

Abstract
Ser/Thr phosphorylation of insulin receptor substrate (IRS) proteins negatively modulates insulin signaling. Therefore, the identification of serine sites whose phosphorylation inhibit IRS protein functions is of physiological importance. Here we mutated seven Ser sites located proximal to the phosphotyrosine binding domain of insulin receptor substrate 1 (IRS-1) (S265, S302, S325, S336, S358, S407, and S408) into Ala. When overexpressed in rat hepatoma Fao or CHO cells, the mutated IRS-1 protein in which the seven Ser sites were mutated to Ala (IRS-17A), unlike wild-type IRS-1 (IRS-1WT), maintained its Tyr-phosphorylated active conformation after prolonged insulin treatment or when the cells were challenged with inducers of insulin resistance prior to acute insulin treatment. This was due to the ability of IRS-17A to remain complexed with the insulin receptor (IR), unlike IRS-1WT, which underwent Ser phosphorylation, resulting in its dissociation from IR. Studies of truncated forms of IRS-1 revealed that the region between amino acids 365 to 430 is a main insulin-stimulated Ser phosphorylation domain. Indeed, IRS-1 mutated only at S408, which undergoes phosphorylation in vivo, partially maintained the properties of IRS-17A and conferred protection against selected inducers of insulin resistance. These findings suggest that S408 and additional Ser sites among the seven mutated Ser sites are targets for IRS-1 kinases that play a key negative regulatory role in IRS-1 function and insulin action. These sites presumably serve as points of convergence, where physiological feedback control mechanisms, which are triggered by insulin-stimulated IRS kinases, overlap with IRS kinases triggered by inducers of insulin resistance to terminate insulin signaling.
The insulin receptor (IR) mediates insulin action through the phosphorylation of substrate proteins on tyrosine residues. IR substrates include the three isoforms of Shc, insulin receptor substrate (IRS) proteins (IRS-1 to IRS-4), p60dok, Cbl, APS, and Gab-1 (reviewed in references 16, 20, 29, and 40). IRS proteins contain a conserved pleckstrin homology (PH) domain, located at their amino termini, that serves to anchor the IRS proteins to membrane phosphoinositides in close proximity to the insulin receptor (35). The PH domain of IRS proteins is flanked by a phosphotyrosine binding (PTB) domain. The PTB domain, present in a number of signaling molecules (24), shares 75% sequence identity between IRS-1 and IRS-2 (31) and functions as a binding site to the NPXY motif at the juxtamembrane (JM) domain of the insulin receptor (7, 36). The C-terminal regions of IRS proteins are poorly conserved. The C-terminal region contains multiple Tyr phosphorylation motifs that serve as a signaling scaffold, providing a docking interface for SH2 domain-containing proteins, such as the p85α regulatory subunit of phosphatidylinositol 3-kinase (PI3K), Grb2, Nck, Crk, Fyn, and SHP-2, which further propagate the metabolic and growth-promoting effects of insulin (16, 20, 29, 40).
IRS-1 contains more than 70 potential Ser/Thr phosphorylation sites with homologies to consensus phosphorylation sites for casein kinase II, protein kinase B (PKB), protein kinase C (PKC), mitogen-activated protein kinases (MAPKs), CDC2, and cyclic-AMP- and cyclic-GMP-dependent protein kinase (33). Phosphorylation of Ser/Thr residues of IRS proteins has a dual function and positively or negatively modulates insulin signal transduction. Serine phosphorylation within the PTB domain of IRS-1 by insulin-stimulated PKB protects IRS proteins from the rapid action of protein tyrosine phosphatases and enables them to maintain their Tyr-phosphorylated active conformation, implicating PKB as a positive regulator of IRS-1 functions (26). In contrast, Ser/Thr phosphorylation of IRS proteins by other insulin-stimulated Ser/Thr kinases, such as PKCζ (22), serves as a physiological negative-feedback control mechanism utilized by insulin to uncouple IR-IRS complexes, inhibit further Tyr phosphorylation of IRS proteins, and terminate insulin signaling. Furthermore, inducers of insulin resistance, such as free fatty acids (FFA), take advantage of this physiological shutoff mechanism and activate Ser/Thr kinases that phosphorylate IRS-1 at the same inhibitory sites (reviewed in reference 40).
Ser/Thr phosphorylation can induce the dissociation of IRS proteins from the IR (14, 22, 25), hinder Tyr phosphorylation sites (23), release the IRS proteins from intracellular complexes that maintain them in close proximity to the receptor (34), induce IRS protein degradation (27), or turn IRS proteins into inhibitors of the IR kinase (15). These multiple effects suggest that the Ser sites subjected to phosphorylation play a key role in regulating IRS-1 function. Several such Ser residues were identified. Ser307, the phosphorylation of which is catalyzed by a number of kinases (1, 10, 39), negatively regulates IRS functions. Because Ser307 is adjacent to the PTB domain of IRS-1, its phosphorylation might disrupt the interaction between the JM domain of the IR and the PTB domain of IRS-1 and thus inhibit insulin-stimulated Tyr phosphorylation of IRS-1. Similarly, conventional members of the PKC family that are activated by phorbol esters or endothelin 1 stimulate members of the MAPK pathway to phosphorylate IRS-1 at Ser612 and at additional sites in its COOH tail (5). Such phosphorylation inhibits the interactions of IRS-1 both with IR and with downstream effectors of IRS-1, such as PI3K.
Still, phosphorylation of IRS-1 at the above sites cannot account for all the effects of Ser kinases on IRS proteins; therefore, in this study, we set out to identify novel Ser sites that undergo insulin-dependent phosphorylation by IRS kinases that negatively regulate IRS-1 function. Our results indicate that mutation of seven Ser residues, six located at putative PKC phosphorylation sites (RXXS) proximal to the PTB domain of IRS-1, renders the mutated IRS-1 protein resistant to the inhibitory effects of IRS-1 kinases stimulated either by insulin or by agents that induce insulin resistance. Utilizing truncated forms of IRS-1, we show that Ser408 is a prime inhibitory phosphorylation site, located within a region (amino acids [aa 365 to 430]) subjected to Ser phosphorylation in response to insulin or inducers of insulin resistance. These findings suggest that S408 and additional Ser sites among the seven mutated Ser sites (7S sites), are targets for IRS-1 kinases that play a key negative regulatory role in IRS-1 function and insulin action. These sites could therefore serve as novel potential targets for therapeutic intervention in cases of insulin resistance and diabetes.
MATERIALS AND METHODS
Materials.
Human insulin, wheat germ agglutinin (WGA) coupled to agarose beads, glutathione-agarose beads, protease inhibitor cocktail, protein A-Sepharose CL-4B, goat anti-mouse antibodies coupled to agarose beads, wortmannin, and phorbol 12-myristate 13-acetate (TPA) were purchased from Sigma. Lipofectamine and OptiMem were from GIBCO-BRL (Grand Island, N.Y.). Alkaline phosphatase was from Boehringer GmbH (Mannheim, Germany). T4 ligase, gel extraction kit, and pGEM-T were purchased from Promega. Tri-Reagent was purchased from Molecular Research Center, Inc. SeaPlaque agarose was purchased from Bio Whittaker Molecular Applications (Rockland, Maine). Rapamycin, PD98059, and SB203580 were from Calbiochem (La Jolla, Calif.). Jet-PEI was purchased from Poly Transfection.
Antibodies.
Monoclonal phosphorylated-Tyr (PY-20) and polyclonal IRβ antibodies were obtained from Transduction Labs (Lexington, Ky.). Polyclonal IRS-1 antibodies were prepared as described previously (11). Polyclonal Myc antibodies were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, Calif.). Polyclonal antibodies to phosphorylated-Ser307 of IRS-1 were from BioSource, Inc. (Camarillo, Calif.). Rabbit polyclonal serum directed against phosphorylated Ser408 was generated using a synthetic peptide designed to contain phosphorylated Ser408 and surrounding amino acids CLFPRRSS(-PO4)ASVSG, with an additional Cys residue at the N-terminal end of the peptide.
Plasmid construction. (i) Myc-tagged IRS-1.
The cDNA coding for the mouse IRS-1 in the pCNA-3 expression plasmid (35) was digested with HindIII and BspE1. This deleted a 9-bp fragment from the 5′ IRS-1 cDNA, which was replaced by a double-stranded synthetic oligonucleotide encoding the Myc tag and the missing amino acids of IRS-1 (shown in bold type) MEQKLISEEDLNMASP.
(ii) Myc-tagged truncated IRS-1 (aa 1 to 430) (extended PH/PTB domain of IRS-1 [PH/PTB-L]).
Myc-tagged mouse IRS-1 cDNA (N terminus, nucleotides [nt]1 to 1290), coding for the first 430 aa of IRS-1, was PCR amplified with primers 5′-CAG GAT CCG CAT ATG GAA CAA AAG CTC-3′ (sense) and 5′-GAG AAT TCA TCA GGG ACT AGA ACC ATA-3′ (antisense), using the pcDNA3-Myc-IRS-1 plasmid as a template. The PCR product was ligated into pGEM-T. The insert was excised from pGEM-T with BamHI and EcoRI (shown in italics) and then ligated into pcDNA3 at the same restriction sites to generate pcDNA3-Myc-IRS-1PH/PTB-L.
(iii) Myc-tagged truncated IRS-1 (aa 1 to 365) (shorter truncated form of IRS-1 [PH/PTB-S]).
