

Abstract
A detailed understanding of the functional mechanism of a protein entails the characterization of its energy landscape. Achieving this ambitious goal requires the integration of multiple approaches including determination of high resolution crystal structures, uncovering conformational sampling under distinct biochemical conditions, characterizing the kinetics and thermodynamics of transitions between functional intermediates using spectroscopic techniques, and interpreting and harmonizing the data into novel computational models. With increasing sophistication in solution-based and ensemble-oriented biophysical approaches such as electron paramagnetic resonance (EPR) spectroscopy, atomic resolution structural information can be directly linked to conformational sampling in solution. Here, we detail how recent methodological and technological advances in EPR spectroscopy have contributed to the elucidation of membrane protein mechanisms. Furthermore, we aim to assist investigators interested in pursuing EPR studies by providing an introduction to the technique, a primer on experimental design, and a description of the practical considerations of the method towards generating high quality data.
Keywords: Site directed spin labeling, EPR, DEER, membrane proteins, structure, dynamics
1. Introduction
Structural biology is at the cusp of a fundamental transition in its focus. As the number of structures in the protein data bank (PDB) surpassed 100,000 protein structures, it has shown a steady, but perhaps predictable, decline in the number of novel folds added each year (Levitt, 2007). Within this vast trove of predominantly atomic resolution structures, we may already have access to the overwhelming majority of protein folds sampled by nature (Sillitoe et al., 2015). Structural redundancy has emerged as the rule of protein evolution rather than the exception. Recurrent folds have been found in functionally distinct protein superfamilies suggesting that elements of this conserved architecture are central to the mechanism (Forrest, Kramer, & Ziegler, 2011; ter Beek, Guskov, & Slotboom, 2014). However, it has become increasingly apparent that although proteins adopt similar folds, they can have different conformational dynamics necessitated by the evolution of the underlying mechanisms presumably in adaptation to their specific functional contexts (Bhabha et al., 2013; Faham et al., 2008; Kazmier, Sharma, Islam, Roux, & McHaourab, 2014; Krishnamurthy, Piscitelli, & Gouaux, 2009; Ma et al., 2012; Perez, Koshy, Yildiz, & Ziegler, 2012). We contend that revealing mechanistic commonalities and differences and the underlying interplay between sequence, structure and dynamics will catalyze a transition in focus for structural biology from the collection of static structures to the characterization of energy landscapes. This will necessitate uncovering intermediate protein states and the pathways of the transitions between them by combining atomistic models with spectroscopic, biochemical, thermodynamic and kinetic studies with a central role for computational biology in guiding experimental investigation and integrating data from diverse techniques.
This vision is particularly pertinent to membrane proteins which are implicated in a spectrum of diseases and represent 50% of pharmaceutical drug targets (Nigam, 2015; Overington, Al-Lazikani, & Hopkins, 2006; Yildirim, Goh, Cusick, Barabasi, & Vidal, 2007). Membrane proteins are often involved in cellular signaling and signal transduction pathways enabling cells to respond to their environments and carry out regulatory processes vital to the physiology of the organism (Lin, Yee, Kim, & Giacomini, 2015). Despite their clinical importance, membrane proteins have posed significantly greater challenges for structural analysis due to their large size, intrinsic dynamic properties, and the inherent complications arising from the necessity of detergent solubilization and formation of stable crystal contacts. While various developments in stabilization and conformational selection have extended the reach of x-ray crystallography into the realm of membrane proteins, such as hyperthermophilic target selection (Wiener, 2004; Yamashita, Singh, Kawate, Jin, & Gouaux, 2005), detergent optimization (Sonoda et al., 2011), antibody chaperones (Griffin & Lawson, 2011; Pardon et al., 2014) and mutagenic thermostabilization (Penmatsa, Wang, & Gouaux, 2013; Serrano-Vega, Magnani, Shibata, & Tate, 2008), the degree to which these modifications alter the energy landscapes of these protein remains largely undetermined.
Thus, membrane proteins pose two distinct challenges in the transition from static structures to mechanism. The first challenge is that membrane protein crystal structures, while they are generally accurate representations of the tertiary fold, are often crystallized under conditions that may obscure the position of these structures in the functional energy landscape (Cross, Sharma, Yi, & Zhou, 2011; Cuello, Cortes, & Perozo, 2004; Freed, Horanyi, Wiener, & Cafiso, 2010; Kazmier, Sharma, Quick et al., 2014). Therefore, these structures require rigorous validation to assign their mechanistic identities. The second challenge arises from the fact that mechanistic descriptions of membrane proteins, like all dynamic proteins, require an understanding of conformational sampling under biochemical conditions that mimic the in vivo context. For example, how are the relative populations of intermediate states altered in response to ligand or drug binding, or energy input? It is only with this comprehensive view that we can move toward an understanding of the energy landscape that underlies the full mechanistic description of function.
Describing energy landscapes requires methodologies to measure thermodynamics (ΔG) and kinetics (ΔGǂ) of conformational changes at a resolution adequate to enable comparisons between solution conformations and atomic-scale models. Spectroscopic approaches like nuclear magnetic resonance (NMR), electron paramagnetic resonance (EPR), and fluorescence resonance energy transfer (FRET) are well suited for this task with access to different time scales and amplitudes of structural changes (Figure 1). Liquid state NMR describes solution structure and heterogeneity of proteins at high resolution and monitors conformational changes from the local backbone to the domain levels with high sensitivity (Mittermaier & Kay, 2009). Unfortunately, most membrane proteins are not amenable to NMR analysis primarily due to size restrictions. Single molecule (SM) FRET has the distinct advantage of monitoring the kinetics of conformational change at the level of individual molecules (Akyuz, Altman, Blanchard, & Boudker, 2013; Akyuz et al., 2015; Zhao et al., 2010; Zhao et al., 2011), which is key for identifying transition state free energies and rate limiting steps. The application of SM-FRET requires relatively large probes which limit their placement in the sequence and may compromise the identification of the nature and magnitude of conformational changes. Extracting distances from FRET efficiencies is nontrivial and therefore data-driven computational modeling of intermediates using FRET data remains challenging (Brunger, Strop, Vrljic, Chu, & Weninger, 2011).
Figure 1. Biophysical methods to study protein structure and dynamics.

Whereas x-ray crystallography is the most robust method to determine high resolution structures of small and large proteins, cryoelectron microscopy is best suited for large proteins and protein complexes. Despite its utility to investigate dynamic properties, current molecular size constraints limit the applicability of liquid state NMR to <50,000 MW. In contrast, EPR and fluorescence spectroscopies can interrogate dynamic processes regardless of size or complexity. The application of these probe-based methods to proteins of known structure amplifies the interpretation of data toward understanding mechanism.
EPR analysis of spin-labeled proteins enables direct observation of triggered movements in domains and secondary structural elements as well as equilibrium fluctuations of these units arising from the isomerization of the protein between conformations (Figure 2) (McHaourab, Steed, & Kazmier, 2011).. Although EPR suffers from low throughput and its structural resolution is moderate, it is no longer unusual to see EPR datasets to describe conformational intermediates, assign mechanistic identity, and discover novel intermediate states in conjunction with computational modeling (Cuello et al., 2010; Durr et al., 2014; Freed, Lukasik, Sikora, Mokdad, & Cafiso, 2013; Georgieva, Borbat, Ginter, Freed, & Boudker, 2013; Hanelt, Wunnicke, Bordignon, Steinhoff, & Slotboom, 2013; Jao, Hegde, Chen, Haworth, & Langen, 2008; Kazmier, Sharma, Islam et al., 2014; Masureel et al., 2014; Wen, Verhalen, Wilkens, McHaourab, & Tajkhorshid, 2013). EPR has a number of advantages that are well-suited for the investigation of membrane proteins. In addition to the lack of size limitations on protein targets, experiments can be conducted in a variety of conditions including detergent micelles, proteoliposomes and Nanodiscs, which yield unique insights into conformational dynamics. Furthermore, limited quantities of protein are sufficient for experimentation in EPR due to relatively high signal-to-noise ratios compared to NMR. Importantly, spin labeling at appropriate sites produces limited structural and functional perturbations due to small probe size.
Figure 2. EPR methods report on the ensemble of conformations in solution.
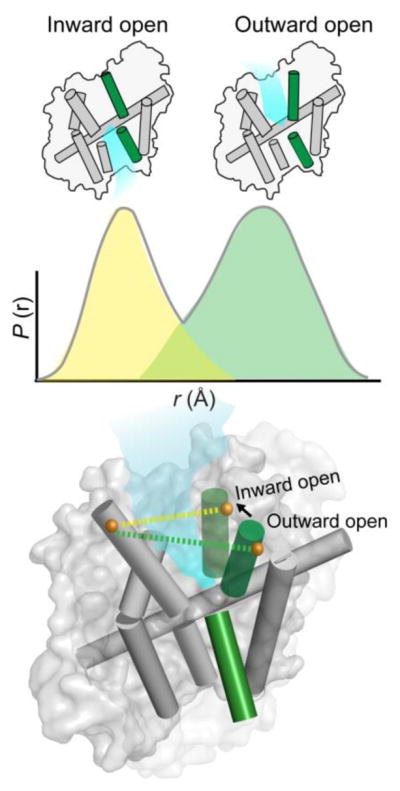
In a DEER experiment, each molecule in solution reports a characteristic inter-probe distance consistent with its conformation. The distance distribution reports these distances as a function of their frequency within the ensemble. Thus, discrete conformations undergoing equilibrium fluctuations in solution (inward-facing and outward-facing) at ambient temperatures are represented as distinct distance populations in the DEER distance distribution (yellow and green) in the solid state. Therefore, individual conformations can be described using distance parameters generated from ensemble-based measurements.
With the emerging recognition of the need for a multifaceted view of protein structure and dynamics, structural biology and biochemistry laboratories are becoming more interested in adding EPR spectroscopy to complement other approaches. The goal of this review is to provide information on the practical aspects of conducting EPR spectroscopy and interpreting EPR data from the perspective of a structural biologist. We will also discuss how EPR data can be effectively incorporated with data from other approaches to generate mechanistic descriptions of protein function.
2. An EPR Primer
2.1 Strategy of site directed spin labeling
Due to the rarity of stable unpaired electrons in nature, applications of EPR have historically been limited to biological systems that naturally incorporate EPR active transition metals, such as photosynthetic reaction centers (Britt et al., 2004; Calvo, Passeggi, Isaacson, Okamura, & Feher, 1990), organic radicals including biradical and triplet state molecules (Weil & Bolton, 2007), and oxidation/reduction reactions (Bhattacharjee et al., 2011; Zielonka et al., 2014). For these reasons, development of methodologies that allow site-specific incorporation of spin labels bearing stable unpaired electrons into protein systems was a highly sought advancement in the field. The introduction of sulfhydryl-specific spin probes combined with site-directed cysteine mutagenesis ushered the ability to selectively attach spin labels at essentially any site along the polypetide chain. This was coined as site-directed spin labeling (SDSL, Figure 3A) (Altenbach, Marti, Khorana, & Hubbell, 1990; W. L. Hubbell, Altenbach, C., 1994).
