate. Nuclear and cytosolic extracts were made, and the localization of Raf-1 and Rb was probed by immunoblotting. Figure 2E shows that serum-starved cells contain a low amount of Raf-1 and that it is localized in the cytosol. Serum stimulation of U2OS cells causes an increase in Raf-1 levels and induces the translocation of a subset of Raf-1 to the nucleus. The treatment of cells with penetratin-Raf-1 conjugate does not affect the levels of Raf-1 in cell, nor does it have any impact on the nuclear translocation of Raf-1. Rb remains in the nuclear fraction both in quiescent and in serum-stimulated cells. A Western blot for PARP was done to verify the purity and integrity of the nuclear extracts.
FIG. 2.

The penetratin-Raf-1 conjugate can abolish Rb-Raf-1 interaction in vivo. U2OS cells were immunostained with an anti-Raf-1 polyclonal antibody and an anti-Rb mouse monoclonal antibody, and the proteins were visualized by confocal microscopy. (A) Serum-starved U2OS cells show no association of Rb (in green) and Raf-1 (in red). (B) Serum stimulation induces a subset of Raf-1 to translocate to the nucleus where it colocalizes with Rb. Areas of colocalization can be seen in yellow. (C and D) The presence of 1 μM penetratin-Raf-1 peptide conjugate markedly inhibits the binding of Raf-1 to Rb (C), whereas a scrambled peptide conjugate is unable to do so (D). (E) Penetratin-Raf-1 peptide conjugate does not affect the nuclear translocation of Raf-1. U2OS cells were serum stimulated in the presence or absence of the peptide conjugate. Subsequently, cells were harvested, and cytosolic and nuclear extracts were prepared and immunoblotted for Raf-1 and Rb. Serum stimulation of cells induces Raf-1 expression and causes its translocation to the nucleus. Treatment with 1 μM penetratin-Raf-1 peptide conjugate does not affect the levels of Raf-1 in the nucleus. Rb is localized in the nucleus of both quiescent and serum-stimulated cells. A Western blot for PARP was done as a nuclear marker.
The disruption of the interaction in vivo was further confirmed by an immunoprecipitation-Western blot experiment. Quiescent U937 cells were serum stimulated for 2 h in the presence or absence of the 1 μM concentrations of the penetratin-Raf-1 peptide conjugate, and the Raf-1-Rb association was probed by immunoprecipitation-Western blot experiments. Serum-starved cells showed negligible Rb-Raf-1 binding, whereas there is a considerable amount of Rb bound to Raf-1 after 2 h of serum stimulation (Fig. 3A). The presence of 1 μM penetratin-Raf-1 peptide conjugate caused a significant reduction in the amount of Rb associated with Raf-1. There was no reduction in Rb associated with E2F1 in the same samples, suggesting that the penetratin-Raf-1 peptide conjugate specifically disrupts the Rb-Raf-1 interaction. The Rb-Raf-1 association was not affected by the presence of 1 μM penetratin or the scrambled peptide conjugate (Fig. 3B). Similarly, the Rb-Raf-1 binding remained unaffected by a conjugate of penetratin and a control peptide from the C terminus region of Raf-1, suggesting that the disruption was due to the specific peptide moiety. In addition, the penetratin-Raf-1 peptide conjugate had no detectable effect on the Raf-MEK1 interaction (Fig. 3C) or the B-Raf-Rb interaction in vivo (Fig. 3D).
FIG. 3.

(A). Disruption of Rb-Raf-1 interaction by a 1 μM concentration of penetratin-Raf-1 conjugate in vivo. Quiescent U937 cells stimulated with serum for 2 h in the presence or absence of a 1 μM concentration of the penetratin-Raf-1 conjugate or a 1 μM concentration of unconjugated penetratin as the control. Whole-cell extracts (WCE) were made and immunoprecipitated with Raf-1 (top panel) or E2F1 (bottom panel) antibody. WCE lane shows an equivalent amount of U937 extracts used for immunoprecipitation. (B and C) The Raf-1 Rb interaction is abolished by a 1 μM concentration of the penetratin-Raf-1 conjugate (B) but is not affected by the scrambled peptide conjugate or control peptide conjugate in a similar experiment (C). The penetratin-Raf-1 peptide conjugate does not affect the binding of Raf-1 to MEK1 in vivo. U937 cells were serum starved and subsequently serum stimulated as indicated, and immunoprecipitation-Western blots were done. (D) B-Raf binds to Rb in vivo, and this interaction is unaffected by 1 μM penetratin-Raf-1 peptide conjugate. A 1 μM concentration of the carrier peptide penetratin was used as a control.
The binding of Raf-1 to Rb precedes the binding of cyclin D.
We next assessed the kinetics of the Rb-Raf-1 interaction upon serum stimulation. The binding of Raf-1 and cyclin D to Rb at different time points after serum stimulation of quiescent U937 cells was assessed by immunoprecipitation-Western blot experiments with E2F1 as a control. Figure 4A shows that Raf-1 could bind to Rb as early as 30 min after serum stimulation; the interaction persisted up to 4 h (Fig. 4A). Analysis of the same extracts showed that the earliest point at which cyclin D associates with Rb was 4 h after stimulation. There was no change in the amount of E2F1 associated with Rb at these time points. This experiment shows that the binding of Raf-1 to Rb precedes the binding of cyclin D.
FIG. 4.

(A) Raf-1 and cyclin D associate with Rb according to different kinetics. Whole-cell extracts from U937 cells serum stimulated for the indicated periods of time were immunoprecipitated with the Raf-1, cyclin D, or E2F1 antibodies, and the associated Rb was detected by Western blotting. (B) Rb phosphorylation can be detected in U937 cells serum stimulated for 2 h. (C) The penetratin-Raf-1 peptide conjugate can inhibit Rb phosphorylation in U937 cells. Serum-starved U937 cells were stimulated with serum for 16 h in the presence or absence of 1 μM penetratin-Raf-1 peptide conjugate or a 1 μM concentration of scrambled peptide conjugate or the control peptide conjugate. Whole-cell extracts were made, and the phosphorylation status of the Rb protein was checked by Western blotting. (D) The Raf-1 conjugate can inhibit Rb phosphorylation in primary human fibroblast HSF-8 cells.
Previous studies in our laboratory had shown that Raf-1 could phosphorylate Rb in vitro. Since it is known that Raf-1 is activated early after serum stimulation, we examined whether Rb phosphorylation could be observed in early G1 phase as well. Quiescent U937 cells were stimulated with serum for 2 h, and the phosphorylation status of Rb was ascertained by Western blotting. As shown in Fig. 4B, a certain amount of Rb phosphorylation could be detected after 2 h of serum stimulation, when only Raf-1, but not cyclin D, is bound to Rb. It thus appears that a minimal amount of Rb phosphorylation occurs prior to the binding of cyclin D.
Penetratin-Raf-1 peptide conjugate inhibits serum-induced Rb phosphorylation.
Since we found a limited amount of Rb phosphorylation when cells were serum stimulated for 2 h, we tested whether disrupting the Rb-Raf-1 interaction by using the penetratin-Raf-1 peptide conjugate affected Rb phosphorylation. We found that Rb phosphorylation was increased upon serum stimulation of quiescent U937 cells for 16 h (Fig. 4C); this was reduced when the stimulation was done in the presence of the penetratin-Raf-1 peptide conjugate. The C-terminal peptide conjugate or scrambled peptide conjugate had no effect, suggesting that the observed effects are specific to the Raf-1 peptide. The effect on Rb phosphorylation was more pronounced on human diploid fibroblast cell line HSF-8 (Fig. 4D). However, the penetratin-Raf-1 peptide conjugate had no effects on the levels of E2F1 in these cells.
