

Abstract
The N-terminal domain (NTD) of NIP1/eIF3c interacts directly with eIF1 and eIF5 and indirectly through eIF5 with the eIF2-GTP-Met-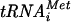 ternary complex (TC) to form the multifactor complex (MFC). We investigated the physiological importance of these interactions by mutating 16 segments spanning the NIP1-NTD. Mutations in multiple segments reduced the binding of eIF1 or eIF5 to the NIP1-NTD. Mutating a C-terminal segment of the NIP1-NTD increased utilization of UUG start codons (Sui− phenotype) and was lethal in cells expressing eIF5-G31R that is hyperactive in stimulating GTP hydrolysis by the TC at AUG codons. Both effects of this NIP1 mutation were suppressed by eIF1 overexpression, as was the Sui− phenotype conferred by eIF5-G31R. Mutations in two N-terminal segments of the NIP1-NTD suppressed the Sui− phenotypes produced by the eIF1-D83G and eIF5-G31R mutations. From these and other findings, we propose that the NIP1-NTD coordinates an interaction between eIF1 and eIF5 that inhibits GTP hydrolysis at non-AUG codons. Two NIP1-NTD mutations were found to derepress GCN4 translation in a manner suppressed by overexpressing the TC, indicating that MFC formation stimulates TC recruitment to 40S ribosomes. Thus, the NIP1-NTD is required for efficient assembly of preinitiation complexes and also regulates the selection of AUG start codons in vivo.
ternary complex (TC) to form the multifactor complex (MFC). We investigated the physiological importance of these interactions by mutating 16 segments spanning the NIP1-NTD. Mutations in multiple segments reduced the binding of eIF1 or eIF5 to the NIP1-NTD. Mutating a C-terminal segment of the NIP1-NTD increased utilization of UUG start codons (Sui− phenotype) and was lethal in cells expressing eIF5-G31R that is hyperactive in stimulating GTP hydrolysis by the TC at AUG codons. Both effects of this NIP1 mutation were suppressed by eIF1 overexpression, as was the Sui− phenotype conferred by eIF5-G31R. Mutations in two N-terminal segments of the NIP1-NTD suppressed the Sui− phenotypes produced by the eIF1-D83G and eIF5-G31R mutations. From these and other findings, we propose that the NIP1-NTD coordinates an interaction between eIF1 and eIF5 that inhibits GTP hydrolysis at non-AUG codons. Two NIP1-NTD mutations were found to derepress GCN4 translation in a manner suppressed by overexpressing the TC, indicating that MFC formation stimulates TC recruitment to 40S ribosomes. Thus, the NIP1-NTD is required for efficient assembly of preinitiation complexes and also regulates the selection of AUG start codons in vivo.
Translation initiation is a multistep process culminating in formation of the 80S initiation complex containing methionyl initiator tRNA (Met-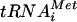 ) base paired with the AUG start codon in the P site of the ribosome. A large number of soluble eukaryotic translation initiation factors (eIFs) have been identified that stimulate the partial reactions of this process (reviewed in reference 12 and 13). A critical step early in the pathway is the binding of Met-
) base paired with the AUG start codon in the P site of the ribosome. A large number of soluble eukaryotic translation initiation factors (eIFs) have been identified that stimulate the partial reactions of this process (reviewed in reference 12 and 13). A critical step early in the pathway is the binding of Met-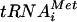 to the 40S ribosomal subunit in a ternary complex (TC) comprised of Met-
to the 40S ribosomal subunit in a ternary complex (TC) comprised of Met-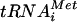 , GTP, and eIF2. The recruitment of TC to 40S subunits is promoted in vitro by eIF1, eIF1A, and the eIF3 complex. The 43S preinitiation complex thus formed interacts with mRNA in a manner stimulated by eIF4F (eIF4A-eIF4E-eIF4G), poly(A)-binding protein, and eIF3, and the 43S complex scans the mRNA until the Met-
, GTP, and eIF2. The recruitment of TC to 40S subunits is promoted in vitro by eIF1, eIF1A, and the eIF3 complex. The 43S preinitiation complex thus formed interacts with mRNA in a manner stimulated by eIF4F (eIF4A-eIF4E-eIF4G), poly(A)-binding protein, and eIF3, and the 43S complex scans the mRNA until the Met-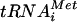 base pairs with an AUG triplet. AUG recognition triggers GTP hydrolysis by eIF2 in a reaction stimulated by eIF5, and the eIF2-GDP and other eIFs are ejected from the ribosome. The eIF1, eIF1A, and eIF4G have been implicated in the scanning process in vitro (23, 24). In the final reaction, eIF5B bound to GTP promotes joining of the 60S subunit with the 40S-Met-
base pairs with an AUG triplet. AUG recognition triggers GTP hydrolysis by eIF2 in a reaction stimulated by eIF5, and the eIF2-GDP and other eIFs are ejected from the ribosome. The eIF1, eIF1A, and eIF4G have been implicated in the scanning process in vitro (23, 24). In the final reaction, eIF5B bound to GTP promotes joining of the 60S subunit with the 40S-Met-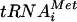 -mRNA complex to produce the 80S initiation complex (15, 25). To begin a new round of initiation, the ejected eIF2-GDP complex must be recycled to eIF2-GTP by the guanine nucleotide exchange factor eIF2B (13).
-mRNA complex to produce the 80S initiation complex (15, 25). To begin a new round of initiation, the ejected eIF2-GDP complex must be recycled to eIF2-GTP by the guanine nucleotide exchange factor eIF2B (13).
From extensive biochemical analysis of the mammalian initiation factors, it was proposed that eIF3 binds to the 40S ribosome independently of other factors and promotes the recruitment of TC and mRNA in a manner stimulated by eIF1 and eIF1A (reviewed in references 12 and 13). There is also evidence, however, that eIF2 stimulates 40S-binding by eIF3 and that eIF3 enhances 40S binding of eIF1 and eIF1A (18). Furthermore, eIF1 and eIF1A cooperate with one another in binding to the 40S ribosome (17, 18) and in promoting TC recruitment (1, 18). In addition to these functional interactions, work in yeast has shown that eIF3, eIF5, and eIF1 are physically associated with one another and with the TC in a multifactor complex (MFC) that can exist free of ribosomes (2, 3, 26, 28, 29) (Fig. 1A). We have proposed that the physical contacts among the factors in the MFC, coupled with their intrinsic ribosome-binding activities, could underlie cooperative binding of the MFC components to the 40S subunit, enhancing assembly of the 43S complex. Formation of the MFC might also coordinate the functions of eIF1, eIF5, and TC in AUG recognition during scanning (7).
FIG. 1.

Phenotypic analysis of the NIP1-NTD mutants. (A) Three-dimensional model of the MFC based on a comprehensive analysis of subunit interactions (29). The labeled protein subunits are shown roughly in proportion to their molecular weights. The degree of overlap between two different subunits depicts the extent of their interacting surfaces. The boundaries of the N terminus of NIP1 (NIP1-NTD) subjected to mutagenesis are indicated by dotted white lines. ntd, N-terminal domain; ctd, C-terminal domain; hld, HCR1-like domain; rrm, RNA recognition motif; TC, ternary complex. (B) The sequence of the first 160 amino acid residues of NIP1 is shown as numbered circles (boxes 1 to 16), each of them composed of 10 residues that were substituted by a stretch of 10 alanines. Different shades of gray indicate the degree of identities between the NIP1-NTD and the N termini of its Caenorhabditis elegans, Human sapiens, Arabidopsis thaliana, and Schizosaccharomyces pombe homologues that were aligned by using the GCG Sequence Analysis Program (version 8; Genetics Computer Group, Inc., Madison, Wis.) (white, <20%; light gray, 20 to 40%; medium gray, 40 to 60%; dark gray, 60 to 80%). Color-coded bars above the circles indicate the phenotypes associated with amino acid substitutions in the corresponding boxes: Ssu− (suppressor of Sui−), Gcd− (general control derepressed), and Sui− (suppressor of initiation codon). Blown-up segments in blue, green, and yellow indicate the amino acid sequences, a consensus sequence derived from sequence alignments, and the substitutions made in the corresponding boxes of the NIP1-NTD. (C) Summary of the growth phenotypes of the NIP1 mutants in a SUI1 strain (second row) and their genetic interactions with sui1-1 (third row). The indicated plasmid-borne hc NIP1-Box alleles (row 1) were introduced into strains HLV04 (nip1Δ SUI1) and HLV05 (nip1Δ sui1-1), both carrying WT NIP1 on a single-copy (sc) URA3 plasmid that was subsequently evicted by growth on 5-FOA medium. Synthetic lethality with sui1-1 was identified by the failure to grow on 5-FOA plates. Growth of the viable strains was analyzed by determining the sizes of colonies formed from single cells streaked on yeast extract-peptone-dextrose (YPD) plates. In the second row, “+” and Slg− designate WT and slow-growth phenotypes, respectively, in the SUI1 strain. In the third row, “+”, “exa”, and “−” indicate no effect, exacerbation of the Slg- phenotype, and synthetic lethality in the sui1-1 strain, respectively. The asterisks in row 1 designate mutants that are lethal when expressed from a single-copy plasmid. (D) Growth phenotypes of selected NIP1 mutants. The strains derived from HLV04 (nip1Δ SUI1 his4-303) containing the indicated NIP1 alleles on high-copy-number (hc) plasmids were streaked for single colonies on yeast extract-peptone-dextrose medium at 30°C (left-hand sectors) and 37°C (right-hand sectors).
To test these hypotheses, we set out to isolate mutations that disrupt connections between eIF3 subunit c/NIP1 and other initiation factors in the MFC and determine whether these lesions impair preinitiation complex assembly or the stringency of AUG selection. Such defects can be recognized in vivo by using two well-established genetic assays. Mutations that reduce the rate of TC binding to 40S ribosomes derepress translation of GCN4 mRNA, encoding a transcriptional activator of amino acid biosynthetic enzymes. GCN4 translation is regulated by the four upstream open reading frames (uORFs) 1 to 4 in its mRNA leader. After uORF1 is translated, many 40S subunits remain attached to the mRNA, resume scanning, and reinitiate translation downstream. When TC recruitment is efficient, all of the rescanning 40S subunits rebind the TC before reaching uORF4, translate this sequence, and dissociate from the mRNA, leaving GCN4 repressed. In cells starved for amino acids, TC levels are reduced by phosphorylation of eIF2 by protein kinase GCN2, and this enables a fraction of rescanning 40S ribosomes to rebind TC only after bypassing uORF4 and reinitiate at GCN4 instead. Mutations in eIF2B that reduce formation of the TC lead to constitutive derepression of GCN4 translation in gcn2Δ cells. This Gcd− (for general control derepressed) phenotype was also identified for a mutation in eIF1A that appears to reduce 40S-binding by the TC (22).
Mutations that reduce the stringency of AUG selection in yeast have been isolated on the basis of restoring translation of a his4 allele lacking an AUG start codon by increasing initiation at an in-frame UUG triplet at the 5′ end of the gene. Mutations with this Sui− (for suppressor of initiation codon) phenotype were isolated in initiator tRNA, eIF5, eIF1, and all three subunits of eIF2 (reviewed in reference 7). Biochemical analysis of dominant Sui− mutants in eIF2 subunits and eIF5 suggests that selection of UUG start codons can be enhanced by increasing the GTPase activity of eIF2, either directly by alterations in eIF2 or by stimulating the GTPase activating (GAP) function of eIF5. It was proposed that this biochemical defect increases the probability of inappropriate GTP hydrolysis and release of eIF2-GDP from Met-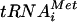 base paired with a UUG triplet (14). The biochemical basis for the Sui- phenotypes of mutations in eIF1 is unknown. However, it was shown recently that mammalian eIF1 enables 48S complexes to reject mismatches between near-cognate start codons and Met-
base paired with a UUG triplet (14). The biochemical basis for the Sui- phenotypes of mutations in eIF1 is unknown. However, it was shown recently that mammalian eIF1 enables 48S complexes to reject mismatches between near-cognate start codons and Met-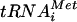 independently of eIF5 (24). Interestingly, physical interaction between eIF1 and eIF4G, a subunit of the eIF4F complex, seems to enhance the accuracy of AUG selection during scanning (11). Thus far, no mutations in eIF3 have been described with a Sui− phenotype, but we found recently that the prt1-1 mutation in eIF3b decreases selection of UUG as a start codon at HIS4, rendering initiation hyperaccurate (21).
independently of eIF5 (24). Interestingly, physical interaction between eIF1 and eIF4G, a subunit of the eIF4F complex, seems to enhance the accuracy of AUG selection during scanning (11). Thus far, no mutations in eIF3 have been described with a Sui− phenotype, but we found recently that the prt1-1 mutation in eIF3b decreases selection of UUG as a start codon at HIS4, rendering initiation hyperaccurate (21).