Figure 3. Site-directed spin labeling and correlation of the EPR spectrum with local structure.

(A) Targeted cysteine mutagenesis introduces a sulfhydryl moiety for the attachment of a nitroxide spin label, such as MTSSL. Rotational isomerization of MTSSL predominantly around the bonds highlighted in gray is reflected in the EPR spectral lineshape. (B) The degree of rotational freedom of the label is determined by the local packing environment. Fast rotational correlation times (~1ns) correspond to spin labels attached to surface-exposed sites. Tertiary contact interactions or buried sites that restrict spin label motion reduce the rate and amplitude of isomerization leading to broadening of the lineshape. The dashed line emphasizes the progressive appearance of a slow motion component associated with restricted rotation.
SDSL requires initial mutagenesis to remove endogenous, labile cysteine residues followed by reintroduction of cysteines only at selected sites of interest typically as single or double mutants. Sites for cysteine replacement are selected to avoid structural perturbation and are typically located on the protein surface at non-conserved residues. Importantly, native residue substitution and spin labeling have been found to have little effect on protein structural and functional properties at these locations (Mchaourab, Lietzow, Hideg, & Hubbell, 1996). Spin probes are covalently attached to the protein backbone through thiol reactive functional groups including methanethiolsulfonate, maleimide, and iodoacetamide moieties (Klare & Steinhoff, 2009).
The most commonly used spin label is the methanethiosulfonate nitroxide label ((1-Oxyl-2,2,5,5-tetramethylpyrroline-3-methyl) methanethiosulfonate, or MTSSL, Figure 3A) (Berliner, Grunwald, Hankovszky, & Hideg, 1982). In this molecule, the free electron is located in a π-like orbital along the N-O bond. The radical is stable due to steric shielding provided by the proximal set of dimethyl groups of the pyrrole ring. Unlike maleimide and iodoacetamide derivatives, the thiol moiety of MTSSL is highly specific to cysteine modification creating a disulfide linkage that can be easily cleaved with reducing agents for control experiments. Furthermore, MTSSL is theoretically well characterized (Columbus & Hubbell, 2002; Columbus, Kalai, Jeko, Hideg, & Hubbell, 2001; Mchaourab, Kalai, Hideg, & Hubbell, 1999; Mchaourab et al., 1996) and possesses the molecular flexibility to label most sites. Because the protein is modified, it is imperative to establish the structural and functional integrity of mutant proteins through activity assays. From this information, an accurate assessment of the effect of the mutation and labeling can be made and spin-labeled mutants in which perturbations that are judged to be too severe can be removed from the dataset.
2.2 The EPR tool kit
EPR analysis of spin labeled proteins yields a number of parameters that describe the local environment of the label as well as its distance from a second site-specifically introduced paramagnetic center (Figure 4). The structural interpretation of these parameters has been established in model systems and a number of reviews present a detailed description of their applications (W. L. Hubbell, Cafiso, & Altenbach, 2000; W. L. Hubbell, Lopez, Altenbach, & Yang, 2013; W. L. Hubbell, McHaourab, Altenbach, & Lietzow, 1996; Klare, 2013; McHaourab et al., 2011). Briefly, the local steric environment of the spin label determines to a large extent its dynamics as reflected in the EPR lineshape. Solvent exposure is a function of the topological location of the spin label, whether buried in the hydrophobic core or at the interface of helices, or in direct contact with solvent. Accessibility and mobility are parameters which reflect the local structure. In contrast, distance-dependent dipolar interactions report a more global perspective enabling the determination of the spatial relationships between secondary structures or domains. The selection of which parameter to measure depends on the questions to be addressed but, more often than not, informative spin labeling analysis requires the integration of all three parameters. Systematic and exhaustive investigations are critical to permit an unequivocal interpretation of the data.
Figure 4. EPR spectroscopy at a glance.

Summary of the methods in EPR highlighting structural interpretation and caveats for each method.
2.3 Spin labels as molecular spies of local structure
The EPR lineshape uncovers essential features of the local environment by describing the properties of spin label motion. The rotation of the spin label about internal bonds (Figure 3) and local dynamic fluctuations of the backbone to which the nitroxide is attached contribute to the overall “mobility” observed in the EPR lineshape (W. L. Hubbell et al., 1996). For large macromolecules (>50,000 MW), such as membrane proteins, the overall rate of protein tumbling is too slow (τc ≅ 10−8 s or slower depending on solution viscosity) to have an effect on the EPR spectrum (10−11-10−9 s timescale). The lineshape displays a range of dynamic motion depending on the degree of steric interaction experienced by the spin label due to side and main chain atoms. A detailed motional analysis of MTSSL suggested that intrinsic spin label rotation is largely limited to the Cε—Cζ and the Cε—Sδ bonds (Figure 3A, Figure 4) (Mchaourab et al., 1996). Rotation about the disulfide linkage is restricted by a sufficiently large energy barrier (≥ 7 kcal mol−1) (Fraser, Boussard, Saunders & Lambert, 1971; Jiao, Barfield, Combariza & Hruby, 1992). Rotation about the Sγ—Cβ is severely hindered by interaction of the Sγ atom with the Cα hydrogen, as was supported by crystallographic analysis of spin labeled T4 lysozyme (Langen, Oh, Cascio, & Hubbell, 2000). Importantly, the chemical structure of the nitroxide side chain impacts the EPR lineshape (Mchaourab et al., 1999) and provides a way to increase motional sensitivity to backbone dynamics (Columbus et al., 2001). Furthermore, Mchaourab and coworkers showed that spin label dynamics are contingent on the local molecular structure of the protein (Mchaourab et al., 1996).
Although deconvolution of the dynamic modes requires full simulation of the EPR lineshape (Columbus et al., 2001; Columbus & Hubbell, 2002), in most cases a phenomenological analysis of the EPR spectrum can qualitatively differentiate between spin labels attached to buried, surface or loop sites as well as those in tertiary interaction (Figure 3B). (Mchaourab et al., 1996) and (Columbus et al., 2001) established the use of the inverse linewidth from the central resonance line as a measure of spin label mobility. A remarkable correlation of this parameter with the structural class of the spin label, i.e. buried, exposed, or in tertiary contact, has been demonstrated. In favorable cases, it is possible to extract a measure of the flexibility of the backbone from analysis of the EPR lineshape. Although these calibration studies were carried out on the water soluble protein T4 Lysozyme, the parametrization of the lineshape analysis can be extended to membrane proteins in general. It was noted however that spin label interactions with local side chains are modified at sites located on membrane-exposed α-helices relative to α-helices on water-soluble proteins (Kroncke, Horanyi, & Columbus, 2010).
Solvent accessibility is a complementary parameter to the spectroscopic signature of local structure reported in the EPR spectrum. Although pulsed-based methods have been developed (Subczynski, Mainali, Camenisch, Froncisz, & Hyde, 2011), spin label solvent accessibility (Π) is commonly determined by monitoring the peak-to-peak EPR central line amplitude as a function of increasing microwave power in the presence and absence of a small, paramagnetic fast relaxing agent (PRA, Figure 5) (Altenbach, Froncisz, Hemker, McHaourab, & Hubbell, 2005). These power saturation experiments produce multi-point curves in which their shapes are informed by the collisional frequency of the nitroxide with a PRA. Due to slow nitroxide relaxation times (T1 ≈ 1μs), the EPR signal intensity decreases at high powers as a result of equalized spin state populations (saturation) in the absence of a PRA (Figure 5C). Direct collision of the nitroxide with a PRA shifts the saturation curve to the right by means of enhanced T1 relaxation rates incurred through Heisenberg spin exchange (Figure 5D). Curves generated in the absence and presence of PRAs are characterized by a P1/2 value, which is the microwave power required to achieve half of the unsaturated EPR signal amplitude and used to determine the dimensionless Π parameter relative to a standard sample. Although limited by the PRA diffusion, this method is advantageous over other accessibility methods such as cysteine alkylation, which is inferred from chemical reactivity profiles that are dependent upon thiol acid dissociation, steric constraints and the charge or size of the modifying reagent (Zhu & Casey, 2007). Importantly, the spin label accessibility parameter reflects the steady state exposure as opposed to the reactivity-based methods which could represent rare excursions of protein to the trapped state.
Figure 5. Spin label solvent accessibility and the correlation with local environment.

(A) The differential solubility of fast-relaxing PRAs (NiEDDA and O2) allows the determination of spin label environment. Nitroxide scanning of an α-helix that is asymmetrically solvated between aqueous and hydrocarbon milieu will report the gradient of oxygen accessibility toward the center of the bilayer in accordance with helix periodicity. The dotted line highlights the site of expected maximum in O2 accessibility. The NiEDDA accessibility profile, which probes water exposure, is 180° out-of-phase with the O2 profile. (B) Two spin labeled sites are shown on a model of LeuT (PDB 2A65), which are used to probe the membrane-water interface. An approximate position for this interface is outlined by an orange box. (C) Power saturation experiments showing the reduction in signal intensity as a function of microwave power. (D) The high NiEDDA accessibility at site 480 in LeuT relative to O2 and N2 determined from power saturation curves (inset) suggests a water exposed position of the spin label. (D) In contrast, the high O2 accessibility at site 488 indicates that the spin label samples the lipid bilayer.
Paramagnetic Ni(II)ethylenediaminediacetic acid (NiEDDA) and molecular oxygen (O2) possess ideal characteristics to serve as PRAs, including small size and fast T1 relaxation (Altenbach et al., 2005). Especially valuable for membrane protein studies, these compounds display disparate solubility profiles in which O2 demonstrates a finite concentration in water, but a concentration gradient into the center of the membrane bilayer. By contrast NiEDDA is almost exclusively water soluble and diffuses into the membrane-water interface. Differential accessibility to the aqueous and lipid phase defines the membrane-water interface, topological organization and independently assigns secondary structure (Figure 5A) (Altenbach, Greenhalgh, Khorana, & Hubbell, 1994; W. L. Hubbell et al., 1996).
Mobility and accessibility can be used as a readout of conformational changes. However, characterization of the underlying structural arrangements can be problematic owing to the local nature of these parameters. They are most informative when used in conjunction with long range distance measurements which are more conducive to assessing the nature and amplitude of movements. In our laboratory, these parameters were utilized to outline the ion and substrate-dependent formation and collapse of a water-permeable pathway within the protein interior of the Na+-coupled leucine transporter, LeuT (Claxton et al., 2010), and to map the ATP-dependent structural transition of the lipid flippase MsbA which drives alternating access to a hydrated central cavity (Dong, Yang, & McHaourab, 2005).
Distance information can be extracted from pairs of spin labels by exploiting the distance-dependence of dipolar interactions and utilized to establish the spatial relationships of structural elements within or between proteins in solution. The energy of the dipolar interaction is inversely proportional to the cube of the distance (r3). When r<20Å, spin-spin coupling significantly alters the EPR lineshape through broadening of the spectrum. The strength of the interaction can be assessed qualitatively from the degree of line broadening or measured directly by a variety of lineshape analysis techniques to extract distance information (Hustedt et al., 2006; McHaourab, Oh, Fang, & Hubbell, 1997; Rabenstein & Shin, 1995). For r>20Å, the intrinsic width of the absorption lines in the EPR spectrum (6–8 Gauss) obscures the line broadening due to dipolar interactions (3–5 Gauss). The introduction of pulse methods, particularly double electron electron resonance (DEER, or PELDOR), allows detection of distances up to 70Å under favorable conditions (Borbat, McHaourab, & Freed, 2002; G. Jeschke & Polyhach, 2007).