Since we observed that disruption of the Rb-Raf-1 interaction by the penetratin-Raf-1 peptide conjugate prevented the phosphorylation of Rb (Fig. 4C and D), we sought to determine whether this was due to a fortuitous inhibition of the kinase activity associated with cyclins D and E. U937 cells serum stimulated in the presence or absence of the penetratin-Raf-1 peptide conjugate were analyzed for cyclin D-associated kinase activity by an in vitro assay. Full-length human Rb protein was used as the substrate for cdk4. Studies from the Sherr laboratory (40) have shown that conventional lysates containing NP-40 do not support cyclin D/cdk4 activity. Therefore, lysates were made by using a modified protocol and probed for cdk4 activity (40). As shown in Fig. 5A and B, kinase activity associated with cyclin D was induced to comparable levels 4 h after serum stimulation, in the presence or absence of the penetratin-Raf-1 peptide conjugate. Similarly, an induction of cyclin E-associated kinase activity was also found (data not shown). However, interestingly, there was no hyperphosphorylation of Rb when the serum stimulation was done in the presence of Raf-1 peptide conjugate, even though cdk activity was induced (Fig. 5B). Western blotting was done to examine whether the Raf-1 peptide conjugate affected the levels of cyclins D and E. As shown in Fig. 5D, serum stimulation in the presence of the peptide conjugate led to the expression of cyclins D and E; the levels were comparable to control serum-stimulated cells (Fig. 5C).
FIG. 5.

The penetratin-Raf-1 peptide conjugate does not affect the phosphorylation of Rb by cyclin D, the cyclin D-Rb interaction, or the kinase activity associated with cyclin D, but Rb phosphorylation is not observed when the Raf-1 peptide conjugate is present during serum stimulation. Quiescent U937 cells were serum stimulated in the presence (B) or absence (A) of 1 μM penetratin-Raf-1 peptide conjugate for the indicated time points. Lysates were made and the phosphorylation status of Rb was examined by Western blotting (top panel). Lysates were immunoprecipitated with cyclin D antibody and an in vitro kinase assay was done by using 1 μg of full-length human Rb as the substrate (middle panel). The presence of Rb in the immunoprecipitates was detected by Western blotting (bottom panel). (C and D) The penetratin-Raf-1 peptide conjugate does not affect the expression of cyclin D or cyclin E in serum-stimulated cells. (E) Raf-1 can phosphorylate Rb before the appearance of cdk activity. Quiescent U937 cells were serum stimulated for the indicated time points. Lysates were made and subsequently immunoprecipitated with Raf-1 antibody. An in vitro kinase assay was performed with 1 μg of human Rb as a substrate (top panel). The gel was rehydrated and stained for Rb protein (bottom panel).
An in vitro kinase assay was done to analyze whether Raf-1 from early time points of serum stimulation could phosphorylate Rb. It was found that Raf-1 from cells serum stimulated for 30 min to 4 h could phosphorylate Rb (Fig. 5E); this was before cyclins or cdk's were activated. It has been proposed that Rb is phosphorylated sequentially by cyclin D- and E-associated kinase activities during normal cell cycle progression (26). Our results raise the possibility that Raf-1-mediated phosphorylation is an event that initiates this sequence and precedes the phosphorylation mediated by cdk's.
Raf-1 mediated inactivation of Rb is independent of the MAP kinase cascade.
Our earlier data had shown that Raf-1 could functionally inactivate Rb and reverse the Rb-mediated repression of E2F1 transcriptional activity. Since this required a direct binding of Raf-1 to Rb, we wanted to determine whether Raf-1-mediated inactivation of Rb involved the MAP kinase cascade. A transient transfection experiment was done in Saos-2 cells, where transfection of Rb repressed E2F1-mediated transcription of a CAT reporter (Fig. 6A). Cotransfection of Raf-1 could relieve Rb-mediated repression; Raf-1Δ28, which could not bind Rb, was unable to do so. Interestingly, cotransfection of RKIP, which disrupts the binding of Raf-1 to MEK1 and inhibits the MAP kinase cascade (65), had no effect on Raf-1 mediated inactivation of Rb function. Similarly, a kinase dead Raf-1(kin−) mutant did not affect Rb either, as we had shown previously. On the contrary, RKIP could totally abolish Raf-1-mediated activation of AP1CAT (Fig. 6B). The Raf-1 deletion mutant Raf-1Δ28 had no effect on Raf-1-mediated activation of AP1CAT, showing that, whereas the N terminus of Raf-1 plays a role in the mediating the Rb-Raf-1 interaction, it does not affect the MAP kinase pathway. Similar results were obtained when dominant-negative MEK (dn-MEK) was used to inhibit Raf-1 activity (Fig. 6C and D). dn-MEK1 could abolish Raf-1-mediated activation of AP1CAT in a dose-dependent manner (Fig. 6D). In contrast, the same doses of dn-MEK1 had no effect on Raf-1-mediated inhibition of Rb repression of E2F1 activity (Fig. 6C). These data suggest that Raf-1 inactivates Rb directly, independent of the MEK/ERK pathway.
FIG. 6.

Raf-1-mediated inactivation of Rb is independent of its MEK1 kinase activity. (A) Saos-2 cells were transiently transfected with 2 μg each of E2CAT, E2F, and Rb. Transfection with wild-type Raf-1 reversed Rb-mediated repression of E2F activity, whereas Raf-1Δ28 or a kinase-deficient Raf-1 mutant were unable to do so. The presence of RKIP had no effect on the Raf-1-mediated inactivation of Rb. (B) RKIP can inhibit Raf-1-mediated activation of AP1CAT. Rb deficient Saos-2 cells were transiently transfected with an AP1CAT reporter plasmid. Raf-1 wild type (WT) in the presence or absence of RKIP, Raf-1Δ28, or a kinase-deficient Raf-1 mutant was cotransfected as indicated. (C) Increasing amounts of dn-MEK1 had no effect on Raf-1-mediated reversal of the transcriptional repressive activity of Rb. (D) Increasing amounts of dn-MEK1 could completely abolish Raf-1-mediated activation of AP1CAT.
Raf-1 can inactivate Rb independent of cdk activity.
The next question we asked was whether Raf-1-mediated inactivation of Rb required cyclin or cdk activity. A transient-transfection experiment was done in Saos-2 cells as described above in which transfection of Rb repressed E2F1-mediated transcription (Fig. 7A). Cotransfection of Raf-1 effectively inactivated Rb, relieving Rb-mediated repression of E2F1; but cotransfection of cdk inhibitor p16INK4 (35) or p21Waf-1/CIP1 (42) had no effect on Raf-1-mediated inactivation of Rb function. On the contrary, p16 and p21 could prevent cyclin D- or cyclin E-mediated inactivation of Rb effectively. Similar results were obtained when dominant-negative cdk2 (dn-cdk2), -4, or -6 was used to inhibit cdk activity (data not shown). Since the cdk inhibitors had no effect on Raf-1 mediated inactivation of Rb, it appears that Raf-1 is inactivating Rb directly, independent of cyclins and cdk's.
FIG. 7.

(A) Raf-1 inactivates Rb independent of cdk's. Saos-2 cells were transiently transfected with 2 μg each of E2CAT, E2F1 and the indicated vectors. A CAT assay reveals that, whereas Raf-1 reverses Rb-mediated repression of E2F1, cotransfection of cdk inhibitors p16INK4 or p21Waf-1/CIP1 did not affect Raf-1-mediated inactivation of Rb, but inactivation mediated by cyclin D and cyclin E was inhibited. (B) Raf-1 is able to inhibit cell cycle arrest by p16, Rb, or dn-cdk2, whereas Raf-1 Δ28 is unable to do so. A549 cells were transfected with the indicated vectors, along with GFP. BrdU incorporation was assessed after 48 h. The numbers of cells positive for both GFP and BrdU were determined and are expressed as percentages of GFP-positive cells. (C) Colony formation assays to show that suppression of colony growth by p16, Rb, or dn-cdk2 can be reversed by Raf-1. A549 cells were grown in six-well plates and transfected with the indicated plasmids. G418-resistant colonies with more than 20 cells were counted after 21 days. The number of colonies obtained in cells transfected with empty pCDNA3 vector was taken as 100%, and the rest of the data were calculated as percentages of the colony formation observed in pCDNA3 transfected cells.