The eIF5 C-terminal domain (eIF5-CTD) mediates many of the known interactions that stabilize the MFC, since it is capable of interacting simultaneously with eIF1, eIF2β/SUI3, and the N-terminal domain of NIP1 (NIP1-NTD) (2-4). Thus, eIF5-CTD mediates an indirect contact between eIF2 and eIF3 in the MFC (Fig. 1A). The CTD of eIF3a/TIF32 mediates a second, direct contact with eIF2β, and it also interacts with eIF1 (29). A multiple-alanine substitution in conserved residues of the eIF5-CTD, tif5-7A, leads to temperature-sensitive (Ts-) cell growth that is partially suppressed by overexpressing all three subunits of eIF2 and 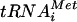 , i.e., the macromolecules comprising the TC (3). The overproduction of TC (hc TC) also partially suppressed the slow-growth phenotype (Slg−) conferred by overexpressing a dominant-negative TIF32 allele lacking the C-terminal binding domain for eIF2β (hc TIF32-Δ6-His). Combining hc TIF32-Δ6-His with tif5-7A produces a synthetic growth defect and impairment of translation initiation (29) and leads to reduced binding of eIF2 to 40S subunits in vivo (21). These findings, together with the fact that tif5-7A impairs Met-
, i.e., the macromolecules comprising the TC (3). The overproduction of TC (hc TC) also partially suppressed the slow-growth phenotype (Slg−) conferred by overexpressing a dominant-negative TIF32 allele lacking the C-terminal binding domain for eIF2β (hc TIF32-Δ6-His). Combining hc TIF32-Δ6-His with tif5-7A produces a synthetic growth defect and impairment of translation initiation (29) and leads to reduced binding of eIF2 to 40S subunits in vivo (21). These findings, together with the fact that tif5-7A impairs Met-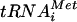 binding to 40S subunits in yeast extracts (4), support the notion that MFC formation enhances recruitment of TC to 40S subunits.
binding to 40S subunits in yeast extracts (4), support the notion that MFC formation enhances recruitment of TC to 40S subunits.
Surprisingly, the tif5-7A hc TIF32-Δ6-His double mutant does not exhibit the Gcd− phenotype that would be expected for a defect in TC recruitment (29). It appears that tif5-7A additionally impairs postassembly functions of the MFC (4), most likely the rate of scanning between uORFs 1 and 4, and thus compensates for the delay in TC recruitment that results from destabilizing the MFC. The same mechanism probably explains the failure to induce GCN4 translation (Gcn− phenotype) we observed recently in prt1-1 cells (21), despite the deleterious effect of this eIF3b mutation on 43S assembly in cell extracts (6, 26).
Considering that the NIP1-NTD interacts with eIF5-CTD, which in turn binds to eIF2β, we set out to isolate substitution mutations in this segment of NIP1 that would disrupt the physical connection between eIF3 and the eIF5-CTD/eIF2 module of the MFC without impairing the postassembly functions of eIF5. Such mutations should decrease TC recruitment to 40S subunits and produce a Gcd− phenotype that can be suppressed by hc TC. Because NIP1-NTD also interacts with eIF1, it seemed possible that other mutations in this segment would alter the stringency of AUG selection and produce a Sui− phenotype or suppress known Sui− mutations in eIF1 or eIF5. In this report we describe clustered alanine substitutions in NIP1-NTD that fulfill both of these predictions and provide in vivo evidence that the connections between eIF3 and other MFC components mediated by the NIP1-NTD enhance the assembly of 43S complexes and regulate the selection of AUG codons during scanning.
MATERIALS AND METHODS
Yeast strain constructions.
All of the strains used here are listed in Table 1. Strains HLV04 and HLV05 were constructed by tetrad analysis of a diploid strain HLV03 (MATa/α ura3-52/ura3-52 trp1-63/trp1-63 leu2-3,112/leu2-3,112 his4-303[ATT]/his4-303[ATT] SUI1/sui1-1 NIP1/nip1Δ [pNIP1+ URA3]) that was created as follows. HLV01a (MATa ura3-52 trp1-63 leu2-3,112 his4-303[ATT]) and TD301-8D (MATα leu2-3 leu2-112 ura3-52 his4-303[ATT] sui1-1) (provided by T. Donahue) were crossed, and the resulting hybrid was transformed with the nip1Δ::hisG-URA3-hisG cassette contained on pLV10 to delete one chromosomal copy of NIP1. Uracil auxotrophy was regained by growing the cells on 5-fluoro-orotic acid (5-FOA) plates, and the resulting strain was transformed with pNIP1+ (10) carrying wild-type (WT) NIP1 to produce HLV03. HLV01a was constructed by tetrad analysis of a cross involving H1515 (MATa ura3-52 leu2-3,112 trp1-63) and 76-3D (MATα his4-303[ATT] ura3-52 leu2-3) (provided by T. Donahue). To produce HLV04-f, HLV04 was transformed with a fun12Δ::KanMX deletion cassette, and the deletion was verified by PCR analysis of chromosomal DNA. Strain HKN06 was created in two steps. First, strains H2881a and H2881α (21) were crossed, and the resulting hybrid was transformed with the nip1Δ::hisG-URA3-hisG to delete one chromosomal copy of NIP1. Uracil auxotrophy was regained on 5-FOA plates, and the resulting strain was then transformed with pNIP1+ to produce HKN05. The strain HKN06 was produced by tetrad dissection of HKN05.
TABLE 1.
Yeast strains used in this study
| Strain | Description | Source or reference |
|---|---|---|
| HLV04 | MATα ura3-52 trp1-63 leu2-3,112 his4-303[ATT] SUI1 nip1Δ [pNIP1+URA3] | This study |
| HLV05 | MATα ura3-52 trp1-63 leu2-3,112 his4-303[ATT] sui1-1 nip1Δ[pNIP1+URA3] | This study |
| HKN06 | MATα ura3-52 trp1-63 leu2-3,112 gcn2Δ nip1Δ[pNIP1+URA3] | This study |
| 76-3D | MATα his4-303[ATT] ura3-52 leu2-3 | T. Donahue |
| TD301-8D | MATα leu2-3 leu2-112 ura3-52 his4-303[ATT] sui1-1 | T. Donahue |
| H2880 | MATatrp1-1 leu2-3,112 ura3-52 | 21 |
| H2881 | MATatrp1-1 leu2-3,112 ura3-52 gcn2Δ | 21 |
| HLV04-f | MATα ura3-52 trp1-63 leu2-3,112 his4-303[ATT] nip1Δ fun12Δ::KanMX [pNIP1+URA3] | This study |
Plasmid constructions and site-directed mutagenesis of the NIP1-NTD.
YCpNIP1-His-help3 is a derivative of YCpNIP1-His-help2 (29) created to facilitate construction of various NIP1 His-tagged mutant alleles by replacing the BsgI-AatII URA3 fragment from YCpNIP1-His-help2 with the BsgI-AatII LEU2-containing fragment from YCplac11 (Table 2). YCpNIP1-His-L was constructed by inserting the AvaI-XbaI 0.6-kb fragment from YCpNIP1-His-U (29) into YCpNIP1-His-help3 that was digested with AvaI and XbaI. YCpNIP1-Box1-His was constructed by insertion of the appropriate AvaI/XbaI-digested PCR product amplified from pNIP1+ (10) by using primers LVBOX1 and LV22 (Table 3) into AvaI/XbaI-digested YCpNIP1-His-help3.
TABLE 2.
Plasmids used in this study
| Plasmid | Description | Source or reference |
|---|---|---|
| YCpNIP1-His-U | Single-copy NIP1-His, URA3 plasmid from YCplac33 | 29 |
| YEpNIP1-His | High-copy-number NIP1-His, LEU2 plasmid from YEplac181 | 29 |
| YCpNIP1-His-L | Single-copy NIP1-His, LEU2 plasmid from YCplac11 | This study |
| YCpNIP1-Box1 to YCpNIP1-Box16-His | Single-copy NIP1-His containing 10 Ala substitutions in Box1 to Box16, respectively, LEU2 plasmid from YCplac11 | This study |
| YCpNIP1-Box6R-His | Single-copy NIP1-His containing nine Arg substitutions in Box6, LEU2 plasmid from YCplac11 | This study |
| YEpNIP1-Box1-His to YEpNIP1-Box16-His | High-copy-number NIP1-His containing 10 Ala substitutions in Box1 to Box16, respectively, LEU2 plasmid from YEplac181 | This study |
| YEpNIP1-Box6R-His | High-copy-number NIP1-His containing nine Arg substitutions in Box6, LEU2 plasmid from YEplac181 | This study |
| YEpNIP1-N′-His-X | High-copy-number NIP1-N′-His [1-205], LEU2 plasmid from YEplac181 | This study |
| YEpNIP1-N′-Box2, -4, -6, -12, -14, and -15-His | High-copy-number NIP1-N′-His[1-205] containing 10 Ala substitutions in Box2, -4, -6, -12, -14, and -15, respectively, LEU2 plasmid from YEplac181 | This study |
| YEpNIP1-N′-Box6R-His | High-copy-number NIP1-N′-His[1-205] containing Box6R (9-Arg substitutions), LEU2 plasmid from YEplac181 | This study |
| pT7-NIP1-N′ | NIP1[1-205] ORF under T7 promoter | This study |
| pT7-NIP1-N′-Box2, -4, -12, -14, and -15 | NIP1[1-205] ORF containing ten Ala substitutions in Box2, -4, -12, -14, and -15, respectively, under T7 promoter | This study |
| pT7-NIP1-N′-Box6R | NIP1[1-205] ORF containing nine Arg substitutions in Box6 under T7 promoter | This study |
| pLV10 | nip1Δ::hisG::URA3::hisG | This study |
| YEpTIF32-Δ6-His-U | High-copy-number TIF32-Δ5-His[1-790], LEU2 plasmid from YEplac181 | 29 |
| YEpTIF5-U | High-copy-number TIF5-FLAG, URA3 plasmid from YEplac195 | This study |
| YEpTIF5-7A-U | High-copy-number tif5-7A-FLAG, URA3 plasmid from YEplac195 | This study |
| YEpSUI1-U | High-copy-number SUI1, URA3 plasmid from YEplac195 | This study |
| YEpTIF5+SUI1 | High-copy-number TIF5-FLAG and SUI1, URA3 plasmid from YEplac195 | 29 |
| pRSSUI3-S264Y-U | Low-copy SUI3-S264Y, URA3 plasmid from pRS316 | This study |
| YCpTIF5-G31R-U | Single-copy TIF5-G31R, URA3 plasmid from YCplac33 | This study |
| YCpSUI3-S264Y-W | Single-copy SUI3-S264Y, TRP1 plasmid from YCplac22 | This study |
| YCpTIF5-G31R-W | Single-copy TIF5-G31R, TRP1 plasmid from YCplac22 | This study |
| p1780-IMT | High-copy-number SUI2, SUI3, GCD11, IMT4, URA3 plasmid from YEp24 | 3 |
| pGEX-TIF5 | GST-TIF5 fusion plasmid from pGEX-4T-1 | 26 |
| pGEX-SUI1 | GST-SUI1 fusion plasmid from pGEX-5X-3 | K. Asano |
| pGEX-sui1-1 | GST-SUI1-D83G fusion plasmid from pGEX-5X-3 | K. Asano |
| p367 | Low-copy-number URA3 vector containing HIS4-ATG-lacZ fusion | 5 |
| p391 | Low-copy-number URA3 vector containing HIS4-TTG-lacZ fusion | 5 |
| p2042 | Low-copy-number URA3 vector containing HIS4-ATT-lacZ fusion, third codon replaced with TTA | 14 |
| p180 | Low-copy-number URA3 vector containing GCN4-lacZ fusion | 20 |
| YCplac22 | Single-copy cloning vector, TRP1 | 9 |
| YCplac33 | Single-copy cloning vector, URA3 | 9 |
| YEplac112 | High-copy-number cloning vector, TRP1 | 9 |
| YEplac195 | High-copy-number cloning vector, URA3 | 9 |
TABLE 3.