DEER spectroscopy employs a four-pulse sequence to selectively interrogate the dipolar interaction between spin labels (Figure 6A) (Pannier, Veit, Godt, Jeschke, & Spiess, 2000). In this experiment, the first set of pulses generates a spin echo, which contains the dipolar information. Extraction of this information is achieved by a second pulse at a slightly different frequency which modulates the dipolar interaction and is reported by a change in the spin echo intensity. The second pulse is varied along specified time intervals within a defined data collection window, which leads to oscillations in the intensity of the spin echo decay. The period of the oscillatory frequency (or frequencies) that describes this time-dependent decay reflects the distance between probes.
Figure 6. Monitoring global conformational changes with DEER spectroscopy.

(A) The four-pulse protocol for DEER spectroscopy is designed to interrogate distance-dependent dipolar interactions between spin A and spin B. The inversion Π pulse on spin B modulates the echo decay of spin A as a function of time t, and the frequency of the resulting oscillation is inversely proportional to the average distance. The decay rate of the spin echo modulation is informed by the distribution of distances in the sample. (B) A simulated conformational change between two states of discrete energies as shown in the spin echo decay (top panels) is manifested by distinct rav and σ in the unimodal distance distribution. The middle panels illustrate an equilibrium between two states, which is the sum of contributions from each conformation. For simplicity, each conformation is equally populated. In the bottom panels, a shift in the equilibrium (induced by ligand binding, for instance) toward an increase in population of the short distance component is visualized in the spin echo decay. The dotted lines in P(r) show the position of rav.
3. Principles of DEER spectroscopy to uncover conformational dynamics
3.1 The distance distribution
Data analysis of the dipolar interaction between spin labels yields a probability distribution, P(r), of distances defined by an average distance (rav) and width or standard deviation (σ) (Figure 6B). The shape of the distribution, whether unimodal or multimodal, is informed by the collective dynamic processes associated with protein motion ranging from spin label side chain isomerization to backbone fluctuations, as well as the ensemble of distinct protein conformers. Because DEER experiments are carried out in the solid state, dynamic equilibria at ambient temperatures lead to broad or multicomponent distance distributions. Thus, not only can transitions between distinct states be detected from the change in average distance but also shifts in preexisting conformational equilibria are manifested in the width and shape of the distribution.
As shown in the upper panels of Figure 6B, P(r) with a specific rav and σ arises from a spin echo decay characterized by the oscillation frequency and rate of signal decay. Transitions between discrete energetic states of a protein, as reported by unimodal distributions, are reflected by changes in rav. In an ensemble undergoing equilibrium transitions between conformers of different energies, the DEER signal reflects a composite of multiple frequencies each with oscillation amplitude related to the relative population of the associated distance component. In the context of unresolved multimodal distributions (Figure 6B, middle and lower panels), rav is inadequate in the global description of protein movement since it does not correspond to a particular distance component and hence it does not describe a specific conformation of the protein. Instead, the relative populations of the distance components uncover conformational preference in the molecular ensemble under a defined set of conditions. A shift in the ensemble toward another equilibrium conformer under a new set of conditions (i.e. addition of ligands) will lead to a change in rav, but more importantly the change in the relative amplitude of the distance component implies a change in the energetic preference of the protein.
The result of DEER experiments between numerous nitroxide pairs is a web of distances that can be used to evaluate crystal structures, describe structural features of intermediate states or used as constraints into computational structure determination. Variations in rav and σ reveal unique properties of structural states induced by different biochemical conditions, such as changes in absolute distance between conformers or increased or decreased conformational sampling, often rationalizing site-specific changes in mobility and accessibility. This has been demonstrated exceptionally well in MsbA where systematic analysis of distance changes correlated with the pattern of spin label accessibility under conditions that define the power stroke of its transport mechanism (Figure 7) (Borbat et al., 2007; Dong et al., 2005; Zou, Bortolus, & McHaourab, 2009; Zou & McHaourab, 2009).
Figure 7. Correlation of global structural rearrangements with local helix packing in MsbA.

(A) Model of the MsbA homodimer in the open, Apo (PDB 3B5W) and the closed, AMP-PNP-bound (PDB 3B60) states showing symmetry-related sites for spin label incorporation. Individual monomers are identified by the color scheme. (B) EPR spectra of spin labels at these positions and the corresponding distance distributions (C) in the Apo and ADP-Vi-bound states (trapped post-hydrolysis). Labels at 561 and 162 show opposite distance changes between states, consistent with rigid body movement of helices in an alternating access mechanism. Although separated by ~50Å in the Apo state (C), spin labels at site 121 are within 20Å in the ADP-Vi-bound state as indicated by broadening of the EPR lineshape (arrow in B). (D) Formation of a closed conformation on the intracellular side according to distance analysis is consistent with changes in the NiEDDA accessibility profile of transmembrane helix 3 induced by ADP-Vi.
3.2 Factors influencing DEER data analysis and interpretation
A number of experimental factors contribute to the generation of a reliable P(r) from DEER measurements. The spin echo dephasing time, Tm, defines the time interval that the dipolar interaction can be observed and sets limits to the detection time of the experiment, which in turn imposes upper boundaries on the distance range and determines confidence levels in the width of the distribution. Notably, the Tm of MTSSL (and other nitroxides) is too short at room temperature to capture the spin echo decay. Since Tm is primarily a function of T2 relaxation mechanisms, echo coherence can be improved by decreasing the absolute temperature. For MTSSL, a complete characterization of the echo decay requires lowering the temperature substantially, typically between 50–80 K. Samples for DEER experiments that are performed under cryogenic conditions are supplemented with a cryoprotectant such as 20–25% (v/v) glycerol. Below 80 K, Tm can also be increased by deuteration of the sample buffer and glycerol which reduces proton-mediated T2 relaxation (Jeschke & Polyhach, 2007).
The low-temperature acquisition of DEER data potentially hampers the interpretation of resulting distance distributions. Because the short Tm of nitroxides at ambient temperatures is a consequence of rotation of the dimethyl moieties of the ring, alteration of the nitroxide design could conceivably remove this source of spin echo dephasing and increase the accessible temperature range. This elegant approach has recently been shown effective in measuring distances between sites on T4 lysozyme (Meyer et al., 2015; Yang et al., 2012). In one case, exchange of the dimethyl groups for spirocyclohexyl moieties in combination with protein immobilization in a trehalose glass matrix permitted accurate distance measurements of ~30 Å at room temperature (Meyer et al., 2015).
In addition to Tm, the impact of intermolecular dipolar coupling on the DEER signal cannot be overstated. The oscillating spin echo decay that characterizes the DEER spectrum is a product of an intramolecular term arising from dipolar coupling between spin labels within the same protein and an intermolecular term, referred to as the background, which describes the randomized spatial distribution of neighboring proteins. The background component is therefore directly proportional to the spin labeled protein concentration. During processing, this background must be removed to isolate the intramolecular term. Without explicit determination, the background is generally assumed a priori to be a stretched exponential decay that dampens the intramolecular dipolar interaction and effectively reduces the distance range and sensitivity of DEER measurements (Brandon, Beth, & Hustedt, 2012; G. Jeschke et al., 2007). At sufficiently high spin concentrations (above 200 μM), the background signal can dominate the DEER signal by forming a steep sloping baseline that is difficult to remove. Furthermore, an ill-defined background as a result of a short data collection window will introduce artifacts in the distance distribution, such as the appearance of an artificial long distance component or alteration of σ. Defining the background often introduces a tradeoff since longer collection windows result in a decrease in signal-to-noise as a practical consequence of Tm, necessitating more signal averaging. To practically account for these limiting factors, we suggest designing spin label pairs not exceeding 50Å based on an estimate from the Cα-Cβ projection or simulations. This conservative approach recognizes that spin labels can add more than 10Å to the predicted distance (based on the Cα-Cβ projection) due to the length of the nitroxide linker and relative orientation between nitroxide rotamers.
In the absence of a unique spin label orientation relative to the backbone, the inherent flexibility of the probe sets a lower boundary on σ. Thus, interpretation of σ requires a quantitative understanding of rotamer populations. Although still being evaluated, the development and application of rotamer libraries derived from spin labeled T4 lysozyme crystal structures and molecular dynamics (MD) simulations appears to be a valid approach to predict distance distributions which account for spin label flexibility at specific sites (Polyhach, Bordignon, & Jeschke, 2011). Experimentally, we have found that σ varies as an approximate Gaussian function from 1–5Å for surface exposed sites (McHaourab et al., 2011). Thus, broad distance distributions indicated by σ values larger than this likely reflect dynamic fluctuations of the protein backbone.
Since the spin echo decay is a convolution of P(r) with the ensemble average of dipolar coupling, isolating P(r) from DEER data is an ill-posed mathematical problem. As a result, a number of approaches have emerged that impose additional constraints in order to obtain a tractable P(r). The DeerAnalysis suite developed by Jeschke employs Tikhonov regularization, which is contingent on the identification of an appropriate regularization parameter defined by the L-curve criterion (Chiang, Borbat, & Freed, 2005; G. Jeschke, Chechik V., Ionita P., Godt A., Zimmermann H., Banham J., Timmel CR., Hilger D., Jung H., 2006; G. Jeschke, Wegener, Nietschke, Jung, & Steinhoff, 2004). This approach generally assumes a priori knowledge of the intermolecular background. Although “model free”, the regularization enforces a degree of smoothness onto P(r) resulting in Gaussian-like distributions. Another approach to identify P(r) utilizes a parameterized model to fit the DEER signal and describes all distance components as Gaussian in shape (Fajer, Brown & Song, 2007). Recently, a method for direct fitting of DEER data without a priori background correction has been developed which demonstrates the capacity to analyze multiple datasets simultaneously to identify global changes in P(r) (Brandon et al., 2012; Mishra et al., 2014) and see “A Straightforward Approach to the Analysis of DEER Data” by Stein, Beth and Hustedt (this volume).