Myc-tagged mouse IRS-1 cDNA (N terminus, nt 1 to 1095), coding for the first 365 aa of IRS-1, was PCR amplified with primers 5′-AAGCTTAAGATATCGATCATATG-3′ (sense) and 5′-TTAGTTGAGTGGGGGGTGCAGCCT 3′ (antisense), using pcDNA3-Myc-IRS-1 as a template. The PCR product was ligated into pGEM-T. The insert was excised from pGEM-T with EcoRV (shown in italics) and NotI (in the pGEM-T construct) and ligated into pcDNA3 at the same restriction sites.
Generation of IRS-1 mutants.
Site-directed mutagenesis was performed with the primers given below using a QuikChange site-directed mutagenesis kit (Stratagene) according to the manufacturer's instructions. pcDNA3-IRS-1 served as a template. Mutated nucleotides are shown in bold type, and mutated amino acid codons are shown in italic type. The restriction sites introduced are underlined. The mutations were verified by restriction mapping and sequencing. (i) The S336A primers were 5′-C ATG TCC CGT CCA GCT GCA GTG GAT GGC AG-3′ (sense) and 5′-CT GCC ATC CAC TGC AGC TGG ACG GGA CAT G-3′ (antisense). A PstI site was introduced. (ii) The S407A primers were 5′-C TTC CCG AGG CGC GCT AGC GCT TCC GTG TCC GG-3′ (sense) and 5′-CC GGA CAC GGA AGC GCT AGC GCG CCT CGG GAA G-3′ (antisense). NheI/Eco47III sites were introduced. (iii) The S408A primers were 5′-C TTC CCG AGG CGC TCAGCT GCT TCC GTG TCC GG-3′ (sense) and 5′-CC GGA CAC GGA AGC AGC TGA GCG CCT CGG GAA G-3′ (antisense). A PvuII site was introduced. (iv) The S407/408A IRS-1 primers were 5′-C TTC CCG AGG CGC GCT GCA GCT TCC GTG TCC GG-3′ (sense) and 5′-CC GGA CAC GGA AGC TGC AGC GCG CCT CGG GAA G-3′ (antisense). A PstI site was introduced. (v) The IRS-1 protein in which the seven serines were mutated to alanine (IRS-17A) was generated on the basis of IRS-14A, the IRS-1 protein in which four Ser residues, S265, S302, S325, and S358, were mutated to Ala as we described previously (26). The other three Ser sites, S336, S407, and S408, were mutated into Ala sequentially using the above two sets of overlapping primers.
Transient and stable transfections of CHO-T cells.
Chinese hamster ovary (CHO) cells that overexpress the insulin receptor (CHO-T cells) (26) were transiently transfected with plasmid pcDNA3-Myc-IRS-1PH/PTB-L or pcDNA3-Myc-IRS-1PH/PTB-S, using Lipofectamine according to the protocol provided by Life Technologies. The transfected cells were cultured in F12 medium supplemented with 10% fetal calf serum (FCS). After 24 h, the cells were starved for 16 h and then stimuli were applied. To generate stable clones, CHO-T cells were cotransfected with plasmid pcDNA3-IRS-1wt or pcDNA3-IRS-17A, encoding wild-type IRS-1 (IRS-1WT) or the mutant IRS-1 (IRS-17A), respectively, together with pBabe-puro, encoding puromycin resistance. After 24 h, the transfected cells were subjected to selection with 10 μg of puromycin per ml. Stable clones expressing proper amounts of the target protein were selected and further propagated in a medium containing 10 μg of puromycin per ml.
Generation of adenovirus-based IRS-1 constructs.
Adenoviruses harboring the genes of interest were generated according to the protocol provided with the AdEasy vector system (Quantum) (13). Briefly, the target cDNAs, encoding Myc-tagged mouse IRS-1 (wild-type [WT], 7A mutant, and the S408A mutant) were ligated into the shuttle plasmid pAdTrack-CMV at an EcoRV restriction site. The correct orientation was confirmed by restriction mapping with XhoI or KpnI, which yielded a ∼200- or ∼300-bp fragment, respectively. This plasmid also contains a green fluorescent protein cassette whose expression is driven by an independent promoter, which serves as a tracing marker. The pAdTrack-CMV carrying the above target genes was cotransformed with pAdEasy-1, containing the adenovirus genome, into Escherichia coli strain BJ5183, where homologous recombination took place. Positive colonies were identified by restriction analysis. The recombinant pAdEasy-1-IRS-1 plasmid (WT, 7A, or S408A) was linearized with PacI and transfected into 293 cells using the Jet-PEI transfection reagent. Viruses, amplified in the 293 cell line according the manufacturer's instructions, were stored at −80°C at a viral titer of ∼1010 PFU/ml.
Infection of Fao cells with adenovirus.
Fao cells were grown in RPMI medium containing 10% FCS. When the cells reached 70% confluence, they were infected with adenoviruses harboring the gene of interest at a titer of 7 × 107 PFU/ml. After incubation for 2 h at 37°C, the virus-containing medium was diluted 1:5 in fresh RPMI medium. After 24 h, the medium was replaced with fresh RPMI medium containing 10% FCS. At 48 h postinfection, the Fao cells were starved in serum-free RPMI medium for 16 h and then subjected to the different treatments described below.
Preparation of FFA solution.
Solutions containing FFA complexed to bovine serum albumin (BSA) were prepared essentially as described previously (38). Briefly, a 100 mM palmitic acid stock solution was prepared in 0.1 M NaOH at 70°C. The appropriate amount was then complexed with a FFA-free 10% BSA solution at 55°C for 30 min. The FFA-BSA complex was cooled down to room temperature and filtered in a sterile manner prior to its addition to the cell culture.
Treatment of cells and preparation of extracts.
Virally infected Fao cells were grown as described above. Naïve rat hepatoma (Fao) cells or CHO cells were grown in RPMI medium supplemented with 10% FCS as described previously (22). When the cells had reached 70 to 80% confluence, they were deprived of serum for 16 h prior to each experiment and then incubated with inducers of insulin resistance and/or insulin. Treated cells were washed three times with ice-cold phosphate-buffered saline (PBS) and harvested in buffer B (25 mM Tris-HCl, 2 mM sodium orthovanadate, 0.5 mM EGTA, 10 mM NaF, 10 mM sodium pyrophosphate, 80 mM β-glycerophosphate, 25 mM NaCl, 1% Triton X-100, protease inhibitor cocktail [diluted 1:1,000] [pH 7.4]). Cell extracts were centrifuged at 12,000 × g for 20 min at 4°C, and the supernatants were collected. Samples of 50 to 150 μg were mixed with 5× Laemmli sample buffer (17), boiled, and resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) under reducing conditions. The proteins were transferred to nitrocellulose membrane for Western blotting with various antibodies.
Immunoprecipitation.
Protein A Sepharose beads (10 to 20 μl of packed beads/point) or goat anti-mouse antibodies, coupled to agarose beads (10 μl packed beads/point) were washed three times with ice-cold 0.1 M Tris-HCl (pH 8.0) and were incubated with various antibodies in 0.1 M Tris-HCl (pH 8.0) for 1 h at 4°C. Supernatants (centrifuged at 12,000 × g) of cell extracts in buffer B, containing 0.5 to 1.5 mg of protein, were incubated for 2 h at 4°C with the immobilized antibodies. Immunocomplexes were washed twice with buffer B and twice with ice-cold PBS and mixed with 5× Laemmli sample buffer (17). The samples were boiled and then resolved by SDS-PAGE under reducing conditions.
Assay of IR-IRS-1 complex formation.
Fao cells at 80% confluence were harvested in buffer IR (50 mM Tris-HCl, 1% Nonidet P-40 [NP-40], 0.25% sodium deoxycholate, 150 mM NaCl, 1 mM EGTA, 1 mM sodium orthovanadate, 1 mM NaF, protease inhibitor cocktail [diluted 1:1,000] [pH 7.4]). The cell extracts were centrifuged at 12,000 × g for 15 min at 4°C, and the supernatants were collected. Aliquots (1.0 to 1.5 mg) were incubated with 20 μl of packed WGA-coupled agarose beads for 1 h at 4°C. The immobilized IR was washed twice with buffer IR and three times with buffer A (25 mM Tris-HCl, 2 mM sodium orthovanadate, 0.5 mM EGTA, 10 mM NaF, 10 mM sodium pyrophosphate, 80 mM β-glycerophosphate, 25 mM NaCl, protease inhibitor cocktail [diluted 1:1,000] [pH 7.4]). Samples (20 μl) of the IR-WGA-coupled agarose beads were then incubated at 4°C with 12,000 × g supernatants of cytosolic extracts (1.0 to 1.5 mg), made in buffer A, derived from Fao cells treated with insulin for different periods of time. The beads were washed twice with buffer A and twice with ice-cold PBS and then boiled in Laemmli sample buffer (17). Samples were resolved by SDS-PAGE and were immunoblotted with IRS-1 antibodies.
In vitro Tyr kinase assay.