Given this finding, experiments were performed to evaluate whether Raf-1 could overcome growth arrest mediated by p16 or dn-cdk2. We sought to determine whether whether Raf-1 or Raf-1Δ28 (which is deficient in binding to Rb) or could reverse cell cycle arrest induced by ectopic expression of p16 or dn-cdk2. To do this, A549 cells were transfected with Raf-1 or Raf-1Δ28, along with Rb, p16, or dn-cdk2, in the presence of GFP. The cells were fixed and stained for BrdU at 48 h after transfection. The cells that stained positive for both GFP and BrdU were counted, and the percentage of transfected cells in S phase was assessed. Figure 7B shows that Raf-1 could efficiently reverse the cell cycle arrest mediated by Rb, p16, and dn-cdk2. However, Raf-1Δ28, which lacks the Rb-binding domain, was unable to do so, suggesting that overexpression of Raf-1 can overcome cell cycle arrest mediated by inhibitors of cyclins or cdk's. These observations were further confirmed by a colony formation assay on plastic. A549 cells were stably transfected with Raf-1 wild type or Raf-1Δ28, in the presence of p16 or dn-cdk2, and G418-resistant colonies with more than 20 cells were counted after 21 days. We observed that p16 and dn-cdk2 could greatly inhibit the number of G418-resistant colonies, whereas wild-type cdk2 and Raf-1 could increase the number of colonies more than twofold (Fig. 7C). Interestingly, wild-type Raf-1 could abolish the growth inhibitory effects of both p16 and dn-cdk2, whereas Raf-1Δ28 was unable to do so. Our observations raise the possibility that overexpression of Raf-1 can overcome growth arrest mediated by cdk inhibitors, probably through a direct phosphorylation of Rb.
Dissociation of Brg-1 from Rb is prevented by the Raf-1 peptide conjugate.
Our earlier studies had shown that Raf-1 does not cause a direct dissociation of E2F1 from Rb; further, Raf-1 could be detected in complexes containing Rb and E2F1. This scenario suggested that Raf-1 is not inducing E2F-mediated transcription by dissociating E2F1 from Rb-like viral oncoproteins but probably by dissociating a corepressor from Rb; subsequent inactivation of Rb leads to the dissociation of E2F1 at later stages in the cell cycle (13, 21). The possibility that Raf-1 is leading to the dissociation of a corepressor from Rb was examined by ChIP assays. Quiescent U937 cells were serum stimulated for 2 h in the presence or absence of 1 μM concentrations of penetratin-Raf-1 peptide conjugate or the scrambled peptide conjugate. ChIP lysates were made, and immunoprecipitations were performed with antibodies to E2F1, Raf-1, Rb, HDAC1, HP1, and Brg1 and an irrelevant antibody. After reversal of cross-linking, the bound DNA was isolated, and PCR was performed to probe the association of these proteins with E2F1-responsive proliferative promoters such as cdc6 and cdc25A. As shown in Fig. 8A, quiescent cells had considerable amounts E2F1 and Rb bound to both the promoters. In addition, Brg1, HP1, and HDAC1 were also associated with the promoters in serum-starved cells. There was no Raf-1 bound to the promoters. Serum stimulation for 2 h caused a robust association of Raf-1 with the cdc6 and cdc25A promoters. The amount of HP1 and HDAC1 binding to the promoter remained unchanged. However, the amount of Brg1 associated with both cdc6 and cdc25A promoters was considerably reduced in cells serum stimulated for 2 h, suggesting that this corepressor might be dissociated as a result of serum stimulation. At the same time, serum stimulation in the presence of 1 μM penetratin-Raf-1 peptide conjugate prevented the dissociation of Brg-1 from the promoters. It appears that there is more Brg-1 associated with both cdc25A and cdc6 promoters when the serum stimulation was done in the presence of the peptide conjugate than in serum-starved cells; the reason for this is not known. The penetratin-Raf-1 peptide conjugate also prevented the association of Raf-1 with the promoters, as expected. Serum stimulation in the presence of a scrambled peptide conjugate led to a dissociation of Brg1 from the promoter. Corepressors such as HDAC1 and HP1, which are also known to associate with Rb, were not affected at the early points in cell cycle progression; this suggests that Brg-1 could be the first corepressor to dissociate from promoters during cell cycle progression. All of the proteins except E2F1 are dissociated from both the promoters in U937 cells stimulated with serum for 16 h. This could be because, after 16 h of serum stimulation, most of the cells are entering S phase and the dissociation of these corepressors is necessary for the expression of these proliferative promoters. PCR analysis of the c-Fos promoter was done as a control in all of the experiments.
FIG. 8.

Serum stimulation for 2 h dissociates Brg1 from Rb, as well as from proliferative promoters. (A) ChIP assays show that Brg1, but not Raf-1, is present on cdc6 and cdc25A promoters on quiescent U937 cells. Upon serum stimulation, Brg1 is dissociated from both the promoters, whereas Raf-1 associates with them. Serum stimulation in the presence of penetratin-Raf-1 peptide conjugate causes the dissociation of Raf-1 and retention of Brg1 on E2F1-responsive promoters. A scrambled peptide conjugate did not alter the binding of Brg-1 or Raf-1. Serum stimulation for 16 h causes dissociation of Rb, Raf-1, HP1, Brg1, and HDAC1 from the promoters. An irrelevant antibody was used as a control for immunoprecipitations; c-fos promoter was amplified as a negative control. (B) Immunoprecipitation-Western blot analysis showing the dissociation of Brg-1 from Rb upon serum stimulation for 2 h. This is inhibited by the penetratin-Raf-1 peptide conjugate. IP, immunoprecipitation.
Experiments were done to verify whether the reduction in the association of Brg1 with cdc25A and cdc6 promoters upon serum stimulation correlated with its dissociation from Rb. Quiescent U937 cells were stimulated for 2 h in the presence of 1 μM concentrations of the penetratin-Raf-1 conjugate. Lysates were made and the interaction of Rb with E2F1, Raf-1, Brg1, HP1, and HDAC1 were probed by immunoprecipitation-Western blot analysis. Rb could be detected in association with E2F1, HP1, Brg1, and HDAC1, but not Raf-1, in quiescent cells (Fig. 8B). As seen earlier, serum stimulation led to the binding of Raf-1 to Rb; at the same time, the binding of Brg1 to Rb was significantly inhibited. Serum stimulation of cells in the presence of 1 μM penetratin-Raf-1 peptide conjugate prevented the binding of Raf-1 to Rb; furthermore, the dissociation of Brg1 from Rb was inhibited. These results seem to suggest that binding of Raf-1 might contribute to the dissociation of Brg1 from Rb; preventing the binding of Raf-1 to Rb leads to Brg1 being retained on Rb and Rb-bound proliferative promoters.
Disruption of the Rb-Raf-1 interaction could inhibit S-phase entry of cells.
The effect of disrupting the Rb-Raf-1 interaction on cell proliferation was next examined. Quiescent HSF-8 fibroblast cells were serum stimulated for 16 h in the presence or absence of a 1 μM concentration of penetratin-Raf-1 peptide conjugate, and the number of S-phase cells was quantitated by BrdU staining. As shown in Fig. 9A, the S-phase entry of HSF-8 cells stimulated with serum or EGF and of HAECs stimulated with VEGF was considerably reduced (48 to 73%) in the presence of the penetratin-Raf-1 peptide conjugate. However, the scrambled peptide conjugate or the control peptide conjugate did not inhibit S-phase entry. Similar results were obtained on a variety of human cancer cell lines stimulated with serum (data not shown). We compared the effect of the Raf-1 peptide conjugate on the proliferation of two osteosarcoma cell lines, U2OS and Saos-2, which differ in the status of their Rb gene. It was found that while the proliferation of U2OS cells was reduced significantly by the Raf-1 peptide conjugate (Fig. 9A), it had negligible effect on the proliferation of Rb-negative Saos-2 cells. Thus, the ability of the penetratin-Raf-1 peptide conjugate to inhibit cell proliferation depends on the presence of a functional Rb gene.
