

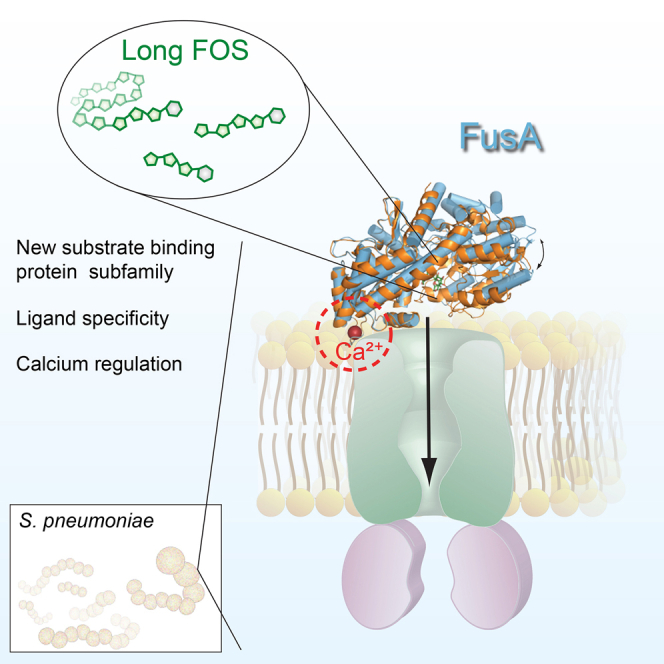
Summary
Streptococcus pneumoniae is dependent on carbohydrate uptake for colonization and pathogenesis, and dedicates over a third of its transport systems to their uptake. The ability of the pneumococcus to utilize fructooligosaccharides (FOSs) is attributed to the presence of one of two types of FOS ATP-binding cassette (ABC) transporters. Strains encoding SfuABC are only able to utilize short-chain FOSs, while strains encoding FusABC can utilize both short- and long-chain FOSs. The crystal structures of the substrate-binding protein FusA in its open and closed conformations bound to FOSs, and solution scattering data of SfuA, delineate the structural basis for import of short- and long-chain FOSs. The structure of FusA identifies an EF hand-like calcium-binding motif. This is shown to be essential for translocation of FOSs in FusABC and forms the basis for the definition of a new class of substrate-binding proteins that regulate substrate translocation by calcium.
Keywords: carbohydrate ABC transporters, substrate-binding proteins, Streptococcus pneumoniae, calcium signaling, substrate translocation regulation
Graphical Abstract
Highlights
-
•
A new cluster of substrate-binding proteins is structurally defined
-
•
An EF hand-like calcium-binding site regulates substrate translocation
-
•
Specificity toward short- and long-chain fructooligosaccharides elucidated
Culurgioni et al. define the structural basis for uptake of short- and long-chain fructooligosaccharides in pneumococcus. They describe a new structural cluster of substrate-binding proteins that share a common calcium-binding motif. They propose that regulation of substrate translocation is mediated by calcium for this subfamily.
Introduction
Acquisition of nutrients by bacterial pathogens from their host environment is essential for their survival and subsequent ability to cause disease. To import a wide variety of nutrients bacteria use multiple nutrient-specific transporter systems, of which the most abundant is the ATP-binding cassette (ABC) transporter superfamily (Higgins, 1992). ABC transporters use ATP hydrolysis to transport a large range of small molecules across cellular membranes. Plants, prokaryotes, and archaea have ABC importers and exporters, while eukaryotes only encode exporters. Despite their varied substrates and the ability to both import and export they share a common architecture consisting of two hydrophobic transmembrane domains (TMDs) and two hydrophilic nucleotide-binding domains that interact with the TMDs from the cytosolic side of the membrane (Rees et al., 2009). The family has been classified into four types, on the basis of structural and biochemical data, as either type I, II, or III importers, or as exporters. Type I and II importers rely on a substrate-binding protein (SBP) for import that specifically associates with the ligand to deliver it to the TMDs. Type III importers or energy-coupling factor transporters use an integral membrane protein known as the EcfS or S component (Rodionov et al., 2009). Type I importers are the best characterized to date (Gerber et al., 2008, Hollenstein et al., 2007, Kadaba et al., 2008, Maruyama et al., 2015, Oldham et al., 2007, Rice et al., 2014, Yu et al., 2015). However, the structural data have highlighted the high level of diversity in the substrate translocation mechanism and much remains unknown because of the large range of substrates transported by SBPs (Hopfner, 2016).
Structural analysis of the SBPs has led to the identification of six distinct clusters which have been assigned based solely on structural similarity and independently of substrate specificity (Berntsson et al., 2010). Although having little sequence similarity and ranging in size from approximately 25 to 70 kDa, SBPs share a common core structural fold: two α/β domains formed by a central β sheet flanked by α helices; a hinge region connects the two domains and the ligand-binding site resides between them. In the substrate-free state the two domains are able to freely move and rotate around a flexible hinge region (open configuration), whereas on ligand-binding the protein is stabilized in a closed conformation with the two domains closely packed around the ligand (Tang et al., 2007). This binding mode is known as the “Venus Fly-trap” mechanism (Mao et al., 1982).
Streptococcus pneumoniae (pneumococcus) typically asymptomatically colonizes the upper airway of humans, but can migrate to normally sterile regions and cause diseases including pneumonia, meningitis, otitis media, sinusitis, and conjunctivitis (Ispahani et al., 2004). The pneumococcus relies exclusively on carbohydrates as a carbon source and commits at least a third of all its transport mechanisms to their import (Tettelin et al., 2001). Nearly half of these carbohydrate transporters have been implicated in pneumococcal disease but much work remains to fully understand the role of carbohydrate import in pneumococcal pathogenesis (Buckwalter and King, 2012). The carbohydrate transporters encoded by pneumococci include at least eight ABC transporters (Bidossi et al., 2012). One of these transporters was initially described as part of a sucrose utilization system (sus) (TIGR4; SP_1796-8) (Iyer and Camilli, 2007). A transporter mutant had a mild phenotype in the lung, but any role in colonization was not examined. Subsequent data revealed that this transporter does not contribute to sucrose utilization, but transports fructooligosaccharides (FOSs) including inulin and as a result was renamed the Fus (FOS utilization system) transporter (Linke et al., 2013). FOSs consist of a variable number (n) of D-fructose units linked by β2-1 bonds and a terminal D-glucose connected through an α2-1 bond. While the majority of pneumococcal strains can utilize long-chain FOSs as a sole carbon source, some strains are only able to grow on shorter chain FOSs (Bidossi et al., 2012, Langvad-Nielsen, 1944, Linke et al., 2013, Morch-Lund, 1949). Examination of 33 clinical isolates demonstrated that all six strains that were able to use GF3 but not inulin as a sole carbon source encoded an alternative transporter named Sfu (short-chain FOS utilization) (Linke et al., 2013). The SBP of this transporter shares little sequence similarity with that encoded by the fus locus and the structural basis of the substrate specificity is unclear. It is currently unknown where pneumococci encounter these polysaccharides, but our hypothesis is that they are encountered as dietary carbohydrates during colonization of the oropharynx or as extracellular polysaccharides produced by other normal flora. The fact that one of these transporters is present in every pneumococcal strain screened supports the hypothesis that the ability to utilize FOSs is important during pneumococcal colonization.