3.3 From distance distributions to structure
The first step in interpretation of P(r) is to link medium-resolution EPR data to the available high-resolution structural models including crystal structures or homology models. To accomplish this task, computationally-calculated distributions are generated using MD simulations of the distance between spin labels (Islam, Stein, McHaourab, & Roux, 2013). These simulations can be directly compared to experimentally-derived P(r) to assign distance populations to structural models. Overlap between experimental and simulated distributions suggests that the structure or model is sampled in solution. Disagreements that manifest consistently across the dataset can often be used to suggest specific differences between solution intermediates and structural models, though this approach requires caution and rigorous validation. While explicit rendering of spin labels is possible for a small number of mutants (Alexander, Bortolus, Al-Mestarihi, McHaourab, & Meiler, 2008), it becomes computationally expensive at the level of global EPR datasets that commonly include 20–100 mutants. Therefore, programs have been developed that leverage rotamer libraries to approximate spin label flexibility and simplify the underlying calculations. Two of the most successful approaches have been Jeschke’s MMM (Multiscale Modeling of Macromolecules) program (Polyhach et al., 2011) and Roux’s dummy label approach (Islam et al., 2013; Roux & Islam, 2013). We recently compared DEER data and available crystal structures for LeuT and the Na+/hydantoin transporter Mhp1 using MMM and the dummy label approach (Kazmier, Sharma, Islam et al., 2014; Kazmier, Sharma, Quick et al., 2014). We were able to conclude that, in general, the Mhp1 conformations identified in crystal structures were consistent with those we described in solution by EPR spectroscopy. For LeuT, we concluded that many of the crystal structures were consistent with an outward-facing solution conformation. Furthermore, we proposed and later experimentally verified that a putative inward-facing conformation, crystallized from a heavily mutated construct, was not a major conformation in solution, arguing for a re-evaluation of the proposed mechanism (Kazmier, Sharma, Quick et al., 2014).
Once an accurate model of the protein has been established, relatively few mutants are required to identify the motifs that undergo conformational changes, describe the magnitude and directionality of these transitions, and evaluate established crystallographic or computational mechanistic models. In brief, multi-component distance distributions are most commonly associated with structural motifs undergoing equilibrium fluctuations which can be shifted by changes in biochemical conditions (e.g. ligand binding). To detect conformational changes, however, transitions must result in distance changes between spin labels. Thus, a unimodal distribution or static equilibrium does not itself exclude the possibility of dynamics, as different conformations can report similar or identical distances. Therefore, identifying dynamic motifs requires triangulation, with at least two distance measurements per site (Figure 8). To characterize the movement of motifs in detail, the data density should be increased to three or four distances per site, producing quadrangles and pyramids, respectively. The quality of such investigations is judged primarily through internal consistency; mutants that cannot be made to fit the established lattice of triangles also often display deviations from WT in functional assays. Such mutants are then removed from the composite structural analysis but may be important for identifying functionally important residues.
Figure 8. EPR distance measurements and structure elucidation.
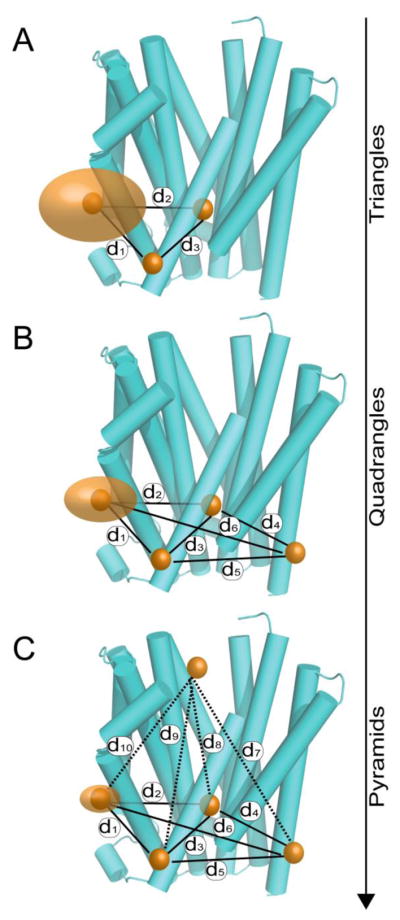
Analysis of protein structure using EPR distance measurements requires triangulation of spin label positions. (A) Triangles represent the least dense labeling strategy that can identify whether a motif is undergoing conformational reorganization. (B) More dense strategies, like quadranges, narrow the possible space that spin label can occupy thereby providing a more detailed description of protein structure and more informative restraints for modeling. (C) To effectively identify positions in three dimensions a pyramid scheme is required.
If it becomes clear that available crystal structures need refinement or that conformations sampled in solution (as reflected in the EPR data set) represent novel states not yet observed, then the data density requirements increase significantly. At this stage, it is advisable to conduct pyramid analysis for each secondary structural element predicted to function as part of a dynamic motif. This can require a substantial commitment of time and resources. However, this resolution is required if intermediate structures are to be described in detail and accurate models are to be generated in conjunction with computational modeling. The long term quest to transcend a qualitative description of conformational changes to structural models that capture the spatial information encoded in the EPR data has been recently achieved. Although fold determination from spin labeling is not a central focus of this review, we note that new methods have been recently developed that have demonstrated the feasibility of the approach. RosettaEPR focuses primarily on de novo modeling of proteins of unknown structures whereas an MD method (Hirst, Alexander, McHaourab, & Meiler, 2011), restrained ensemble molecular dynamics (REMD), emphasizes modeling intermediate states starting from high resolution structures (Islam et al., 2013). These methods are now accessible to users through direct contacts with the Meiler and Roux laboratories respectively.
3.4 Connecting structure and thermodynamics to elucidate mechanism
As described previously, in the solid state conditions under which DEER data is collected, each molecule will possess a conformation that is reported by the distance between the spin labels. Each distance is represented by a specific frequency of oscillation in the DEER decay and leads to a distinct distance component in P(r). Thus, the distribution represents a snapshot of the energy landscape of the protein under the specific set of experimental conditions. Therefore, it is possible to estimate the change in free energy between sampled states from their relative populations (Georgieva et al., 2013). With optimized sample preparation, these values tend to be consistent across mutants, although small perturbations in these equilibria due to label incorporation are not unexpected.
A mechanistic analysis requires an EPR dataset in which each distance population has been linked to a specific conformation, all major conformations have been described structurally and equilibrium information has been generated for all important ligand binding conditions. In LeuT, we compared multi-component distance distributions in apo (ligand-free), Na+-bound, and Na+/Leu-bound conditions for spin label pairs on the intracellular and extracellular sides of the transporter (Figure 9) (Kazmier, Sharma, Quick et al., 2014). In the apo condition, LeuT favored a closed conformation on the extracellular side and an open conformation on the intracellular side. This inward-facing state represented 50–60% of the ensemble under apo conditions. Upon saturation binding of Na+, the equilibrium shifted to favor the open conformation on the extracellular side and the closed conformation on the inside. This outward-facing intermediate represented a majority (60–70%) of the molecular ensemble in the Na+-bound state. In contrast, addition of Leu drove an equilibrium shift toward a closed conformation favored on both sides of the transporter (70–90%), which was evidence of a doubly-occluded intermediate. Notably, the population ratios in all conditions implied that conformational sampling of the extracellular and intracellular sides is governed by unique equilibrium constants, suggesting a degree of structural independence in the functional cycle. The composite analysis underscores the role of conformational dynamics evident in distance distributions to identify intermediate states sampled in solution and to assign their relative positions in the energy landscape.
Figure 9. EPR reveals equilibria that can be used to describe energy landscapes and mechanism.

LeuT is a Na+-coupled amino acid transporter. We conducted DEER experiments that monitored the conformational transitions on the extracellular and intracellular sides, shown here for helix 6/intracellular loop 3 (orange). We observed conformational equilibria between inward-facing, outward-facing and occluded conformations associated with apo (ligand-free, black), Na+-bound (blue) and Na+/Leu-bound (red) conditions. These were used along with structural characterization of intermediate states (numerically identified) and a biochemical description of transport to produce a novel description of alternating access in LeuT.
Through linking global EPR data to structural and biochemical descriptions, we generated a description of the mechanistic cycle of LeuT(Figure 9). In our EPR-derived model, the apo equilibrium favors a previously uncharacterized inward-facing conformer (1), but a minor outward-facing state (2) must be sampled to bind ligand from the extracellular side. Upon Na+ binding, the outward-facing conformation (3) becomes favored so that the majority of Na+-bound LeuT is primed to bind the co-transported Leu substrate. The Na+-bound equilibrium also suggests the presence of a minor inward-facing population (4). Upon binding of Leu, a new equilibrium is adopted between an occluded conformation (5) and rarely-sampled inward-facing conformation (6). Upon sampling of this conformation (6), ion and substrate will dissociate due to the presence of cellular electrochemical gradients (7). Thus we described a mechanistic cycle of alternating access in LeuT that introduced a novel ligand-coupling mechanism and previously unidentified conformational intermediates.
4. Practical considerations in sample preparation
4.1 Criteria for selection of labeling sites
Although not absolutely required, the availability of crystal structures greatly improves the experimental design and interpretation of EPR data by providing a high resolution reference to generate mechanistic hypotheses and rationalize findings concerning protein dynamics, ligand binding or protein-protein interactions. These structures are initially used as a guide for appropriate site selection of single and double mutants, the goal of which is to avoid sites that would interfere with structural integrity or function. In general, mutation of conserved residues, which are often buried within the protein core or contribute to ligand binding interactions, should be avoided. Due to the relatively low throughput of the approach, it is critical to design experiments that will monitor predicted conformational movements and evaluate specific mechanistic models. Secondary structural elements that form or contribute to ion or substrate binding sites, or those anticipated to participate in pathways relevant to ligand binding are ideal candidates for labeling. Incorporation of biochemical or computational studies in conjunction with crystal structures provides an additional reference for locating appropriate sites for spin labeling (Claxton et al., 2010). If more than one crystal structure is available, identification of domains or helices that undergo the largest displacements between structures are likely to yield the greatest information content for data interpretation. Subsequent incorporation of background mutations that interfere with functional activity (disruption of ion or substrate binding for instance) may uncover novel roles for these structural elements in a functional cycle. If such mutations have been used to stabilize and trap the membrane protein in a specific conformation for subsequent crystallization experiments, elucidating their effects is critical to evaluating the mechanistic relevance of the crystal structures (Kazmier, Sharma, Quick et al., 2014).
Generating a model of the protein and its conformational changes requires implementation of a spin label network that fingerprints the 3D fold. Initially, experiments are conducted to generate a system of overlapping triangles within a plane parallel to membrane (Figure 8). This triangulation of spin labeled pairs defines the relative spatial distribution between nitroxide centers in two dimensions and is often an information-rich approach when dealing with topological restriction associated with membrane-embedded proteins. However, a more sophisticated matrix of distance pyramids between labeled sites is required to outline the 3D fold (Figure 8). Although pyramids substantially increase the number of DEER measurements, the results will distinguish between many types of conformational movements, including horizontal and vertical translation, rotation or tilt and decreases the error in the resulting model. The impact of these movements on local packing interactions can be determined using site-specific changes in mobility and accessibility.
In order to obtain optimal information content, spin labeling sites for DEER studies should be reserved to exposed sites in secondary structural elements (Kazmier, Alexander, Meiler, & McHaourab, 2011). Labeling of unstructured loops increases conformational entropy of the spin label leading to featureless time-dependent spin echo decays and broad distance distributions that are not informative for computational modeling and may obscure relevant structural movements. If possible, the mobility of spin labeled sites reported in the EPR spectrum should be insensitive to the biochemical conditions of the DEER experiment as this information can be used to exclude changes in the rotameric freedom of the label as the origin of distance changes. Changes in the spin label mobility may indicate selection of specific nitroxide rotamer populations that may induce shifts in the distance distributions confouding the interpretation of conformational changes.