FIG. 9.

(A) Disruption of the Rb-Raf-1 interaction by 1 μM penetratin-Raf-1 blocks S-phase entry. Although the Rb-positive U2OS cells were growth arrested by the penetratin-Raf-1 peptide conjugate, it had no effect on the Rb-negative Saos-2 cells. (B) The penetratin-Raf-1 conjugate potently inhibits S-phase entry in Rb+/+ MEFs, whereas it has no effect on the proliferation of Rb−/− cells. A 1 μM concentration of the scrambled peptide conjugate was used as a control in the experiment. (C) Cell cycle arrest of asynchronous U2OS cells by the penetratin-Raf-1 peptide conjugate. (D) The antiproliferative activity of penetratin-Raf-1 conjugate precedes activation of cdk's. Quiescent U2OS cells were serum stimulated for 16 h, and then a 1 μM concentration of penetratin-Raf-1 peptide conjugate was added either at the time of serum stimulation or 2, 4, 6, and 8 h after the onset of serum stimulation (the peptide treatment was for 16, 14, 12, 10, and 8 h, respectively). Penetratin-Raf-1 peptide conjugate can inhibit S-phase entry of U2OS cells when it is added within 2 h of serum stimulation, prior to activation of the cdk pathway, but addition at a later time points did not inhibit S-phase entry.
The above findings were further confirmed by repeating the BrdU incorporation experiments in Rb+/+ and Rb−/− mouse embryo fibroblasts (MEFs). As can be seen in Fig. 9B, treatment of Rb+/+ MEFs with 1 μM of the penetratin-Raf-1 conjugate potently inhibits S-phase entry. On the other hand, the penetratin-Raf-1 conjugate displayed no antiproliferative activity in Rb−/− MEFs, which conclusively proves that the presence of functional Rb protein in the cell, is crucial for the growth-inhibitory effects of the penetratin-Raf-1 conjugate.
The experiments described above were done on quiescent cells that were induced to proliferate by serum stimulation; additional experiments were done to assess whether the penetratin-Raf-1 peptide conjugate could affect the proliferation of asynchronous cells. Asynchronously growing U2OS cells were treated with a 1 μM concentration of penetratin-Raf-1 conjugate for 36 h. The peptide was added every 12 h, and the percentage of cells in S phase was quantitated by BrdU staining. As shown in Fig. 9C, the penetratin-Raf-1 peptide conjugate suppressed S-phase entry in asynchronous cells by ca. 35%.
The data presented in Fig. 4A show that the interaction of Rb with Raf-1 occurs prior to activation of the cyclin-cdk's. We wanted to examine whether addition of the penetratin-Raf-1 conjugate peptide to cells after cyclin-cdk's are activated would inhibit cell cycle progression. To do this, quiescent U2OS cells were serum stimulated for 16 h; the penetratin-Raf-1 conjugate was variously added at the same time as the serum or added 2, 4, 6, and 8 h after the addition of serum. A BrdU assay showed that cells entered S phase upon serum stimulation. The addition of the penetratin-Raf-1 peptide conjugate at the same time as the serum inhibited S-phase entry considerably (Fig. 9D). There was a certain amount of repression even when the conjugate was added 2 h after the serum stimulation began. However, when the penetratin-Raf-1 conjugate peptide was added 4, 6, or 8 h after serum stimulation subsequent to activation of cdk's, no antiproliferative activity was observed. We believe that these results suggest that the penetratin-Raf-1 peptide conjugate is ineffective in arresting cell proliferation once cyclin D/cdk's are activated.
Cell cycle arrest mediated by disrupting Rb-Raf-1 interaction can be overcome by E2F1.
Our results indicate that penetratin-Raf-1 peptide conjugate blocked cell cycle progression in G1 phase of the cell cycle and that the conjugate might function upstream of cyclins and cdk's. We had also found that overexpression of Raf-1 could overcome growth arrest mediated by p16 and dn-cdk2. We next wanted to examine whether cyclin D or E2F1 could overcome the cell cycle arrest mediated by the penetratin-Raf-1 peptide conjugate. A549 and U2OS cells were infected with Ad-E2F1 or Ad-cyclin D, in the presence of 1 μM of penetratin-Raf-1 peptide conjugate for 36 h. Cell treated with Ad-GFP were used as the control for the assay. Subsequently, a BrdU incorporation assay was done to quantitate S-phase entry. Figures 10A and B show that ectopic expression of E2F1 is able to efficiently inhibit the antiproliferative activity of penetratin-Raf-1, whereas overexpression of cyclin D has only a partial effect in both A549 and U2OS cells. This may be reflective of the fact that complete inactivation of Rb requires the function of cyclin E and associated kinases as well. Figure 10C and D shows the overexpression of E2F1 and cyclin D in A549 and U2OS cells infected with the respective Ads, relative to controls. This result suggests that E2F1, which functions as a downstream target of Rb, can overcome the growth suppressive effects of the penetratin-Raf-1 peptide conjugate.
FIG. 10.

The ectopic expression of E2F1, but not cyclin D, can override the antiproliferative effects of penetratin-Raf-1 peptide conjugate. A549 and U2OS cells were infected with Ad-E2F1 or Ad-cyclin D in the presence or absence of 1 μM penetratin-Raf-1 peptide conjugate or scrambled peptide conjugate for 36 h. Cells infected with Ad-GFP were used as the control. (A and B) The S-phase entry of cells was quantitated by BrdU staining. (C and D) Western blot analysis of A549 cells and U2OS cells infected with the indicated adenoviral constructs show overexpression of E2F1 and cyclin D compared to control cells.
Penetratin-Raf-1 peptide conjugate inhibits VEGF-induced capillary tube formation.
Since we found that the Raf-1 peptide conjugate could inhibit VEGF-induced endothelial cell proliferation and since Raf-1 kinase functions in VEGF signaling (22, 53), we examined whether it had any effect on VEGF-induced angiogenic tubule formation. To do this, HAECs were grown in Matrigel and treated with VEGF for 24 h (15). As shown in Fig. 11A, VEGF treatment led to the formation of discrete angiogenic tubules; this was not affected by the presence of penetratin or the scrambled peptide conjugate. However, VEGF treatment in the presence of a 1 μM concentration of penetratin-Raf-1 peptide conjugate caused a significant inhibition of tubule formation. This experiment suggests that Raf-1-mediated inactivation of Rb contributes to the angiogenic cascade and disruption of this interaction can inhibit neovascularization. Additional experiments showed that the penetratin-Raf-1 peptide conjugate inhibits steps of angiogenesis, including the adhesion, invasion, and migration (59) of the endothelial cells in vitro (data not shown).
FIG. 11.