Here we describe the crystal structure of the SBP (SP_1796, FusA) of the ABC carbohydrate transporter encoded by the fus locus of S. pneumoniae TIGR4 in the absence of carbohydrate and in complex with kestose, nystose, and fructofuranosyl-nystose. The structural data also enabled a new cluster of SBPs to be proposed with the notable presence of an EF hand calcium-binding site, which we show is essential for substrate translocation by the fus importer. The solution structure of the SBP SfuA of the ABC importer encoded by the sfu locus in the S. pneumoniae D39 strain is also presented revealing a significantly smaller substrate-binding cavity compared with FusA. Characterization of binding of FOSs to both FusA and SfuA together with analysis of mutagenesis of the substrate-binding site of FusA provide the structural basis for FOS uptake in S. pneumoniae.
Results
Specificity of FusA and SfuA for FOSs
Previous work has shown that each pneumococcal strain encodes one of two ABC transporters that enable uptake of short-chain FOSs encoded by genes within the fus and sfu loci (Linke et al., 2013). However, only the ABC transporter encoded by the fus locus enables uptake of long-chain FOS such as inulin. The fus locus has also been implicated in sucrose uptake (Iyer and Camilli, 2007), thus we set out to provide a biochemical and structural characterization of the SBPs FusA and SfuA encoded by the fus and sfu loci, respectively.
We assessed binding specificities of the SBPs FusA and SfuA for varying chain length FOSs. The purified proteins were screened by thermal shift assay in the presence of FOSs to identify their impact on protein stabilities: FusA showed an increased melting temperature (ΔTm > 4°C–7°C) with fructofuranosyl-nystose (GF4), nystose (GF3), and inulin (GFn, where n ∼ 36), while SfuA was stabilized by kestose (GF2) (ΔTm = 6.4°C) and to a lower extent by nystose (GF3) (ΔTm = 3.6°C); neither SBP was stabilized by sucrose (GF1) supporting the work by Linke et al. (2013), which showed growth of an S. pneumoniae fusABC deletion mutant with sucrose as a sole source of carbon was unaltered with respect to wild-type (Figure 1A). Pull-down analysis on beads covalently linked to the different sugars showed the same trend: FusA bound longer-chain FOSs with higher affinity than SfuA, which preferentially bound kestose (Figure 1B). Binding affinities were determined by measuring the dissociation constant (KD) using isothermal titration calorimetry (ITC). FusA had submicromolar affinity for fructofuranosyl-nystose and nystose (Figures 1D, 1E, and S1) and SfuA bound kestose with a similar KD range (Figures 1H and 1I). The binding affinities for inulin, a heterogeneous mixture of FOSs with as many as 40 fructose units, decreased binding to FusA by a factor of 5 but to SfuA by a factor of 100 (Figures 1F and 1J). These data show that SfuA preferentially binds shorter FOSs (tri- or tetrasaccharides), while FusA has a higher affinity for longer FOSs (tetrasaccharides and larger).
Figure 1.

FusA and SfuA Preference for Different Chain Length FOSs
(A) Thermal shift stabilization assay performed on FusA and SfuA with and without sucrose, kestose, nystose, fructofuranosyl-nystose, and inulin. The displayed values represent the melting temperature (Tm) of the samples and the error bars correspond to the SE. The SEM was calculated as σ/√n, where σ is the SD.
(B) Pull-down analysis on fructooligosaccharide-conjugated resin: FusA, SfuA, and SP1690 (as negative control) were incubated with each FOS-conjugated resin and after washing, the bound fraction was analyzed by SDS-PAGE (Inulin resins were loaded on a separate gel).
(C–J) ITC experiments between FusA and SfuA toward sucrose, kestose, fructofuranosyl-nystose, and inulin. All reactions are exothermic and exhibit a 1:1 stoichiometry except for inulin, the heterogeneity of which affects the experimental stoichiometry. FusA exhibits a clear binding preference for fructofuranosyl-nystose and SfuA for kestose.
Structure of FusA in Ligand-Free Form and in Complex with FOSs
To understand the structural determinants for FOS specificity and mode of binding, we determined the high-resolution crystallographic structure of FusA in substrate-free form (Apo-FusA, PDB: 5G5Y) and in complex with kestose (PDB: 5G5Z), nystose (PDB: 5G60), and fructofuranosyl-nystose (PDB: 5G61). Crystals of FusA grown in the presence or absence of carbohydrates diffracted to high-resolution (1.9–2.4 Å). Crystallographic data collection and refinement statistics are summarized in Table 1.
Table 1.
Data Collection and Refinement Statistics
| FusA-Apo | FusA-GF2 | FusA-GF3 | FusA-GF4 | FusAD223A+E224A-GF4 | |
|---|---|---|---|---|---|
| Data Collection | |||||
| Beamline | DLS-I03 | DLS-I03 | DLS-I24 | DLS-I03 | DLS-I03 |
| Wavelength (Å) | 1.2552 | 0.97625 | 0.96861 | 0.97625 | 0.97625 |
| Resolution (Å) | 70.76–1.84 (1.89–1.84) | 70.14–2.01 (2.06–2.01) | 57.37–1.99 (2.04–1.99) | 90.37–2.40 (2.46–2.40) | 86.86–1.99 (2.04–1.99) |
| Space group | P1 | P21 | P21 | P21 | P43 212 |
| Cell Dimensions | |||||
| a, b, c (Å) | 66.07, 72.17, 77.66 | 70.61, 127.82, 120.55 | 71.03, 129.74, 120.28 | 76.37, 135.91, 121.19 | 122.58, 122.58, 169.47 |
| α, β, γ (°) | 108.02, 104.99, 92.49 | 90, 96.58, 90 | 90, 96.34, 90 | 90, 93.28, 90 | 90, 90, 90 |
| Total observations | 35,3275 | 800,887 | 522,806 | 352,795 | 130,4158 |
| Unique reflections | 104,848 | 140,592 | 144,038 | 95,973 | 88,883 |
| Completeness (%) | 92.5 (60.6) | 99.7 (99.4) | 97.3 (77.2) | 99.6 (99.8) | 100 (100) |
| Redundancy | 3.4 (3.0) | 5.7 (5.8) | 3.6 (2.7) | 3.7 (3.8) | 14.7 (14.6) |
| I/σI | 12.5 (2.2) | 8.0 (1.4) | 7.1 (1.1) | 5.1 (1.4) | 12.2 (1.5) |
| Rmerge | 0.055 (0.520) | 0.133 (1.206) | 0.125 (0.833) | 0.209 (0.891) | 0.197 (1.937) |
| CC1/2 | 1.0 (0.6) | 1.0 (0.6) | 1 (0.5) | 1 (0.5) | 1 (0.6) |
| Refinement | |||||
| Resolution (Å) | 70.76–1.73 (1.77–1.73) | 70.14–2.01 (2.08–2.01) | 48.32–1.99 (2.06–1.99) | 90.37–2.4 (2.49–2.4) | 77.17–1.99 (2.06–1.99) |
| No. of reflections | 108,465 (3,228) | 140,533 (13,942) | 143,983 (11,744) | 95,932 (9,543) | 88,785 (8,717) |
| Rwork/Rfree | 0.1624/0.1932 | 0.1681/0.2221 | 0.1771/0.2209 | 0.1856/0.2513 | 0.1710/0.2080 |
| No. of atoms | 9,261 | 17,104 | 17,584 | 17,125 | 8,917 |
| Protein | 7,932 | 15,803 | 15,685 | 15,789 | 7,915 |
| Ligand | 35 (SO4 and GOL) | 137 (GF2) | 205 (GF3, Cl, and GOL) | 224 (GF4) | 112 (GF4) |
| Ca | 4 | 8 | 8 | 8 | 3 |
| Zn | 10 | 0 | 0 | 0 | 0 |
| Water | 1,280 | 1,156 | 1,686 | 1,112 | 887 |
| B factors | 25.79 | 38.71 | 30.05 | 34.00 | 35.92 |
| Protein | 23.90 | 38.39 | 29.10 | 33.74 | 33.75 |
| Ligand/ion | 38.91 | 33.00 | 25.64 | 47.99 | 64.12 |
| Water | 37.03 | 43.80 | 39.64 | 34.86 | 51.68 |
| RMSD | |||||
| Bond lengths (Å) | 0.012 | 0.010 | 0.015 | 0.009 | 0.012 |
| Bond angles (°) | 1.47 | 1.020 | 1.68 | 1.000 | 1.100 |
| Ramachandran (%) | |||||
| Most favored | 98.4 | 98.4 | 98 | 97 | 98 |
| Allowed | 1.6 | 1.6 | 1.9 | 2.9 | 1.8 |
| Outliers | 0 | 0 | 0.1 | 0.1 | 0.2 |
| PDB: | 5G5Y | 5G5Z | 5G60 | 5G61 | 5G62 |
Values in parentheses are for highest-resolution shell.