Stable cell lines of CHO-TWT (CHO-T cells stably overexpressing IRS-1WT) and CHO-T7A (CHO-T cells stably overexpressing IRS-17A) were grown in 90-mm-diameter plates in F12 medium supplemented with 10% FCS and 10 μg of puromycin per ml. Cells were washed three times with PBS and harvested in 100 μl of buffer A containing 0.5% NP-40. Cell extracts were centrifuged at 12,000 × g for 20 min at 4°C, and the supernatants were collected. The supernatants were divided into aliquots (90 μl, ∼1 mg). These samples were incubated with 10−6 M insulin for 10 min at 22°C. Then, 90 μl of 2× buffer E (50 mM HEPES, 10 mM Mg acetate, 4 mM Mn acetate, 1 mM ATP, protease inhibitor cocktail [diluted 1:1,000] [pH 7.4]) was added to each sample, and the samples were incubated for another 15 min at room temperature with vigorous shaking. The reaction in each sample was stopped by adding 200 μl of buffer A, and the samples were cooled to 4°C. The reaction mixture was subjected to immunoprecipitation with IRS-1 antibodies. Immunocomplexes were resolved by SDS-PAGE and immunoblotted with antiphosphotyrosine (anti-PY) antibodies.
RESULTS
Mutation of selected Ser residues of IRS-1 enhances its ability to complex with IR and for its Tyr residues to remain phosphorylated after chronic insulin treatment.
We have previously demonstrated that incubation of Fao cells with 10−7 M insulin rapidly stimulates transient Tyr phosphorylation of IRS proteins, which declines after 60 min of incubation with the hormone (22, 25). This decline is preceded by a decrease in the electrophoretic mobility of IRS-1 as a result of Ser/Thr phosphorylation that causes a marked (>40%) reduction in its ability to interact with the insulin receptor (22, 25). These findings, illustrated in Fig. 1A, suggest that prolonged insulin treatment results in activation of insulin-stimulated Ser/Thr kinases that phosphorylate IRS-1, uncouple it from the receptor, and inhibit further Tyr phosphorylation of the protein. Because the PTB domain of IRS proteins is the major region that forms contacts with the JM region of IR, phosphorylation of Ser residues located at or in close proximity to the PTB domain might inhibit IR-IRS-1 interactions, thus negatively regulating insulin signaling.
FIG. 1.

Tyr phosphorylation of IRS-17A and its interactions with IR. (A) CHO-T cells stably overexpressing either WT IRS-1 (CHO-TWT) or IRS-17A (CHO-T7A) at 80% confluence were deprived of serum for 16 h prior to the experiment. The cells were then incubated with insulin for the indicated times at 37°C. Cell extracts (100 μg) were resolved by SDS-PAGE (7.5% polyacrylamide) and were immunoblotted with the indicated antibodies (anti-PY [α-PY] or anti-IRS-1 [α-IRS-1] antibodies). The results of one experiment, which were representative of six experiments, are shown. (B) IRS-1 was isolated from CHO-TWT or CHO-T7A cells. IRS-1WT and IRS-17A were phosphorylated in vitro by IR as described in Materials and Methods. The reaction mixture was subjected to immunoprecipitation with anti-IRS-1 antibodies. Immunocomplexes were resolved by SDS-PAGE (7.5% polyacrylamide) and were immunoblotted with anti-pY or anti-IRS-1 antibodies. (C) CHO-TWT and CHO-T7A cells were deprived of serum for 16 h prior to the experiment. The cells were stimulated with 100 nM insulin for the indicated times at 37°C. Cell extracts were prepared, and samples (1 mg) were bound to immobilized IR. IR-IRS-1 complexes were resolved by SDS-PAGE (7.5% polyacrylamide) and immunoblotted with the indicated antibodies. In parallel, samples of total cell extracts (100 μg) were resolved by SDS-PAGE (7.5% polyacrylamide) and immunoblotted with anti-IRS-1 antibodies. The results of one experiment, which were representative of two experiments performed on duplicate samples, are shown.
The extended PH/PTB domain of IRS-1, termed PH/PTB-L (aa 1 to 430), contains 89 Ser/Thr residues, making the identification of selected phosphorylation sites within this region a serious task. To narrow down the options, we began by mutating seven Ser residues to Ala. The seven Ser residues were located at potential phosphorylation sites for PKC, a family of Ser kinases whose conventional and atypical isoforms (e.g., PKCα and PKCζ) have been implicated as potential IRS-1 kinases that negatively modulate IRS-1 function (6, 21, 22, 25). The amino acids flanking the seven Ser sites that were mutated are Ser265-RPRSKSQ, Ser302-RSRTESI, Ser325-RVRASSD, Ser336-MSRPASV, Ser358-RHRGSSR, Ser407-LFPRRSSASV, and Ser408-FPRRSSA (conserved amino acids are shown in bold type). The seven mutated sites contain four residues (S265, S302, S325, and S358 [numbering based on the mouse IRS-1 sequence]) within PKB and PKC phosphorylation sites (RXRXXS), two residues (S336 and S408) within PKC phosphorylation sites (RXXS), and one site (S407) that conforms to an initiation site for phosphorylation by glycogen synthase kinase 3beta (GSK-3β) (SXXS). The IRS-1 mutant was named IRS-17A.
In vitro Tyr phosphorylation of IRS-17A by partially purified IR revealed that IRS-17A was phosphorylated to a level comparable to that of WT IRS-1 (Fig. 1B), indicating that the overall structure of IRS-17A is not altered due to the 7A mutation. When transfected into CHO-T cells, IRS-17A underwent rapid Tyr phosphorylation, similar to IRS-1WT, in response to acute (3-min) insulin treatment (Fig. 1A, compare lanes b and e), which was consistent with the results of the in vitro study. However, while the phosphorylated-Tyr (P-Tyr) content of IRS-1WT rapidly declined upon prolonged (60-min) insulin treatment, IRS-17A was significantly more resistant to Tyr dephosphorylation (Fig. 1A, compare lanes c and f). Furthermore, as a consequence of its increased Ser phosphorylation, the mobility of IRS-1WT treated with insulin for 60 min was decreased compared to its mobility at the 3-min time point (Fig. 1A, compare lanes c and b). In contrast, IRS-17A showed only a slight decrease in its mobility at 60 min compared to its mobility after 3 min of insulin treatment (Fig. 1A, compare lanes f and e).
To study the mechanism underlying the sustained Tyr phosphorylation of IRS-17A, in vitro binding assays of IRS-1 to IR were performed. IRS-1WT and IRS-17A showed comparable levels of binding to IR after acute (3 min) insulin treatment (Fig. 1C, compare lanes a and c). Consistent with our previous studies (22, 25), binding of IRS-1WT to IR was reduced after prolonged (60-min) insulin treatment (Fig. 1C, lane b); however this reduction was largely abolished in IRS-17A (Fig. 1C, lane d). These results indicate that insulin-induced phosphorylation of serine residues mutated in IRS-17A serves to negatively regulate IRS-1 function by impairing its ability to interact with IR. Note that four of the serines mutated in IRS-17A conform to potential PKB (positive regulatory) sites (26), while others are potential PKC (negative regulatory) sites (22). Our results indicate that when simultaneously mutated (as in 7A), the sites associated with negative regulation are functionally dominant over the serine sites that positively regulate IRS function.
Adenoviral infections enable quantitative expression of Myc-IRS in Fao cells.
To study the effect of mutation of IRS-1 in insulin responsive cells, the cDNAs encoding Myc-tagged IRS-1 (Myc-IRS-1) (wild type) or mutant IRS-17A were incorporated into adenoviral constructs that enables their expression in rat hepatoma Fao cells that are otherwise refractory to quantitative introduction of foreign genes by conventional transfection methods. Myc-IRS-1 (wild type or mutant) was readily detected in Fao cells 48 h postinfection at increasing amounts that correlated with the virus titer (Fig. 2A). Note that infection of Fao cells with control insert-free adenoviruses did not affect insulin-induced Tyr phosphorylation or the protein level of the endogenous IRS-1 (Fig. 2B), indicating that infection with adenoviruses per se does not impair insulin signaling in these cells.
FIG. 2.

Overexpression of Myc-IRS-1WT and Myc-IRS-17A in Fao cells. Fao cells at 70 to 80% confluence were infected with Ad-Myc-IRS-1WT or Ad-Myc-IRS-17A at the indicated PFU (A) or with an insert-free adenoviral construct at 7 × 107 PFU/ml (B). After 48 h, cells were left untreated (A) or treated with insulin for the indicated times (B). Cell extracts were prepared, samples were resolved by SDS-PAGE (7.5% polyacrylamide) and immunoblotted with the indicated antibodies (anti-IRS-1 [α-IRS-1], anti-myc [α-myc], or anti-PY [α-PY] antibodies). The results of one experiment, which were representative of three experiments (B) or two experiments (A), are shown.
IRS-17A is protected from the reduction in its P-Tyr content after chronic insulin treatment.
Next, Fao cells were infected with adenoviral constructs expressing either Myc-IRS-1WT or Myc-IRS-17A, and their insulin-induced Tyr phosphorylation was compared. Myc-IRS-17A underwent rapid Tyr phosphorylation, similar to wild-type IRS-1, in response to acute (1-min) insulin treatment (Fig. 3A, compare lanes a and c). However, while wild-type IRS-1 underwent significant (∼55%) Tyr dephosphorylation after prolonged (60-min) insulin treatment (Fig. 3A, compare lanes a and b, and Fig. 3B), IRS-17A maintained its P-Tyr content that was reduced only ∼15% after 60-min insulin treatment (Fig. 3A, compare lanes c and d, and Fig. 3B). These results further indicate that mutations of potential inhibitory Ser phosphorylation sites protect IRS-17A from the action of insulin-stimulated IRS-1 kinases. This prevents the dissociation of IRS-1 from the IR (Fig. 1C) and enables it to maintain its Tyr-phosphorylated active conformation. To determine whether IRS-17A is more resistant to proteolytic cleavage, the effects of the proteosome inhibitor MG-132 were studied. The presence of the inhibitor did not alter significantly the protein content and Tyr phosphorylation patterns of IRS-17A and IRS-1WT (not shown). These findings lead us to conclude that there is no massive proteolysis of IRS-1 in our model system in the time frame of our experiments.