4.2 General principles for avoiding erroneous interpretation of EPR data
The most significant and potentially dooming source of experimental error is found in the quality of sample preparation. Isolation of stable and functionally-optimized protein devoid of unattached spin label, impurities, degradation products, and aggregation should be viewed as a prerequisite for EPR analysis. Not only does compromised sample integrity impact experimental design and throughput, but it also invariably leads to compounded errors in data analysis and subsequent structural interpretation. The intrinsic spin label sensitivity to the local environment will report altered packing arrangements and tertiary contacts relative to the structure or the model as a consequence of direct or indirect structural perturbations, which then may obfuscate pertinent conformational changes associated with function. Furthermore, the relative thermodynamic relationship between biochemical intermediates can be perturbed even under mildly destabilizing conditions.
A number of factors should be considered to control the potential for aggregation induced by compromised membrane protein stability. Site directed mutagenesis designed to interrogate putative transport mechanisms can generate a reduction in protein expression and/or increase heterogeneity by promoting aggregation. Although classified as a “negative” result, such observations may help to identify previously unknown key residues associated with activity. Separate from other buffer components, detergent selection for extraction from the native membrane environment and subsequent purification should be carefully considered for maintaining global homogeneity and functional activity (Roy, 2015). Harsh detergents, even those that are routinely used in crystallization, should be avoided. In our experience, the mild non-ionic dodecyl-β-D-maltoside (DDM) efficiently solubilizes a variety of proteins without altering structural or functional integrity. However, it should be noted that removal of functionally-relevant biomolecules through the purification process, including lipids and substrates, can reduce membrane protein stability or compromise function. In such cases, purification in the presence of lipids or substrates may be necessary.
4.3 Labeling and handling of purified membrane proteins in detergent
We express proteins of interest fused with an N- or C-terminal poly-histidine tag to facilitate purification by standard methods in metal affinity chromatography with either Ni2+ or Co2+ resin. The histidine tag is not removed by proteolytic cleavage unless it interferes with analysis. Samples are labeled immediately after elution from the resin with two consecutive rounds of sulfhydryl-reactive MTSSL (dissolved in dimethylformamide at 100–200 mM) for two hours each round at room temperature with 10–20 fold molar excess reagent per labeling site. We note that reducing the temperature to 4 °C may be necessary if cloudiness in the solution appears during the reaction or if protein stability dictates purification at low temperatures. To further drive label attachment, another round of spin label is added and the solution placed on ice overnight. Generally reactivity can be site and protein specific. Due to the nature of the reaction, labeling efficiency will theoretically increase above pH 7. However, MTSSL forms dimers under these conditions, effectively reducing the pool of reactive species. Since affinity purification is typically performed above pH 7 to increase sample binding to the resin, titration with a weak acid (i.e. 1M Mes hydrate) can be used to decrease sample pH to neutral for labeling purposes. Any precipitation that forms during overnight incubation is removed by centrifugation, syringe filtration (0.45 μm pore size) or both.
To ensure good quality and consistent sample preparation, we subsequently employ size exclusion chromatography to remove unreacted spin label and assess global structural properties and purity. This step is especially important to reduce contamination from sample aggregation that can confound global structural analysis by introducing artificial distance components (Figure 10). Determination of labeling efficiency is a necessary means of evaluating the tractability of a sample for EPR analysis and to establish a correlation between labeling and functional activity. Labeling efficiency can be approximated by quantifying the absolute number of spins in the sample through double integration of the EPR spectrum and comparing this value to the concentration of available labeling sites. For singly labeled proteins, the concentration of labeling sites is equal to the protein concentration.
Figure 10. Impact of protein aggregates on the spin echo decay and the resulting distance distribution.

(A) Preparative size exclusion chromatography demonstrating different levels of protein aggregation for the same mutant as indicated by the leading shoulder. The traces were normalized by area. The peak fractions pooled for DEER analysis are highlighted by a gray rectangle. Changes in the intermolecular background of the DEER experiment tracked with the presence of aggregated species (B), introducing a long distance artifact in the distance distributions (C).
If sample limitations require concentrating the protein prior to EPR analysis, centrifugal filters can be used with caution. Recently, a systematic analysis of ligand binding in DDM-purified LeuT indicated that binding of radiolabeled substrate was significantly impaired upon an increase in DDM-to-protein ratio with a concomitant loss of phospholipid content (Quick, Shi, Zehnpfennig, Weinstein, & Javitch, 2012). Further binding studies and subsequent computational experiments correlated the increase in DDM concentration with detergent occupation of the second substrate binding site (Khelashvili et al., 2013), an observation consonant with functional impairment imposed through binding of the crystallization detergent octyl-glucoside in a similar location (Quick et al., 2009).
At a practical level, these studies emphasize the need for monitoring detergent-induced effects on membrane protein function by optimizing and maintaining an appropriate detergent-to-protein ratio. Furthermore, one must choose suitable filtration units when concentrating membrane protein samples by taking account of the molecular weight of the protein and the physicochemical properties of the detergent. For instance, the aggregation number (which depends on buffer composition) and molecular weight of DDM monomers suggests a micelle molecular weight of approximately 50,000 on average. LeuT, which is a 60,000 molecular weight monomer in solution, would be expected to form proteomicelles with DDM that are greater than a molecular weight of 100,000. Indeed, DDM concentration increased when LeuT proteomicelles were subjected to concentration in centrifugal filters with a molecular weight pore size of 50,000 (Quick et al., 2012). In preparation of LeuT in DDM micelles, we conservatively estimate a transporter loss of 20% when using a 100,000 molecular weight cut off.
Additionally, subjecting membrane proteins to concentrating procedures also introduces the possibility of decreasing the detergent-to-protein ratio. Over-concentrating samples using ideal centrifugal filters can shift the ratio through loss of empty micelles and detergent monomers in equilibrium with proteomicelles. The loss of detergent promotes formation of proteomicelles with more than one protein per micelle, or non-specific multimerization. We find that this phenomenon rationalizes the appearance of a broad range of distance components as a result of increased intermolecular dipolar interactions between neighboring proteins in DEER experiments. The population distribution of these components will reflect the relative orientation between individual proteins in the micelle, potentially confounding distance information associated with unique structural intermediates. Although this behavior is expected to be dependent on detergent and protein properties, we suggest that final protein concentrations not exceed 100 μM in samples prepared for DEER analysis to reduce this effect.
4.4 Lipid environments
We have found that EPR analysis of membrane proteins in non-destabilizing detergents provides an appropriate starting point for assessing conformational dynamics. In general, EPR spectra of membrane-exposed sites demonstrate similar lineshapes in detergent and lipid environments (Claxton et al., 2010; Dong et al., 2005). Additionally, local and global changes in structure have been shown to be comparable (Claxton et al., 2010). However, reconstitution into more native-like lipid environments adds a new dimension to investigate the role of specific lipids on conformational equilibria and to map changes in the packing and orientation of structural elements within a membrane (Figure 11). Although liposomes are a traditional platform for such experiments, reconstitution into Nanodiscs is a viable and promising alternative. Nanodiscs are discoidal phospholipid bilayers in which the size and water solubility are determined by an annulus of amphipathic membrane scaffold protein (MSP) from an engineered fragment of apolipoprotein A-1 (Bayburt & Sligar, 2010). Like liposomes, the phospholipid content can be controlled to reflect the conditions for optimal activity of the protein.
Figure 11. Detergent and lipid environments and the consequence on the DEER signal background.

Solvation of membrane protein in (A) detergent micelles, (B) liposomes and (C) Nanodiscs. Liposome reconstitution often introduces more than one protein copy per liposome. As a result, the higher effective spin concentration increases the contribution of the background decay in the DEER signal relative to proteomicelles and nanodiscs (D).
The use of Nanodiscs for DEER sample preparation and subsequent analysis using Q-band (34 GHz) pulse spectrometers yielded an order of magnitude greater sensitivity despite reduced sample requirements. The origin of this striking result lies at the convergence of the intrinsic increase in signal-to-noise achieved with higher microwave frequencies relative to X-band (9.5 GHz) and the reduced contribution of background intermolecular dipolar coupling in the Nanodisc environment (Ghimire, McCarrick, Budil, & Lorigan, 2009; Zou & McHaourab, 2010). In proteoliposomes, multiple proteins can incorporate into a single liposome, which compresses the bulk spatial distribution of spins from three dimensions to approximately two (Figure 11) (Hilger et al., 2005). This change in protein distribution increases the effective spin concentration leading to steep intermolecular backgrounds that are difficult to remove during processing and reduce the measurable distance range. In contrast, a one-to-one incorporation of membrane protein into Nanodics can be achieved, maintaining the bulk spatial distribution in three dimensions. Given this benefit, we encourage the use of Nanodiscs for EPR studies in lipids. The primary disadvantage of Nanodiscs is the inherent inability to generate gradients that are often necessary for functional studies of transporters.
Our detailed protocols for reconstitution into either liposomes (Claxton et al., 2010; Zou, Bortolus et al., 2009; Zou & McHaourab, 2009) or Nanodiscs (Mishra et al., 2014; Zou & Mchaourab, 2010) are readily available elsewhere and we make additional suggestions here. For both environments, efficient reconstitution requires careful manipulation of component molar ratios. Furthermore, optimization of Nanodisc reconstitution is dependent on the target protein and the use of the appropriate size MSP. In our hands using the MSP1E3D1 construct (Bayburt & Sligar, 2010), the appropriate molar ratios for Nanodisc reconstitution appear to correlate with molecular weight of the membrane protein. We have found the following molar ratios to be good initial conditions for smaller proteins (50–60,000 MW, e.g. LeuT): protein-to-MSP, 1:5; MSP-to-lipid, 1:60; lipid-to-DDM, 1:5. Further decreasing the MSP-to-lipid ratio to ~1:120 was necessary for larger proteins (over 100,000 MW), such as MsbA and BmrCD. Following reconstitution, performing size exclusion chromatography on an HPLC equipped with a dual UV/fluorescence detector will help to resolve the membrane protein-Nanodisc complex from unincorporated target protein, lipids and MSP, which tend to run in the void volume of a Superdex200 10/300 column. Subsequent SDS-PAGE will confirm the presence of the transporter and scaffold protein. Stoichiometry between MSP and membrane protein can be corroborated by combining densitometry measurements from SDS-PAGE for the scaffold protein with the absolute spin concentration from the protein.
5. Perspective
Following the seminal work of Hubbell and coworkers on Rhodopsin, the application of spin labeling and EPR spectroscopy has expanded to include virtually every class of membrane protein. While an exhaustive account is beyond the scope of this review, we note some of the major contributions and the unique insight that emerged from them. SDSL studies of Rhodopsin preceded the determination of its crystal structures revealing local dynamics and structures in transmembrane helices (W. L. Hubbell, Altenbach, Hubbell, & Khorana, 2003; Palczewski et al., 2000; Salom et al., 2006). Importantly, distance measurements between spin labels were instrumental in uncovering the movement of helix 6 upon photoexcitation (Altenbach, Kusnetzow, Ernst, Hofmann, & Hubbell, 2008), which proved to be a conserved functional feature in GPCRs (Choe et al., 2011).