The overall structure of FusA is formed by two α/β domains linked by three loops, which make up the hinge region of the protein and follows the general architecture of SBPs (Figures 2A–2C). The first domain (residues 48–165 and 350–440) is formed by two central β sheets, of four and three strands, surrounded by nine α helices and an additional two-stranded β sheet. The second domain (residues 166–349 and 441–535) consists of a four-stranded β sheet enclosed by eight α helices and an extra β sheet. FusA has several 310-helices, two in the first domain and five in the second, which could contribute to the intrinsic flexibility of the protein (Figures 2C, 3D and S2). A notable structural feature is the presence of two calcium-binding sites located in the second domain of FusA, one of which is an EF hand-like motif. This motif is formed by a solvent-exposed loop encompassing residues 216-224 that connects helix α8 to β strand 6 (β6) (Figure 2D). While the canonical EF hand structural signature is formed by a 12-residue calcium-binding loop between two helices, several atypical motifs diverging in length and in the secondary structure elements composition have been found and called EF hand-like motifs (Feng et al., 2011, Gifford et al., 2007, Rigden and Galperin, 2004). These motifs also deviate from the sequence pattern of the calcium-binding site, where the Glu/Asp/Asn residues involved in ion interaction are not necessarily located in positions 1, 3, 5, 7, 9, and 12 as in the classical EF hand motif. The second calcium-binding site is located in the region encompassing β strand β8 and the central buried η4 310-helix (residues 263-268), where it may play a structural role in the stabilization of the second domain; in particular Phe-264 on β strand β8 is directed toward a hydrophobic patch formed by the C-terminal helices α15, α16, and α17 (Figure 2E).
Figure 2.

FusA-apo and FusA-FOS Complex Structures
(A–C) Ribbon models of the FusA-apo viewed at the indicated orientations.
(D and E) Close-up of the first and second calcium-binding sites; water molecules are shown as red spheres: w1 = 76, w2 = 918, w3 = 816 in PDB: 5G5Y. Calcium ions are shown as purple spheres.
(F and G) Structural comparison of FusA-apo (cylindrical helices, light blue) and FusA:nystose complexes (ribbon helices, orange), using the server Rapido (Mosca and Schneider, 2008). The superposition showed a global RMSD = 2.78 Å and the presence of two invariant rigid bodies: domain I (residues 48–165, 350–440) local RMSD = 0.43 Å, domain II (residue 166–349, 441–535) local RMSD = 0.48 Å. Two views are shown.
Figure 3.

Identification of a New SBP Protein Subfamily
(A) Close-up of the superposition of the EF hand-like motif shown in sticks, highlighting the polar residue oriented toward the permease.
(B and C) Superposition of FusA, AlgQ1 (PDB: 1Y3N), AlgQ2 (PDB: 1J1N), and Blon_2351 (PDB: 3OMB) structures: the ribbon models, viewed at the indicated orientations, are colored using rainbow colors (blue to red from the N to C termini). FusA is shown with cylindrical helices.
(D) Sequence and secondary structure alignment of FusA, AlgQ1, AlgQ2, and Blon_2351 colored according to their conservation using ESPript (Robert and Gouet, 2014).
The structures of FusA in complex with kestose (PDB: 5G5Z), nystose (PDB: 5G60), and fructofuranosyl-nystose (PDB: 5G61) have a similar overall topology to FusA. All sugar-bound structures undergo the expected large change in conformation on binding of substrate. This entails a rigid-body reorientation of the two α/β domains on binding of FOSs, which rotate 23° closer and trap the FOS substrates at the interdomain cleft (Figures 2F and 2G, Movie S1).
Structures of all of the SBPs determined up to 2010, except for three, were assigned to six clusters on the basis of structural similarities (Berntsson et al., 2010). Each cluster has a distinguishing structural feature but other trends were derivable; for example, most carbohydrate-binding SBPs were found in clusters B and D. Comparison of the structure of FusA failed to assign it to one of the six classes, but it aligns with two of the previously unclassified SBPs that are involved in alginate import in Sphingomonas sp. A1: AlgQ1 (PDB: 1Y3N) and AlgQ2 (PDB: 1J1N). These structures are similar and exhibit additional structural elements including a β sheet in the first domain and an extra C-terminal region formed by four helices in the second domain (Figures 3B–3D and S2). Another distinguishing structural feature is the presence of an EF hand-like calcium-binding site in domain II. Further analysis of the PDB identified another uncharacterized SBP from Bifidobacterium logum, Blon_2351 (PDB: 3OMB) with an EF hand-like calcium-binding site that also structurally aligned with this cohort of SBP structures (Figure 3A). Thus, we propose an extension of the structural classification to a seventh cluster, characterized by a larger size (∼70 kDa), the presence of additional structural elements that define a larger ligand-binding cavity, with respect to the canonical SBPs, and an EF hand-like calcium-binding site.
Structural Determinants for FOS Binding in FusA
The longest oligosaccharide chain length in the FusA-FOS complex structures elucidated here was the pentasaccharide fructofuranosyl-nystose (GF4), which consists of four D-fructose units and a terminal D-glucose unit. To aid description of the FusA-FOS complexes we have annotated each FOS as F1-F2 … Fn-G1 (F, fructose; G, glucose); fructofuranosyl-nystose is thus represented by F1-F2-F3-F4-G1 (Figure 4F). In the FusA-GF4 complex the first fructose, F1, is deeply inserted into the substrate-binding cavity with four amino acids interacting directly with it through hydrogen bounds (Glu167, Trp314, Trp384, and Arg419; Figure 4A and Table S2). His177 and Glu167 form the base of the substrate-binding pocket contributing to the overall polarity of the site and, along with Arg419, anchor the first fructose to FusA. The F2 fructose hydroxyl groups form hydrogen bonds with Asn235, Asn318, Arg419, and ordered waters; this sugar moiety is sandwiched between Trp314, which stacks against the fructopyranose ring, and Arg419. The F3 and F4 fructose moieties interact with Glu423 and well-defined water molecules, respectively. The terminal glucose G1 makes no direct contacts with FusA, and the binding cavity is sufficiently large to allow a large degree of flexibility that is reflected in the higher B factors for G1. Arg419 plays a pivotal role in defining the binding conformation of fructopyranose by interacting through hydrogen bonds to the first and the second fructose units and the oxygens of the glycosidic bonds between F1, F2, and F3.
Figure 4.

FusA Structural Determinants for FOS Binding
(A–C) Enlarged views of the FusA substrate-binding pocket (magenta) (A), (orange) (B), and (yellow) (C), respectively, in complex with fructofuranosyl-nystose (pink), nystose (light orange), and kestose (light green); water molecules are shown as red spheres.
(D) Structural superposition of the binding pockets.
(E) Pull-down assay of FusA mutants.
(F) Chemical structures of FOSs.