FIG. 3.

Insulin-induced Tyr phosphorylation of IRS-17A in Fao cells. (A) Fao cells were infected with Ad-Myc-IRS-1WT or Ad-Myc-IRS-17A at 7 × 107 PFU/ml. After 48 h, the cells were starved in serum-free RPMI medium for 16 h and then treated with insulin for the indicated times. Cell extracts were prepared, and samples (1 mg) were subjected to immunoprecipitation (IP) with anti-Myc (α-Myc) antibodies. Immunocomplexes were resolved by SDS-PAGE (7.5% polyacrylamide) and immunoblotted (IB) with anti-PY (α-PY) antibodies. In parallel, samples (100 μg) of total cell extracts were resolved by SDS-PAGE (7.5% polyacrylamide) and immunoblotted with anti-Myc antibodies. (B) The bands corresponding to anti-PY/total Myc-IRS-1 were quantitated by densitometry, and the results are shown in a bar graph. Results of a representative experiment performed with duplicate samples are shown.
PKB and its downstream effectors, but not MAPK, are activated to a greater extent in Fao cells infected with Myc-IRS-17A than in Fao cells infected with Myc-IRS-1WT.
To study the effects of the 7A mutation on downstream effectors of IRS-1, PKB activity was compared in Fao cells infected with either Myc-IRS-1WT or Myc-IRS-17A. PKB underwent rapid phosphorylation (on Ser408) in response to insulin, which slightly decreased after 60 min of insulin treatment in Fao cells infected with Myc-IRS-1WT. As shown in Fig. 4A, this activation was potentiated 1.5- to 2-fold in cells infected with an equal amount of Myc-IRS-17A. In contrast, there was no difference in the extent of insulin-stimulated activation of MAPK in Fao cells infected with either IRS-1 construct (Fig. 4B). These results suggest that the ability of IRS-1 to maintain its Tyr-phosphorylated active conformation results in more sustained activation of PKB, a downstream effector of PI3K, while MAPK activity, which is activated along the Shc/Grb2/Sos pathway is largely unaffected by the introduction of the different IRS-1 constructs. The enhanced activation of PKB was translated into higher phosphorylation of its downstream effectors. As shown in Fig. 4C, GSK-3β underwent significantly higher (approximately twofold) phosphorylation in cells infected with Myc-IRS-17A, and similar results were observed when the phosphorylation and activation of Forkhead were monitored (not shown).
FIG. 4.

Effects of insulin on activation of PKB, MAPK, and GSK-3β in Fao cells infected with IRS-1WT or IRS-17A. Fao cells at 70% confluence were infected with Ad-Myc-IRS-1WT or Ad-Myc-IRS-17A at 6 × 107 PFU/ml. After 48 h, the cells were starved in serum-free medium for 16 h and incubated with 100 nM insulin for the indicated times (5 min [5′] to 60 min [60′]) at 37°C (−, not treated with insulin). Cytosolic extracts were prepared, and the samples (100 μg) were resolved by SDS-PAGE (7.5% polyacrylamide) and immunoblotted (IB) with the indicated antibodies. Antibodies directed against phosphorylated PKB (α-p-PKB), total PKB (α-total PKB), phosphorylated MAPK (α-p-MAPK), and total MAPK (α-total MAPK) were used. The results of four independent experiments were quantified and normalized relative to the total cellular content of the protein under study (A and C). Results of a representative experiment are shown in panel B.
Inducers of insulin resistance are less potent in inhibiting insulin-stimulated Tyr phosphorylation of IRS-17A.
The effects of inducers of insulin resistance on Tyr phosphorylation of IRS-1WT and IRS-17A were compared. Consistent with the results presented in Fig. 3, Myc-IRS-17A underwent rapid Tyr phosphorylation similar to Myc-IRS-1WT in response to acute insulin treatment (Fig. 5A, compare lanes a and c). Pretreatment with TPA significantly inhibited (∼40%) the insulin-stimulated Tyr phosphorylation of Myc-IRS-1WT, while Tyr phosphorylation of IRS-17A was affected to a much lower extent (Fig. 5A). Similar results were observed in anisomycin-treated cells (Fig. 5B). Note that the introduction of the 7A mutation did not impair the ability of IRS-1 to undergo phosphorylation on Ser307, either as a result of insulin treatment or after treatment with inducers of insulin resistance, such as anisomycin (Fig. 5C).
FIG. 5.

Effects of TPA and anisomycin on insulin-induced Tyr phosphorylation of IRS-17A in Fao cells. Fao cells were infected with Ad-Myc-IRS-1WT or Ad-Myc-IRS-17A at 7 × 107 PFU/ml. After 48 h, the cells were deprived of serum for 16 h and treated (+) for 60 min with 200 nM TPA (A), 50 ng of anisomycin per ml (B), or 1 μg of anisomycin per ml (C) or not treated (−). This procedure was followed by treatment with 100 nM insulin for 2 min (A) or for the indicated times (2, 10, or 15 min [2′, 10′, or 15′], respectively) (B and C). Cell extracts were prepared, and samples (1 mg) were subjected to immunoprecipitation (IP) with anti-Myc (α-Myc) antibodies (A). Immunocomplexes were resolved by SDS-PAGE (7.5% polyacrylamide) and immunoblotted (IB) with the indicated antibodies (e.g., anti-myc antibody [α-myc], anti-phosphorylated Ser307 antibody [α-pSer307]). Alternatively, total cell extracts (B and C) were resolved by SDS-PAGE (7.5% polyacrylamide) and immunoblotted with the indicated antibodies. The results of one experiment, which were representative of four experiments, are shown. The results of two independent experiments performed in duplicate are quantified in the bar graph in panel A.
IRS-17A also conferred protection against the action of FFA, the underlying cause of obesity-induced insulin resistance (32). As shown in Fig. 6, pretreatment of Fao cells with FFA (0.5 to 0.75 mM) reduced (∼35%) their subsequent response to insulin, as manifested by the reduction in insulin-stimulated Tyr phosphorylation of IRS-1WT. However, IRS-17A was significantly more resistant to the inhibitory effects of FFA, as evident by the smaller reduction in its Tyr phosphorylation state after treatment with FFA. These findings implicate Ser residues among the 7A as being targets for IRS kinases activated by inducers of insulin resistance. They further suggest that mutations of these sites might confer protection against the adverse effects of these agents.
FIG. 6.
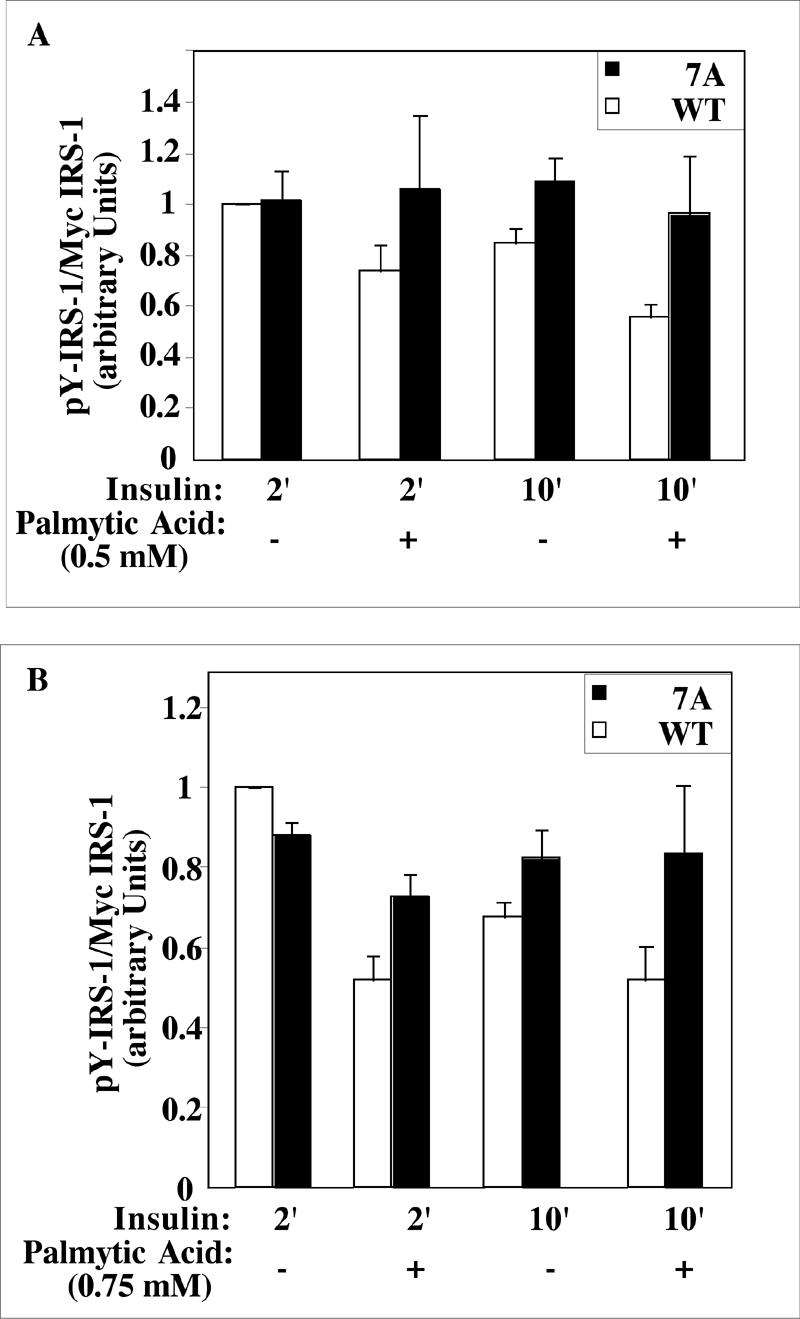
Effect of palmitic acid on insulin-induced Tyr phosphorylation of IRS-1 mutants in Fao cells. Fao cells were infected with Ad-Myc-IRS-1WT or Ad-Myc-IRS-17A at 7 × 107 PFU/ml. After 24 h, the cells were deprived of serum for 12 h and incubated for 12 h with (+) or without (−) the indicated concentration of palmitic acid in serum-free medium before being stimulated with 100 nM insulin for the indicated times (2 or 10 min [2′ or 10′], respectively). Cell extracts (100 μg) were resolved by SDS-PAGE (7.5% polyacrylamide) and immunoblotted with anti-PY and anti-Myc antibodies. The results of two independent experiments done in duplicate were quantified.