One of the most extensive spin labeling studies targeted MsbA combining nitroxide scanning of five of its transmembrane segments and the determination of at least 60 distances in the context of known but mechanistically-suspect crystal structures (Borbat et al., 2007; Dong et al., 2005; Zou, Bortolus et al., 2009; Zou & McHaourab, 2009). The data set revealed that ATP energy input changes the hydration of a transmembrane chamber, evident in changes in local accessibility and dynamics of spin labels, concomitant with large amplitude distance changes between the two halves of the transporter (Figure 7). These dramatic conformational changes were interpreted to reflect the ATP-coupled transition from inward-facing to outward-facing states. The degree to which this blueprint of conformational changes is conserved across ABC transporters is being explored in other members of the family (Mishra et al., 2014).
Similar questions regarding mechanistic commonalities and divergence that focus on the patterns of ion- and substrate-dependent conformational changes have been posed in ion-coupled transporters of the LeuT-fold class. Here, crystal structures have left the question of the structural mechanics and the ion dependence of the inward to outward-facing transition clouded with ambiguity (Krishnamurthy & Gouaux, 2012; Shimamura et al., 2010; Weyand et al., 2008; Yamashita et al., 2005). Spin labeling studies of LeuT and Mhp1, two members of this fold class, have illuminated aspects of the mechanistic divergence between these transporters (Kazmier, Sharma, Islam et al., 2014). Finally, spin labeling investigation of an ion-coupled multidrug transporter from the major facilitator superfamily is a recent example of extensive DEER analysis in the context of a homology model (Masureel et al., 2014). Despite the lack of a high resolution structures, the authors were able to identify a protonation sensitive switch critical for the antiporter mechanism of this transporter.
A large body of work from the Perozo lab illustrates the application of spin labeling and EPR spectroscopy to ion channels. Extensive analysis of accessibility, mobility, and short range distances in conjunction with modeling enabled insight into the conformational changes associated with gating of multiple classes of ion channels(Cordero-Morales et al., 2007; Cortes, Cuello, & Perozo, 2001; Perozo, Cortes, & Cuello, 1999; Perozo, Cortes, Sompornpisut, Kloda, & Martinac, 2002; Vasquez, Sotomayor, Cordero-Morales, Schulten, & Perozo, 2008). A methodologically novel contribution was the combination of EPR spectroscopy and crystallography to define aspects of channel function such as inactivation (Cuello et al., 2010).
One remarkable example of the application of spin labeling is the studies of Cafiso and coworkers on the E. coli vitamin B12 outer membrane transporter BtuB. In a collection of experiments, EPR spectroscopy was used to show that a short, yet conserved N-terminal segment (the Ton box) that couples BtuB to the inner membrane protein TonB undergoes a conformational equilibrium between folded and unfolded states (Fanucci, Coggshall et al., 2003; Xu, Ellena, Kim, & Cafiso, 2006). Importantly, crystal lattice interactions and osmolytes in the crystallization milieu were found to inhibit this structural transition, shifting the free energy difference between the unfolded and folded forms of the Ton box by ~3 kcal/mol (Fanucci, Lee, & Cafiso, 2003; Freed et al., 2010).
While these examples and others unequivocally highlight the contribution of spin labeling to understanding the mechanisms of all classes of membrane proteins, the next frontier is the integration of EPR parameters with other techniques. There are early examples of the combined use of crystallography cryoEM and EPR to characterize the conformational changes in ligand-gated ion channels (Durr et al., 2014). In combination with computational tools, such multi-pronged investigations hold the promise for elucidation of energy landscapes.
Acknowledgments
The authors wish to thank Dr. Richard A Stein and other members of the Mchaourab laboratory for helpful comments during the preparation of this chapter. Research in the Mchaourab laboratory is funded by NIH grants U54-GM087519 and R01-GM077659.
References
- Akyuz N, Altman RB, Blanchard SC, Boudker O. Transport dynamics in a glutamate transporter homologue. Nature. 2013;502(7469):114–118. doi: 10.1038/nature12265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akyuz N, Georgieva ER, Zhou Z, Stolzenberg S, Cuendet MA, Khelashvili G, et al. Transport domain unlocking sets the uptake rate of an aspartate transporter. Nature. 2015;518(7537):68–73. doi: 10.1038/nature14158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander N, Bortolus M, Al-Mestarihi A, McHaourab H, Meiler J. De novo high-resolution protein structure determination from sparse spin-labeling EPR data. Structure. 2008;16(2):181–195. doi: 10.1016/j.str.2007.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altenbach C, Froncisz W, Hemker R, McHaourab H, Hubbell WL. Accessibility of nitroxide side chains: absolute Heisenberg exchange rates from power saturation EPR. Biophys J. 2005;89(3):2103–2112. doi: 10.1529/biophysj.105.059063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altenbach C, Greenhalgh DA, Khorana HG, Hubbell WL. A collision gradient method to determine the immersion depth of nitroxides in lipid bilayers: application to spin-labeled mutants of bacteriorhodopsin. Proc Natl Acad Sci U S A. 1994;91(5):1667–1671. doi: 10.1073/pnas.91.5.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altenbach C, Kusnetzow AK, Ernst OP, Hofmann KP, Hubbell WL. High-resolution distance mapping in rhodopsin reveals the pattern of helix movement due to activation. Proc Natl Acad Sci U S A. 2008;105(21):7439–7444. doi: 10.1073/pnas.0802515105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altenbach C, Marti T, Khorana HG, Hubbell WL. Transmembrane protein structure: spin labeling of bacteriorhodopsin mutants. Science. 1990;248(4959):1088–1092. doi: 10.1126/science.2160734. [DOI] [PubMed] [Google Scholar]
- Bayburt TH, Sligar SG. Membrane protein assembly into Nanodiscs. FEBS Lett. 2010;584(9):1721–1727. doi: 10.1016/j.febslet.2009.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berliner LJ, Grunwald J, Hankovszky HO, Hideg K. A novel reversible thiol-specific spin label: papain active site labeling and inhibition. Anal Biochem. 1982;119(2):450–455. doi: 10.1016/0003-2697(82)90612-1. [DOI] [PubMed] [Google Scholar]
- Bhabha G, Ekiert DC, Jennewein M, Zmasek CM, Tuttle LM, Kroon G, et al. Divergent evolution of protein conformational dynamics in dihydrofolate reductase. Nat Struct Mol Biol. 2013;20(11):1243–1249. doi: 10.1038/nsmb.2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharjee S, Deterding LJ, Chatterjee S, Jiang J, Ehrenshaft M, Lardinois O, et al. Site-specific radical formation in DNA induced by Cu(II)-H(2)O(2) oxidizing system, using ESR, immuno-spin trapping, LC-MS, and MS/MS. Free Radic Biol Med. 2011;50(11):1536–1545. doi: 10.1016/j.freeradbiomed.2011.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borbat PP, McHaourab HS, Freed JH. Protein structure determination using long-distance constraints from double-quantum coherence ESR: study of T4 lysozyme. J Am Chem Soc. 2002;124(19):5304–5314. doi: 10.1021/ja020040y. [DOI] [PubMed] [Google Scholar]
- Borbat PP, Surendhran K, Bortolus M, Zou P, Freed JH, McHaourab HS. Conformational motion of the ABC transporter MsbA induced by ATP hydrolysis. PLoS Biol. 2007;5(10):e271. doi: 10.1371/journal.pbio.0050271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandon S, Beth AH, Hustedt EJ. The global analysis of DEER data. J Magn Reson. 2012;218:93–104. doi: 10.1016/j.jmr.2012.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britt RD, Campbell KA, Peloquin JM, Gilchrist ML, Aznar CP, Dicus MM, et al. Recent pulsed EPR studies of the photosystem II oxygen-evolving complex: implications as to water oxidation mechanisms. Biochim Biophys Acta. 2004;1655(1–3):158–171. doi: 10.1016/j.bbabio.2003.11.009. [DOI] [PubMed] [Google Scholar]
- Brunger AT, Strop P, Vrljic M, Chu S, Weninger KR. Three-dimensional molecular modeling with single molecule FRET. J Struct Biol. 2011;173(3):497–505. doi: 10.1016/j.jsb.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo R, Passeggi MC, Isaacson RA, Okamura MY, Feher G. Electron paramagnetic resonance investigation of photosynthetic reaction centers from Rhodobacter sphaeroides R-26 in which Fe2+ was replaced by Cu2+. Determination of hyperfine interactions and exchange and dipole-dipole interactions between Cu2+ and QA. Biophys J. 1990;58(1):149–165. doi: 10.1016/S0006-3495(90)82361-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang YW, Borbat PP, Freed JH. The determination of pair distance distributions by pulsed ESR using Tikhonov regularization. J Magn Reson. 2005;172(2):279–295. doi: 10.1016/j.jmr.2004.10.012. [DOI] [PubMed] [Google Scholar]
- Choe HW, Kim YJ, Park JH, Morizumi T, Pai EF, Krauss N, et al. Crystal structure of metarhodopsin II. Nature. 2011;471(7340):651–655. doi: 10.1038/nature09789. [DOI] [PubMed] [Google Scholar]
- Claxton DP, Quick M, Shi L, de Carvalho FD, Weinstein H, Javitch JA, et al. Ion/substrate-dependent conformational dynamics of a bacterial homolog of neurotransmitter:sodium symporters. Nat Struct Mol Biol. 2010;17(7):822–829. doi: 10.1038/nsmb.1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Columbus L, Hubbell WL. A new spin on protein dynamics. Trends Biochem Sci. 2002;27(6):288–295. doi: 10.1016/s0968-0004(02)02095-9. [DOI] [PubMed] [Google Scholar]
- Columbus L, Kalai T, Jeko J, Hideg K, Hubbell WL. Molecular motion of spin labeled side chains in alpha-helices: analysis by variation of side chain structure. Biochemistry. 2001;40(13):3828–3846. doi: 10.1021/bi002645h. [DOI] [PubMed] [Google Scholar]
- Cordero-Morales JF, Jogini V, Lewis A, Vasquez V, Cortes DM, Roux B, et al. Molecular driving forces determining potassium channel slow inactivation. Nat Struct Mol Biol. 2007;14(11):1062–1069. doi: 10.1038/nsmb1309. [DOI] [PubMed] [Google Scholar]
- Cortes DM, Cuello LG, Perozo E. Molecular architecture of full-length KcsA: role of cytoplasmic domains in ion permeation and activation gating. J Gen Physiol. 2001;117(2):165–180. doi: 10.1085/jgp.117.2.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross TA, Sharma M, Yi M, Zhou HX. Influence of solubilizing environments on membrane protein structures. Trends Biochem Sci. 2011;36(2):117–125. doi: 10.1016/j.tibs.2010.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuello LG, Cortes DM, Perozo E. Molecular architecture of the KvAP voltage-dependent K+ channel in a lipid bilayer. Science. 2004;306(5695):491–495. doi: 10.1126/science.1101373. [DOI] [PubMed] [Google Scholar]