Analysis of the structure of FusA in complex with kestose (GF2) and nystose (GF3) reveals that the protein-FOS interactions are conserved at the F1 and F2 fructose-binding sites. The shorter chain length FOSs position the terminal G1 glucose deeper in the binding cleft where it directly interacts with FusA through interactions not found in the GF4 complex with Lys353 and Gln376 in the case of the nystose and kestose complexes, respectively (Figures 4B–4D).
Based on the analysis of the three FusA-FOS complexes we probed the importance of Glu167, His177, Trp314, Asn318, Trp384, Arg419, and Glu423 by site-directed mutagenesis and binding analysis of the mutants. Pull-down experiments on beads covalently conjugated with GF3, GF4, and inulin showed that all mutants except His177Ala failed to bind FOS, although this mutation did reduce the binding affinity for FOS (Figure 4E). ITC analysis confirmed the pull-down data and showed a decrease in the binding affinities of His177Ala and Glu423Ala mutants by approximately 2- and 10-fold, respectively, a considerable reduction to the millimolar range for Asn318Ala and Trp384Ala mutants, and the abolishment of binding for all others mutants (Table S1). In summary, the data show that binding of FOSs by FusA is dependent on multiple amino acids, which directly interact with the fructose units inserted in the inner cavity of the binding site (F1-F3) of FusA. In particular, mutation of Glu167 and Arg419, which make strong hydrogen-bonding interactions with the fructose units F1-F3, and Trp314, which is involved in π-π stacking interactions with the F3 fructose ring, each play an essential role in binding of FOSs (Figure 4D).
To assess the impact of an altered substrate-binding affinity for FOSs in S. pneumoniae we tested the ability of all FusA mutants to grow using nystose (GF3) as their sole carbon source and compared this with the parental strain. As a control the genes encoding the fus transporter were deleted, which led, as reported previously, to no growth, while maximal growth is observed at 40 hr when wild-type FusA is expressed (Figure 5A). Mutation of Glu167, Trp314, and Arg419 to alanine prevented any growth on nystose, while the generation time and maximal optical density are considerably impaired and altered for mutation of His177, Asn318, and Trp384 to alanine (Table S3). These data align with the structural data emphasizing the importance of the hydrogen-bonding interactions of Glu167, Trp314, Asn318, and Arg419 and of the stacking of Trp314. Growth on nystose with the His177Ala mutant is greatly impaired indicating the importance of the substrate cleft floor polarity for directing binding of FOS to FusA. Mutation of Glu423 also perturbs the growth on nystose: in each experiment, growth of the Glu423 mutant starts earlier than that of the parental strain, but reaches a lower maximal optical density. The structural data show that the carboxylate of Glu423 forms stereochemically less-favorable hydrogen bonds to the hydroxyl group of the fructose F3 and reduces the binding affinity by an order of magnitude. Why the absence of this interaction leads to the observed growth profile is unclear, but all data are consistent with a role for Glu423 in carbohydrate binding and transport.
Figure 5.

FusA Structural Determinants for Carbohydrate Binding Are Involved in FOS Import
(A) Growth curves of S. pneumoniae on defined medium containing nystose as a sole carbon source. Endogenous FusA was mutated as described for each panel. An optical density at 600 nm (OD600) for wild-type and every mutant was measured every 20 min for 96 hr, showing that most of the mutants have impaired growth on nystose.
(B) Schematic representation of the FusA-FOS-binding site, highlighting the residues involved in FOS binding and import: dotted lines indicate hydrogen bonds or polar interactions, while the gray ring represents the stacking interaction of Trp314 on F2. Mutation of these amino acids affect strongly (red), weakly (yellow), or do not affect (green) binding (inner rectangle) or import (outer circle).
Functional Role of the EF Hand-like Calcium-Binding Site
As EF hand-like calcium-binding proteins have been implicated in a variety of cellular processes in bacteria, we probed the importance of calcium-binding by this motif for FOS uptake by FusA. A double mutation of two of the protein calcium metal ligands Asp223 and Glu224 to alanine was designed to disrupt binding of calcium to the EF hand-like motif. The pull-down assay and ITC measurements revealed that this double alanine mutation of the protein calcium ligands does not perturb FOS binding (Figure 4E and Table S1). Strikingly, the ability of the EF hand-like mutant to grow on nystose as the sole carbon source was abolished (Figure 6B). The structure of the Asp223Ala:Glu224Ala mutant in complex with fructofuranosyl-nystose has been determined to a resolution of 1.99 Å (PDB: 5G62) and reveals the overall structure to be identical to the wild-type protein (global root-mean-square deviation [RMSD] of 0.7 Å for all protein atoms, Figure 6A). While one of the chains of the asymmetric unit is completely lacking the calcium ion in this EF hand-like loop, the other subunit shows calcium bound but displaced by 6.1 Å from its position in the wild-type. This perturbs the calcium-binding site in the Asp223Ala:Glu224Ala mutant from an octahedral to a less-favorable pentagonal bipyramidal coordination. The structural rearrangements result in the metal coordinating with the carboxylate of Asp215, the main chain oxygen of Lys212, and five water molecules, instead of the seven protein ligands as in the wild-type (Figures 2D and 6C). We propose the presence of this calcium ion in one of the subunits of FusA to be a crystal artifact arising from the high concentration of calcium in the crystallization conditions (20 mM) and the intermolecular interactions provided by the crystal packing in this subunit. Nevertheless, the mutations in the EF hand-like motif result in the EF hand-like loop being shifted by 6 Å (Figure 6A) and significant disorder introduced (Figure 6D).
Figure 6.

Functional Role of the Calcium-Binding Site
(A) Superposition of the structures of FusAwt:fructofuranosyl-nystose (orange ribbons) and FusAD223A−E224A:fructofuranosyl-nystose (green and yellow ribbons) complexes, indicating that the mutation affects only the EF hand-like motif reorienting Glu221.
(B) Growth curves of S. pneumoniae on nystose-supplemented medium. Endogenous FusA was mutated as described for each panel. No growth for the double mutant is observed on nystose.
(C) Details of the EF hand-like calcium-binding site of chain A of the FusAD223A−E224A:fructo-nystose complex (yellow ribbons and sticks, with mutated residues highlighted in magenta; calcium and waters in white and red spheres, respectively); water molecules w1 = 95, w2 = 412, w3 = 284, w4 = 599, and w5 = 238 in PDB: 5G62.
(D) Normalized mean B factors for FusA:GF2 (green), FusA:GF3 (cyan), FusA:GF4 (blue), and FusAD223A−E224A:GF4 (magenta-red) complexes as a function of amino acid residue, highlighting the effect on the EF-hand-like motif for FusAD223A−E22.
These data indicate that the conformation and integrity of the EF hand-like calcium-binding site plays an essential role in substrate translocation as binding of substrate to the SBP is unperturbed. Analysis of a recently determined structure of a bacterial alginate ABC transporter in complex with its SBP provides a high-resolution structure of the SBP:permease interface. The SBPs of this alginate ABC transporter, AlgQ1 and AlgQ2, are structurally homologous to FusA (Figure 3) and are grouped in the identified seventh class of SBPs proposed here, based on structural similarity and in particular on the presence of an EF hand-like calcium-binding site. The EF hand-like motif of AlgQ2 interacts with the permease (AlgM1) at the complex interface and contributes to a proposed substrate tunnel between AlgQ2 and AlgM1 (Maruyama et al., 2015) (Figure 7A). The EF hand-like bound calcium restrains the loop conformation and directs Lys201 of AlgQ2 toward Gln60 of AlgM1 to form a salt bridge. Superposition of FusA onto AlgQ2 bound to the AlgM1M2SS transporter shows the EF hand-like calcium-binding site is comparable and we propose a similar salt bridge to be formed with Glu221 of FusA and potentially Arg59 of FusB derived from a structure-based sequence alignment of AlgM1 and FusB (Figure 7B). Disruption of the calcium-binding site as observed in the Asp223Ala:Glu224 mutant would affect the SBP permease interactions, which are directed by binding of calcium in the EF hand-like motif. Moreover we propose that these interactions are essential to sense conformational changes in the nucleotide-binding domains of the ABC transporter that form part of the alternating access model of substrate translocation.