Primers used for site-directed mutagenesis of NIP1
| Primer | Sequence (5′ to 3′) |
|---|---|
| LVNIP1-AVAI | GCC TGC CAA GCC CGA GAT CTA |
| LVNIP1MUT | GCC AAG CCC GAG ATC TAC GCA TAT G |
| LV22 | CAT TTT CTC TAG AAA CTT |
| LVBOX1 | CCA AGC CCG AGA TCT ACG CAT ATG GCA GCT GCT GCA GCA GCA GCA GCA GCA TAC GAT GTA GCC AGT TCT TCA TCC GAA GAA |
| LVBOX2 | CCA AGC CCG AGA TCT ACG CAT ATG TCC CGT TTC TTT TCG TCT AAT TAC GAA GCA GCT GCT GCA GCA GCA GCA GCA GCA GCA |
| LVBOX2C | GCA GCT GCT GCA GCA GCA GCA GCA GCA GCA GAT CTT TTA TCT TCG TCT GAA |
| LVRBOX3B | TTC TTC GGA TGA AGA ACT GGC |
| LVBOX3C | GCC AGT TCT TCA TCC GAA GAA GCA GCT GCT GCA GCA GCA GCA GCA GCA GCA TTA AGC TCT TCC TCC TCT GAG |
| LVRBOX4B | CAA ATC TTC TTC AGA CGA AGA |
| LVBOX4C | TCT TCG TCT GAA GAA GAT TTG GCA GCT GCT GCA GCA GCA GCA GCA GCA GCA GAC CAA GAA TCT GAC GAC TCC |
| LVRBOX5B | CAA TTC AGA CTC AGA GGA GGA |
| LVBOX5C | TCC TCC TCT GAG TCT GAA TTG GCA GCT GCT GCA GCA GCA GCA GCA GCA GCA GAA AGT GAA AGT GAA AGT GAA |
| LVRBOX6B | ATT GAA AAA GGA GTC GTC AGA |
| LVBOX6C | TCT GAC GAC TCC TTT TTC AAT GCA GCT GCT GCA GCA GCA GCA GCA GCA GCA GAC TCT GAT GAT TCT GAT GCA |
| LVNBOX61-R | TCT GAC GAC TCC TTT TTC AAT CGT CGA CGA CGA CGA CGA CGA CGA CGA GTA GAC TCT GAT GAT TCT GAT |
| LVNBOXA2 | ATT GAA AAA GGA GTC GTC AGA |
| LVRBOX7B | TAC ATC AGC TTC ACT TTC ACT |
| LVBOX7C | AGT GAA AGT GAA GCT GAT GTA GCA GCT GCT GCA GCA GCA GCA GCA GCA GCA GGT CCT GAC TGG TTC AAG AAA |
| LVRBOX8B | ATA AGG CTT TGC ATC AGA ATC |
| LVBOX8C | GAT TCT GAT GCA AAG CCT TAT GCA GCT GCT GCA GCA GCA GCA GCA GCA GCA AGA AAA CAA GGT GGA GGT TCA |
| LVRBOX9B | GAA CTC AGA TTT CTT GAA CCA |
| LVBOX9C | TGG TTC AAG AAA TCT GAG TTC GCA GCT GCT GCA GCA GCA GCA GCA GCA GCA TTG AAA AGC TCT AAC TAT GAT |
| LVRBOX10B | AAA TTT ATT TGA ACC TCC ACC |
| LVBOX10C | GGT GGA GGT TCA AAT AAA TTT GCA GCT GCT GCA GCA GCA GCA GCA GCA GCA GAA GAA TCC GAT GAA GAA GAT |
| LVRBOX11B | ATC ACT GGA ATC ATA GTT AGA |
| LVBOX11C | TCT AAC TAT GAT TCC AGT GAT GCA GCT GCT GCA GCA GCA GCA GCA GCA GCA GTA GTC AAG TCT GCC AAA GAA |
| LVRBOX12B | CTT CTT GCC ATC TTC TTC ATC |
| LVBOX12C | GAT GAA GAA GAT GGC AAG AAG GCA GCT GCT GCA GCA GCA GCA GCA GCA GCA GAT GAA ATG CAA GAC GTT TAT |
| LVRBOX13B | CAA TAG TTT TTC TTT GGC AGA |
| LVBOX13C | TCT GCC AAA GAA AAA CTA TTG GCA GCT GCT GCA GCA GCA GCA GCA GCA GCA TCT CAA GCT GAG AAC TCT GAT |
| LVRBOX14B | GAT CTT ATT ATA AAC GTC TTG |
| LVBOX14C | CAA GAC GTT TAT AAT AAG ATC GCA GCT GCT GCA GCA GCA GCA GCA GCA GCA ACT ATT TCT AAT GAG TTT GAT |
| LVRBOX15B | CAA CCA GTC ATC AGA GTT CTC |
| LVBOX15C | GAG AAC TCT GAT GAC TGG TTG GCA GCT GCT GCA GCA GCA GCA GCA GCA GCA CGT CTC TTA GTT AGG GCT CAA |
| LVRBOX16B | CGA GAT CAA ATC AAA CTC ATT |
| LVBOX16C | AAT GAG TTT GAT TTG ATC TCG GCA GCT GCT GCA GCA GCA GCA GCA GCA GCA TGG GGG ACT CCA AAT ATT TTC |
| LV22-HIII | AAT ACA AGC TTA ACT TTC TTG ACT CTT TGC TT |
| LV91 | ATA TAG AGC TCT GAA AGG AAT GAA AAA TTA |
| LV92 | ATA TAG GTA CCT TTC GTA GAT CTC GGG CTT |
| LV101 | ATA TAG TCG ACC TAA GCA GGA GAG TAT AA |
| LV102 | ATA TAG CAT GCT GCA ACT GTT GTA TCT TG |
YCpNIP1-Box2-His was created in the following three steps. First, primers LV22 and LVBOX2c were used to PCR amplify 0.6-kb of pNIP1+. Second, the resulting PCR product was used as a template for a second round of PCR with primers LV22 and LVBOX2. Third, the final PCR product was cleaved with AvaI and XbaI and then ligated with AvaI-XbaI-digested YCpNIP1-His-help3. To construct the 14 plasmids YCpNIP1-Box3-His to YCpNIP1-Box16-His, the following two pairs of primers were used for separate PCR amplifications with pNIP1+ as a template: (i) LVNIP1-AVAI and LVBOX3b to LVBOX16b, respectively, and (ii) LV22 and LVBOX3c to LVBOX16c, respectively. The PCR products thus obtained were used in a 1:1 ratio as templates for a third PCR amplification with primers LVNIP1-AVAI and LV22. The resulting PCR products were digested with AvaI and XbaI and ligated with AvaI/XbaI-cleaved YCpNIP1-His-help3. To make YCpNIP1-Box6R-His, the following two pairs of primers were used for separate PCR amplifications with pNIP1+ as a template: (i) LVNIP1-AVAI and LVNBOXA2 and (ii) LV22 and LVNBOX61-R. The PCR products thus obtained were used in a 1:1 ratio as templates for a third PCR amplification with LVNIP1-AVAI and LV22 as primers. The resulting PCR products were digested with AvaI and XbaI and then ligated with AvaI/XbaI-cleaved YCpNIP1-His-help3.
YEpNIP1-Box1-His and YEpNIP1-Box2-His were constructed by insertion of a 1.1-kb HindIII-BbvCI fragment from YCpNIP1-Box1-His and YCpNIP1-Box2-His, respectively, into HindIII/BbvCI-cleaved YEpNIP1-His. YEpNIP1-Box6R-His was constructed by insertion of a 1.05-kB HindIII-NdeI fragment from YCpNIP1-Box6R into HindIII/NdeI-cleaved YEpNIP1-His. The 14 plasmids from YEpNIP1-Box3-His to YEpNIP1-Box16-His were constructed analogously by insertion of the 1.05-kb HindIII-NdeI fragments from YCpNIP1-Box3-His to YCpNIP1-Box16-His, respectively, into HindIII/NdeI-cleaved YEpNIP1-His.
YEpNIP1-N′-His-X is a derivative of YEpNIP1-N′-His that lacks XbaI in the multiple cloning site and was created to facilitate insertion of various NIP1-NTD mutations into the NIP1-N′-His construct. To produce YEpNIP1-N′-His-X, YEpNIP1-N′-His was cut with SalI, treated with Klenow Fragment, cut with SmaI, and then self-ligated. To create YEpNIP1-N′-Box2, -Box4, -Box6R, -Box12, -Box14, and -Box15-His, the 0.8-kb HindIII-XbaI fragments from YCpNIP1-Box2, -Box4, -Box6R, -Box12, -Box14, and -Box15-His, respectively, were inserted into HindIII/XbaI-cut YEpNIP1-N′-His-X.
pT7-NIP1-N′ and the six related plasmids containing -Box2, -Box4, -Box6R, -Box12, -Box14, and -Box15 were constructed by insertion of the appropriate BamHI/HindIII-digested PCR product amplified from YEpNIP1-His, YEpNIP1-Box2, -Box4, -Box6R, -Box12, -Box14, and -Box15, respectively, into BamHI/HindIII-digested pT7-7 (27). The terminal restriction sites on the PCR fragments were introduced by the primers LVN-BHI-ATG (29) and LV22-HIII during PCR amplification.
To generate the NIP1 deletion plasmid pLV10, 0.16- and 0.87-kb fragments corresponding to the 5′ and 3′ ends of the NIP1 insert in pNIP1+ were amplified by PCR with pNIP1+ as a template and the primer pairs LV91-LV92 and LV101-LV102, respectively. The amplified 5′ fragment was digested at its termini with HindIII and KpnI (both PCR incorporated) and subcloned between the HindIII and KpnI sites of a pUC18 derivative containing the hisG::URA3::hisG cassette to produce pLV09. Subsequently, the amplified 3′ fragment was digested at its termini with SalI and SphI (both PCR incorporated) and subcloned between the SalI and SphI sites of pLV09 to produce pLV10. When digested with SacI and SphI, pLV10 yields a 4.9-kb fragment that can be used in yeast transformations to delete chromosomal NIP1.
To produce YEpTIF5-U and YEpTIF5-7A-U, we first digested YEpTIF5+SUI1 and YEpTIF5-7A+SUI1 (29) with HindIII and NaeI to remove an 827-bp fragment containing SUI1. The 5′ overhangs of the remaining DNA were filled in with T4 DNA polymerase and self-ligated.YEpSUI1-U was created by excision of an ∼1.4-kb TIF5 fragment from YEpTIF5+SUI1 (29) by digestion with SalI and NruI, followed by end-filling with T4 DNA polymerase and self-ligation. To produce YCpTIF5-G31R-U, pKA235 (3) was cut with KpnI and NruI, and the resulting ∼1.3-kb fragment bearing nearly the entire TIF5 ORF was replaced with a KpnI-NruI fragment from p2187 (14) carrying TIF5-G31R.
To construct pRSSUI3-S264Y-U, plasmid p6-4 (provided by T. Dever) carrying WT SUI3 on a low-copy-number URA3 plasmid was cut with BglII and AgeI, and the resulting 261-bp fragment bearing the C terminus of SUI3 was replaced with a BglII-AgeI fragment from pBE66 (14) carrying SUI3-S264Y. To produce YCpTIF5-G31R-W, YCpTIF5-G31R-U was cut with SalI and EcoRI, and the resulting ∼2.2-kb fragment bearing TIF5-G31R was ligated with SalI/EcoRI-digested YCplac22. To generate YCpSUI3-S264Y-W, pRSSUI3-S264Y-U was digested with BamHI and SalI and the resulting ∼1.9-bp fragment bearing SUI3-S264Y was inserted into BamHI/SalI-cut YCplac22.
Biochemical techniques.
Glutathione S-transferase (GST) pull-down experiments with bacterially expressed GST fusions and in vitro-synthesized 35S-labeled NIP1-NTD polypeptides, Ni2+ chelation chromatography of eIF3 complexes containing His-tagged proteins from yeast whole-cell extracts (WCEs), and Western blot analysis were all conducted as described previously (29). Formaldehyde cross-linking and fractionation of extracts by sedimentation through sucrose gradients were carried out according to (21). Measurements of β-galactosidase in WCEs were conducted according to (19).
RESULTS
Clustered alanine mutagenesis of the NIP1-NTD reveals genetic interaction with eIF1.
The NIP1-NTD interacts directly with eIF1 and the eIF5-CTD, and the eIF5-CTD mediates an indirect contact between the NIP1-NTD and eIF2β (Fig. 1A) (29). We sought to demonstrate that these interactions have functional significance in vivo by making mutations in the NIP1-NTD and examining their effects on the efficiency and fidelity of translation initiation in yeast cells. NIP1 residues 1 to 160 are depicted schematically in Fig. 1B as a string of 16 10-residue segments, referred to below as boxes 1 to 16. (Thus, box 1 [Box1] contains residues 1 to 10, Box2 contains residues 11 to 20, and so on.) Boxes 1 to 4 lack the NTDs of eIF3c from human, Caenorhabditis elegans, or Arabidopsis thaliana sources, although these boxes occur in the eIF3c-NTD from Schizosaccharomyces pombe A relatively high level of sequence similarity between S. cerevisiae NIP1 and the other eIF3c homologs is evident in the region spanning boxes 6 to 16 of the NTD (depicted by shading in Fig. 1B), with an average of 31.5% identity and 41% similarity. There is a preponderance of acidic residues in boxes 2 to 7, 11, and 13 to 15, whereas boxes 9, 12, and 16 of the S. cerevisiae NTD are basic in character (Fig. 1B). Considering that an acidic motif in the eIF5-CTD is required for binding to the NIP1-NTD (2) and that mammalian eIF1 contains multiple acidic and basic clusters on its surface (8), we reasoned that the charged clusters in the NIP1-NTD might mediate ionic interactions with the charged residues in eIF5 and eIF1.