- Cuello LG, Jogini V, Cortes DM, Pan AC, Gagnon DG, Dalmas O, et al. Structural basis for the coupling between activation and inactivation gates in K(+) channels. Nature. 2010;466(7303):272–275. doi: 10.1038/nature09136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J, Yang G, McHaourab HS. Structural basis of energy transduction in the transport cycle of MsbA. Science. 2005;308(5724):1023–1028. doi: 10.1126/science.1106592. [DOI] [PubMed] [Google Scholar]
- Durr KL, Chen L, Stein RA, De Zorzi R, Folea IM, Walz T, et al. Structure and dynamics of AMPA receptor GluA2 in resting, pre-open, and desensitized states. Cell. 2014;158(4):778–792. doi: 10.1016/j.cell.2014.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faham S, Watanabe A, Besserer GM, Cascio D, Specht A, Hirayama BA, et al. The crystal structure of a sodium galactose transporter reveals mechanistic insights into Na+/sugar symport. Science. 2008;321(5890):810–814. doi: 10.1126/science.1160406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fajer PG, Brown L, Song L. Biological Magnetic Resonance: ESR Spectroscopy in Membrane Biophysics. Vol. 27. New York: Kluwer Academic/Plenum Publ; 2007. Practical Pulsed Dipolar ESR (DEER) pp. 95–128. [Google Scholar]
- Fanucci GE, Coggshall KA, Cadieux N, Kim M, Kadner RJ, Cafiso DS. Substrate-induced conformational changes of the periplasmic N-terminus of an outer-membrane transporter by site-directed spin labeling. Biochemistry. 2003;42(6):1391–1400. doi: 10.1021/bi027120z. [DOI] [PubMed] [Google Scholar]
- Fanucci GE, Lee JY, Cafiso DS. Spectroscopic evidence that osmolytes used in crystallization buffers inhibit a conformation change in a membrane protein. Biochemistry. 2003;42(45):13106–13112. doi: 10.1021/bi035439t. [DOI] [PubMed] [Google Scholar]
- Forrest LR, Kramer R, Ziegler C. The structural basis of secondary active transport mechanisms. Biochim Biophys Acta. 2011;1807(2):167–188. doi: 10.1016/j.bbabio.2010.10.014. [DOI] [PubMed] [Google Scholar]
- Fraser RR, Boussard G, Saunders JK, Lambert JB. Barriers to rotation about the sulfur-sulfur bond in acyclic disulfides. Journal of the American Chemical Society. 1971;93(15):3822–3823. [Google Scholar]
- Freed DM, Horanyi PS, Wiener MC, Cafiso DS. Conformational exchange in a membrane transport protein is altered in protein crystals. Biophys J. 2010;99(5):1604–1610. doi: 10.1016/j.bpj.2010.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freed DM, Lukasik SM, Sikora A, Mokdad A, Cafiso DS. Monomeric TonB and the Ton box are required for the formation of a high-affinity transporter-TonB complex. Biochemistry. 2013;52(15):2638–2648. doi: 10.1021/bi3016108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgieva ER, Borbat PP, Ginter C, Freed JH, Boudker O. Conformational ensemble of the sodium-coupled aspartate transporter. Nat Struct Mol Biol. 2013;20(2):215–221. doi: 10.1038/nsmb.2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghimire H, McCarrick RM, Budil DE, Lorigan GA. Significantly improved sensitivity of Q-band PELDOR/DEER experiments relative to X-band is observed in measuring the intercoil distance of a leucine zipper motif peptide (GCN4-LZ) Biochemistry. 2009;48(25):5782–5784. doi: 10.1021/bi900781u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin L, Lawson A. Antibody fragments as tools in crystallography. Clin Exp Immunol. 2011;165(3):285–291. doi: 10.1111/j.1365-2249.2011.04427.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanelt I, Wunnicke D, Bordignon E, Steinhoff HJ, Slotboom DJ. Conformational heterogeneity of the aspartate transporter Glt(Ph) Nat Struct Mol Biol. 2013;20(2):210–214. doi: 10.1038/nsmb.2471. [DOI] [PubMed] [Google Scholar]
- Hilger D, Jung H, Padan E, Wegener C, Vogel KP, Steinhoff HJ, et al. Assessing oligomerization of membrane proteins by four-pulse DEER: pH-dependent dimerization of NhaA Na+/H+ antiporter of E. coli. Biophys J. 2005;89(2):1328–1338. doi: 10.1529/biophysj.105.062232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirst SJ, Alexander N, McHaourab HS, Meiler J. RosettaEPR: an integrated tool for protein structure determination from sparse EPR data. J Struct Biol. 2011;173(3):506–514. doi: 10.1016/j.jsb.2010.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbell WL, Altenbach C, Hubbell CM, Khorana HG. Rhodopsin structure, dynamics, and activation: a perspective from crystallography, site-directed spin labeling, sulfhydryl reactivity, and disulfide cross-linking. Adv Protein Chem. 2003;63:243–290. doi: 10.1016/s0065-3233(03)63010-x. [DOI] [PubMed] [Google Scholar]
- Hubbell WL, Altenbach C. Investigation of structure and dynamics in membrane proteins using site-directed spin labeling. Curr Opin Struct Biol. 1994;4(4):566–573. [Google Scholar]
- Hubbell WL, Cafiso DS, Altenbach C. Identifying conformational changes with site-directed spin labeling. Nat Struct Biol. 2000;7(9):735–739. doi: 10.1038/78956. [DOI] [PubMed] [Google Scholar]
- Hubbell WL, Lopez CJ, Altenbach C, Yang Z. Technological advances in site-directed spin labeling of proteins. Curr Opin Struct Biol. 2013;23(5):725–733. doi: 10.1016/j.sbi.2013.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbell WL, McHaourab HS, Altenbach C, Lietzow MA. Watching proteins move using site-directed spin labeling. Structure. 1996;4(7):779–783. doi: 10.1016/s0969-2126(96)00085-8. [DOI] [PubMed] [Google Scholar]
- Hustedt EJ, Stein RA, Sethaphong L, Brandon S, Zhou Z, Desensi SC. Dipolar coupling between nitroxide spin labels: the development and application of a tether-in-a-cone model. Biophys J. 2006;90(1):340–356. doi: 10.1529/biophysj.105.068544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam SM, Stein RA, McHaourab HS, Roux B. Structural refinement from restrained-ensemble simulations based on EPR/DEER data: application to T4 lysozyme. J Phys Chem B. 2013;117(17):4740–4754. doi: 10.1021/jp311723a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jao CC, Hegde BG, Chen J, Haworth IS, Langen R. Structure of membrane-bound alpha-synuclein from site-directed spin labeling and computational refinement. Proc Natl Acad Sci U S A. 2008;105(50):19666–19671. doi: 10.1073/pnas.0807826105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeschke G, Chechik V, Ionita P, Godt A, Zimmermann H, Banham J, Timmel CR, Hilger D, Jung H. DeerAnalysis2006--a comprehensive software package for analyzing pulsed ELDOR data. Applied Magnetic Resonance. 2006;30:473–498. [Google Scholar]
- Jeschke G, Polyhach Y. Distance measurements on spin-labelled biomacromolecules by pulsed electron paramagnetic resonance. Phys Chem Chem Phys. 2007;9(16):1895–1910. doi: 10.1039/b614920k. [DOI] [PubMed] [Google Scholar]
- Jeschke G, Wegener C, Nietschke M, Jung H, Steinhoff HJ. Interresidual distance determination by four-pulse double electron-electron resonance in an integral membrane protein: the Na+/proline transporter PutP of Escherichia coli. Biophys J. 2004;86(4):2551–2557. doi: 10.1016/S0006-3495(04)74310-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao D, Barfield M, Combariza JE, Hruby VJ. Ab initio molecular orbital studies of the rotational barriers and the sulfur-33 and carbon-13 chemical shieldings for dimethyl disulfide. Journal of the American Chemical Society. 1992;114(10):3639–3643. [Google Scholar]
- Kazmier K, Alexander NS, Meiler J, McHaourab HS. Algorithm for selection of optimized EPR distance restraints for de novo protein structure determination. J Struct Biol. 2011;173(3):549–557. doi: 10.1016/j.jsb.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazmier K, Sharma S, Islam SM, Roux B, McHaourab HS. Conformational cycle and ion-coupling mechanism of the Na+/hydantoin transporter Mhp1. Proc Natl Acad Sci U S A. 2014;111(41):14752–14757. doi: 10.1073/pnas.1410431111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazmier K, Sharma S, Quick M, Islam SM, Roux B, Weinstein H, et al. Conformational dynamics of ligand-dependent alternating access in LeuT. Nat Struct Mol Biol. 2014;21(5):472–479. doi: 10.1038/nsmb.2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khelashvili G, LeVine MV, Shi L, Quick M, Javitch JA, Weinstein H. The membrane protein LeuT in micellar systems: aggregation dynamics and detergent binding to the S2 site. J Am Chem Soc. 2013;135(38):14266–14275. doi: 10.1021/ja405984v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klare JP. Site-directed spin labeling EPR spectroscopy in protein research. Biol Chem. 2013;394(10):1281–1300. doi: 10.1515/hsz-2013-0155. [DOI] [PubMed] [Google Scholar]
- Klare JP, Steinhoff HJ. Spin labeling EPR. Photosynth Res. 2009;102(2–3):377–390. doi: 10.1007/s11120-009-9490-7. [DOI] [PubMed] [Google Scholar]
- Krishnamurthy H, Gouaux E. X-ray structures of LeuT in substrate-free outward-open and apo inward-open states. Nature. 2012;481(7382):469–474. doi: 10.1038/nature10737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamurthy H, Piscitelli CL, Gouaux E. Unlocking the molecular secrets of sodium-coupled transporters. Nature. 2009;459(7245):347–355. doi: 10.1038/nature08143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroncke BM, Horanyi PS, Columbus L. Structural origins of nitroxide side chain dynamics on membrane protein alpha-helical sites. Biochemistry. 2010;49(47):10045–10060. doi: 10.1021/bi101148w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langen R, Oh KJ, Cascio D, Hubbell WL. Crystal structures of spin labeled T4 lysozyme mutants: implications for the interpretation of EPR spectra in terms of structure. Biochemistry. 2000;39(29):8396–8405. doi: 10.1021/bi000604f. [DOI] [PubMed] [Google Scholar]
- Levitt M. Growth of novel protein structural data. Proc Natl Acad Sci U S A. 2007;104(9):3183–3188. doi: 10.1073/pnas.0611678104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L, Yee SW, Kim RB, Giacomini KM. SLC transporters as therapeutic targets: emerging opportunities. Nature Reviews Drug Discovery. 2015;14(8):543–560. doi: 10.1038/nrd4626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma D, Lu P, Yan C, Fan C, Yin P, Wang J, et al. Structure and mechanism of a glutamate-GABA antiporter. Nature. 2012;483(7391):632–636. doi: 10.1038/nature10917. [DOI] [PubMed] [Google Scholar]
- Masureel M, Martens C, Stein RA, Mishra S, Ruysschaert JM, McHaourab HS, et al. Protonation drives the conformational switch in the multidrug transporter LmrP. Nat Chem Biol. 2014;10(2):149–155. doi: 10.1038/nchembio.1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mchaourab HS, Kalai T, Hideg K, Hubbell WL. Motion of spin-labeled side chains in T4 lysozyme: effect of side chain structure. Biochemistry. 1999;38(10):2947–2955. doi: 10.1021/bi9826310. [DOI] [PubMed] [Google Scholar]