Figure 7.

Fus/Alg Transporter Superposition
(A) Ribbon diagrams of FusA (orange) superposed to AlgQ2 (purple) in the whole transporter context (PDB: 4TQU), including AlgM1 (cyan) AlgM2 (green), and AlgS (pink and yellow) shows that the position of the EF hand-like motif is conserved at the interface between the SBP and the permease.
(B) Sequence and secondary structure alignment of FusB and AlgM1 colored according to their conservation using the ESPript software (Robert and Gouet, 2014).
Solution Structures of SfuA in Its Apo-Open and Holo-Closed Conformation
As SfuA failed to crystallize, we structurally characterized SfuA-apo and SfuA in complex with kestose using small-angle X-ray scattering (SAXS). As expected, the scattering curves and the pair distribution functions P(r) for SfuA in complex with kestose point to a more rigid and compact globular structure than for SfuA-apo, with a radius of gyration (Rg) of 21.8 Å and a maximal intraparticle dimension (Dmax) of 68 Å; while the apo-protein shows a bimodal curve with two distinctive shoulders in the scattering curve at q values of 0.25 and 0.35, an Rg of 22.4 Å and Dmax of 72 Å, reflecting the larger interdependent flexibility of the two characteristic domains in the absence of bound FOS (Figures 8A and 8B). We used the extensive structural data available for the SBP family in the PDB to generate homology models of SfuA in its apo- and holo-forms using automated homology-modeling servers (Biasini et al., 2014, Kelley et al., 2015). Comparison of the SAXS experimental profiles to the theoretical curves generated with the homology models showed good agreement, with χ2free values of 1.26 and 1.21 for the apo- and holo-forms, respectively (Figures 8E and 8H). Furthermore, the ab initio SAXS envelopes generated by the experimental curves fit well with the homology models used in our analyses (Figures 8C–8I). The SfuA homology model fits to the cluster B class of SBPs and presents a substrate-binding cavity with dimensions of approximately 8.9 × 4.0 Å and a volume of ∼391 Å3, which is in line with the other SBPs in this subclass (cavity volumes ranging from 115 to 590 Å3). The SfuA cavity is significantly smaller when compared with that of FusA, which extends in length by 21.5 Å and has a radius varying between 2.4 and 6.5 Å (volume ∼2,218 Å3). The significantly reduced size of the SfuA-binding cavity aligns with the higher binding affinities of short-chain FOSs and the inability of pneumococcal strains encoding the SfuABC transporter to grow on inulin as a sole carbon source (Figures 8J and 8K).
Figure 8.

SfuA-apo and SfuA:kestose Complex in Solution Structures
(A) SAXS profiles for SfuA-apo (dark green circles) and SfuA:kestose complex (purple circles) overlaid with fits derived from the indirect Fourier transform of the shown data points (light green and pink lines, respectively).
(B) P(r) plots calculated with scatter and normalized at peak height exhibit a bimodal pattern for SfuA-apo with a radius of gyration (Rg) of 22.4 Å and a maximal intraparticle dimension (Dmax) of 72 Å; the SfuA:kestose complex structure shows a lower Rg and Dmax (21.8 and 68 Å, respectively).
(C, D, F, G, and I) Ab initio models calculated with DAMMIF (Petoukhov et al., 2012) of SfuA-apo (light green envelope) and SfA:kestose complex (pale pink envelope) superposed with SfuA homology models with (pink ribbon) and without ligand (green ribbon) in two orientations.
(E and H) SAXS profiles for SfuA-apo (light green) and SfuA:kestose complex (pale pink) overlaid with the calculated profiles (FOXS server; Schneidman-Duhovny et al., 2010) from the structures of SfuA-apo and SfuA-holo models (red lines).
(J and K) Ribbon diagram of the structure of SfuA:kestose complex (pink) and FusA:nystose complex (orange), highlighting the different substrate-binding cavity volumes (gray mesh) obtained with POCASA (Zhang et al., 2011).
Discussion
The structures of the SBPs FusA and SfuA of the FOS ABC transporters FusABC and SfuABC from S. pneumoniae shows the substrate-binding cleft of FusA to be at least three times greater in volume than SfuA and provides the structural basis for the observation that only S. pneumoniae strains encoding FusABC are able to utilize inulin as a carbon source. High-resolution structural and mutational analysis of the FusA SBP complexed to a range of FOSs revealed that three amino acid residues (Glu167, Trp314, and Arg419) are essential for FOS binding and pneumococcal growth on nystose (Figures 5A and 5B). The structures of FusA complexed to fructofuranosyl-nystose, the longest FOS studied here, identified another four amino acid residues that either directly interact with bound FOS or formed part of the inner wall of the binding cavity. Mutation of each of these amino acid residues to alanine (His177, Asn318, Trp384, and Glu423) reduced the affinity of FusA to FOS and substantially affected pneumococcal growth of nystose (Figures 5A and 5B). FusABC had previously been proposed to transport sucrose (Bidossi et al., 2012), but here we show conclusively that both FusA and SfuA have no affinity for sucrose which agrees with earlier evidence of involvement in FOS transport (Linke et al., 2013). Analysis of the structures of the FusA:FOS complexes point to Trp314 and Arg419 endowing strict specificity for a fructose ring in the second position at this FOS subsite binding. FusA is unable to accommodate the glucose ring of sucrose at this subsite due to the stereochemical restraints of the binding site that prevent favorable stacking and hydrogen-bonding interactions with Trp314 and Arg419, respectively. A similar level of fine-tuning of substrate specificity has been observed for the SBP of the GlnPQ transporter, where several residues in the ligand cavity contribute the binding interaction and where substrate affinity is determined by a subset of these interacting residues (Fulyani et al., 2013).