PH/PTB-L undergoes Ser phosphorylation in vivo in response to chronic insulin treatment.
To better analyze the Ser residues that are subjected to insulin-stimulated phosphorylation, PH/PTB-L, a truncated form of IRS-1 (aa 1 to 430) encoding an extended PH/PTB domain, was generated, and CHO-T cells that overexpress Myc-PH/PTB-L were studied. As expected, Myc-PH/PTB-L failed to undergo Tyr phosphorylation in response to insulin (Fig. 7A), because all Tyr phosphorylation sites of IRS-1 are confined to its C-terminal region, which was deleted in PH/PTB-L. However, we demonstrated that PH-PTB-L was subjected to insulin-induced Ser phosphorylation, exemplified by its mobility shift, when CHO cells were treated with insulin for 60 min (Fig. 7A).
FIG. 7.

In vivo phosphorylation of PH/PTB-L in response to insulin. (A) CHO-T cells were transiently transfected with pcDNA3-Myc-PH/PTB (nt 1 to 1290) or with an insert-free plasmid (control). Twenty-four hours posttransfection, the cells were deprived of serum for 16 h and treated with insulin for the indicated time periods. Cell extracts (100 μg) were resolved by SDS-PAGE (10% polyacrylamide) and immunoblotted with anti-PY (α-PY) or anti-Myc (α-Myc) antibodies. (B) In parallel, samples (100 μg, 40 μl) were incubated (+) with 3 U of calf intestine phosphatase (CIP) at 37°C for 1 h. The samples were resolved by SDS-PAGE (10% polyacrylamide) and immunoblotted with anti-Myc antibodies. The results of one experiment, which were representative of two experiments, are shown. (C) CHOPH/PTB-L cells were starved in serum-free F12 medium for 16 h. The cells were treated for 30 min with the indicated inhibitors or not treated with an inhibitor (−) before being treated with 100 nM insulin for 30 min. Cell extracts were prepared, and samples were resolved by SDS-PAGE (10% polyacrylamide) and immunoblotted with anti-Myc antibodies. The results of one experiment, which were representative of three experiments, are shown.
To ensure that the mobility shift was due to Ser phosphorylation, Myc-PH/PTB-L, isolated from insulin-treated cells, was subjected to in vitro dephosphorylation with calf intestine alkaline phosphatase. Such treatment abolished the mobility shift of PH/PTB-L, indicating that it did indeed result from Ser phosphorylation (Fig. 7B). Pretreatment with wortmannin, a PI3K inhibitor, completely blocked the mobility shift induced by chronic insulin treatment (Fig. 7C), whereas inhibitors of mTOR (rapamycin), MEK (PD98059), and p38 MAPK (SB203580) were ineffective. These results suggest that Ser residues located within PH-PTB-L undergo phosphorylation by insulin-stimulated wortmannin-sensitive IRS kinases.
To determine whether insulin-stimulated phosphorylation of PH-PTB-L takes place at Ser sites mutated in IRS-17A, a Myc-PH/PTB-L domain harboring the 7A mutation was generated. In contrast to PH/PTB-LWT, PH/PTB-L7A showed no mobility shift (Ser phosphorylation) after prolonged (60-min) insulin treatment (Fig. 8A), indicating that some of the serines mutated in PH/PTB-L7A are subjected to in vivo phosphorylation, resulting in decreased mobility of PH/PTB-LWT in response to prolonged insulin treatment.
FIG. 8.

Effects of insulin on serine phosphorylation of Myc-PH/PTB-L7A. CHO-T cells were transiently transfected with constructs encoding Myc-PH/PTB-L (nt 1 to 1290), either wild type (WT) or the 7A mutant (A). Alternatively, the cells were transfected with the constructs encoding Myc-PH/PTB-L (nt 1 to 1290; aa 1 to 430) or Myc-PH/PTB-S (nt 1 to 1095; aa 1 to 365) (B). After 24 h, the cells were incubated in serum-free F12 medium for 16 h before being treated with insulin for the indicated times. Cell extracts were resolved by SDS-PAGE (10% polyacrylamide) and immunoblotted with anti-Myc (α-Myc) antibodies. The results of one experiment, which were representative of three experiments, are shown.
Truncation of PH/PTB-L, which deletes Ser407 and Ser408, or a selected mutation of Ser408 inhibits the ability of PH/PTB-L to undergo Ser phosphorylation.
To further analyze the Ser sites under study, a shorter truncated form of IRS-1, encoding aa 1 to 365 was generated and named PH/PTB-S. PH/PTB-S contains five of the seven Ser residues mutated in IRS-17A, but it lacks S407 and S408. When PH/PTB-S was introduced into CHO-T cells, there was no decrease in its mobility after prolonged (60-min) insulin treatment (Fig. 8B), indicating that phosphorylation of Ser407 and/or Ser408 presumably contributes to the mobility shift of PH/PTB-L, induced after prolonged insulin treatment. In view of these results, selected mutations of Ser407 and Ser408 to Ala (alone or combined) were introduced into PH/PTB-L. Additional mutation to Ala of Ser336 served as a control. As shown in Fig. 9A, PH/PTB-LS336A and PH/PTB-LS407A manifested mobility shifts identical to that of PH/PTB-LWT after 30-min insulin treatment. In contrast, mutation of Ser408, either alone (PH/PTB-LS408A) or together with Ser407 (PH/PTB-L407/S408A) or the triple mutation of Ser336, Ser407, and Ser408 (PH/PTB-L3A) abolished the mobility shift induced upon 30-min insulin treatment. These results suggest that phosphorylation of Ser408 significantly contributes to the decreased mobility of PH/PTB-L after prolonged insulin treatment.
FIG. 9.

Effects of insulin and calyculin A on Ser phosphorylation of PH/PTB-L mutants. (A) CHO-T cells were transiently transfected with constructs expressing either Myc-PH/PTBWT or the following PH/PTB mutants: S336A, S407A, S408A, S407A/S408A (S407/8A), and S336A/S407A/S408A (S3A). After 24 h, the cells were deprived of serum for 16 h and treated with insulin for the indicated times. Cell extracts were resolved by SDS-PAGE (10% polyacrylamide) and immunoblotted with anti-Myc (α-myc) antibodies. The results of one experiment, which were representative of two or three experiments, are shown. (B) CHO-T cells were transiently transfected with pcDNA3-myc-PH/PTBWT or pcDNA3-myc-PH/PTBS408A. After 24 h, the cells were starved for 16 h and treated either with 100 nM insulin for 30 min or with 25 nM calyculin A for 60 min or left alone (−). Cell extracts were resolved by SDS-PAGE (10% polyacrylamide) and immunoblotted with anti-Myc antibodies. The results of one experiment, which were representative of two experiments, are shown.
Calyculin A, a Ser/Thr phosphatase inhibitor and an inducer of insulin resistance, triggers Ser phosphorylation of the full-length IRS-1 (25), as well as the phosphorylation of Myc-PH/PTB-LWT, exemplified by its mobility shift (Fig. 9B). When Ser408 was mutated to Ala, the mobility shifts induced by insulin or by calyculin A were significantly reduced (Fig. 9B, compare lanes b and e and lanes c and f), suggesting that Ser408 might be a target for Ser kinases activated either by insulin or by selected inducers of insulin resistance.
Ser408 is a target for in vivo phosphorylation promoted either by insulin or by inducers of insulin resistance.
To ensure that Ser408 is indeed an in vivo phosphorylation site of IRS-1, antibodies directed against the phosphorylated form of Ser408 (anti-P-S408) were generated. As shown in Fig. 10A, these antibodies readily reacted with IRS-1WT, but they failed to interact with either IRS-17A or IRS-1S408A, expressed in Fao cells as an adenoviral construct.