To examine this possibility, we individually replaced each of the 16 boxes of the NIP1-NTD with a string of ten alanine residues in the fully functional NIP1-His allele, which is tagged at the C terminus with eight histidine residues to facilitate affinity purification of the mutant proteins (29). In addition, the conserved Glu-rich stretch in Box6 (residues 51 to 59) was substituted with a stretch of nine arginine residues to convert it from a highly acidic to a highly basic segment (Box6R). The resulting NIP1-His alleles were introduced on single-copy (sc) or high-copy-number (hc) plasmids into a yeast strain deleted for chromosomal NIP1 by plasmid shuffling and subjected to phenotypic analysis. NIP1-His on the hc plasmid (hc NIP1-His) gives a level of His8-NIP1 expression that is ∼2-fold higher than that conferred by sc NIP1-His (data not shown). This modest overexpression allowed us to achieve essentially WT expression of the NIP1-Box6R-His and NIP1-Box1-His products which appear to be unstable. As discussed below, overexpression of other mutant NIP1 proteins exacerbated their phenotypes. Because NIP1 interacts independently with TIF32 and PRT1, overexpressing NIP1 leads to the formation of two defective subcomplexes: one containing NIP1, PRT1, TIF34, and TIF35 and the other containing TIF32 and NIP1 (29). Thus, the phenotypes conferred by certain NIP1 mutants were exacerbated by overexpression probably because the deleterious effects of the mutations were compounded by a reduction in the level of intact eIF3.
Only the Box1 and Box6R mutations were lethal on sc plasmids, but this lethality was suppressed when the mutant alleles were introduced on hc plasmids to compensate for the instability of the encoded proteins. The resulting hc Box6R mutant displayed a Slg− phenotype at 16 and 30°C but grew like WT at 37°C, whereas the hc Box1 strain grew like WT at all temperatures. The Box12, Box14, and Box15 alleles conferred Slg− and temperature-sensitive (Ts)− phenotypes when present on sc or hc plasmids (Fig. 1D and data not shown). Western analysis showed that the products of these last three alleles were expressed at essentially WT levels from sc plasmids (data not shown). Thus, it appears that residues in boxes 12, 14, and 15 are important for an essential function of the NIP1-NTD and that replacement of the negatively charged residues in Box6 with arginines is highly deleterious to cell growth. All of the other NIP1-His mutations conferred WT growth at all temperatures on sc or hc plasmids (summarized in Fig. 1C, SUI1 row).
The D83G mutation in eIF1 encoded by the sui1-1 allele produces a Slg- phenotype at 30°C (30) and, as shown below, greatly reduces the steady-state level of eIF1 (Fig. 2D). If any of the NIP1-NTD mutations impair the association of eIF1 with eIF3 in vivo, they might be expected to exacerbate the Slg− phenotype of sui1-1. To test this possibility, we introduced the mutant alleles into a sui1-1 nip1Δ strain by plasmid shuffling and determined the growth phenotypes of the resulting double mutants. (We examined only the hc NIP1-His alleles because we found that WT His8-NIP1 must be expressed from a hc plasmid to match the level of native NIP1 expressed from the chromosome in the sui1-1 strain.) All four NIP1-His mutations that conferred Slg− phenotypes in the SUI1 strain (Box6R, Box12, Box14, and Box15) were synthetically lethal with sui1-1 (summarized in Fig. 1C, sui1-1 row). This interaction was specific since none of the NIP1 mutations were synthetically lethal with a deletion of FUN12 (encoding eIF5B) when introduced by plasmid shuffling into a fun12Δ nip1Δ strain (data not shown). Interestingly, five other NIP1-His alleles that produced no growth defects in the SUI1 strain (Box7, Box8, Box9, Box13, and Box16) also were synthetically lethal with sui1-1 (Fig. 1C). As shown in Fig. 2A (SD+His panel), all of the remaining mutations except Box5 exacerbated the Slg− phenotype of sui1-1 (summarized in Fig. 1C). Importantly, Western analysis of WCEs showed that neither Box2 nor Box4, analyzed in depth below, exacerbated the instability of the sui1-1 product (Fig. 2D). These results are consistent with the idea that the NIP1-NTD promotes one or more functions of eIF1 in translation initiation that is impaired by sui1-1.
FIG. 2.

The Box2, Box4, and Box6R alleles of NIP1 exhibit Ssu− (suppressor of Sui−) phenotypes. (A) Suppression of the Sui− phenotype of sui1-1 by the hc Box2 and hc Box4 alleles. Derivatives of HLV05 (nip1Δ sui1-1 his4-303) containing hc WT NIP1-His on plasmid YEpNIP1-His (lane 2) or the indicated hc NIP1 mutant alleles on the appropriate derivatives of YEpNIP1-His plasmids (lanes 3 to 10), and the parental strain TD301-8D (NIP1 sui1-1 his4-303) transformed with empty vector (lane 1), were spotted in four serial dilutions on SD medium containing histidine (upper panel) or lacking histidine (lower panel) and incubated at 30°C for 7 days. Black circles on the column numbers highlight the mutants displaying Ssu− phenotypes. (B) NIP1-Box6R suppresses the dominant Sui− phenotypes of SUI3-S264Y and TIF5-G31R. Derivatives of HLV04 (nip1Δ SUI1 his4-303) containing hc NIP1-His on YEpNIP1-His (lanes 1 to 3), hc NIP1-Box6R-His on YEpNIP1-Box6R-His (lanes 4 to 6), and hc NIP1-Box2-His on YEpNIP1-Box2-His (lanes 7 to 9) were transformed with empty vector YCplac33 (lanes 1, 4, and 7), lc plasmid pRSSUI3-S264Y-U harboring SUI3-S264Y (lanes 2, 5, and 8), and sc plasmid YCpTIF5-G31R-U harboring TIF5-G31R (lanes 3, 6, and 9). The resulting transformants were spotted in three serial dilutions on SD plates supplemented with histidine (upper panel) or lacking histidine (lower panel) and incubated at 30°C for 3 days. (C) Overexpression of eIF5 but not eIF5-7A suppresses the Slg− phenotype of the hc NIP1-Box6R mutant. The derivative of HLV04 containing hc NIP1-Box6R-His was transformed with empty vector (lane 1), hc plasmid YEpTIF5-U harboring TIF5 (lane 2), or hc plasmid YEpTIF5-7A-U harboring tif5-7A (lane 3) and the resulting transformants were spotted in four serial dilutions on SC medium (containing histidine) and incubated at 30°C for 3 days. (D) The D83G substitution in eIF1 (sui1-1) reduces the steady-state level of eIF1. The derivative of HLV04 containing WT untagged NIP1 on sc plasmid pNIP1+ (lanes 1 to 3) and derivatives of HLV05 containing NIP1-His (lanes 4 to 6), NIP1-Box2-His (lanes 7 to 9), or NIP1-Box4-His (lanes 10 to 12) on hc plasmids were grown in YPD medium, and WCEs were subjected to Western analysis with antibodies against the His8 epitope to detect the NIP1-His proteins, or against GCD6 or eIF1, as indicated to the right of the panels. Three different dilutions of each WCE were loaded in consecutive lanes as indicated by the black triangles.
Genetic evidence that the NIP1-NTD is involved in stringent selection of AUG as start codon in vivo.
The sui1-1 mutation was isolated by its ability to increase translation initiation at a UUG triplet in the 5′ end of the HIS4 open reading frame, suppressing the histidine auxotropy (His- phenotype) conferred by inactivation of the normal AUG start codon by the his4-303 mutation (30). This Sui− phenotype provided the first evidence that eIF1 functions in stringent selection of the start codon. We investigated whether any of the NIP1-NTD mutations suppress the Sui− phenotype of sui1-1. As expected, the sui1-1 his4-303 strain containing hc NIP1-His grew well on synthetic dextrose minimal (SD) medium lacking histidine (Fig. 2A, lane 2, SD-His panel), confirming the Sui− phenotype of sui1-1. Interestingly, the corresponding sui1-1 mutants containing the hc Box2 or Box4 alleles did not grow at all (Box2) or grew more poorly (Box4) on SD-His compared to SD+His medium (Fig. 2A, cf. upper and lower panels for columns 4 and 6). In contrast, the other viable NIP1-His sui1-1 his4-303 mutants grew at nearly equal rates on SD-His and SD+His media (Fig. 2A). These findings suggest that the Sui- phenotype of sui1-1 is at least partly suppressed by the Box2 and Box4 mutations, conferring an Ssu− (for suppressor of Sui-) phenotype (7).
It was of interest to determine whether the Box2 or Box4 mutations would also suppress the Sui− phenotypes conferred by mutations in a subunit of eIF2 or in the GAP eIF5 (7). The dominant Sui− allele SUI3-S264Y, encoding a mutant form of eIF2β, was shown to increase the GTPase activity of the TC independently of eIF5. The dominant Sui− allele SUI5 (now called TIF5-G31R) encodes a mutant form of eIF5 displaying elevated GAP function (14). As expected, introducing SUI3-S264Y on a low-copy-number (lc) plasmid, or TIF5-G31R on an sc plasmid, into a his4-303 hc NIP1+-His strain suppressed the His− phenotype of his4-303, confirming the dominant Sui− phenotypes of these alleles (Fig. 2B, lanes 1 to 3). The dominant His+/Sui− phenotypes conferred by SUI3-S264Y and TIF5-G31R were unaffected by replacement of hc NIP1-His with hc NIP1-Box2-His or hc NIP1-Box4-His (Fig. 2B, lanes 7 to 9, and data not shown). Thus, the Ssu− phenotypes of the Box2 and Box4 mutations appear to be specific for sui1-1 However, it is remarkable that hc NIP1-Box6R-His completely suppressed the His+/Sui− phenotypes conferred by SUI3-S264Y and TIF5-G31R (Fig. 2B, cf. lanes 2 and 3 and lanes 5 and 6). This last finding provides additional evidence that the NIP1-NTD is involved in AUG selection.
One way to account for the Ssu− phenotype conferred by hc Box6R would be to propose that this mutation decreases the GAP function of eIF5, compensating for the elevated GTPase activity of eIF2 conferred by SUI3-S264Y or TIF5-G31R. Consistent with this idea, we found that the Slg− phenotype of the hc Box6R mutant is partly suppressed by overexpression of eIF5 but not by overexpression of eIF5-7A (encoded by tif5-7A), from hc plasmids (Fig. 2C). Because the tif5-7A mutation weakens association of eIF5 with NIP1, eIF2β, and eIF1 (2, 3), it appears that the overexpressed eIF5 must be capable of interactions with other MFC components to suppress the Slg− phenotype conferred by hc Box6R. Thus, we propose that Box6R disrupts the interaction of eIF5 with one or more components of the MFC in a manner that reduces eIF5 GAP function, and this defect can be corrected through mass action by increasing the eIF5 concentration in the cell.
A decrease in eIF5 GAP function could also account for suppression of the Sui- phenotype of sui1-1 by the Box2 and Box4 mutations if we assume that sui1-1 leads to increased GTP hydrolysis at UUG triplets. In accordance with this hypothesis, we found that plasmid-borne TIF5-G31R is synthetically lethal with sui1-1 and that lc SUI3-S264Y partially exacerbates the Slg− phenotype of sui1-1 (data not shown). Given that sui1-1 is recessive for the Sui− phenotype, we propose that one function of eIF1 is to inhibit eIF5-stimulated GTP hydrolysis by the TC positioned at non-AUG triplets. Supporting this idea, we observed that the strong Sui− phenotype of TIF5-G31R is suppressed by overexpressing eIF1 from an hc plasmid (Fig. 3A). This last result suggests that eIF1-eIF5 association contributes to the inhibition of eIF5 GAP function by eIF1.
FIG. 3.

The NIP1-Box12 mutation produces a Sui− phenotype that is suppressed by overexpression of eIF1 and exacerbated by overexpression of eIF5. (A) Overexpression of eIF1 suppresses the dominant Sui− phenotype conferred by TIF5-G31R. The derivative of HLV04 (nip1Δ SUI1 his4-303) containing sc plasmid YCpNIP1-His-L harboring WT NIP1-His was transformed with the empty vectors YCplac22 and YEplac195 (row 1), with sc plasmid YCpTIF5-G31R-W harboring TIF5-G31R and YEplac195 (row 2), or with YCpTIF5-G31R-W and hc SUI1 plasmid YEpSUI1-U (row 3), and the resulting transformants were spotted in three serial dilutions on the SD plates supplemented with histidine (left-hand panel) or lacking histidine (right-hand panel) and incubated at 30°C for 5 days. (B) The HLV04-derivatives containing hc NIP1-His on YEpNIP1-His (lanes 2 to 6) or hc NIP1-Box12-His on YEpNIP1-Box12-His (lanes 7 to 11) were transformed with empty vector YEplac195 (lanes 2 and 7), hc TIF5 plasmid YEpTIF5-U (lanes 3 and 8), hc tif5-7A plasmid YEpTIF5-7A-U (lanes 4 and 9), hc SUI1 plasmid YEpSUI1-U (lanes 5 and 10), or hc plasmid YEpTIF5+SUI1 harboring TIF5 and SUI1 (lanes 6 and 11), and the resulting transformants and the parental strain TD301-8D (NIP1 sui1-1 his4-303) transformed with empty vector (lane 1) were spotted in four serial dilutions on SD medium containing histidine (upper panel) or lacking histidine (lower panel) and incubated at 30°C for 2 days (upper panel) or 7 days (lower panel). (C and D) HLV04 derivatives containing hc NIP1-His or hc NIP1-Box12-His and TD301-8D (NIP1 sui1-1 his4-303) were transformed with p367, p391, and p2042 containing, respectively, HIS4-lacZ reporters harboring ATG, TTG, and ATT start codons. The resulting transformants were grown in SC medium lacking uracil and tryptophan (SC−Ura−Trp) and WCE extracts were prepared and assayed for β-galactosidase activity as described previously (19). Panel C shows the averages and standard deviations from at least six independent measurements with three independent transformants. β-Galactosidase activity was expressed in nanomoles of o-nitrophenyl-β-d-galactopyranoside hydrolyzed per minute per milligram of protein. In panel D, the black bars in the histogram show the mean ratios of expression from the UUG to the AUG reporter, and white bars show the mean ratios of expression from the AUU to the AUG reporter. The fold increases in these two ratios measured in the sui1-1 and hc NIP1-Box12-His mutants versus WT are indicated above the corresponding bars.