Comparison of the FusA structure with other SBPs identified unique structural features that could not be accommodated into previously defined classes, which had successfully assigned all but three known SBPs structures to six clusters (Berntsson et al., 2010). Interestingly FusA aligned with two of the previously unclassified SBPs namely, AlgQ1 and AlgQ2 (Figure 3). The distinguishing features of this cohort of SBPs are that they are significantly larger in size compared with most SBPs with molecular weights of around 70 kDa. Additional structural elements also extend the ligand-binding cavity enabling large substrates to be bound. In particular, the C-terminal region of these SBPs expands to four α helices and the core of the first domain is formed by two coplanar α sheets instead of by only one. This is in contrast to the OppA subfamily, which also are at the larger end of the molecular weight spectrum (cluster C) where the extra structural components that contribute most to the expansion of the binding cavity are assembled as a single domain at the N terminus of the protein (Berntsson et al., 2009, Tame et al., 1994). As in OppA∗, which can potentially bind peptides up to 35 residues, FusA is able to bind a long-chain oligosaccharide, such as inulin, composed of up to 40 fructose moieties, as a consequence of its extended cavity. FusA has seven 310-helices, but the number and the position among the other proteins of this cluster are variable (2, 3, and 11 for AlgQ2, AlgQ1, and Blon_2351, respectively), which does not make them a distinctive feature of the subgroup. All these proteins contain an EF hand-like metal-binding site, which is located between helix α8 and the β6 strand (Figures 3A–3C). This EF hand-like motif is positioned at the interface of the SBP and the permease as revealed by the structure of the alginate ABC transporter from Sphingomonas sp. A1 (Maruyama et al., 2015). These structural features and the overall structural similarities allow the extension of SBP classification to a new class (class G) of SBPs represented by AlgQ1, AlgQ2, FusA, and the uncharacterized protein Blon_2351. Our data have shown that integrity of the EF hand-like motif is essential for substrate translocation. Analysis of the cluster G SBPs shows that in the presence of calcium the EF hand-like domain directs the conserved residue 7 of the motif toward the permease interface, enabling formation of a salt bridge in the complex, which we propose has a structural and sensing role for substrate translocation (Figures 3A, 7A, and 7B). A similar metal-binding site in this location can also be detected in other SBPs not belonging to this class, such as the galactose-binding protein (PDB: 1GCG), the chitin-binding protein (PDB: 4GFR and 4GF8), and the oligopeptide-binding protein (PDB: 4PFY) (Flocco and Mowbray, 1994, Ghimire-Rijal et al., 2014). In the case of the galactose-binding protein, a structural as well as signaling role was hypothesized previously for the calcium-binding site, based on roles defined for calcium EF hand motifs in other systems (Vyas et al., 1987). However, at that time, no experimental data to assign a role in signaling were available. Further evidence for the importance of interactions at this localized site in domain II at the SBP:permease interface has been demonstrated by targeting the same surface of the SBP MntC with an antibody fragment which blocks the interaction with the permease component and prevents substrate import (Ahuja et al., 2015). In conclusion, we propose that the EF hand-like calcium-binding site is essential for substrate translocation and also aids in defining a new class of SBP.
Experimental Procedures
Cloning and Protein Production
The gene fragment encoding the FusA and SfuA SBPs of Streptococcus pneumoniae TIGR4 (locus tag SP_1796) and Streptococcus pneumoniae D39 (locus tag SPD_1585) without their signal peptides (FusA47−538 and SfuA31−436, respectively) were cloned into pOPINF and pOPINE-3C-eGFP vectors (OPPF-UK) (Berrow et al., 2007). The fusion proteins were expressed in BL21 Rosetta E. coli cells by autoinduction using Overnight Express Medium (Millipore) supplemented with 1% glycerol. Cells were lysed in 0.1 M HEPES (pH 7.5), 0.5 M NaCl, 0.02 M imidazole, and 10% glycerol supplemented with EDTA-free protease inhibitors (Roche) and the supernatant clarified by centrifugation (1 hr, 100,000 × g). Proteins were purified by Ni-NTA affinity chromatography followed by size-exclusion chromatography using a Superdex 200 column equilibrated in 0.01 M HEPES (pH 7.5), 0.5 M NaCl, 2.5% glycerol, and 0.25 mM Tris(2-carboxyethy1)phosphine. All the point mutation variants (Asp223Ala-Glu224Ala, Glu167Ala, His177Ala, Trp314Ala, Asn318Ala, Trp384Ala, Arg419Ala, and Glu423Ala) that were generated by PCR using QuikChange II Site-Directed Mutagenesis Kit (Agilent Technologies) were expressed and purified as for the wild-type protein.
Crystallization and Structure Determination
Crystals of apo FusA47−538 were obtained by vapor diffusion from a 1:2 volume ratio of protein at 26 mg/mL to the reservoir (15% poly-ethylene glycol 6000 [PEG 6000], 0.1 M Tris-HCl [pH 8.0], and 0.01 M zinc chloride) at 4°C. Needle-shaped crystals of FusA47−538 in complex with kestose, nystose, and fructofuranosyl-nystose were obtained by co-crystallization of the protein at 24 mg/mL in the presence of each FOS at 10 mM concentration, respectively, using a reservoir solution containing 20% PEG 6000, 0.1 M sodium acetate (pH 5.0), and 0.02 M calcium chloride. Plate-shaped crystals of FusA47−538-eGFP were obtained by sitting-drop vapor diffusion at 4°C mixing the protein at 10 mg/mL with a reservoir solution containing 18% PEG 6000, 0.1 M Tris-HCl (pH 8.3), and 0.01 M zinc chloride. X-Ray data were collected at Diamond Light Source from crystals which were cryoprotected (reservoir buffer supplemented with 25% glycerol) and flash-cooled in liquid nitrogen. All data were processed with xia2 (Winter et al., 2013). A summary of the data collection and processing statistics is given in Table 1. Initial phases were obtained by molecular replacement using the structure of EGFP (PDB: 4N3D) as a search model in Phaser (McCoy et al., 2007) using the GFP-tagged FusA diffraction data. Automatic model building with Buccaneer (Cowtan, 2006) produced a series of partial models of FusA47−538, which were successfully used as search templates for molecular replacement against the 1.9 Å resolution dataset of apo FusA47−538, using Phaser and Shelx (Sheldrick, 2010). Model building and refinement was completed through iterative cycles of automatic and manual model building in Phenix (Adams et al., 2010) and Coot (Emsley et al., 2010), respectively (Table 1). The final model of apo FusA47−538 is a dimer with a well-defined interpretable electron density for the complete polypeptide chain in addition to the GP sequence at the N terminus. The FusA47−538-FOS complex structures were solved by molecular replacement using the two domains of FusA47−538 separately (residues 47-143 and 189-339). Model building and refinement was carried out as described above (Table 1). All FusA47−538-FOS complex structures crystallized with four subunits of FusA in the asymmetric unit. The structures were visualized by PyMOL (www.schrodinger.com/pymol) and Chimera (Pettersen et al., 2004).
Small-Angle-X-Ray Scattering
Size-exclusion chromatography-coupled SAXS data were collected on B21 at Diamond Light Source. SfuA protein samples (100 μL, 10 mg/mL) were loaded on a KW404 Shodex column equilibrated with 20 mM Tris-HCl (pH 8) and 200 mM NaCl buffer, and the elution peak was analyzed in an in-vacuum quartz capillary, 1.6 mm diameter. SAXS images were collected with 1 s exposures on a PILATUS 2 M detector (DECTRIS); for the SfuA FOS complex the protein sample was mixed with 5 mM kestose. Images were corrected for variations in beam current, normalized for exposure time, and processed into 1D scattering curves using in-house software (GDA). Buffer subtractions and all other subsequent analysis were performed with the program SCATTER (http://www.bioisis.net/scatter).
Twenty ab initio models were generated by DAMMIF/DAMMIN and averaged by DAMAVER in the ATSAS program package (Petoukhov et al., 2012). The final model for SfuA-apo and SfuA-kestose were superposed to SfuA-apo and SfuA-holo homology models using SUPCOMB, and the theoretical scattering profile of the models in reciprocal space was calculated by FOXS (Schneidman-Duhovny et al., 2013). The agreement between the experimental SfuA and theoretical SfuA curves was estimated using FOXS and SCATTER (χ2free) (Rambo and Tainer, 2013, Schneidman-Duhovny et al., 2010).
Pull-Down Assays
FOS-conjugated beads were prepared using epoxy-activated Sepharose 6B medium (GE Healthcare) as described by the provider. The FOS-conjugated beads were incubated for 1 hr at 4°C with 1–2 μM purified SfuA, FusA47−538 (wild-type and point mutants), and SP1690 (as a negative control) in a buffer containing 0.01 M HEPES (pH 7.5), 0.25 M NaCl, 5% glycerol, and 0.1% Tween. After extensive washes, the bounded fraction was analyzed by SDS-PAGE.
Thermal Shift Assay
The experiments were performed on a Bio-Rad Opticon2 RT-PCR machine monitoring excitation/emission fluorescence at 500/590 nm. The proteins were diluted to 50 μM in 0.01 M HEPES (pH 7.5), 0.25 M NaCl, and 5% glycerol buffer supplemented with 50 μM of ligand and 2× SYPRO orange (Invitrogen).