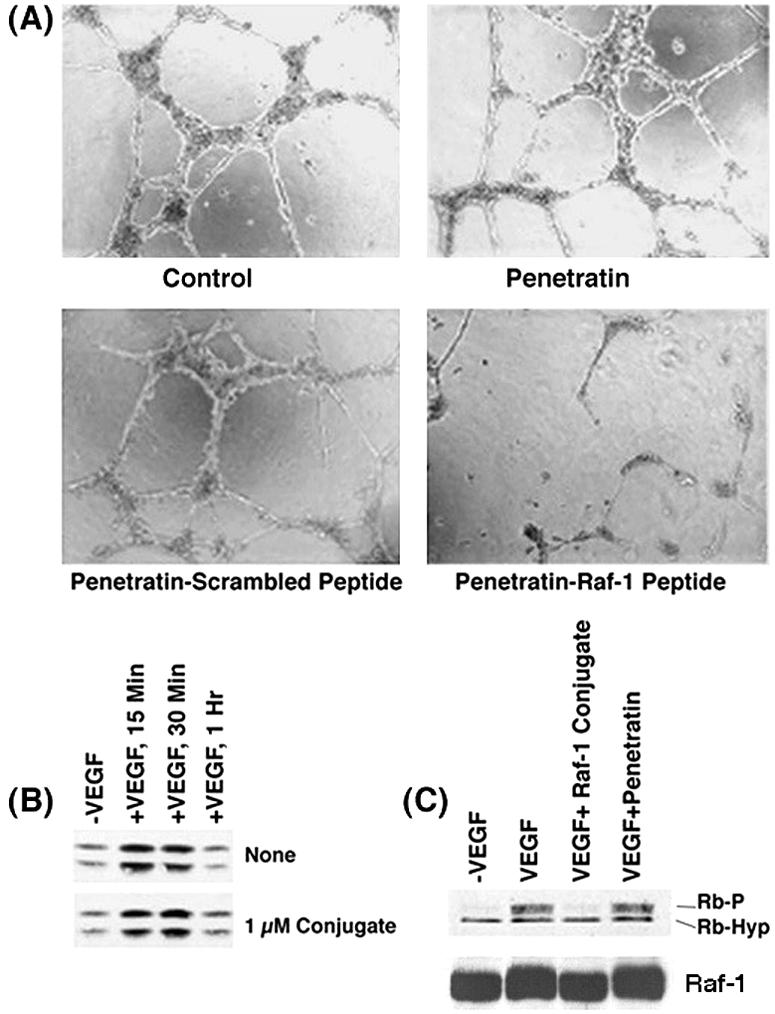
(A) Antiangiogenic activity of penetratin-Raf-1 peptide conjugate in vitro. Quiescent HAECs were stimulated with 100 ng of VEGF/ml and plated on Matrigel, in the presence or absence of 1 μM penetratin-Raf-1 peptide conjugate, penetratin alone, or scrambled peptide conjugate and then incubated for 24 h at 37°C. Capillary tube formation could be observed when the stimulation was done in the presence of a scrambled peptide conjugate,or penetratin alone, but penetratin-Raf-1 peptide conjugate greatly reduced the formation of capillary tubules. (B) Quiescent HAECs stimulated with 100 ng of VEGF/ml for the indicated time points were tested for MAP kinase activation by a phospho-ERK antibody. VEGF stimulation in the presence of the penetratin-Raf-1 peptide conjugate did not reduce MAP kinase activation (bottom panel). (C) The penetratin-Raf-1 peptide conjugate effectively inhibits Rb phosphorylation induced by VEGF stimulation of endothelial cells. Quiescent HAECs were stimulated with 100 ng of VEGF/ml for 16 h. Lysates were prepared and the phosphorylation status of Rb was assessed by Western blotting.
Attempts were made to elucidate the biochemical mechanisms underlying the inhibition. Since VEGF stimulation is known to induce the MAP kinase cascade (22), the effect of the peptide on this pathway was first examined. Quiescent HAECs were stimulated with VEGF for 15 min, 30 min, or 1 h in the presence or absence of the penetratin-Raf-1 peptide conjugate and activation of ERK1 measured by Western blotting for phospho-ERK1. VEGF stimulation induced MAP kinase activation, but the penetratin-Raf-1-peptide conjugate had no discernible effect on the process (Fig. 11B), suggesting that the inhibition of VEGF-induced angiogenesis by penetratin-Raf-1 peptide conjugate occurs independent of the MAP kinase cascade. It was next examined whether there was any change in the phosphorylation status of Rb protein upon VEGF stimulation. Western blot analysis showed that VEGF induced Rb phosphorylation, and the presence of a 1 μM concentration of the penetratin-Raf-1 peptide conjugate significantly inhibited VEGF-induced Rb phosphorylation (Fig. 11C). However, the levels of Raf-1 in the cells remain unaffected. Since the penetratin-Raf-1 peptide conjugate disrupts only the interaction of Raf-1 with Rb, it appears that Raf-1-mediated inactivation of Rb contributes to VEGF-induced angiogenesis.
The penetratin-Raf-1 peptide conjugate inhibits tumor growth in nude mice.
Since disruption of the Rb-Raf-1 interaction could arrest cell proliferation and inhibit angiogenesis, attempts were made to examine whether it had any effect on the growth of A549 human lung tumors in vivo by using a nude mouse model (6). A549 cells were rendered quiescent by serum starvation for 48 h and subsequently restimulated with serum for 2 h in the presence or absence of the penetratin-Raf-1 conjugate peptide or the scrambled peptide conjugate. BrdU incorporation experiments (Fig. 12A) indicate that the penetratin-Raf-1 conjugate significantly inhibits entry of A549 cells into S phase. An immunoprecipitation-Western blot experiment showed that a 1 μM concentration of penetratin-Raf-1 conjugate completely blocks the binding of Raf-1 to Rb, whereas the scrambled peptide conjugate has no effect on Rb-Raf-1 interaction at the same concentration (Fig. 12B). Once the antiproliferative activity of the penetratin-Raf-1 conjugate was demonstrated in A549 cells in vitro, we wanted to examine whether the Raf-1 peptide conjugate possessed antitumor activity in vivo. A549 cells were xenotransplanted into nude mice subcutaneously, and tumors were allowed to develop for 2 weeks bilaterally. At this point, either the unconjugated Raf-1 peptide or the penetratin-Raf-1 peptide conjugate was injected directly into the tumor. After 15 days, it was observed that there was a 79% reduction in the volume of the tumors injected with the Raf-1 peptide conjugate (Fig. 12C). The tumors injected with the unconjugated peptide were comparable in size to the tumors in the control animals. Histopathologic analysis of the tumor section showed a significant (57%) reduction in the number of microvessels in the tumors injected with the penetratin-Raf-1 peptide conjugate, as detected by CD31 staining (Fig. 12D) (6). This experiment shows that disruption of the Rb-Raf-1 interaction could effectively inhibit tumor growth in vivo, and agents that could mimic the function of the Raf-1 peptide might have antitumor activities.
FIG. 12.

(A) The treatment of serum-stimulated A549 cells with 1 μM penetratin-Raf-1 conjugate inhibits S-phase entry, as measured by BrdU incorporation assay. (B) The penetratin-Raf-1 peptide conjugate blocks the association of Raf-1 with Rb in A549 cells, whereas the scrambled peptide conjugate was unable to do so. A549 cells were rendered quiescent by serum starvation for 48 h and subsequently restimulated with serum in the presence of the penetratin-Raf-1 peptide conjugate or the scrambled peptide conjugate. Lysates were prepared, and the association of Rb with Raf-1 was probed by immunoprecipitation-Western blot analysis. (C) The Raf-1 peptide conjugate inhibits tumor growth in nude mice. Tumors were induced by xenografting A549 cells into nude mice. These tumors were injected with 50 mg of Raf-1 peptide or the penetratin-Raf-1 peptide conjugate/kg as described in Materials and Methods. (D) Immunohistochemical analysis of tumor biopsies shows that microvessel formation is reduced in tumors injected with the Raf-1 peptide conjugate as visualized by CD31 staining.
DISCUSSION
The Raf family of serine/threonine protein kinases are integral components of the Ras/Raf/MEK/ERK signaling pathway and are key regulators of cell proliferation and differentiation (27). Growth factor stimulation of cells led to the activation of MEK1 by Raf-1 inducing the MAP kinase cascade, which plays a major role in many cellular functions (27). However, accumulating evidence shows that Raf-1 has several developmental, prosurvival, and antiapoptotic functions independent of its role in the MEK-ERK pathway (12, 29, 50). Raf-1 has been shown to interact with several proteins, such as Bcl-2, cdc25A, and ASK1, independent of MEK1 activation (11, 32). In addition, cells derived from Raf1−/− and Raf1FF/FF (wherein Y340 and Y341 of Raf-1 have been replaced with F) mice show normal ERK activation, strongly suggesting that MEK1 activity is not required for the developmental functions of Raf-1 (12). These results suggest that the Raf protein may have cellular targets outside the MAP kinase cascade. Our results demonstrate another MEK independent function of Raf-1, where it directly targets the Rb protein.