Although none of the other NIP1 mutations had an Ssu− phenotype, we found that the Box12 mutation exhibits a Sui− phenotype in a TIF5 strain, partially suppressing the His− phenotype of the his4-303 allele (Fig. 3B, cf. columns 2 and 7). We quantified the Sui− phenotype of hc Box12 by using HIS4-lacZ fusions containing AUG, UUG, or AUU start codons. The hc Box12 mutation increased expression of the AUG fusion by ∼2-fold, but it produced much larger proportional increases for the corresponding UUG and AUU fusions (Fig. 3C). Consequently, hc Box12 led to 3.2- and 5-fold greater utilization of UUG or AUU, respectively, than in the WT strain, increasing initiation from these triplets to levels of ∼20 and ∼5%, respectively, of that seen for AUG. By comparison, sui1-1 increased UUG or AUU utilization by 9.8- and 5-fold, respectively (Fig. 3D). Thus, hc Box12 is comparable to sui1-1 in boosting initiation at AUU triplets but is somewhat less effective than suil-1 for UUG triplets.
Interestingly, the Sui− and Slg− phenotypes of the Box12 mutation were intensified by overexpression of WT eIF5 but not by overproduction of the mutant eIF5-7A protein (Fig. 3B, cf. rows 7 to 9). Remarkably, exacerbation of the Sui− phenotype of the Box12 mutation produced by overexpressing eIF5 was completely reversed by co-overexpressing eIF1 from an hc SUI1 plasmid (Fig. 3B, cf. columns 8 and 11). (Note that overexpressing eIF1 intensified the growth defect of the Box12 mutant overexpressing eIF5; however, this reduction in growth rate was not sufficient to explain the complete loss of growth on the medium without histidine where the Sui− phenotype is scored.) A similar comparison indicates that overexpressing eIF1 suppresses the Sui− phenotype of Box12 in cells expressing native levels of eIF5 (Fig. 3B, cf. columns 7 and 10). To account for these findings, we suggest that the increased utilization of UUG produced by Box12 results from a defect in eIF1-eIF5 interaction, with attendant activation of eIF5 GAP function, and this defect can be reversed through mass action by overexpressing eIF1. Further support for this hypothesis came from our finding that Box12 is synthetically lethal with the hyperactive TIF5-G31R allele and that this synthetic interaction is also suppressed by overexpressing eIF1 (data not shown). Thus, it appears that Box12 increases the level of GTP hydrolysis at UUG codons to an extent that is intolerable in combination with the activated eIF5-G31R protein and that these defects can be fully corrected by increasing the cellular concentration of eIF1. These genetic findings support the idea that eIF1 negatively regulates eIF5 GAP function at non-AUG codons. Below, we offer an explanation for the fact that overexpressing eIF5 exacerbates the Sui− phenotype of the Box12 mutation.
NIP1 mutations impair physical interactions of eIF5 and eIF1 with the NIP1-NTD in vitro and in vivo.
The genetic results presented thus far suggest that mutations in Box2, Box4, and Box6R in the N-terminal portion of the NIP1-NTD reduce eIF5-stimulated GTP hydrolysis by the ternary complex and thereby suppress the Sui− phenotypes conferred by sui1-1 or TIF5-G31R. In contrast, the Box12 mutation in the C-terminal portion of the NIP1-NTD appears to increase eIF5 GAP function, being lethal in the presence of hyperactive eIF5-G31R and producing a Sui− phenotype on its own (summarized in Fig. 1B and below in Fig. 7). Our findings that sui1-1 is lethal with TIF5-G31R and that overexpression of eIF1 suppresses the Sui− phenotype of TIF5-G31R imply that defects in eIF1 can increase eIF5 GAP function through a defective eIF5-eIF1 interaction. Accordingly, the NIP1-NTD mutations could influence eIF5 activity by altering the interaction of NIP1 with eIF5, eIF1, or both factors. To test this possibility, we examined the effects of Box2 and the four NIP1-NTD mutations that confer Slg− phenotypes on in vitro binding of 35S-labeled NIP1-NTD to GST-eIF5 or GST-eIF1 fusions produced in Escherichia coli.
FIG. 7.

Summary of phenotypes, biochemical defects, and proposed mechanisms for the NIP1 mutants analyzed in the present study. Ssu− (suppressor of Sui−), Slg− (slow-growth), Gcd− (general control derepressed), Sui− (suppressor of initiation codon), Syn. Lethal (synthetic lethal), PIC (preinitiation complex), GAP (GTP activating protein) are indicated. See Results and Discussion for further details.
In agreement with previous results, the WT NIP1-NTD polypeptide bound specifically to GST-eIF5 and GST-eIF1 but not to GST alone (Fig. 4A). Interestingly, GST-eIF1-D83G (harboring the mutation in sui1-1) showed very weak binding to the WT NIP1-NTD, suggesting that impaired association of eIF1 with the NIP1-NTD contributes to the Slg− or Sui− phenotypes of sui1-1 mutants. Importantly, all six NIP1 mutations impaired interaction of the NIP1-NTD with GST-eIF5 or GST-eIF1. The Box2 mutation had a much stronger effect on the binding of NIP1-NTD to GST-eIF5 than to GST-eIF1, reducing the interaction with GST-eIF5 to only ∼10% of that seen for WT NIP1-NTD. A similar result was obtained for Box4, although the binding defect was less severe. In contrast, Box12 and Box6R had greater effects on NIP1-NTD binding to GST-eIF1 versus GST-eIF5, whereas Box14 and Box15 reduced binding to GST-eIF1 and GST-eIF5 by similar amounts (Fig. 4A). The Box6R mutation showed the strongest overall binding defects among the six NIP1 mutations analyzed, in accordance with the fact that it produced the strongest Slg− defect at 30°C in vivo (Fig. 1D).
FIG. 4.

Mutations in the NIP1-NTD impair its binding to eIF1 and eIF5. (A) In vitro binding assays. Full-length eIF5 (lane 3), eIF1 (lane 4), or eIF1-D83G (lane 5) fused to GST, as well as GST alone (lane 2), were expressed in E. coli, immobilized on glutathione-Sepharose beads and incubated with the indicated WT (top panel) or mutant 35S-labeled NIP1-NTD polypeptides synthesized in rabbit reticulocyte lysates. The beads were washed with phosphate-buffered saline, and the bound proteins were eluted, separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, stained with Gelcode Blue Stain Reagent (Pierce) (bottom panel; Coomassie), and subjected to autoradiography (upper panels). Lane 1 shows 20% of the input amounts of in vitro-translated proteins added to each reaction (Input [20%]). The amount of each 35S-labeled NIP1 polypeptide bound to each GST-fusion protein was quantified and is expressed below the corresponding panel as a percentage of the binding observed for WT 35S-labeled NIP1. The mobilities of the 35S-labeled proteins in the input lanes are increased artifactually by the large amount of β-globin present in the reticulocyte lysate. (B) The NIP1-Box2 mutation diminishes binding of eIF5 and eIF2 to the NIP1-NTD in vivo. WCEs were prepared from transformants of strain HLV04 bearing empty vector (lanes 1 to 4), hc plasmid YEpNIP1-N′-His-X (lanes 5 to 8), or hc plasmid YEpNIP1-N′-Box2-His (lanes 9 to 12), with the latter two plasmids encoding WT or Box2 versions of the N-terminal 205 residues of NIP1. WCEs were incubated with Ni2+-nitrilotriacetic acid-silica resin, and the bound proteins were eluted and subjected to Western blot analysis with antibodies to the His8 epitope (to detect the NIP1-NTD polypeptides) or with antibodies to the other factors listed to the right of the blots. Lanes 1, 5, and 9 contained 3% of the input WCEs (In); lanes 2, 6, and 10 contained 15% of the first fractions eluted from the resin (E1); lanes 3, 7, and 11 contained 30% of the same fractions (E2); and lanes 4, 8, and 12 contained 3% of the flowthrough fractions (FT). The Western signals for eIF2, eIF1, and eIF5 in the E1 and E2 fractions for the Box2 mutant (lanes 10 to 11) were quantified, combined, normalized for the amounts of the NIP1-NTD-Box2 fragment in these fractions, and plotted in the histogram on the right as percentages of the corresponding values calculated for the WT NIP1-NTD (fractions 6 to 7). (C) The NIP1-Box2 mutation reduces association of eIF5 and eIF2 with the MFC in vivo. Same as in panel B except that WCEs were prepared from transformants of strain HLV04 bearing empty vector (lanes 1 to 4), hc plasmid YEpNIP1-His (lanes 5 to 8), or YEpNIP1-Box2-His (lanes 9 to 12), with the latter two plasmids encoding WT or Box2 versions of full-length NIP1-His. (D) The NIP1-Box6 mutation diminishes binding of eIF5 and eIF2 to the NIP1-NTD in vivo. Same as in panel B except that WCEs were prepared from transformants bearing empty vector (lanes 1 to 4), hc plasmid YEpNIP1-N′-His-X (lanes 5 to 8), or hc plasmid YEpNIP1-N′-Box6-His (lanes 9 to 12), with the latter two plasmids encoding WT or Box6 versions of the NIP1-NTD. (E) The NIP1-Box6 mutation reduces association of eIF5 and eIF2 with the MFC in vivo. Panel E is the same as panel B except that WCEs were prepared from transformants of HLV04 bearing empty vector (lanes 1 to 4), YEpNIP1-His (lanes 5 to 8), or YEpNIP1-Box6-His (lanes 9 to 12), with the latter two plasmids encoding WT or Box6 versions of full-length NIP1-His.
We showed previously that overexpressing the NIP1-NTD leads to formation of a stable subcomplex in vivo that contains eIF5, eIF1, and eIF2 but is devoid of all eIF3 subunits, in agreement with the structural model for the MFC shown in Fig. 1A (29). By assaying formation of this subcomplex, we confirmed that the Box2 mutation has a relatively stronger effect on the interaction of NIP1-NTD with eIF5 versus eIF1 in vivo. As shown in Fig. 4B, a fraction of eIF2, eIF5, and eIF1 copurified specifically with WT His8-tagged NIP1-NTD in Ni2+ chelation chromatography of WCEs (cf. lanes 6 and 7 and lanes 2 and 3). The Box2 mutation greatly reduced the proportions of eIF5 and eIF2 that copurified with His8-NIP1-NTD without affecting the association with eIF1 (Fig. 4B, cf. lanes 10 and 11 and lanes 6 and 7). Because eIF5 bridges the interaction between the NIP1-NTD and eIF2β, it is not surprising that Box2 reduces the binding of NIP1-NTD to both eIF2 and eIF5. The Box4 mutation had a similar but less severe effect on association of eIF2 and eIF5 with overexpressed NIP1-NTD in vivo (data not shown), a finding consistent with its lesser effect in the in vitro binding assay (Fig. 4A). The Box2 mutation also reduced the copurification of eIF5 and eIF2 with full-length His8-NIP1 from yeast cells, but as expected, had little effect on the association of His8-NIP1 with another eIF3 subunit (TIF35) or with eIF1 (Fig. 4C, cf. lanes 10 and 11 and lanes 6 and 7). Thus, we conclude that Box2 weakens the interactions of eIF5 and eIF2 with the NIP1-NTD, destabilizing the association of these factors with the eIF3/eIF1 subcomplex of the MFC. Because the Box2 mutation does not produce a Slg− phenotype, we presume that the dissociation of the MFC shown in Fig. 4C is more extreme than occurs in vivo and probably reflects the dilute concentrations of the MFC that prevail during Ni2+ chromatography of WCEs.
We could not conduct similar Ni2+ binding assays on Box6R or the other two NIP1 mutations with interesting phenotypes (Box12 and Box15) because they either destabilized the His8-NIP1-NTD or prevented its binding to nickel resin. However, we succeeded in analyzing the effects of the alanine substitutions in Box6 (the Box6 mutation). Unlike Box6R or Box2, the Box6 mutation does not produce an Ssu− phenotype, but it resembles Box2 in reducing the binding of NIP1-NTD to eIF5 and eIF2 in WCEs, with little effect on the interaction of NIP1-NTD with eIF1 (Fig. 4D). It also produces a modest, but reproducible, reduction in the amount of eIF5 and eIF2 associated with the eIF3/eIF1 subcomplex of the MFC in vivo (Fig. 4E). The effects of Box6 on these interactions are less pronounced than observed for Box2 (cf. Fig. 4B and C to Fig. 4D and E), a finding consistent with the absence of an Ssu− phenotype for the Box6 mutant. However, these results provide biochemical evidence that Box6 residues contribute to the association of eIF5 with the NIP1-NTD in vivo. Additional support for this conclusion is provided in the next section.