Isothermal Titration Calorimetry
Binding of FOSs to FusA47−538 was measured at 25°C using a MicroCal iTC200 microcalorimeter (Malvern Instruments). Buffer conditions were 0.01 M HEPES (pH 7.4) and 0.15 M NaCl (ITC buffer). Protein and the respective FOS were dispatched into the cell and syringe, respectively. Specific heat as a result of dilution of FOS in the protein buffer was taken into account. Data were analyzed using MicroCal Origin software (version 7) fitting to a single-site binding model. Detailed methods are given in Supplemental Information.
Pneumococcal Strains, Culture Media, and Chemicals
Growth studies were conducted using S. pneumoniae strain TIGR4 and mutants generated in that background (Tettelin et al., 2001). Broth cultures were routinely grown at 37°C in Todd-Hewitt Broth (Becton Dickinson) supplemented with 0.2% (w/v) yeast extract (Becton Dickinson) (THY). C medium with 5% yeast extract (pH 8; C + Y) was used for transformations (Lacks and Hotchkiss, 1960). S. pneumoniae were also grown at 37°C and 5% CO2 on tryptic soy agar (TS; Becton Dickinson) spread with 5,000 U of catalase (Worthington Biochemical) and TS plates supplemented with 5% sheep blood (Becton Dickinson). Transformants were selected on TS plates containing streptomycin (200 μg/mL) or kanamycin (500 μg/mL).
Growth Assays
Growth assays were conducted in chemically defined media essentially as described previously, except that the concentrations of glucose (6 mM) and nystose (1.5 mM) were modified and the assays were conducted for 96 hr (Linke et al., 2013). All strains were tested for growth in triplicate independently. The data presented are the mean data points from one representative growth experiment. While the data from individual experiments could not be compiled due to day-to-day differences in growth, the reduced or lack of growth on nystose for individual mutants was reproducible in all experiments. No difference in the ability of any strain to grow on glucose was observed. Generation of mutants are described in Supplemental Information.
Author Contributions
Conceptualization, S.C., S.J.K., and M.A.W.; Methodology, S.C., G.H., and A.K.S.; Structural Biology Investigation, S.C.; Microbiology Investigation, A.K.S; Writing – Original Draft, S.C. and M.A.W.; Writing – Review & Editing, S.C., M.A.W., G.H., A.K.S., and S.J.K.; Visualization, S.C.; Resources, G.H. and A.K.S.; Funding Acquisition, M.A.W. and S.J.K.; Supervision, M.A.W. and S.J.K.
Acknowledgments
We are grateful to OPPF-UK for technical support in cloning and protein production. We thank the scientists of the I03, I24, and B21 beamlines at Diamond Light Source for their support and provision of excellent facilities.
Published: December 8, 2016
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, two figures, three tables, and one movie and can be found with this article online at http://dx.doi.org/10.1016/j.str.2016.11.008.
Supplemental Information
{kind=link}
References
- Adams P.D., Afonine P.V., Bunkoczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahuja S., Rouge L., Swem D.L., Sudhamsu J., Wu P., Russell S.J., Alexander M.K., Tam C., Nishiyama M., Starovasnik M.A. Structural analysis of bacterial ABC transporter inhibition by an antibody fragment. Structure. 2015;23:713–723. doi: 10.1016/j.str.2015.01.020. [DOI] [PubMed] [Google Scholar]
- Berntsson R.P., Doeven M.K., Fusetti F., Duurkens R.H., Sengupta D., Marrink S.J., Thunnissen A.M., Poolman B., Slotboom D.J. The structural basis for peptide selection by the transport receptor OppA. EMBO J. 2009;28:1332–1340. doi: 10.1038/emboj.2009.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berntsson R.P., Smits S.H., Schmitt L., Slotboom D.J., Poolman B. A structural classification of substrate-binding proteins. FEBS Lett. 2010;584:2606–2617. doi: 10.1016/j.febslet.2010.04.043. [DOI] [PubMed] [Google Scholar]
- Berrow N.S., Alderton D., Sainsbury S., Nettleship J., Assenberg R., Rahman N., Stuart D.I., Owens R.J. A versatile ligation-independent cloning method suitable for high-throughput expression screening applications. Nucleic Acids Res. 2007;35:e45. doi: 10.1093/nar/gkm047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biasini M., Bienert S., Waterhouse A., Arnold K., Studer G., Schmidt T., Kiefer F., Gallo Cassarino T., Bertoni M., Bordoli L. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014;42:W252–W258. doi: 10.1093/nar/gku340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bidossi A., Mulas L., Decorosi F., Colomba L., Ricci S., Pozzi G., Deutscher J., Viti C., Oggioni M.R. A functional genomics approach to establish the complement of carbohydrate transporters in Streptococcus pneumoniae. PLoS One. 2012;7:e33320. doi: 10.1371/journal.pone.0033320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckwalter C.M., King S.J. Pneumococcal carbohydrate transport: food for thought. Trends Microbiol. 2012;20:517–522. doi: 10.1016/j.tim.2012.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowtan K. The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr. D Biol. Crystallogr. 2006;62:1002–1011. doi: 10.1107/S0907444906022116. [DOI] [PubMed] [Google Scholar]
- Emsley P., Lohkamp B., Scott W.G., Cowtan K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng H., Wang X., Sun Y., Chen X., Guo J., Duan Y., Huang L., Kang Z. Cloning and characterization of a calcium binding EF-hand protein gene TaCab1 from wheat and its expression in response to Puccinia striiformis f. sp. tritici and abiotic stresses. Mol. Biol. Rep. 2011;38:3857–3866. doi: 10.1007/s11033-010-0501-8. [DOI] [PubMed] [Google Scholar]
- Flocco M.M., Mowbray S.L. The 1.9 A x-ray structure of a closed unliganded form of the periplasmic glucose/galactose receptor from Salmonella typhimurium. J. Biol. Chem. 1994;269:8931–8936. [PubMed] [Google Scholar]
- Fulyani F., Schuurman-Wolters G.K., Zagar A.V., Guskov A., Slotboom D.J., Poolman B. Functional diversity of tandem substrate-binding domains in ABC transporters from pathogenic bacteria. Structure. 2013;21:1879–1888. doi: 10.1016/j.str.2013.07.020. [DOI] [PubMed] [Google Scholar]
- Gerber S., Comellas-Bigler M., Goetz B.A., Locher K.P. Structural basis of trans-inhibition in a molybdate/tungstate ABC transporter. Science. 2008;321:246–250. doi: 10.1126/science.1156213. [DOI] [PubMed] [Google Scholar]
- Ghimire-Rijal S., Lu X., Myles D.A., Cuneo M.J. Duplication of genes in an ATP-binding cassette transport system increases dynamic range while maintaining ligand specificity. J. Biol. Chem. 2014;289:30090–30100. doi: 10.1074/jbc.M114.590992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gifford J.L., Walsh M.P., Vogel H.J. Structures and metal-ion-binding properties of the Ca2+-binding helix-loop-helix EF-hand motifs. Biochem. J. 2007;405:199–221. doi: 10.1042/BJ20070255. [DOI] [PubMed] [Google Scholar]
- Higgins C.F. ABC transporters: from microorganisms to man. Annu. Rev. Cell Biol. 1992;8:67–113. doi: 10.1146/annurev.cb.08.110192.000435. [DOI] [PubMed] [Google Scholar]
- Hollenstein K., Dawson R.J., Locher K.P. Structure and mechanism of ABC transporter proteins. Curr. Opin. Struct. Biol. 2007;17:412–418. doi: 10.1016/j.sbi.2007.07.003. [DOI] [PubMed] [Google Scholar]