FIG. 10.

Phosphorylation of S408 of IRS-1 in cultured Fao cells. (A) Fao cells were infected with Adeno-myc-IRS-1 (wild type [WT]), the 7A mutant, or the S408A mutant at a titer of 4 × 107 to 6 × 107 PFU/ml in order to obtain comparable expression levels of the proteins under study (compare with Fig. 11). After 48 h, the cells were starved for 16 h. The medium was removed, and the cells were incubated with 100 nM insulin or without insulin (−) for the indicated times (5, 15, or 60 min [5′, 15′, or 60′], respectively). (B) Alternatively, the cells were incubated with insulin for 2 or 60 min or incubated with sphingomyelinase (SMase) or calyculin A as indicated (+). Cytosolic extracts (100 μg) were resolved by SDS-PAGE (7.5% polyacrylamide) and immunoblotted (IB) with anti-phospho-Ser408 (α-p-Ser408) or anti-PY (α-PY) antibodies as indicated.
To determine whether S408 is subjected to in vivo phosphorylation triggered by inducers of insulin resistance, Fao cells were infected with IRS-1WT and then incubated with different inducers of insulin resistance. As shown in Fig. 10B, sphingomyelinase readily induced in vivo phosphorylation of S408, comparable to that induced by 60-min insulin treatment. Similarly, calyculin A induced phosphorylation of S408 that was accompanied by a significant mobility shift of the protein. These findings clearly indicate that S408 is an in vivo target for phosphorylation induced either by prolonged insulin treatment or by inducers of insulin resistance.
Note that insulin-induced phosphorylation of Ser408 occurred at a rate lower than that of Tyr phosphorylation of IRS-1. While maximal Tyr phosphorylation of IRS-1 was already detected after 2-min insulin treatment (Fig. 1), the maximal phosphorylation of Ser408 required 30- to 60-min incubation with the hormone (Fig. 10A). These findings are consistent with our hypothesis that negative-feedback control mechanisms that involve Ser408 phosphorylation are triggered by insulin only subsequent to the induction of its signaling cascades, which involves Tyr phosphorylation of IRS proteins.
Mutation of Ser408 confers upon IRS-1 partial protection from the inhibitory action of inducers of insulin resistance.
To study the effects of the S408A mutation on IRS-1 function, Fao cells were infected with adenoviral constructs expressing either Myc-IRS-1WT, Myc-IRS-17A, or Myc-IRS-1408A, and their insulin-induced Tyr phosphorylation was compared. Myc-IRS-1408A underwent rapid Tyr phosphorylation similar to IRS-1WT or IRS-17A in response to acute insulin treatment (Fig. 11). However, prolonged (60-min) insulin treatment reduced the Tyr phosphorylation level of wild-type IRS-1 or its 408A mutant to a similar extent, while the Tyr phosphorylation state of the 7A mutant remained elevated. However, the inhibitory effects of TPA on insulin-stimulated Tyr phosphorylation of IRS-1 could be partially prevented in the IRS-1408A mutant (Fig. 11A), although IRS-1408A could not protect against the inhibitory effects of anisomycin (Fig. 11B and C). These results suggest that mutation of Ser408 might confer protection against selected inducers of insulin resistance, while mutation of additional sites among the 7S sites is required to confer protection from the inhibitory action of prolonged insulin treatment and a wider spectrum of inducers of insulin resistance.
FIG. 11.

Effects of TPA and anisomycin on insulin-stimulated Tyr phosphorylation of IRS-1WT, IRS-17A, and IRS-1408A. Fao cells were infected with Adeno-myc-IRS-1 (wild type [WT]), the 7A mutant, or the S408A mutant at a titer of 4 × 107 to 6 × 107 PFU/ml. After 48 h, the cells were deprived of serum for 16 h and treated for 60 min (60′) with 200 nM TPA (A) or 50 ng of anisomycin per ml (B), followed by treatment with 100 nM insulin for the indicated times (2 min [2′] or 60 min [60′]). Cell extracts were prepared, and samples (1 mg) were subjected to immunoprecipitation (IP) with anti-Myc antibodies. Immunocomplexes were resolved by SDS-PAGE (7.5% polyacrylamide) and immunoblotted (IB) with anti-PY (α-PY) or anti-IRS-1 (α-IRS-1) antibodies. (C) Quantitation of the results of two independent experiments done in duplicate is presented.
DISCUSSION
IRS proteins are key players in propagating insulin signaling and are therefore subjected to feedback regulatory systems that inhibit their action. Feedback regulation involves phosphatase-mediated dephosphorylation (8) or Ser/Thr phosphorylation of functionally active Tyr-phosphorylated IRS proteins (40). Ser/Thr phosphorylation can induce, for example, the dissociation of the IRS proteins from the IR or from downstream effectors or could lead to their degradation (40). In the present study, we focused on Ser sites that upon phosphorylation might interfere with the association of IRS-1 with IR and negatively regulate IRS-1 function. Because the PTB domain of IRS-1 mediates its interactions with the JM region of IR, our hypothesis was that phosphorylation of a distinct set of Ser residues located within or proximal to the PTB domain might interrupt with IR-IRS-1 complex formation. We have previously shown that phosphorylation of IRS-1, induced by PKCζ, impairs its ability to interact with IR and undergo Tyr phosphorylation (22); therefore, seven Ser residues at or in close proximity to the PTB domain, six being at potential PKC phosphorylation sites, were mutated to Ala. We demonstrated that the IRS-1 mutant IRS-17A fails to undergo Ser phosphorylation after prolonged insulin treatment. The resistance to Ser phosphorylation enables the mutant IRS-1 to remain tightly complexed with the IR and maintain its active Tyr-phosphorylated state that is otherwise impaired in wild-type IRS-1 as a result of Ser phosphorylation after prolonged insulin treatment. As a consequence of its sustained Tyr phosphorylation, IRS-17A can better propagate insulin signaling, which is manifested by its ability to maintain sustained activation of PKB and its downstream effectors, GSK-β and Forkhead (3). These results therefore suggest that at least some of the mutated Ser residues are subjected to phosphorylation by insulin-stimulated IRS kinases that negatively regulate IRS-1 function. We showed that IRS-17A is also protected from inducers of insulin resistance, such as TPA, anisomycin, and FFA that utilize this physiological feedback control mechanism to promote phosphorylation of IRS-1 (wild type) at the same Ser sites, and in such a way inhibits its action. Hence, the 7S sites, the seven Ser residues subjected to mutation, are key sites for the regulation of IRS-1 function because they serve as a point of convergence, where physiological feedback control mechanisms, triggered by insulin-stimulated IRS kinases, overlap with IRS kinases triggered by inducers of insulin resistance that phosphorylate the very same sites, and in such a way terminate insulin signaling.
Several lines of evidence support this conclusion. First, we could demonstrate that mutation to Ala of the seven Ser sites (S265, S302, S325, S336, S358, S407, and S408) of IRS-1 obliterates its mobility shift, a characteristic feature of Ser-phosphorylated IRS-1. Failure of IRS-17A to undergo insulin-stimulated Ser phosphorylation cannot be attributed to a major conformational change introduced by the mutations per se, because IRS-17A undergoes in vitro Tyr phosphorylation by IR to an extent similar to that of the wild-type protein. Furthermore, both wild-type IRS-1 and its 7A mutant are subjected to comparable levels of in vivo Tyr phosphorylation after acute insulin treatment, again indicating that the 7A mutation does not impair the ability of IRS-17A to localize in close proximity to the IR.
In an attempt to identify the Ser residues subjected to phosphorylation, truncated forms of IRS-1 that either contain all (PH/PTB-L, aa 1 to 430) or only five (PH/PTB-S, aa 1 to 365) of the seven serines under study were generated. Like the full-length IRS-1, PH/PTB-L was subjected to insulin-stimulated Ser/Thr phosphorylation that was evident by its reduced mobility after chronic insulin treatment. The contribution of the 7S sites to the mobility shift became evident when we showed that PH/PTB-L7A fails to undergo a mobility shift after insulin stimulation. Furthermore, only PH/PTB-L, not PH/PTB-S, demonstrated a mobility shift, suggesting that the Ser residues involved are S407 or/and S408. Indeed, introduction of single mutations into PH/PTB-L indicated that Ser408 is the prime site within the PH/PTB-LWT that contributes to the mobility shift in response to prolonged insulin treatment.
Ser408 is a genuine in vivo phosphorylation site. This conclusion is based upon the fact that its phosphorylation after insulin treatment can be detected using peptide-specific antibodies that selectively recognize the phosphorylated form of S408. Note that phosphorylation of S408 occurs at a rate lower than the Tyr phosphorylation rate of IRS-1. This finding is in accordance with the concept that Ser phosphorylation of IRS proteins, being a means to terminate insulin action, should commence subsequent to Tyr phosphorylation of the IRS proteins, which triggers insulin signaling.
Insulin-stimulated phosphorylation of S408 is sensitive to wortmannin, suggesting that the S408 kinase is a downstream effector of PI3K. This kinase presumably differs from mTOR, because rapamycin fails to inhibit phosphorylation of S408. Because S408 is located at a PKC phosphorylation site (RXXS), a potential S408 kinase could be PKCζ. Indeed, previous studies by us (22) and by others (28) have indicated that IRS-1 is subjected to Ser phosphorylation by PKCζ, which is a downstream effector of insulin along the PI3K pathway (22). Several inducers of insulin resistance, such as tumor necrosis factor alpha (30) and fatty acids (37), can also promote the activation of PKCζ, turning it into a common kinase activated by physiological and pathological signals to promote Ser phosphorylation of IRS-1.