- Mchaourab HS, Lietzow MA, Hideg K, Hubbell WL. Motion of spin-labeled side chains in T4 lysozyme. Correlation with protein structure and dynamics. Biochemistry. 1996;35(24):7692–7704. doi: 10.1021/bi960482k. [DOI] [PubMed] [Google Scholar]
- McHaourab HS, Oh KJ, Fang CJ, Hubbell WL. Conformation of T4 lysozyme in solution. Hinge-bending motion and the substrate-induced conformational transition studied by site-directed spin labeling. Biochemistry. 1997;36(2):307–316. doi: 10.1021/bi962114m. [DOI] [PubMed] [Google Scholar]
- McHaourab HS, Steed PR, Kazmier K. Toward the fourth dimension of membrane protein structure: insight into dynamics from spin-labeling EPR spectroscopy. Structure. 2011;19(11):1549–1561. doi: 10.1016/j.str.2011.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer V, Swanson MA, Clouston LJ, Boratynski PJ, Stein RA, McHaourab HS, et al. Room-temperature distance measurements of immobilized spin-labeled protein by DEER/PELDOR. Biophys J. 2015;108(5):1213–1219. doi: 10.1016/j.bpj.2015.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra S, Verhalen B, Stein RA, Wen PC, Tajkhorshid E, McHaourab HS. Conformational dynamics of the nucleotide binding domains and the power stroke of a heterodimeric ABC transporter. Elife. 2014;3:e02740. doi: 10.7554/eLife.02740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittermaier AK, Kay LE. Observing biological dynamics at atomic resolution using NMR. Trends Biochem Sci. 2009;34(12):601–611. doi: 10.1016/j.tibs.2009.07.004. [DOI] [PubMed] [Google Scholar]
- Nigam SK. What do drug transporters really do? Nat Rev Drug Discov. 2015;14(1):29–44. doi: 10.1038/nrd4461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overington JP, Al-Lazikani B, Hopkins AL. How many drug targets are there? Nat Rev Drug Discov. 2006;5(12):993–996. doi: 10.1038/nrd2199. [DOI] [PubMed] [Google Scholar]
- Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, et al. Crystal structure of rhodopsin: A G protein-coupled receptor. Science. 2000;289(5480):739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- Pannier M, Veit S, Godt A, Jeschke G, Spiess HW. Dead-time free measurement of dipole-dipole interactions between electron spins. J Magn Reson. 2000;142(2):331–340. doi: 10.1006/jmre.1999.1944. [DOI] [PubMed] [Google Scholar]
- Pardon E, Laeremans T, Triest S, Rasmussen SG, Wohlkonig A, Ruf A, et al. A general protocol for the generation of Nanobodies for structural biology. Nat Protoc. 2014;9(3):674–693. doi: 10.1038/nprot.2014.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penmatsa A, Wang KH, Gouaux E. X-ray structure of dopamine transporter elucidates antidepressant mechanism. Nature. 2013;503(7474):85–90. doi: 10.1038/nature12533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez C, Koshy C, Yildiz O, Ziegler C. Alternating-access mechanism in conformationally asymmetric trimers of the betaine transporter BetP. Nature. 2012;490(7418):126–130. doi: 10.1038/nature11403. [DOI] [PubMed] [Google Scholar]
- Perozo E, Cortes DM, Cuello LG. Structural rearrangements underlying K+-channel activation gating. Science. 1999;285(5424):73–78. doi: 10.1126/science.285.5424.73. [DOI] [PubMed] [Google Scholar]
- Perozo E, Cortes DM, Sompornpisut P, Kloda A, Martinac B. Open channel structure of MscL and the gating mechanism of mechanosensitive channels. Nature. 2002;418(6901):942–948. doi: 10.1038/nature00992. [DOI] [PubMed] [Google Scholar]
- Polyhach Y, Bordignon E, Jeschke G. Rotamer libraries of spin labelled cysteines for protein studies. Phys Chem Chem Phys. 2011;13(6):2356–2366. doi: 10.1039/c0cp01865a. [DOI] [PubMed] [Google Scholar]
- Quick M, Shi L, Zehnpfennig B, Weinstein H, Javitch JA. Experimental conditions can obscure the second high-affinity site in LeuT. Nat Struct Mol Biol. 2012;19(2):207–211. doi: 10.1038/nsmb.2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quick M, Winther AM, Shi L, Nissen P, Weinstein H, Javitch JA. Binding of an octylglucoside detergent molecule in the second substrate (S2) site of LeuT establishes an inhibitor-bound conformation. Proc Natl Acad Sci U S A. 2009;106(14):5563–5568. doi: 10.1073/pnas.0811322106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabenstein MD, Shin YK. Determination of the distance between two spin labels attached to a macromolecule. Proc Natl Acad Sci U S A. 1995;92(18):8239–8243. doi: 10.1073/pnas.92.18.8239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux B, Islam SM. Restrained-ensemble molecular dynamics simulations based on distance histograms from double electron-electron resonance spectroscopy. J Phys Chem B. 2013;117(17):4733–4739. doi: 10.1021/jp3110369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A. Membrane preparation and solubilization. Methods Enzymol. 2015;557:45–56. doi: 10.1016/bs.mie.2014.11.044. [DOI] [PubMed] [Google Scholar]
- Salom D, Lodowski DT, Stenkamp RE, Le Trong I, Golczak M, Jastrzebska B, et al. Crystal structure of a photoactivated deprotonated intermediate of rhodopsin. Proc Natl Acad Sci U S A. 2006;103(44):16123–16128. doi: 10.1073/pnas.0608022103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano-Vega MJ, Magnani F, Shibata Y, Tate CG. Conformational thermostabilization of the beta1-adrenergic receptor in a detergent-resistant form. Proc Natl Acad Sci U S A. 2008;105(3):877–882. doi: 10.1073/pnas.0711253105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimamura T, Weyand S, Beckstein O, Rutherford NG, Hadden JM, Sharples D, et al. Molecular basis of alternating access membrane transport by the sodium-hydantoin transporter Mhp1. Science. 2010;328(5977):470–473. doi: 10.1126/science.1186303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sillitoe I, Lewis TE, Cuff A, Das S, Ashford P, Dawson NL, et al. CATH: comprehensive structural and functional annotations for genome sequences. Nucleic Acids Res. 2015;43(Database issue):D376–381. doi: 10.1093/nar/gku947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonoda Y, Newstead S, Hu NJ, Alguel Y, Nji E, Beis K, et al. Benchmarking membrane protein detergent stability for improving throughput of high-resolution X-ray structures. Structure. 2011;19(1):17–25. doi: 10.1016/j.str.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subczynski WK, Mainali L, Camenisch TG, Froncisz W, Hyde JS. Spin-label oximetry at Q- and W-band. J Magn Reson. 2011;209(2):142–148. doi: 10.1016/j.jmr.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ter Beek J, Guskov A, Slotboom DJ. Structural diversity of ABC transporters. J Gen Physiol. 2014;143(4):419–435. doi: 10.1085/jgp.201411164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasquez V, Sotomayor M, Cordero-Morales J, Schulten K, Perozo E. A structural mechanism for MscS gating in lipid bilayers. Science. 2008;321(5893):1210–1214. doi: 10.1126/science.1159674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weil JA, Bolton JR. Electron paramagnetic resonance: Elementary theory and practical applications. 2. Hoboken, NJ: Wiley-Interscience; 2007. [Google Scholar]
- Wen PC, Verhalen B, Wilkens S, McHaourab HS, Tajkhorshid E. On the origin of large flexibility of P-glycoprotein in the inward-facing state. J Biol Chem. 2013;288(26):19211–19220. doi: 10.1074/jbc.M113.450114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyand S, Shimamura T, Yajima S, Suzuki S, Mirza O, Krusong K, et al. Structure and molecular mechanism of a nucleobase-cation-symport-1 family transporter. Science. 2008;322(5902):709–713. doi: 10.1126/science.1164440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiener MC. A pedestrian guide to membrane protein crystallization. Methods. 2004;34(3):364–372. doi: 10.1016/j.ymeth.2004.03.025. [DOI] [PubMed] [Google Scholar]
- Xu Q, Ellena JF, Kim M, Cafiso DS. Substrate-dependent unfolding of the energy coupling motif of a membrane transport protein determined by double electron-electron resonance. Biochemistry. 2006;45(36):10847–10854. doi: 10.1021/bi061051x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita A, Singh SK, Kawate T, Jin Y, Gouaux E. Crystal structure of a bacterial homologue of Na+/Cl--dependent neurotransmitter transporters. Nature. 2005;437(7056):215–223. doi: 10.1038/nature03978. [DOI] [PubMed] [Google Scholar]
- Yang Z, Liu Y, Borbat P, Zweier JL, Freed JH, Hubbell WL. Pulsed ESR dipolar spectroscopy for distance measurements in immobilized spin labeled proteins in liquid solution. J Am Chem Soc. 2012;134(24):9950–9952. doi: 10.1021/ja303791p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yildirim MA, Goh KI, Cusick ME, Barabasi AL, Vidal M. Drug-target network. Nat Biotechnol. 2007;25(10):1119–1126. doi: 10.1038/nbt1338. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Terry D, Shi L, Weinstein H, Blanchard SC, Javitch JA. Single-molecule dynamics of gating in a neurotransmitter transporter homologue. Nature. 2010;465(7295):188–193. doi: 10.1038/nature09057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Terry DS, Shi L, Quick M, Weinstein H, Blanchard SC, et al. Substrate-modulated gating dynamics in a Na+-coupled neurotransmitter transporter homologue. Nature. 2011;474(7349):109–113. doi: 10.1038/nature09971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Q, Casey JR. Topology of transmembrane proteins by scanning cysteine accessibility mutagenesis methodology. Methods. 2007;41(4):439–450. doi: 10.1016/j.ymeth.2006.08.004. [DOI] [PubMed] [Google Scholar]
- Zielonka J, Cheng G, Zielonka M, Ganesh T, Sun A, Joseph J, et al. High-throughput assays for superoxide and hydrogen peroxide: design of a screening workflow to identify inhibitors of NADPH oxidases. J Biol Chem. 2014;289(23):16176–16189. doi: 10.1074/jbc.M114.548693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou P, Bortolus M, McHaourab HS. Conformational cycle of the ABC transporter MsbA in liposomes: detailed analysis using double electron-electron resonance spectroscopy. J Mol Biol. 2009;393(3):586–597. doi: 10.1016/j.jmb.2009.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou P, McHaourab HS. Alternating access of the putative substrate-binding chamber in the ABC transporter MsbA. J Mol Biol. 2009;393(3):574–585. doi: 10.1016/j.jmb.2009.08.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou P, McHaourab HS. Increased sensitivity and extended range of distance measurements in spin-labeled membrane proteins: Q-band double electron-electron resonance and nanoscale bilayers. Biophys J. 2010;98(6):L18–20. doi: 10.1016/j.bpj.2009.12.4193. [DOI] [PMC free article] [PubMed] [Google Scholar]