- Hopfner K.P. Invited review: architectures and mechanisms of ATP binding cassette proteins. Biopolymers. 2016;105:492–504. doi: 10.1002/bip.22843. [DOI] [PubMed] [Google Scholar]
- Ispahani P., Slack R.C., Donald F.E., Weston V.C., Rutter N. Twenty year surveillance of invasive pneumococcal disease in Nottingham: serogroups responsible and implications for immunisation. Arch. Dis. Child. 2004;89:757–762. doi: 10.1136/adc.2003.036921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer R., Camilli A. Sucrose metabolism contributes to in vivo fitness of Streptococcus pneumoniae. Mol. Microbiol. 2007;66:1–13. doi: 10.1111/j.1365-2958.2007.05878.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadaba N.S., Kaiser J.T., Johnson E., Lee A., Rees D.C. The high-affinity E. coli methionine ABC transporter: structure and allosteric regulation. Science. 2008;321:250–253. doi: 10.1126/science.1157987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley L.A., Mezulis S., Yates C.M., Wass M.N., Sternberg M.J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015;10:845–858. doi: 10.1038/nprot.2015.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacks S., Hotchkiss R.D. A study of the genetic material determining an enzyme in Pneumococcus. Biochim. Biophys. Acta. 1960;39:508–518. doi: 10.1016/0006-3002(60)90205-5. [DOI] [PubMed] [Google Scholar]
- Langvad-Nielsen A. Fermentation power of pneumococci. Acta Pathol. Microbiol. Scand. 1944;21:370–373. [Google Scholar]
- Linke C.M., Woodiga S.A., Meyers D.J., Buckwalter C.M., Salhi H.E., King S.J. The ABC transporter encoded at the pneumococcal fructooligosaccharide utilization locus determines the ability to utilize long- and short-chain fructooligosaccharides. J. Bacteriol. 2013;195:1031–1041. doi: 10.1128/JB.01560-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao B., Pear M.R., McCammon J.A., Quiocho F.A. Hinge-bending in L-arabinose-binding protein. The “Venus's-flytrap” model. J. Biol. Chem. 1982;257:1131–1133. [PubMed] [Google Scholar]
- Maruyama Y., Itoh T., Kaneko A., Nishitani Y., Mikami B., Hashimoto W., Murata K. Structure of a bacterial ABC transporter involved in the import of an acidic polysaccharide alginate. Structure. 2015;23:1643–1654. doi: 10.1016/j.str.2015.06.021. [DOI] [PubMed] [Google Scholar]
- McCoy A.J., Grosse-Kunstleve R.W., Adams P.D., Winn M.D., Storoni L.C., Read R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morch-Lund E. The fermenting power of pneumococci. Acta Pathol. Microbiol. Scand. 1949;26:709–714. doi: 10.1111/j.1699-0463.1949.tb00772.x. [DOI] [PubMed] [Google Scholar]
- Mosca R., Schneider T.R. RAPIDO: a web server for the alignment of protein structures in the presence of conformational changes. Nucleic Acids Res. 2008;36:W42–W46. doi: 10.1093/nar/gkn197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldham M.L., Khare D., Quiocho F.A., Davidson A.L., Chen J. Crystal structure of a catalytic intermediate of the maltose transporter. Nature. 2007;450:515–521. doi: 10.1038/nature06264. [DOI] [PubMed] [Google Scholar]
- Petoukhov M.V., Franke D., Shkumatov A.V., Tria G., Kikhney A.G., Gajda M., Gorba C., Mertens H.D., Konarev P.V., Svergun D.I. New developments in the program package for small-angle scattering data analysis. J. Appl. Crystallogr. 2012;45:342–350. doi: 10.1107/S0021889812007662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF Chimera – a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Rambo R.P., Tainer J.A. Accurate assessment of mass, models and resolution by small-angle scattering. Nature. 2013;496:477–481. doi: 10.1038/nature12070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees D.C., Johnson E., Lewinson O. ABC transporters: the power to change. Nat. Rev. Mol. Cell Biol. 2009;10:218–227. doi: 10.1038/nrm2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice A.J., Park A., Pinkett H.W. Diversity in ABC transporters: type I, II and III importers. Crit. Rev. Biochem. Mol. Biol. 2014;49:426–437. doi: 10.3109/10409238.2014.953626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigden D.J., Galperin M.Y. The DxDxDG motif for calcium binding: multiple structural contexts and implications for evolution. J. Mol. Biol. 2004;343:971–984. doi: 10.1016/j.jmb.2004.08.077. [DOI] [PubMed] [Google Scholar]
- Robert X., Gouet P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014;42:W320–W324. doi: 10.1093/nar/gku316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodionov D.A., Hebbeln P., Eudes A., ter Beek J., Rodionova I.A., Erkens G.B., Slotboom D.J., Gelfand M.S., Osterman A.L., Hanson A.D. A novel class of modular transporters for vitamins in prokaryotes. J. Bacteriol. 2009;191:42–51. doi: 10.1128/JB.01208-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneidman-Duhovny D., Hammel M., Sali A. FoXS: a web server for rapid computation and fitting of SAXS profiles. Nucleic Acids Res. 2010;38:W540–W544. doi: 10.1093/nar/gkq461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneidman-Duhovny D., Hammel M., Tainer J.A., Sali A. Accurate SAXS profile computation and its assessment by contrast variation experiments. Biophys. J. 2013;105:962–974. doi: 10.1016/j.bpj.2013.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldrick G.M. Experimental phasing with SHELXC/D/E: combining chain tracing with density modification. Acta Crystallogr. D Biol. Crystallogr. 2010;66:479–485. doi: 10.1107/S0907444909038360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tame J.R., Murshudov G.N., Dodson E.J., Neil T.K., Dodson G.G., Higgins C.F., Wilkinson A.J. The structural basis of sequence-independent peptide binding by OppA protein. Science. 1994;264:1578–1581. doi: 10.1126/science.8202710. [DOI] [PubMed] [Google Scholar]
- Tang C., Schwieters C.D., Clore G.M. Open-to-closed transition in apo maltose-binding protein observed by paramagnetic NMR. Nature. 2007;449:1078–1082. doi: 10.1038/nature06232. [DOI] [PubMed] [Google Scholar]
- Tettelin H., Nelson K.E., Paulsen I.T., Eisen J.A., Read T.D., Peterson S., Heidelberg J., DeBoy R.T., Haft D.H., Dodson R.J. Complete genome sequence of a virulent isolate of Streptococcus pneumoniae. Science. 2001;293:498–506. doi: 10.1126/science.1061217. [DOI] [PubMed] [Google Scholar]
- Vyas N.K., Vyas M.N., Quiocho F.A. A novel calcium binding site in the galactose-binding protein of bacterial transport and chemotaxis. Nature. 1987;327:635–638. doi: 10.1038/327635a0. [DOI] [PubMed] [Google Scholar]
- Winter G., Lobley C.M., Prince S.M. Decision making in xia2. Acta Crystallogr. D Biol. Crystallogr. 2013;69:1260–1273. doi: 10.1107/S0907444913015308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J., Ge J., Heuveling J., Schneider E., Yang M. Structural basis for substrate specificity of an amino acid ABC transporter. Proc. Natl. Acad. Sci. USA. 2015;112:5243–5248. doi: 10.1073/pnas.1415037112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z., Li Y., Lin B., Schroeder M., Huang B. Identification of cavities on protein surface using multiple computational approaches for drug binding site prediction. Bioinformatics. 2011;27:2083–2088. doi: 10.1093/bioinformatics/btr331. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.