NIP1-NTD mutations impair 43S/48S preinitiation complex assembly in vivo.
We proposed above that the Box2 mutation suppresses the Sui− phenotype of sui1-1 by reducing eIF5-stimulated GTP hydrolysis by the ternary complex. The biochemical results just described show that Box2 weakens the association of eIF5 with both the isolated NIP1-NTD and the eIF3/eIF1 subcomplex of the MFC in cell extracts. Accordingly, we sought to determine whether Box2 reduces the association of eIF5 with 43S preinitiation complexes in sui1-1 cells. To answer this question, we used a recently devised technique in which living yeast cells are treated with formaldehyde to cross-link native preinitiation complexes in vivo. The composition of these complexes is then examined by Western analysis after resolving the WCEs by sedimentation through sucrose density gradients (21). As shown in Fig. 5A, we observed the expected cosedimentation of a proportion of eIF2, -3, -5, and -1A with the 40S subunit in the sui1-1 strain, a behavior indicative of 43S or 48S preinitiation complexes. We failed to observe 40S binding of the sui1-1 product itself despite its presence in the WCEs (Fig. 5A and B, input lane), whereas 40S-binding of WT eIF1 in SUI1 strains is detectable by this technique (Fig. 5C). Interestingly, the Box2 mutation led to a marked depletion of eIF5 in the 40S fraction from sui1-1 cells (Fig. 5A and B). We did not observe this defect in SUI1 cells expressing the NIP1-Box2-His product (data not shown), indicating that loss of eIF5 from the 40S ribosomes occurs only when Box2 is combined with sui1-1. Presumably, the defect in association of eIF5 with the NIP1-NTD conferred by Box2 is compensated in SUI1+ cells by the direct interactions of eIF5 with eIF1 and eIF2 (2), thus permitting efficient recruitment of eIF5 to 40S subunits. These findings support the idea that the Ssu- phenotype of Box2 in sui1-1 cells arises from a reduction in eIF5 GAP function resulting from impaired binding of eIF5 to preinitiation complexes. The fact that eIF5 association with 43S-48S complexes is not reduced by Box2 in otherwise WT cells is consistent with the fact that Box2 does not suppress the Sui− phenotype of TIF5-G31R.
FIG. 5.

NIP1-NTD mutations impair the assembly of preinitiation complexes in vivo. (A and B) eIF5 binds poorly to 40S subunits in sui1-1 cells expressing NIP1-Box2-His. Derivatives of HLV05 (nip1Δ sui1-1 his4-303) containing hc NIP1-His (A) or hc NIP1-Box2-His (B) were grown in YPD medium and cross-linked with formaldehyde. WCEs were prepared and separated by velocity sedimentation on a sucrose gradient. The gradients were collected and scanned at 254 nm to visualize the ribosomal species. Proteins were subjected to Western analysis with antibodies to the proteins listed to the right of the blots or with antibodies to the His8 epitope to detect the NIP1-His proteins. The first lane contains a sample of the starting WCEs resolved on the gradients (In). The Western signals in fractions 9 and 10 containing the 43S-48S complexes were quantified, and the amounts of each factor from the NIP1-Box2-His extract are plotted in the histogram on the right of panel B as percentages of the corresponding amounts measured for the WT extract analyzed in panel A. (C to E) The Box6R and Box15 mutations reduce the amounts of eIF5 and eIF2 associated with 40S ribosomes in vivo. Same as panels A and B except that derivatives of HLV04 (nip1Δ SUI1 his4-303) expressing WT NIP1-His (C), NIP1-Box6R-His (D), or NIP1-Box15-His (E), respectively, were analyzed after growth in SD medium. Short and long (long e.) exposures are shown for the Western analysis of NIP1-His proteins in the gradient factions. In addition, samples of the input WCEs were subjected to Western analysis of the NIP1-His proteins immediately before (bef.) or after (aft.) incubation for 7 h on ice (lane 1, In).
By cross-linking analysis, we found that Box6R also reduces the amount of eIF5 associated with 40S ribosomes and additionally lowers the amount of 40S-bound eIF2. Western analysis of rapidly prepared extracts from cross-linked cells showed that NIP1-Box6R-His is expressed at nearly WT levels in vivo but is subject to degradation during incubation on ice and is observed at low levels on 40S ribosomes after fractionation of the WCEs on sucrose gradients. Nevertheless, TIF35/eIF3g, TIF32/eIF3a, and eIF1 showed essentially WT binding to 40S subunits in the Box6R mutant (cf. Fig. 5D and C), indicating that this mutation does not impair binding of the eIF3/eIF1 subcomplex of the MFC to 40S ribosomes in vivo. However, binding of eIF5 and eIF2 to 40S subunits was reduced to ∼20% of WT levels (cf. Fig. 5C and D). Note that the total amounts of eIF5 and eIF2 are reduced in the Box6R extract compared to WT (cf. “In” lanes in Fig. 5C and D). Hence, we cannot eliminate the possibility that the impaired binding of eIF5 and eIF2 to 40S subunits produced by Box6R at least partly reflects the decreased amounts of these factors in the extract rather than a decrease in their binding to 40S subunits. However, quantification of the input lanes shows that the decrease in eIF5 and eIF2 levels is not sufficient to explain their reduced association with 40S subunits in Box6R cells.
The NIP1-NTD is required for efficient recruitment of the ternary complex to 40S ribosomes and GCN4 translational control.
The β-subunit of eIF2 makes an indirect contact with the NIP1-NTD that is bridged by eIF5-CTD (Fig. 1A). Mutations in the NIP1-NTD that impair its interaction with eIF5 may weaken the association of TC with the MFC and reduce the rate of TC binding to 40S subunits in vivo. If diminished recruitment of TC is the primary defect resulting from such mutations, they should constitutively derepress GCN4 translation in cells lacking the kinase GCN2 (Gcd− phenotype). Because gcn2Δ mutants cannot induce GCN4 and amino acid biosynthetic enzymes under its control, they fail to grow on medium containing the inhibitor of histidine biosynthesis, 3-aminotriazole (3-AT). Interestingly, the hc Box6R, Box14, and Box15 mutations all permit robust growth of a gcn2Δ strain on medium containing 30 mM 3-AT (Gcd− phenotypes), even though they confer Slg− phenotypes on medium lacking 3-AT (Fig. 6A, lanes 7, 9, and 11, and data not shown). We quantified the Gcd− phenotypes of the hc Box6R and hc Box15 mutants by assaying expression of a GCN4-lacZ reporter containing all four uORFs in the mRNA leader (p180). As expected, addition of 3-AT to a GCN2 NIP1 strain produced ∼9-fold induction of GCN4-lacZ expression, whereas the gcn2Δ NIP1 strain showed constitutively low GCN4-lacZ expression (Fig. 6B, lanes 1 to 4). Importantly, the hc Box6R and hc Box15 mutations led to ∼5- and ∼6-fold derepression of GCN4-lacZ expression in the gcn2Δ background (Fig. 6B, lanes 4 to 7). A control construct lacking all four uORFs (for which translational control is destroyed) showed similar high levels of GCN4-lacZ expression in all of the strains, indicating that mRNA stability was unaffected by the NIP1 mutations (data not shown). Thus, the NIP1 mutations led to the derepression of GCN4 translation independently of eIF2α phosphorylation by GCN2.
FIG. 6.

Genetic evidence that the Box6R and Box15 NIP1-NTD mutations reduce the rate of TC recruitment to 40S ribosomes and thereby impair GCN4 translational control. (A) Derivatives of strain HKN06 (gcn2Δ nip1Δ) containing sc NIP1-His (lanes 3 and 4), hc NIP1-His (lanes 5 and 6), and the indicated hc NIP1-Box-His mutants (lanes 7 to 12) were transformed with empty vector YEplac195 (lanes with odd numbers beginning with lane 3) or hc plasmid p1780-IMT encoding all three subunits of eIF2 and tRNAiMet (hc TC) (lanes with even numbers beginning with lane 4). The resulting transformants and isogenic strains H2880 (GCN2) (lane 1) and H2881 (gcn2Δ) (lane2) were spotted in four serial dilutions on SC (upper panel) or SC containing 30 mM 3-AT (lower panel) and then incubated at 30°C for 5 days. (B) The strains described in panel A containing hc WT NIP1-His (lane 5), hc NIP1-Box6R-His (lane 6), or hc NIP1-Box15-His (lane 7), as well as GCN2 NIP1strain H2880 (lanes 1 and 2) and gcn2Δ NIP1 strain H2881 (lanes 3 and 4), were transformed with p180 containing the GCN4-lacZ fusion with all four uORFs present. The transformants were grown in SC−Ura in the presence of 10 mM 3-AT (white bars, lanes 1 and 3) or without 3-AT (black bars; lanes 2 and 4 to 7), and the β-galactosidase activities were measured in the WCEs. The histogram shows the mean values and standard deviations obtained from at least six independent measurements with three independent transformants. The fold increases observed in the hc NIP1-His mutants versus hc WT NIP1-His are given above the relevant bars. (C) The HLV04 derivatives (nip1Δ SUI1 his4-303) containing the indicated NIP1-Box-His mutants were transformed with empty vector (lanes 1 and 4), p1780 (lanes 2 and 5), or hc plasmid YEp-TIF32-Δ6-His-U containing TIF32-Δ6-His (lanes 3 and 6), and the resulting transformants were spotted in three serial dilutions on SC plates and incubated at 30°C for 3 days.
We hypothesized that the Gcd− phenotypes of the NIP1 mutations result from defects in TC binding to 40S subunits, allowing a fraction of the 40S ribosomes that have translated uORF1 to rebind the TC only after scanning past uORF4 and then reinitiate at the GCN4 start codon instead. If so, the Gcd− phenotypes should be suppressed by increasing the concentration of TC. In agreement with this prediction, the hc TC plasmid partially suppressed the 3ATR/Gcd− phenotypes of the Box6, Box6R, and Box15 mutations (Fig. 6A). The Slg− phenotype of the hc Box15 mutation also was diminished by hc TC (Fig. 6A and C), suggesting that impaired TC binding to 40S ribosomes is the rate-limiting defect in this mutant. Consistent with this conclusion, overexpressing the dominant-negative hc TIF32-Δ6 allele, lacking the C-terminal binding domain for eIF2β in the eIF3a/TIF32 subunit (29), exacerbated the Slg− and Gcd− phenotypes of the Box15 mutation (Fig. 6C and data not shown). Thus, combining mutations in the NIP1-NTD and TIF32-CTD that weaken the independent contacts between eIF3 and eIF2 in the MFC appears to produce an additive reduction in TC binding to 40S subunits in vivo. Consistent with this interpretation, we found that hc Box15 reduces the binding of eIF2 to 40S subunits in cross-linked cells (Fig. 5E).
In contrast to our findings on hc Box15, the Slg− phenotype of hc Box6R was not suppressed by hc TC and was only slightly exacerbated by TIF32-Δ6 (Fig. 6C). Based on the Ssu- phenotype of hc Box6R and the suppression of its Slg− phenotype by hc TIF5, we concluded above that Box6R reduces eIF5 GAP activity. Thus, all of our genetic findings on hc Box6R can be explained by proposing that the rate-limiting defect in this mutant is the impairment of eIF5 GAP function resulting from a defective interaction of the NIP1-NTD with eIF5 (producing the Slg− and Ssu- phenotypes) and that a reduction in TC recruitment is a secondary consequence of this mutation that is responsible for its Gcd− phenotype.
DISCUSSION
We showed previously that deletion of the NIP1-NTD is lethal even though an otherwise intact eIF3 complex is formed by the N-terminally truncated NIP1 protein in vivo. Deleting the NIP1-NTD eliminates a physical connection of the eIF3 complex with eIF1, eIF5, and eIF2, suggesting that one or more of these interactions in the MFC is essential for translation initiation in vivo (29). One possibility is that MFC formation stimulates the assembly of 43S preinitiation complexes through the cooperative binding of its constituent components to 40S subunits. Another possibility is that the NIP1-NTD plays a critical role in coordinating the functions of eIF1 and eIF5 in recognition of the AUG start codon and triggering hydrolysis of GTP in the TC. We have tested these hypotheses by genetic and biochemical analyses of an array of clustered-alanine substitutions in the N-terminal 160 amino acids of NIP1. Our results indicate that the Box2, Box6R, and Box12 mutations of the NIP1-NTD impair the functions of eIF1 and eIF5 in regulating AUG selection and that Box6R and Box15 decrease the rate of TC binding to 40S subunits and disrupt translational control of GCN4. The salient features of these four mutations are summarized in Fig. 7.