Previous studies in our laboratory have shown that Raf-1 could act as a direct link connecting extracellular growth factor signaling to the cell cycle machinery (61). The interaction of Raf-1 family kinases with Rb family members show a certain degree of specificity: Raf-1 was found to physically interact with the pocket proteins Rb and p130, but not p107, in vitro and in vivo. A-Raf did not bind to Rb at all; B-Raf could bind to Rb in vitro and in vivo, but the interaction is not disrupted out by the Raf-1 peptide. This may be because, although the N terminus of B-Raf is homologous to c-Raf, it is not identical. Recent evidence has shown that B-Raf is mutated at a very high frequency in melanomas and to a lesser extent in several other human cancers (16). It is an intriguing possibility that that the binding of B-Raf to Rb might contribute to the oncogenic process in these cancers.
Recent studies have shown that S-phase entry in cells can occur independent of the cyclin E/cdk2 pathway (58). In addition, cell proliferation has been found to be independent of cyclin D/cdk4 pathway in Drosophila melanogaster and mouse models (38, 41). Our studies are also in agreement with previous data showing that Rb can be phosphorylated and inactivated even in the absence of cdk's (56, 63). Although the kinase activity of Raf-1 is essential for inactivation of Rb, the Rb-Raf-1 interaction was found to be independent of the MAP kinase pathway. Our results are in agreement with several studies suggesting a prosurvival function of Raf-1, independent of the MEK/ERK pathway. For example, Raf-1 can bind to and positively regulate the activity of Bcl-2 and cdc25A, whereas it negatively regulates the activity of apoptotic proteins such as ASK1 to promote cell survival (32). Our results showing the direct binding and inactivation of Rb is another example of a MAP kinase-independent function of Raf-1.
Our present data also show that Raf-1 can inactivate Rb, independent of cyclins and cdk's, in transient-transfection experiments. We believe that Raf-1 can phosphorylate Rb and inactivate it directly as well. This view is based on the following facts. (i) A residual amount of Rb phosphorylation is observed in cells prior to the activation of cyclin-cdk's. (ii) Rb phosphorylation can be observed when Raf-1, but not cyclin-cdk's, are bound to Rb. (iii) Raf-1 can phosphorylate Rb in vitro efficiently. (iv) When binding of Raf-1 to Rb is inhibited, there is no Rb phosphorylation even after 16 h of serum stimulation, even though cyclin-cdk's are activated. (v) Finally, kinase activity of Raf-1 is needed to inactivate Rb. It has been proposed that Rb is phosphorylated sequentially by cyclin D- and E-associated kinases during normal cell cycle progression (26). Our results raise the possibility that Raf-1 binding might be initiating this sequence of phosphorylation events leading to Rb inactivation. Further, it appears that the initial phosphorylation by Raf-1 may be essential for the subsequent phosphorylation steps mediated by cdk's. Whereas overexpressed Raf-1 can inactivate Rb by itself, it might be facilitating the downstream phosphorylation events mediated by cyclins and cdk's during normal cell cycle progression. This is also reflected in the observation that Brg-1 dissociates from Rb, as well as from proliferative promoters, prior to the binding of cyclin D to Rb; binding of Raf-1 and the initial phosphorylation of Rb might lead to the dissociation of this transcriptional corepressor. This would be followed by the dissociation of other corepressors, such as HDAC1, which requires further phosphorylation of Rb mediated by cyclin and cdk's (21, 54, 57).
The results presented here also show that growth arrest induced by the overexpression of p16 or dn-cdk2 can be overcome by an excess of Raf-1 that could bind to Rb. This lends support to the notion that overexpressed Raf-1 can target Rb in a manner analogous to that of cyclins or cdk's. This is also supported by the observation that excess E2F1, which is the major downstream target of Rb, can overcome growth arrest mediated by the Raf-1 peptide conjugate, whereas cyclin D has only a partial effect. The latter result also show that overexpression of cyclin D can overcome Rb-mediated growth suppression even when Raf-1 does not contribute to the process. Overall, it appears that inactivation of Rb can be achieved by excess amounts of Raf-1 or cyclin and/or cdk's to facilitate cell cycle progression.
Disruption of the Rb-Raf interaction led to an inhibition of cell proliferation, an inhibition of S-phase entry, and Rb phosphorylation in a variety of cell lines. Interestingly, the Raf-1 peptide conjugate demonstrated potent antiangiogenic activity in vitro and in vivo. Raf-1 function is known to be important for neovascularization induced by proangiogenic mediators such as bFGF and VEGF (22). Raf-1 mutants lacking the ability to bind ATP have been shown to block angiogenesis both in CAM (chicken embryo chorioallantoic membrane), as well as in mouse models (1, 28). In fact mice lacking Raf-1 die early in development, with high levels of vascular defects in the yolk sac and placenta. However, cells derived from Raf-1−/− embryos manifest normal ERK activity, and Raf-1FF/FF mutant mice do not show any defects in vasculogenesis (12). Such studies suggest that the proangiogenic effect of Raf-1 is, at least in part, independent of its role in the MAP kinase cascade. It may be possible that Raf-1 is targeting Rb in such situations to facilitate angiogenesis.
The studies described here demonstrate that Raf-1 kinase is a key regulator of Rb function and the physical interaction between these two molecules facilitate mitogenic response. Our results also show that disrupting the binding of Raf-1 to Rb results in a reduction of tumor growth and angiogenesis in vivo. It may be that agents that are capable of disrupting the Rb-Raf-1 interaction have potential therapeutic value in controlling proliferative disorders.
Acknowledgments
We thank John Sedivy, Ron Prywes, W. D. Cress, E. Harlow, I. Cozar-Castellano, K. L. Guan, D. Anderson, Ann Vojtek, Paul Robbins, and Martin McMahon for generous gifts of constructs and reagents. We also thank E. Seijo and Mark Lloyd of Analytical Microscopy and Sandy Livingston of Histopathology Core Facilities at the Moffitt Cancer Center for help with this study.
This study was funded by a grant from the NCI to S.P.C. (CA63136).