Still, phosphorylation of S408 might be necessary, but it is certainly insufficient to inhibit Tyr phosphorylation of IRS-1 and uncouple IR-IRS-1 complexes. This conclusion is based upon the fact that introduction of an adenoviral construct that harbors the S408A mutation into Fao cells fails to protect IRS-1S408A from undergoing Tyr dephosphorylation after chronic insulin treatment. This contrasts with the full protection against Tyr dephosphorylation provided by the IRS-17A mutant. Similarly, while IRS-1S408A is protected from the inhibitory effects exerted by TPA, it fails to resist other inhibitors of insulin-induced Tyr phosphorylation. This differs from the almost complete protection against several inducers of insulin resistance (e.g., TPA, FFA, and anisomycin) provided by the 7A mutant. These findings suggest that S408 might be a target for phosphorylation by IRS kinases activated only by selected inducers of insulin resistance. Hence, additional sites among the 7S sites, acting in concert with S408, may function as inhibitory Ser phosphorylation sites for IRS function. Their phosphorylation could be catalyzed by PKCζ or by other IRS kinases, which are downstream effectors of PI3K. IKKβ is a potential candidate. IKKβ is a Ser/Thr kinase that is part of the IKK complex that phosphorylates the inhibitor of NF-κB, IκB. IKKβ can bind PKCζ both in vitro and in vivo, it serves as an in vitro substrate for PKCζ, and it is activated by a functional PKCζ (18). IKKβ is activated by insulin in Fao rat hepatoma cells (Y.-F. Liu and Y. Zick, unpublished data); furthermore, insulin-stimulated Ser phosphorylation of IRS-1 is inhibited by salicylates, implicating IKKβ as an insulin-stimulated IRS kinase (Liu et al., unpublished). Indeed, IRS-1 is a direct substrate of stress-activated IKKβ, which phosphorylates IRS-1 on Ser312 (the human homologue of mouse Ser307) (9) and could phosphorylate additional sites as well. Therefore, it appears that PI3K controls a few Ser/Thr kinases to negatively regulate IRS-1 function, namely, mTOR acting at the C-terminal tail and PKCζ or IKKβ acting at the N-terminal region.
We have previously shown that mutations of four Ser residues (4S) (S265, S302, S325, and S358) among the 7S sites, which are located at PKB and PKC phosphorylation sites (RXRXXS), renders IRS-1 prone to the action of protein Tyr phosphatases, thus implicating some of these 4S residues as positive regulators of IRS-1 function (26). This conclusion is supported by the fact that overexpression of PKB significantly attenuates the rate of Tyr dephosphorylation of IRS-1 after 60 min of treatment with insulin (26). Here we show that mutation of seven Ser sites, which includes the 4S positive sites, exerts an overall protective effect on IRS-1 functions, suggesting that mutation of the negative sites among the 7S sites, as in IRS-17A, functionally dominates the mutation of the positive sites, as in IRS-14A. Hence, distinct arrays of Ser residues might negatively or positively regulate IRS protein function. Under physiological conditions, upon insulin stimulation, the positive sites must be phosphorylated prior to the negative sites to enable the insulin signal to be first potentiated before it is being attenuated as part of a negative-feedback loop. Under pathological condition, inducers of insulin resistance presumably trigger the phosphorylation of only the negative sites, with no effects on the positive sites, thus preventing the propagation of insulin signals mediated by IRS proteins and thus causing insulin resistance.
Several other Ser sites, sites altogether different from the 7S sites, have already been implicated in the negative regulation of IRS-1 function. Conventional members of the PKC family, which are activated by phorbol esters or endothelin 1, activate members of the MAPK pathway to phosphorylate IRS-1 at Ser612 and at additional sites in its COOH tail (5). Such phosphorylation inhibits the interactions of IRS-1 both with IR and with downstream effectors of IRS-1, such as PI3K. Hence, phosphorylation of Ser residues at the COOH tail of IRS-1 could act synergistically with phosphorylation at its N-terminal regions. This is in accordance with the idea that increased levels of regulatory inputs can provide a more subtle and powerful regulation. Still, most relevant to this study is Ser307 within the PTB domain, the phosphorylation of which negatively regulates IRS functions (1, 10, 39). Phosphorylation of S307 is catalyzed by a number of kinases, some of which, like JNK, are activated by insulin (1, 2, 19). IRS-17A contains an intact Ser307, yet it is resistant to the inhibitory effects of chronic insulin treatment or to the action of inducers of insulin resistance. Our findings rule out the possibility that introduction of the 7A mutation impairs the phosphorylation of Ser307; therefore, we suggest that phosphorylation of Ser307 is not sufficient to impair IRS-1 function and that phosphorylation of additional Ser sites, Ser408 and others among the 7S sites, is required to uncouple IR-IRS-1 complexes and inhibit IRS-1 functions.
Ser/Thr phosphorylation can inhibit IRS-1 function in a number of ways (4, 12, 15, 22, 23, 25, 27, 34). However, regardless of mechanism, increased Ser phosphorylation of IRS-1 at inhibitory sites underlies a key mode to inhibit IRS protein function, utilized either by insulin itself as a physiological negative-feedback control mechanism or by different agents that induce insulin resistance and type 2 diabetes. Given the large number of stimuli, pathways, kinases, and potential sites involved, it appears that Ser/Thr phosphorylation of IRS proteins represents a combinatorial consequence of several kinases activated by different pathways acting in concert to phosphorylate multiple sites. Devising effective means to prevent the phosphorylation of the inhibitory sites could be beneficial in attempts to promote insulin action and protect against the adverse effects of inducers of insulin resistance.
Acknowledgments
We thank Ronit Sagi-Eisenberg and Kenneth Siddle for helpful comments and discussions.
This work was supported in part by research grants from the Juvenile Diabetes Foundation International, the European Foundation for the Study of Diabetes, the United States-Israel Binational Fund, the Israel Science Foundation (founded by the Israel Academy of Sciences and Humanities), and the Mitchel Kaplan Fund for Diabetes Research. Y.Z. is an incumbent of the Marte R. Gomez Professorial Chair.
REFERENCES
- 1.Aguirre, V., T. Uchida, L. Yenush, R. Davis, and M. F. White. 2000. The c-Jun NH2-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser307. J. Biol. Chem. 275:9047-9054. [DOI] [PubMed] [Google Scholar]
- 2.Aguirre, V., E. D. Werner, J. Giraud, Y. H. Lee, S. E. Shoelson, and M. F. White. 2002. Phosphorylation of Ser307 in insulin receptor substrate-1 blocks interactions with the insulin receptor and inhibits insulin action. J. Biol. Chem. 277:1531-1537. [DOI] [PubMed] [Google Scholar]
- 3.Brunet, A., A. Bonni, M. J. Zigmond, M. Z. Lin, P. Juo, L. S. Hu, M. J. Anderson, K. C. Arden, J. Blenis, and M. E. Greenberg. 1999. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96:857-868. [DOI] [PubMed] [Google Scholar]
- 4.Clark, S. F., J. C. Molero, and D. E. James. 2000. Release of insulin receptor substrate proteins from an intracellular complex coincides with the development of insulin resistance. J. Biol. Chem. 275:3819-3826. [DOI] [PubMed] [Google Scholar]
- 5.De Fea, K., and R. A. Roth. 1997. Modulation of insulin receptor substrate-1 tyrosine phosphorylation and function by mitogen-activated protein kinase. J. Biol. Chem. 272:31400-31406. [DOI] [PubMed] [Google Scholar]
- 6.De Fea, K., and R. A. Roth. 1997. Protein kinase C modulation of insulin receptor substrate-1 tyrosine phosphorylation requires serine 612. Biochemistry 36:12939-12947. [DOI] [PubMed] [Google Scholar]
- 7.Eck, M. J., P. S. Dhe, T. Trub, R. T. Nolte, and S. E. Shoelson. 1996. Structure of the IRS-1 PTB domain bound to the juxtamembrane region of the insulin receptor. Cell 85:695-705. [DOI] [PubMed] [Google Scholar]