Evidence that NIP1-NTD mutations disrupt AUG start codon selection by eIF1 and eIF5.
The sui1-1 mutation in eIF1 increases the utilization of UUG start codons at his4-303 (Sui− phenotype) (30), and we discovered that sui1-1 is synthetically lethal with TIF5-G31R, encoding a hyperactive form of eIF5 (14). These phenotypes are consistent with the idea that sui1-1 leads to an elevated level of eIF5-stimulated GTP hydrolysis by the TC that becomes lethal in cells expressing eIF5-G31R. The sui1-1 allele is recessive for its Sui− phenotype and the encoded eIF1-D83G protein is present at low levels in WCEs and in 43-48S preinitiation complexes (Fig. 2D and 5A). Hence, we propose that eIF1 normally functions to inhibit eIF5 GAP function at non-AUG codons and that this inhibitory activity is diminished in sui1-1 cells to permit increased utilization of UUG as start codon (see model in Fig. 8A and B). This proposal is consistent with the fact that the loss-of-function tif5-G62S (ssu2-1) mutation in eIF5 was isolated as a suppressor of sui1-1 (7). We provided additional support for this model by showing that overexpression of eIF1 suppresses the Sui− phenotype of TIF5-G31R (Fig. 3A). This last finding suggests that eIF1 negatively regulates eIF5 GAP function at non-AUG triplets through a direct interaction between the two proteins, a finding consistent with the fact that eIF1 binds specifically to eIF5 in vitro (2). Interestingly, we found recently that eIF1 overexpression also suppresses the Sui− phenotype of SUI3-S264Y (L. Valášek and A. G. Hinnebusch, unpublished observations), whose product exhibits high intrinsic GTPase activity in vitro. Thus, there is now considerable genetic evidence that eIF1 functions to inhibit GTP hydrolysis by the TC during scanning in the absence of perfect base pairing between Met-tRNAiMet and AUG in the P site.
FIG. 8.

Hypothetical models depicting the proposed functions of eIF1, eIF3, eIF5, and TC in AUG selection and the consequences of sui1-1 and NIP1-NTD mutations on this process. (A to F) The interface side of the 40S ribosomal subunit is depicted with Met-tRNAiMet bound to eIF2 and GTP in the TC and base paired with UUG or AUG triplets in the ribosomal P site. The bulk of eIF3 is bound to the solvent side of the 40S subunit and only the NIP1-NTD and TIF32-CTD are visible as they gain access to the interface side of the ribosome. eIF5 is bound to the NIP1-NTD and to eIF2, and the TIF32-CTD contacts eIF2 directly, as shown in Fig. 1A. The eIF1 has been released from its interactions with NIP1-NTD and TIF32-CTD and is bound near the P site to eIF5. (A) In WT cells, base pairing of tRNAiMet with UUG during scanning (upper schematic) does not elicit GTP hydrolysis by the TC because eIF1 senses the imperfect codon-anticodon interaction and inhibits the GTPase activating function of eIF5. Scanning continues and, upon base pairing of tRNAiMet with AUG (lower schematic), the negative regulation of eIF5 by eIF1 is disabled to permit eIF5-stimulated GTP hydrolysis, release of eIF2-GDP and other eIFs, and joining of the 60S subunit to form an 80S initiation complex. (B) In sui1-1 cells, eIF1 does not bind effectively to 40S subunits, which decreases its ability to inhibit eIF5 GAP function and allows increased rates of GTP hydrolysis and initiation at a UUG codon (Sui− phenotype). (C) The NIP1-Box2 mutation partially suppresses the Sui− phenotype in sui1-1 cells by disrupting a contact between eIF5 and the NIP1-NTD, thus leading to a weaker association of eIF5 with the preinitiation complex and attendant reduction in its GAP function. This partially compensates for the increased rate of GTP hydrolysis at UUG codons produced by sui1-1, yielding an Ssu- phenotype. The compound defects in binding of eIF1 and eIF5 to the 40S ribosome in the sui1-1 NIP1-Box2-His double mutant dramatically decreases the rate of translation initiation at AUG codons and produces a severe growth defect. (D) NIP1-Box12 leads to increased GTP hydrolysis by the TC at UUG triplets by altering the interaction of eIF5 with the 40S ribosome and thereby reducing the ability of eIF1 to interact with eIF5 in the manner required to inhibit its GAP function at UUG codons (Sui− phenotype). Overexpression of eIF1 can restore, by mass action, the interaction between eIF1 and eIF5 needed to prevent GTP hydrolysis at UUG codons and suppress the Sui− phenotype of NIP1-Box12 (not depicted). (E) The TIF5-G31R allele encodes a hyperactive form of eIF5 that escapes strong inhibition of its GAP function by eIF1 at UUG codons (Sui− phenotype). (F) NIP1-Box6R suppresses the Sui− phenotype of TIF5-G31R by weakening the interaction of eIF5 with the NIP1-NTD, leading to reduced association of eIF5 with the 40S ribosome and diminishing the GAP function of eIF5. This offsets the increased rate of GTP hydrolysis at UUG codons produced by TIF5-G31R and results in a Ssu− phenotype. (G) The NIP1-Box6R and -Box15 mutations weaken the association of TC with the MFC and thus decrease the rate of TC binding to 40S subunits scanning downstream from uORF1 in the GCN4 mRNA leader. This allows a fraction of 40S subunits to bypass uORF4 and reinitiate at GCN4 instead of reinitiating at uORF4 and dissociating from the mRNA, even in the absence of eIF2α phosphorylation in gcn2Δ cells, where TC levels are high (Gcd− phenotype).
Recent findings on mammalian eIF1 (24) demonstrate a role for this factor in the rejection of non-AUG codons during the scanning process independently of eIF5, possibly by destabilizing the base pairing of Met-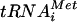 with non-AUG triplets in the P site. In this way, eIF1 increases the processivity of scanning Y3S complexes and facilitates AUG selection. This activity is not incompatible with the regulatory function for eIF1 postulated in Fig. 8A. Base pairing between Met-
with non-AUG triplets in the P site. In this way, eIF1 increases the processivity of scanning Y3S complexes and facilitates AUG selection. This activity is not incompatible with the regulatory function for eIF1 postulated in Fig. 8A. Base pairing between Met-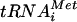 and AUG may trigger a conformational change in the 40S subunit or in eIF1 that stabilizes Met-
and AUG may trigger a conformational change in the 40S subunit or in eIF1 that stabilizes Met-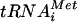 binding in the P site and at the same time disables the inhibitory effect of eIF1 on eIF5 GAP activity. Indeed, high-resolution mapping of the binding site for mammalian eIF1 on the 40S ribosome places it in the vicinity of the P site (16). To accommodate this last finding with current models for binding of eIF3 to the solvent side of the 40S ribosome, we suggested previously that interaction of eIF1 with the NIP1-NTD occurs only during recruitment of eIF1 to the 40S subunit and that eIF1 is subsequently transferred to its location near the P site while maintaining interaction with eIF5 (28). This configuration is depicted in the models in Fig. 8 presented to account for the effects of the Box2, Box6R, and Box12 mutations in NIP1 on AUG selection.
binding in the P site and at the same time disables the inhibitory effect of eIF1 on eIF5 GAP activity. Indeed, high-resolution mapping of the binding site for mammalian eIF1 on the 40S ribosome places it in the vicinity of the P site (16). To accommodate this last finding with current models for binding of eIF3 to the solvent side of the 40S ribosome, we suggested previously that interaction of eIF1 with the NIP1-NTD occurs only during recruitment of eIF1 to the 40S subunit and that eIF1 is subsequently transferred to its location near the P site while maintaining interaction with eIF5 (28). This configuration is depicted in the models in Fig. 8 presented to account for the effects of the Box2, Box6R, and Box12 mutations in NIP1 on AUG selection.
The first genetic evidence implicating NIP1-NTD in AUG recognition came from our finding that the Box2 and Box4 mutations partially suppressed the Sui− phenotype of sui1-1 (Ssu− phenotype) (Fig. 2A). We also found that sui1-1 reduces the amount of eIF1 associated with the 48S complex, which should lead to increased GTP hydrolysis at non-AUG codons (Fig. 8B). Hence, we propose that the Box2 mutation partially compensates for this defect by disrupting a contact between eIF5 and the NIP1-NTD, leading to partial dissociation of eIF5 from preinitiation complexes in sui1-1 cells. This would decrease the efficiency of eIF5 GAP function and lower the rate of GTP hydrolysis by the TC at non-AUG codons, offsetting the increased rate of GTP hydrolysis produced by sui1-1 (Fig. 8C). Supporting this model, the Box2 mutation preferentially reduced association of the NIP1-NTD with eIF5 versus eIF1 in binding assays with recombinant proteins (Fig. 4A), and it had the same effect in yeast extracts for both the NIP1-NTD and full-length NIP1 (Fig. 4B and C). Furthermore, the Box2 mutation reduced binding of eIF5 to 40S subunits in cross-linked sui1-1 cells (Fig. 5). In contrast, in SUI1+ cells Box2 had no effect on association of eIF5 with preinitiation complexes in cross-linked cells, explaining why Box2 does not suppress the Sui− phenotype of TIF5-G31R. The additive effect of sui1-1 and the Box2 mutation in reducing the amount of eIF5 associated with 43S-48S complexes implies that the eIF1-eIF5 interaction promotes eIF5 binding to 40S subunits. Thus, we propose that eIF1 is required for efficient eIF5 GAP function at AUG codons through its stimulatory effect on 40S binding of eIF5, while at the same time inhibiting eIF5 activity at non-AUG triplets. The mechanism just described for the Box2 mutation may also account for the Ssu- phenotype of the Box4 mutation, since the latter also selectively impairs interaction of the NIP1-NTD with eIF5 (Fig. 4A and data not shown).
The Box6R mutation suppressed the Sui− phenotypes of the TIF5-G31R and SUI3-S264Y mutations (Fig. 2C), both of which increase GTP hydrolysis by the TC in vitro (14). According to our model, the G31R substitution in eIF5 activates GAP function to the point where it overcomes the inhibitory effect of eIF1 and permits elevated GTP hydrolysis at non-AUG codons (Fig. 8E). We propose that by weakening the interaction of eIF5 with the NIP1-NTD, Box6R would reduce the GAP function of eIF5 enough to suppress the elevated GTP hydrolysis at UUG codons produced by TIF5-G31R and SUI3-S264Y (Fig. 8F). (This explanation assumes that the GTPase activity of eIF2 harboring the SUI3-S264Y mutation is still stimulated by eIF5.) Supporting this model, the Slg− phenotype of the Box6R mutation was partially suppressed by overexpressing eIF5 but not eIF5-7A (Fig. 2C). In addition, we found that Box6R led to a strong reduction in binding of NIP1-NTD to eIF5 in vitro (Fig. 4A). In vivo, it selectively reduced the binding of eIF5 and eIF2 to the overexpressed NIP1-NTD fragment and also impaired the association of eIF5 and eIF2 with eIF1 and the eIF3 complex (Fig. 4D and E). Finally, Box6R decreased the binding of eIF5, but not eIF1, to 40S subunits in cross-linked yeast cells (Fig. 5D), thus providing direct biochemical support for the model in Fig. 8F. We predict that the Box6R mutation would also suppress the Sui− phenotype of sui1-1 and reduce the association of eIF5 with 40S subunits in sui1-1 cells, as observed for Box2; however, the lethality of Box6R in sui1-1 cells prevented us from testing this prediction.
The Box12 mutation produced a Sui− phenotype (Fig. 3B and C), comparable to that given by lc SUI3-S264Y in SUI3+ cells (Fig. 2B). In addition, Box12 was synthetically lethal with the activated Sui− allele TIF5-G31R. In both respects, Box12 qualitatively resembles sui1-1. Hence, we propose that Box12 leads to increased GTP hydrolysis by the TC at UUG triplets (Fig. 8D). This defect is intensified and becomes lethal when combined with the activating G31R mutation in eIF5. It is intriguing that the Sui− phenotype of Box12 was exacerbated by overexpressing eIF5, but not eIF5-7A, and was suppressed by overexpressing eIF1. In addition, the effect of overexpressing eIF5 on the Sui− phenotype of Box12 was overcome by co-overexpressing eIF1 (Fig. 3B). The simplest way to explain the suppression of the Box12 Sui− phenotype by hc eIF1 is to propose that Box12 weakens the ability of eIF1 to interact with eIF5 in the manner required to inhibit eIF5 activity at non-AUG codons. Increasing the concentration of eIF1 would restore the correct interaction between eIF1 and eIF5 by mass action and reinstate the stringent regulation of eIF5 GAP function. Strongly supporting this interpretation, we found that overexpression of eIF1 eliminated the synthetic lethality produced by combining Box12 with TIF5-G31R.