REFERENCES
- 1.Alavi, A., J. D. Hood, R. Frausto, D. G. Stupack, and D. A. Cheresh. 2003. Role of Raf in vascular protection from distinct apoptotic stimuli. Science 301:94-96. [DOI] [PubMed] [Google Scholar]
- 2.Angus, S. P., A. F. Fribourg, M. P. Markey, S. L. Williams, H. F. Horn, J. DeGregori, T. F. Kowalik, K. Fukasawa, and E. S. Knudsen. 2002. Active RB elicits late G1/S inhibition. Exp. Cell Res. 276:201-213. [DOI] [PubMed] [Google Scholar]
- 3.Avni, D., H. Yang, F. Martelli, F. Hofmann, W. M. ElShamy, S. Ganesan, R. Scully, and D. M. Livingston. 2003. Active localization of the retinoblastoma protein in chromatin and its response to S phase DNA damage. Mol. Cell 12:735-746. [DOI] [PubMed] [Google Scholar]
- 4.Bell, L. A., and K. M. Ryan. 2004. Life and death decisions by E2F-1. Cell Death Differ. 11:137-142. [DOI] [PubMed] [Google Scholar]
- 5.Bernards, R. 1997. E2F: a nodal point in cell cycle regulation. Biochim. Biophys. Acta 1333:M33-M40. [DOI] [PubMed] [Google Scholar]
- 6.Blaskovich, M. A., Q. Lin, F. L. Delarue, J. Sun, H. S. Park, D. Coppola, A. D. Hamilton, and S. M. Sebti. 2000. Design of GFB-111, a platelet-derived growth factor binding molecule with antiangiogenic and anticancer activity against human tumors in mice. Nat. Biotechnol. 18:1065-1070. [DOI] [PubMed] [Google Scholar]
- 7.Bracken, A. P., D. Pasini, M. Capra, E. Prosperini, E. Colli, and K. Helin. 2003. EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. EMBO J. 22:5323-5335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brehm, A., E. A. Miska, D. J. McCance, J. L. Reid, A. J. Bannister, and T. Kouzarides. 1998. Retinoblastoma protein recruits histone deacetylase to repress transcription. Nature 391:597-601. [DOI] [PubMed] [Google Scholar]
- 9.Chau, B. N., and J. Y. Wang. 2003. Coordinated regulation of life and death by RB. Nat. Rev. Cancer 3:130-138. [DOI] [PubMed] [Google Scholar]
- 10.Chellappan, S. P., S. Hiebert, M. Mudryj, J. M. Horowitz, and J. R. Nevins. 1991. The E2F transcription factor is a cellular target for the RB protein. Cell 65:1053-1061. [DOI] [PubMed] [Google Scholar]
- 11.Chen, J., K. Fujii, L. Zhang, T. Roberts, and H. Fu. 2001. Raf-1 promotes cell survival by antagonizing apoptosis signal-regulating kinase1 through a MEK-ERK independent mechanism. Proc. Natl. Acad. Sci. USA 98:7783-7788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chong, H., J. Lee, and K. L. Guan. 2001. Positive and negatve regulation of Raf kinase activity by phosphorylation. EMBO J. 20:3716-3727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dahiya, A., M. R. Gavin, R. X. Luo, and D. C. Dean. 2000. Role of the LXCXE binding site in Rb function. Mol. Cell. Biol. 20:6799-6805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dahiya, A., S. Wong, S. Gonzalo, M. Gavin, and D. C. Dean. 2001. Linking the Rb and polycomb pathways. Mol. Cell 8:557-569. [DOI] [PubMed] [Google Scholar]
- 15.Dasgupta, P., A. T. Singh, and R. Mukherjee. 2000. Lipophilization of somatostatin analog RC-160 with long chain fatty acid improves its antiproliferative activity on human oral carcinoma cells in vitro. Life Sci. 66:1557-1570. [DOI] [PubMed] [Google Scholar]
- 16.Davies, H., G. R. Bignell, C. Cox, P. Stephens, S. Edkins, S. Clegg, J. Teague, H. Woffendin, M. J. Garnett, W. Bottomley, N. Davis, E. Dicks, R. Ewing, Y. Floyd, K. Gray, S. Hall, R. Hawes, J. Hughes, V. Kosmidou, A. Menzies, C. Mould, A. Parker, C. Stevens, S. Watt, S. Hooper, R. Wilson, H. Jayatilake, B. A. Gusterson, C. Cooper, J. Shipley, D. Hargrave, K. Pritchard-Jones, N. Maitland, G. Chenevix-Trench, G. J. Riggins, D. D. Bigner, G. Palmieri, A. Cossu, A. Flanagan, A. Nicholson, J. W. Ho, S. Y. Leung, S. T. Yuen, B. L. Weber, H. F. Seigler, T. L. Darrow, H. Paterson, R. Marais, C. J. Marshall, R. Wooster, M. R. Stratton, and P. A. Futreal. 2002. Mutations of the BRAF gene in human cancer. Nature 417:949-954. [DOI] [PubMed] [Google Scholar]
- 17.De Bruin, A., B. Maiti, L. Jakoi, C. Timmers, R. Buerki, and G. Leone. 2003. Identification and characterization of E2F7, a novel mammalian E2F family member capable of blocking cellular proliferation. J. Biol. Chem. 278:42041-42049. [DOI] [PubMed] [Google Scholar]
- 18.De Gregori, J. 2002. The genetics of the E2F family of transcription factors: shared functions and unique roles. Biochim. Biophys. Acta 1602:131-150. [DOI] [PubMed] [Google Scholar]
- 19.Derossi, D., A. H. Joliot, G. Chassaing, and A. Prochiantz. 1994. The third helix of the Antennapedia homeodomain translocates through biological membranes. J. Biol. Chem. 269:10444-10450. [PubMed] [Google Scholar]
- 20.Diehl, J. A., M. Cheng, M. F. Roussel, and C. J. Sherr. 1998. Glycogen synthase kinase-3b regulates cyclin D1 stability and subcellular localization. Genes Dev. 12:3499-3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dunaief, J. L., B. E. Strober, S. Guha, P. A. Khavari, K. Alin, J. Luban, M. Begemann, G. R. Crabtree, and S. P. Goff. 1994. The retinoblastoma protein and BRG1 form a complex and cooperate to induce cell cycle arrest. Cell 79:119-130. [DOI] [PubMed] [Google Scholar]
- 22.Ferrara, N., H. P. Gerber, and J. LeCouter. 2003. The biology of VEGF and its receptors. Nat. Med. 9:669-676. [DOI] [PubMed] [Google Scholar]
- 23.Fusaro, G., P. Dasgupta, S. Rastogi, B. Joshi, and S. P. Chellappan. 2003. Prohibitin induces the transcriptional activity of p53 and is exported from the nucleus upon apoptotic signaling. J. Biol. Chem. 278:47853-47861. [DOI] [PubMed] [Google Scholar]
- 24.Fusaro, G., S. Wang, and S. Chellappan. 2002. Differential regulation of Rb family and prohibitin during camptothecin-induced apoptosis. Oncogene 21:4539-4548. [DOI] [PubMed] [Google Scholar]
- 25.Harbour, J. W., and D. C. Dean. 2000. Chromatin remodeling and Rb activity. Curr. Opin. Cell Biol. 12:685-689. [DOI] [PubMed] [Google Scholar]
- 26.Harbour, J. W., R. X. Luo, A. Dei Santi, A. A. Postigo, and D. C. Dean. 1999. Cdk phosphorylation triggers sequential intramolecular interactions that progressively block Rb functions as cells move through G1. Cell 98:859-869. [DOI] [PubMed] [Google Scholar]
- 27.Hilger, R. A., M. E. Scheulen, and D. Strumberg. 2002. The Ras-Raf-MEK-ERK pathway in the treatment of cancer. Onkolohie 25:511-518. [DOI] [PubMed] [Google Scholar]
- 28.Hood, J. D., M. Bednarski, R. Frausto, S. Guccione, R. A. Reisfeld, R. Xiang, and D. A. Cheresh. 2002. Tumor regression by targeted gene delivery to the neovasculature. Science 296:2404-2407. [DOI] [PubMed] [Google Scholar]
- 29.Huser, M., J. Luckett, A. Chiloeches, K. Mercer, M. Iwobi, S. Giblett, X. M. Sun, J. Brown, R. Marais, and C. Pritchard. 2001. MEK kinase activity is not necessary for Raf-1 function. EMBO J. 20:1940-1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Joshi, B., D. Ko, D. Ordonez-Ercan, and S. P. Chellappan. 2003. A putative coiled-coil domain of prohibitin is sufficient to repress E2F1-mediated transcription and induce apoptosis. Biochem. Biophys. Res. Commun. 312:459-466. [DOI] [PubMed] [Google Scholar]
- 31.Knudsen, E. S., C. Buckmaster, T. T. Chen, J. R. Feramisco, and J. Y. Wang. 1998. Inhibition of DNA synthesis by RB: effects on G1/S transition and S-phase progression. Genes Dev. 12:2278-2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kolch, W. 2000. Meaningful relationships: the regulation of the Ras/Raf/MEK/ERK pathway by protein interactions. Biochem. J. 351 Pt. 2:289-305. [PMC free article] [PubMed] [Google Scholar]