- 8.Elchebly, M., P. Payette, E. Michaliszyn, W. Cromlish, S. Collins, A. L. Loy, D. Normandin, A. Cheng, H. J. Himms, C. C. Chan, C. Ramachandran, M. J. Gresser, M. L. Tremblay, and B. P. Kennedy. 1999. Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1B gene. Science 283:1544-1548. [DOI] [PubMed] [Google Scholar]
- 9.Gao, Z., D. Hwang, F. Bataille, M. Lefevre, D. York, M. Quon, and J. Ye. 2002. Serine phosphorylation of insulin receptor substrate 1 (IRS-1) by inhibitor κB kinase (IKK) complex. J. Biol. Chem. 277:48115-48121. [DOI] [PubMed] [Google Scholar]
- 10.Gao, Z., A. Zuberi, M. J. Quon, Z. Dong, and J. Ye. 2003. Aspirin inhibits serine phosphorylation of insulin receptor substrate 1 in tumor necrosis factor-treated cells through targeting multiple serine kinases. J. Biol. Chem. 278:24944-24950. [DOI] [PubMed] [Google Scholar]
- 11.Hadari, Y. R., K. Paz, R. Dekel, T. Mestrovic, D. Accili, and Y. Zick. 1995. Galectin-8: a new rat lectin, related to galectin-4. J. Biol. Chem. 270:3447-3453. [DOI] [PubMed] [Google Scholar]
- 12.Haruta, T., T. Uno, J. Kawahara, A. Takano, K. Egawa, P. M. Sharma, J. M. Olefsky, and M. Kobayashi. 2000. A rapamycin-sensitive pathway down-regulates insulin signaling via phosphorylation and proteasomal degradation of insulin receptor substrate-1. Mol. Endocrinol. 14:783-794. [DOI] [PubMed] [Google Scholar]
- 13.He, T. C., S. Zhou, L. T. da Costa, J. Yu, K. W. Kinzler, and B. Vogelstein. 1998. A simplified system for generating recombinant adenoviruses. Proc. Natl. Acad. Sci. USA 95:2509-2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hemi, R., K. Paz, N. Wertheim, A. Karasik, Y. Zick, and H. Kanety. 2002. Transactivation of ErbB2 and ErbB3 by tumor necrosis factor-alpha and anisomycin leads to impaired insulin signaling through serine/threonine phosphorylation of IRS proteins. J. Biol. Chem. 277:8961-8969. [DOI] [PubMed] [Google Scholar]
- 15.Hotamisligil, G. S., P. Peraldi, A. Budavari, R. Ellis, M. F. White, and B. M. Spiegelman. 1996. IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-alpha- and obesity-induced insulin resistance. Science 271:665-668. [DOI] [PubMed] [Google Scholar]
- 16.Khan, A. H., and J. E. Pessin. 2002. Insulin regulation of glucose uptake: a complex interplay of intracellular signalling pathways. Diabetologia 45:1475-1483. [DOI] [PubMed] [Google Scholar]
- 17.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680-685. [DOI] [PubMed] [Google Scholar]
- 18.Lallena, M.-J., M. T. Diaz-Meco, G. Bren, C. V. Paya, and J. Moscat. 1999. Activation of IκB kinase β by protein kinase C isoforms. Mol. Cell. Biol. 19:2180-2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee, Y.-H., J. Giraud, R. J. Davis, and M. F. White. 2003. c-Jun N-terminal kinase (JNK) mediates feedback inhibition of the insulin signaling cascade. J. Biol. Chem. 278:2896-2902. [DOI] [PubMed] [Google Scholar]
- 20.LeRoith, D., and Y. Zick. 2001. Recent advances in our understanding of insulin action and insulin resistance. Diabetes Care 24:588-597. [DOI] [PubMed] [Google Scholar]
- 21.Li, J., K. DeFea, and R. A. Roth. 1999. Modulation of insulin receptor substrate-1 tyrosine phosphorylation by an Akt/phosphatidylinositol 3-kinase pathway. J. Biol. Chem. 274:9351-9356. [DOI] [PubMed] [Google Scholar]
- 22.Liu, Y. F., K. Paz, A. Herschkovitz, A. Alt, T. Tennenbaum, S. R. Sampson, M. Ohba, T. Kuroki, D. LeRoith, and Y. Zick. 2001. Insulin stimulates PKCζ-mediated phosphorylation of insulin receptor substrate-1 (IRS-1). A self-attenuated mechanism to negatively regulate the function of IRS proteins. J. Biol. Chem. 276:14459-14465. [DOI] [PubMed] [Google Scholar]
- 23.Mothe, I., and E. Van Obberghen. 1996. Phosphorylation of insulin receptor substrate-1 on multiple serine residues, 612, 632, 662, and 731, modulates insulin action. J. Biol. Chem. 271:11222-11227. [DOI] [PubMed] [Google Scholar]
- 24.Pawson, T. 1995. Protein modules and signalling networks. Nature 373:573-580. [DOI] [PubMed] [Google Scholar]
- 25.Paz, K., R. Hemi, R. LeRoith, A. Karasik, E. Elhanany, H. Kanety, and Y. Zick. 1997. A molecular basis for insulin resistance: elevated serine/threonine phosphorylation of IRS-1 and IRS-2 inhibits their binding to the juxtamembrane region of the insulin receptor and impairs their ability to undergo insulin-induced tyrosine phosphorylation. J. Biol. Chem. 272:29911-29918. [DOI] [PubMed] [Google Scholar]
- 26.Paz, K., L. Yan-Fang, H. Shorer, R. Hemi, D. LeRoith, M. Quon, H. Kanety, R. Seger, and Y. Zick. 1999. Phosphorylation of insulin receptor substrate-1 (IRS-1) by PKB positively regulates IRS-1 function. J. Biol. Chem. 274:28816-28822. [DOI] [PubMed] [Google Scholar]
- 27.Pederson, T. M., D. L. Kramer, and C. M. Rondinone. 2001. Serine/threonine phosphorylation of IRS-1 triggers its degradation: possible regulation by tyrosine phosphorylation. Diabetes 50:24-31. [DOI] [PubMed] [Google Scholar]
- 28.Ravichandran, L. V., D. L. Esposito, J. Chen, and M. J. Quon. 2001. PKC-ζ phosphorylates IRS-1 and impairs its ability to activate PI 3-kinase in response to insulin. J. Biol. Chem. 276:3543-3549. [DOI] [PubMed] [Google Scholar]
- 29.Saltiel, A. R., and J. E. Pessin. 2002. Insulin signaling pathways in time and space. Trends Cell Biol. 12:65-71. [DOI] [PubMed] [Google Scholar]
- 30.Sanz, L., P. Sanchez, M. J. Lallena, M. M. Diaz, and J. Moscat. 1999. The interaction of p62 with RIP links the atypical PKCs to NF-κB activation. EMBO J. 18:3044-3053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sawka-Verhelle, D., S. Tartare-Deckert, M. F. White, and E. Van Obberghen. 1996. Insulin receptor substrate-2 binds to the insulin receptor through its phosphotyrosine-binding domain and through a newly identified domain comprising amino acids 591-786. J. Biol. Chem. 271:5980-5983. [DOI] [PubMed] [Google Scholar]
- 32.Shulman, G. I. 1999. Cellular mechanisms of insulin resistance in humans. Am. J. Cardiol. 84:3J-10J. [DOI] [PubMed] [Google Scholar]
- 33.Sun, X. J., P. Rothenberg, C. R. Kahn, J. M. Backer, E. Araki, P. A. Wilden, D. A. Cahill, B. J. Goldstein, and M. F. White. 1991. Structure of the insulin receptor substrate IRS-1 defines a unique signal transduction protein. Nature 352:73-77. [DOI] [PubMed] [Google Scholar]
- 34.Tirosh, A., R. Potashnik, N. Bashan, and A. Rudich. 1999. Oxidative stress disrupts insulin-induced cellular redistribution of insulin receptor substrate-1 and phosphatidylinositol 3-kinase in 3T3-L1 adipocytes. A putative cellular mechanism for impaired protein kinase B activation and GLUT4 translocation. J. Biol. Chem. 274:10595-10602. [DOI] [PubMed] [Google Scholar]
- 35.Voliovitch, H., D. Schindler, Y. R. Hadari, S. I. Taylor, D. Accili, and Y. Zick. 1995. The pleckstrin-homology (PH) domain of insulin receptor substrate-1 (IRS-1) is required for proper interaction of IRS-1 with the insulin receptor. J. Biol. Chem. 270:18083-18087. [DOI] [PubMed] [Google Scholar]
- 36.Wolf, G., T. Trüb, E. Ottinger, L. Groninga, A. Lynch, M. F. White, M. Miyazaki, J. Lee, and S. E. Shoelson. 1995. PTB domains of IRS-1 and Shc have distinct but overlapping binding specificities. J. Biol. Chem. 270:27407-27410. [DOI] [PubMed] [Google Scholar]
- 37.Wrede, C. E., L. M. Dickson, M. K. Lingohr, I. Briaud, and C. J. Rhodes. 2003. Fatty acid and phorbol ester-mediated interference of mitogenic signaling via novel protein kinase C isoforms in pancreatic beta-cells (INS-1). J. Mol. Endocrinol. 30:271-286. [DOI] [PubMed] [Google Scholar]
- 38.Wrede, C. E., L. M. Dickson, M. K. Lingohr, I. Briaud, and C. J. Rhodes. 2002. Protein kinase B/Akt prevents fatty acid-induced apoptosis in pancreatic beta-cells (INS-1). J. Biol. Chem. 277:49676-49684. [DOI] [PubMed] [Google Scholar]
- 39.Yu, C., Y. Chen, H. Zong, Y. Wang, R. Bergeron, J. K. Kim, G. W. Cline, S. W. Cushman, G. J. Cooney, B. Atcheson, M. F. White, E. W. Kraegen, and G. I. Shulman. 2002. Mechanism by which fatty acids inhibit insulin activation of IRS-1 associated phosphatidylinositol 3-kinase activity in muscle. J. Biol. Chem. 277:50230-50236. [DOI] [PubMed] [Google Scholar]
- 40.Zick, Y. 2001. Insulin resistance: a phosphorylation-based uncoupling of insulin signaling. Trends Cell Biol. 11:437-441. [DOI] [PubMed] [Google Scholar]