To explain why overexpressing eIF5 exacerbates the Sui− phenotype of the Box12 mutation (Fig. 3B), we suggest that excess eIF5 titrates a fraction of eIF1 from the MFC and sequesters it in inactive subcomplexes. This would stimulate GTP hydrolysis at non-AUG codons by reducing the inhibitory effect of eIF1 on eIF5, intensifying the Sui− phenotype of the Box12 mutation. Co-overexpressing eIF1 with eIF5 would restore WT levels of eIF1 in the MFC. Because overexpressing eIF5 does not produce a Sui− phenotype in WT cells (Fig. 3A), its ability to titrate eIF1 from the MFC would depend on a weakened interaction between eIF1 and the NIP1-NTD containing the Box12 mutation. Indeed, we found that Box12 impaired binding of eIF1 to the NIP1-NTD in vitro (Fig. 4A). It should be noted that we did not detect a reduction in eIF1 association with 40S subunits in cross-linked Box12 mutant cells (data not shown). Thus, it appears that Box12 alters the interaction of eIF1 with eIF5 on the 40S ribosome without diminishing eIF1 recruitment. We suggest that by increasing the residence time of eIF1 on the 40S subunit by overexpressing this factor we restored the eIF5-eIF1 interaction required for stringent regulation of eIF5 GAP function in Box12 mutant cells.
Evidence that mutations in the NIP1-NTD decrease the recruitment of TC to 40S ribosomes.
The Box6R and Box15 mutations produced Gcd− phenotypes in cells lacking protein kinase GCN2, and these phenotypes were partially suppressed by overexpressing the TC (Fig. 6A and B). This provides strong genetic evidence that these mutations decrease the rate of TC binding to 40S ribosomes in vivo (Fig. 8G). The Slg− phenotype of the Box15 mutant also was partially suppressed by hc TC (Fig. 6A and C), suggesting that TC recruitment is a rate-limiting defect in Box15 cells. Consistent with this last conclusion, the Slg− and Gcd− phenotypes of Box15 were exacerbated by overexpressing the TIF32-Δ6 product (Fig. 6C and data not shown). TIF32-Δ6 lacks the binding domain for eIF2β and its overexpression in otherwise WT cells produces a Slg− phenotype that is diminished by hc TC (29). Hence, we propose that overexpressing TIF32-Δ6 in the Box15 mutant leads to an additive reduction in TC binding and exacerbation of the Gcd− phenotype of Box15.
Analysis of eIF2 binding to 40S subunits in cross-linked cells supports our conclusion that the Box6R and Box15 mutations impair the recruitment of TC to 40S subunits in vivo (Fig. 5C to E). Since both mutations also reduce the level of 40S-bound eIF5, it could be proposed that the decreased TC recruitment observed in these mutants reflects the loss of eIF5 adaptor function in bridging association of eIF2 with eIF3 in the MFC. Ostensibly at odds with this interpretation, we found that eIF2 binding to 40S subunits was not reduced in cross-linked sui1-1 NIP1-Box2-His cells despite the strong reduction in eIF5 binding to the 40S in this double mutant. One way to explain this discrepancy is to note that the TC can interact with the 40S ribosome independently of other initiation factors under certain in vitro conditions and thus appears to have intrinsic 40S-binding activity (reviewed in reference 13). Hence, the extremely slow rate of translation initiation displayed by the sui1-1 NIP1-Box2-His double mutant (doubling time of ∼12 h) may allow eIF2 to reach a high level of 40S occupancy with the assistance of the CTD of eIF3a/TIF32 and eIF1A. In contrast, the reduced rate of TC recruitment produced by impaired association of eIF5 with 43S complexes would lead to lower steady-state levels of 40S-bound TC in the faster-growing Box6R and Box15 mutants.
In conclusion, we have provided a combination of genetic and biochemical data indicating that the NIP1-NTD plays an important role in promoting the binding of both eIF5 and the TC to 40S subunits through its direct interaction with eIF5 and its indirect contact with eIF2β in the MFC. This function is critical for proper GCN4 translational control, demonstrating that the MFC participates in TC recruitment during the reinitiation events on GCN4 mRNA. Interaction of the NIP1-NTD with eIF5 is also required for proper regulation of AUG selection during the scanning process, and we obtained several mutations in the NIP1-NTD that appear to reduce or elevate eIF5 GAP function. We also provided evidence that eIF1 negatively regulates eIF5 GAP activity at non-AUG codons, while promoting association of eIF5 with the preinitiation complex, most likely through physical contact between these two proteins in the preinitiation complex. Thus, the NIP1-NTD interacts with both eIF1 and eIF5 (4) and plays an important role in linking the function of eIF1 in recognizing correct codon-anticodon interactions in the P site to the GTPase-activating function of eIF5, both key determinants of stringent AUG selection (7, 14, 16, 24).
Acknowledgments
We are grateful to Věra Valášková for technical assistance and Katsura Asano and Thomas Donahue for timely gifts of plasmids. We thank Thomas Dever and Jiří Hašek for critical reading of the manuscript and the members of the Hinnebusch and Dever laboratories for helpful discussions and suggestions during the course of this work.
L.V. was partly funded by a J. E. Purkyně Fellowship from the Academy of Sciences of the Czech Republic and by the Institutional Research Concept no. AV0Z5020903.
REFERENCES
- 1.Algire, M. A., D. Maag, P. Savio, M. G. Acker, S. Z. Tarun, Jr., A. B. Sachs, K. Asano, K. H. Nielsen, D. S. Olsen, L. Phan, A. G. Hinnebusch, and J. R. Lorsch. 2002. Development and characterization of a reconstituted yeast translation initiation system. RNA 8:382-397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Asano, K., J. Clayton, A. Shalev, and A. G. Hinnebusch. 2000. A multifactor complex of eukaryotic initiation factors eIF1, eIF2, eIF3, eIF5, and initiator tRNAMet is an important translation initiation intermediate in vivo. Genes Dev. 14:2534-2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Asano, K., T. Krishnamoorthy, L. Phan, G. D. Pavitt, and A. G. Hinnebusch. 1999. Conserved bipartite motifs in yeast eIF5 and eIF2Bɛ, GTPase-activating and GDP-GTP exchange factors in translation initiation, mediate binding to their common substrate eIF2. EMBO J. 18:1673-1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Asano, K., A. Shalev, L. Phan, K. Nielsen, J. Clayton, L. Valášek, T. F. Donahue, and A. G. Hinnebusch. 2001. Multiple roles for the carboxyl-terminal domain of eIF5 in translation initiation complex assembly and GTPase activation. EMBO J. 20:2326-2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cigan, A. M., E. K. Pabich, and T. F. Donahue. 1988. Mutational analysis of the HIS4 translational initiator region in Saccharomyces cerevisiae. Mol. Cell. Biol. 8:2964-2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Danaie, P., B. Wittmer, M. Altmann, and H. Trachsel. 1995. Isolation of a protein complex containing translation initiation factor Prt1 from Saccharomyces cerevisiae. J. Biol. Chem. 270:4288-4292. [DOI] [PubMed] [Google Scholar]
- 7.Donahue, T. 2000. Genetic approaches to translation initiation in Saccharomyces cerevisiae, p. 487-502. In N. Sonenberg, J. W. B. Hershey, and M. B. Mathews (ed.), Translational control of gene expression. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 8.Fletcher, C. M., T. V. Pestova, C. U. T. Hellen, and G. Wagner. 1999. Structure and interactions of the translation initiation factor eIF1. EMBO J. 18:2631-2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gietz, R. D., and A. Sugino. 1988. New yeast-Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair restriction sites. Gene 74:527-534. [DOI] [PubMed] [Google Scholar]
- 10.Gu, Z., R. P. Moerschell, F. Sherman, and D. S. Goldfarb. 1992. NIP1, a gene required for nuclear transport in yeast. Proc. Natl. Acad. Sci. USA 89:10355-10359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.He, H., T. von der Haar, C. R. Singh, M. Ii, B. Li, A. G. Hinnebusch, J. E. McCarthy, and K. Asano. 2003. The yeast eukaryotic initiation factor 4G (eIF4G) HEAT domain interacts with eIF1 and eIF5 and is involved in stringent AUG selection. Mol. Cell. Biol. 23:5431-5445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hershey, J. W. B., and W. C. Merrick. 2000. Pathway and mechanism of initiation of protein synthesis, p. 33-88. In N. Sonenberg, J. W. B. Hershey, and M. B. Mathews (ed.), Translational control of gene expression. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 13.Hinnebusch, A. G. 2000. Mechanism and regulation of initiator methionyl-tRNA binding to ribosomes, p. 185-243. In N. Sonenberg, J. W. B. Hershey, and M. B. Mathews (ed.), Translational control of gene expression. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 14.Huang, H., H. Yoon, E. M. Hannig, and T. F. Donahue. 1997. GTP hydrolysis controls stringent selection of the AUG start codon during translation initiation in Saccharomyces cerevisiae. Genes Dev. 11:2396-2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee, J. H., T. V. Pestova, B. S. Shin, C. Cao, S. K. Choi, and T. E. Dever. 2002. Initiation factor eIF5B catalyzes second GTP-dependent step in eukaryotic translation initiation. Proc. Natl. Acad. Sci. USA 99:16689-16694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lomakin, I. B., V. G. Kolupaeva, A. Marintchev, G. Wagner, and T. V. Pestova. 2003. Position of eukaryotic initiation factor eIF1 on the 40S ribosomal subunit determined by directed hydroxyl radical probing. Genes Dev. 17:2786-2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maag, D., and J. R. Lorsch. 2003. Communication between eukaryotic translation initiation factors 1 and 1A on the yeast small ribosomal subunit. J. Mol. Biol. 330:917-924. [DOI] [PubMed] [Google Scholar]
- 18.Majumdar, R., A. Bandyopadhyay, and U. Maitra. 2003. Mammalian translation initiation factor eIF1 functions with eIF1A and eIF3 in the formation of a stable 40S preinitiation complex. J. Biol. Chem. 278:6580-6587. [DOI] [PubMed] [Google Scholar]
- 19.Moehle, C. M., and A. G. Hinnebusch. 1991. Association of RAP1 binding sites with stringent control of ribosomal protein gene transcription in Saccharomyces cerevisiae. Mol. Cell. Biol. 11:2723-2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mueller, P. P., and A. G. Hinnebusch. 1986. Multiple upstream AUG codons mediate translational control of GCN4. Cell 45:201-207. [DOI] [PubMed] [Google Scholar]
- 21.Nielsen, K. H., B. Szamecz, L. Valasek, A. Jivotovskaya, B. S. Shin, and A. G. Hinnebusch. 2004. Functions of eIF3 downstream of 48S assembly impact AUG recognition and GCN4 translational control. EMBO J. 23:1166-1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Olsen, D. S., S. E.M., A. Mathew, F. Zhang, T. Krishnamoorthy, L. Phan, and A. G. Hinnebusch. 2003. Domains of eIF1A that mediate binding to eIF2, eIF3 and eIF5B and promote ternary complex recruitment in vivo. EMBO J. 22:193-204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pestova, T. V., S. I. Borukhov, and C. U. T. Hellen. 1998. Eukaryotic ribosomes require initiation factors 1 and 1A to locate initiation codons. Nature 394:854-859. [DOI] [PubMed] [Google Scholar]
- 24.Pestova, T. V., and V. G. Kolupaeva. 2002. The roles of individual eukaryotic translation initiation factors in ribosomal scanning and initiation codon selection. Genes Dev. 16:2906-2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pestova, T. V., I. B. Lomakin, J. H. Lee, S. K. Choi, T. E. Dever, and C. U. T. Hellen. 2000. The joining of ribosomal subunits in eukaryotes requires eIF5B. Nature 403:332-335. [DOI] [PubMed] [Google Scholar]
- 26.Phan, L., X. Zhang, K. Asano, J. Anderson, H. P. Vornlocher, J. R. Greenberg, J. Qin, and A. G. Hinnebusch. 1998. Identification of a translation initiation factor 3 (eIF3) core complex, conserved in yeast and mammals, that interacts with eIF5. Mol. Cell. Biol. 18:4935-4946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tabor, S., and C. C. Richardson. 1987. DNA sequence analysis with a modified bacteriophage T7 DNA polymerase. Proc. Natl. Acad. Sci. USA 84:4767-4771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Valášek, L., A. Mathew, B. S. Shin, K. H. Nielsen, B. Szamecz, and A. G. Hinnebusch. 2003. The yeast eIF3 subunits TIF32/a and NIP1/c and eIF5 make critical connections with the 40S ribosome in vivo. Genes Dev. 17:786-799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Valášek, L., K. H. Nielsen, and A. G. Hinnebusch. 2002. Direct eIF2-eIF3 contact in the multifactor complex is important for translation initiation in vivo. EMBO J. 21:5886-5898. [DOI] [PMC free article] [PubMed] [Google Scholar]
-
30.Yoon, H. J., and T. F. Donahue. 1992. The sui1 suppressor locus in Saccharomyces cerevisiae encodes a translation factor that functions during
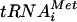 recognition of the start codon. Mol. Cell. Biol. 12:248-260. [DOI] [PMC free article] [PubMed] [Google Scholar]
recognition of the start codon. Mol. Cell. Biol. 12:248-260. [DOI] [PMC free article] [PubMed] [Google Scholar]