- 33.La Thangue, N. B. 2003. The yin and yang of E2F-1: balancing life and death. Nat. Cell Biol. 5:587-589. [DOI] [PubMed] [Google Scholar]
- 34.Lee, K. Y., M. H. Ladha, C. McMahon, and M. E. Ewen. 1999. The retinoblastoma protein is linked to the activation of Ras. Mol. Cell. Biol. 19:7724-7732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lowe, S. W., and C. J. Sherr. 2003. Tumor suppression by Ink4a-Arf: progress and puzzles. Curr. Opin. Genet. Dev. 13:77-83. [DOI] [PubMed] [Google Scholar]
- 36.Lukas, J., J. Bartkova, and J. Bartek. 1996. Convergence of mitogenic signaling cascades from diverse classes of receptors at the cyclin D-cyclin-dependent-kinase-pRb-controled G1 checkpoint. Mol. Cell. Biol. 16:6917-6925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ma, Y., R. Croxton, R. L. Moorer, Jr., and W. D. Cress. 2002. Identification of novel E2F1-regulated genes by microarray. Arch. Biochem. Biophys. 399:212-224. [DOI] [PubMed] [Google Scholar]
- 38.Malumbres, M., and M. Baracid. 2001. To cycle or not to cycle: a critical decision in cancer. Nat. Rev. Cancer 1:222-231. [DOI] [PubMed] [Google Scholar]
- 39.Malumbres, M., and A. Pellicer. 1998. Ras pathways to cell cycle control and cell transformation. Front. Biosci. 3:D887-D912. [DOI] [PubMed] [Google Scholar]
- 40.Matsushime, H., D. E. Quelle, S. A. Shurtleff, M. Shibuya, C. J. Sherr, and J. Y. Kato. 1994. D-type cyclin-dependent kinase activity in mammalian cells. Mol. Cell. Biol. 14:2066-2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Meyer, C. A., H. W. Jacobs, and C. F. Lehner. 2002. CyclinD-cdk4 is not a master regulator of cell multiplication in Drosophila embroyos. Curr. Biol. 12:661-666. [DOI] [PubMed] [Google Scholar]
- 42.Mitra, J., C. Y. Dai, K. Somasundaram, W. S. El-Deiry, K. Satyamoorthy, M. Herlyn, and G. H. Enders. 1999. Induction of p21WAF1/CIP1 and inhibition of Cdk2 mediated by the tumor suppressor p16INK4a. Mol. Cell. Biol. 19:3916-3928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moroni, M. C., E. S. Hickman, E. L. Denchi, G. Caprara, E. Colli, F. Cecconi, H. Muller, and K. Helin. 2001. Apaf-1 is a transcriptional target for E2F and p53. Nat. Cell Biol. 3:552-558. [DOI] [PubMed] [Google Scholar]
- 44.Morris, E. J., and N. J. Dyson. 2001. Retinoblastoma protein partners. Adv. Cancer Res. 82:1-54. [DOI] [PubMed] [Google Scholar]
- 45.Murray, A. W. 2004. Recycling the cell cycle: cyclins revisited. Cell 116:221-234. [DOI] [PubMed] [Google Scholar]
- 46.Narita, M., S. Nunez, E. Heard, A. W. Lin, S. A. Hearn, D. L. Spector, G. J. Hannon, and S. W. Lowe. 2003. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 113:703-716. [DOI] [PubMed] [Google Scholar]
- 47.Nath, N., H. Bian, E. F. Reed, and S. P. Chellappan. 1999. HLA class I-mediated induction of cell proliferation involves cyclin E-mediated inactivation of Rb function and induction of E2F activity. J. Immunol. 162:5351-5358. [PubMed] [Google Scholar]
- 48.Nevins, J. R. 2001. The Rb/E2F pathway and cancer. Hum. Mol. Genet. 10:699-703. [DOI] [PubMed] [Google Scholar]
- 49.Nielsen, S. J., R. Schneider, U. M. Bauer, A. J. Bannister, A. Morrison, D. O'Carroll, R. Firestein, M. Cleary, T. Jenuwein, R. E. Herrera, and T. Kouzarides. 2001. Rb targets histone H3 methylation and HP1 to promoters. Nature 412:561-565. [DOI] [PubMed] [Google Scholar]
- 50.Pearson, G., R. Bumeister, D. O. Henry, M. H. Cobb, and M. A. White. 2000. Uncoupling Raf-1from MEK1/2 impairs only a subset of cellular responses to Raf activation. J. Biol. Chem. 48:37303-37306. [DOI] [PubMed] [Google Scholar]
- 51.Pediconi, N., A. Ianari, A. Costanzo, L. Belloni, R. Gallo, L. Cimino, A. Porcellini, I. Screpanti, C. Balsano, E. Alesse, A. Gulino, and M. Levrero. 2003. Differential regulation of E2F1 apoptotic target genes in response to DNA damage. Nat. Cell Biol. 5:552-558. [DOI] [PubMed] [Google Scholar]
- 52.Pruitt, K., R. G. Pestell, and C. J. Der. 2000. Ras inactivation of the retinoblastoma pathway by distinct mechanisms in NIH 3T3 fibroblasts and RIE-1 epithelial cells. J. Biol. Chem. 275:40916-40924. [DOI] [PubMed] [Google Scholar]
- 53.Seko, Y., N. Takahashi, K. Tobe, K. Ueki, T. Kadowaki, and Y. Yazaki. 1998. Vascular endothelial growth factor (VEGF) activates Raf-1, mitogen-activated protein (MAP) kinases, and S6 kinase (p90rsk) in cultured rat cardiac myocytes. J. Cell Physiol. 175:239-246. [DOI] [PubMed] [Google Scholar]
- 54.Siddiqui, H., D. A. Solomon, R. W. Gunawardena, Y. Wang, and E. S. Knudsen. 2003. Histone deacetylation of RB-responsive promoters: requisite for specific gene repression but dispensable for cell cycle inhibition. Mol. Cell. Biol. 23:7719-7731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stevaux, O., and N. J. Dyson. 2002. A revised picture of E2f transcriptional regulation and Rb funtion. Curr. Opin. Cell Biol. 14:684-691. [DOI] [PubMed] [Google Scholar]
- 56.Stiewe, T., J. Stanelle, C. C. Theseling, B. Pollmeier, M. Beitzinger, and B. M. Putzer. 2003. Inactivation of retinoblastoma (RB) tumor suppressor by oncogenic isoforms of the p53 family member p73. J. Biol. Chem. 278:14230-14236. [DOI] [PubMed] [Google Scholar]
- 57.Strobeck, M. W., K. E. Knudsen, A. F. Fribourg, M. F. DeCristofaro, B. E. Weissman, A. N. Imbalzano, and E. S. Knudsen. 2000. BRG-1 is required for RB-mediated cell cycle arrest. Proc. Natl. Acad. Sci. USA 97:7748-7753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tetsu, O., and F. McCormick. 2003. Proliferation of cancer cells despite CDK2 inhibition. Cancer Cell 3:233-245. [DOI] [PubMed] [Google Scholar]
- 59.Vukanovic, J., A. Passaniti, T. Hirata, R. J. Traystman, B. Hartley-Asp, and J. T. Isaacs. 1993. Antiangiogenic effects of the quinoline-3-carboxamide linomide. Cancer Res. 53:1833-1837. [PubMed] [Google Scholar]
- 60.Wang, S., G. Fusaro, J. Padmanabhan, and S. P. Chellappan. 2002. Prohibitin colocalizes with Rb in the nucleus and recruits N-CoR and HDAC1 for transcriptional repression. Oncogene 21:8388-8396. [DOI] [PubMed] [Google Scholar]
- 61.Wang, S., R. N. Ghosh, and S. P. Chellappan. 1998. Raf-1 physically interacts with Rb and regulates its function: a link between mitogenic signaling and cell cycle regulation. Mol. Cell. Biol. 18:7487-7498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang, S., N. Nath, M. Adlam, and S. Chellappan. 1999. Prohibitin, a potential tumor suppressor, interacts with RB and regulates E2F function. Oncogene 18:3501-3510. [DOI] [PubMed] [Google Scholar]
- 63.Wang, S., N. Nath, A. Minden, and S. Chellappan. 1999. Regulation of Rb and E2F by signal transduction cascades: divergent effects of JNK1 and p38 kinases. EMBO J. 18:1559-1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Weinberg, R. A. 1995. The retinoblastoma protein and cell cycle control. Cell 81:323-330. [DOI] [PubMed] [Google Scholar]
- 65.Yeung, K., T. Seitz, S. Li, P. Janosch, B. McFerran, C. Kaiser, F. Fee, K. D. Katsanakis, D. W. Rose, H. Mischak, J. M. Sedivy, and W. Kolch. 1999. Suppression of Raf-1 kinase activity and MAP kinase signalling by RKIP. Nature 401:173-177. [DOI] [PubMed] [Google Scholar]