

Abstract
Over 1/3 of patients who have undergone oral squamous cell carcinoma (OSCC) surgical resections develop life-threatening and often untreatable recurrences. A variety of drugs, intended for management of recurrent or disseminated cancers, were designed to exploit cancer cells’ reliance upon overexpressed receptors and gratuitous signaling. Despite their conceptual promise, clinical trials showed these agents lacked efficacy and were often toxic. These findings are consistent with evasion of pathway-targeted treatments via extensive signaling redundancies and compensatory mechanisms common to cancers. Optimal secondary OSCC chemoprevention requires long term efficacy with multifaceted, nontoxic agents. Accordingly, this study evaluated the abilities of three complementary chemopreventives i.e. the vitamin A derivative fenretinide (4-HPR, induces apoptosis and differentiation, inhibits signaling proteins and invasion), the estrogen metabolite 2-methoxyestradiol (2-ME, apoptosis-inducing, antiangiogenic) and the humanized monoclonal antibody to the IL-6R receptor tocilizumab (TOC, reduces IL-6 signaling) to suppress OSCC gratuitous signaling and tumorigenesis. Modeling studies demonstrated 4-HPR's high affinity binding at STAT3's dimerization site and c-Abl and c-Src ATP-binding kinase sites. Although individual agents suppressed cancer-promoting pathways including STAT3 phosphorylation, STAT3-DNA binding, and production of the trans-signaling enabling sIL-6R, maximal chemopreventive effects were observed with agent combinations. OSCC tumor xenograft studies showed that locally-delivered TOC, TOC+4-HPR and TOC+4-HPR+2-ME treatments all prevented significant tumor growth. Notably, the TOC+4-HPR+2-ME treatment resulted in the smallest overall increase in tumor volume. The selected agents employ diverse mechanisms to disrupt tumorigenesis at multiple venues i.e. intracellular, tumor cell-ECM and tumor microenvironment; beneficial qualities for secondary chemopreventives.
Keywords: secondary chemoprevention, oral squamous cell carcinoma, STAT3, fenretinide, tocilizumab, 2-methoxyestradiol
Introduction
Five-year survival rates for human papillomavirus-negative OSCCs have only marginally improved over the past 40 years and still hover around 50% [1]. Following management of the primary OSCC tumor (surgical resection often accompanied by radiation and/or chemotherapy) patients are managed by close clinical follow up supplemented with CT, PET, or MRI imaging. Despite vigilant monitoring and well-recognized risk factors for recurrence (close margins, immunosuppression, high histologic grade, deep tumor extension) over one third of patients develop life-threatening and often untreatable recurrent OSCCs [2, 3]. Replacement of this “watchful waiting” strategy with a well-tolerated and effective strategy to prevent OSCC recurrence (secondary chemoprevention) could benefit patients.
In an effort to reduce toxicities and bystander effects, cancer drugs have been designed to exploit cancers’ dependence on overexpressed receptors and signaling pathways [4-9]. While conceptually appealing, the collective clinical data from small molecule receptor antagonists and tyrosine kinase inhibitors have been disappointing [8-10]. The EGFR chimeric monoclonal antibody, cetuximab, was ineffective both as monotherapy and also when combined with platinum-based chemoradiation in patients with advanced OSCC [8]. Small molecular tyrosine kinase receptor inhibitors e.g. EGFR family, VEGF and combinations thereof e.g. Vandetanib have also been ineffective [9]. A recent dose escalation trial to evaluate combined Afatinib (EGFR tyrosine kinase inhibitor) and Vargatef (inhibits VEGFR, PDGFR and FGFR tyrosine kinases) treatment on advanced solid tumors resulted in high rates of disease progression and other trial-terminating severe adverse events [10].
Related cancer-targeted drugs have also been developed to blockade downstream signaling hubs such as STAT3 [11]. STAT3, which is activated in many solid tumors including OSCC, upregulates transcription of several cancer-relevant genes including COX-2, IL-6, VEGF, MMPs and can silence genes by DNA methylation [12,13,14]. STAT3 in conjunction with IL-6 participates in an intracrine “feed forward” loop made possible by IL-6's reciprocal activation of STAT3 [13]. High OSCC tumor levels of IL-6 positively correlate with elevated intratumor pSTAT3 levels and a worse prognosis including higher rates of regional and distant metastases [15]. Furthermore, OSCC recurrences are accompanied by elevated serologic levels of IL-6, along with C-reactive protein and serum amyloid [16]. While STAT3 blockade should theoretically abate effects of inappropriately sustained upstream signaling pathways, STAT3 inhibitor trials closed early due to disease progression [17, 18]. Notably, the referenced studies employed the targeted drugs as chemotherapeutics in advanced clinical stage cohorts [6-10, 17, 18]. Their collective results showed that while targeted treatment may be effective initially, signaling redundancies and other compensatory mechanisms ultimately limit efficacy [9, 19]. In contrast, secondary OSCC chemoprevention in a relatively healthy patient cohort requires a distinctive strategy i.e. one that is effective long term and capable of addressing multiple growth perturbations without marked toxicities.
As opposed to targeted drugs, chemopreventives possess multiple mechanisms of action which include growth state regulation, inhibition of angiogenesis, and suppression of signaling cascades [20]. Our lab recently showed fenretinide binds to and perturbs two proteins i.e. FAK and PYK2 essential for signaling and OSCC-ECM interactions including invasion [21]. This current study evaluated the abilities of three chemopreventives i.e. the vitamin A derivative fenretinide (4-HPR), the estrogen metabolite 2-methoxyestradiol (2-ME) and the humanized monoclonal antibody to the IL-6R receptor tocilizumab (TOC) to modulate OSCC cell gratuitous signaling and tumorigenesis. These agents possess complementary mechanisms of action that include induction of apoptosis (4-HPR, 2-ME) and differentiation (4-HPR), capacity to inhibit intracellular signaling proteins and invasion (4-HPR) and reduce IL-6-mediated signaling (TOC) [15, 21-25]. Corresponding studies on OSCC tumors (from which cell lines were isolated) provided corresponding in situ data while molecular modeling studies depicted 4-HPR-cell target interactions. Our results show that while monotherapy provides therapeutic benefits, chemopreventive combinations provide enhanced in vitro and in vivo efficacy.
Materials and Methods
Cell isolation, validation, culture and characterization
OSCC tumor, perilesional and metastatic tissues and corresponding cell lines (fresh tumor tissue derived) were obtained in accordance with Ohio State University Institutional Review Board approval. JSCC-1, JSCC-2, and JSCC-3 cells which were isolated from OSCCs of tonsil, tongue and floor of mouth, respectively, were cultured in Advanced DMEM supplemented with 1X Glutamax and 5% heat-inactivated FBS (GIBCO; Life Technologies; “complete” medium). All OSCC tumors from which the JSCC cell lines were derived represented primary resections and had therefore not been exposed to chemotherapy. For experiments to assess endogenous or growth factor stimulated effects, sera was omitted (“base” medium). Cell lines were authenticated via short tandem repeats profiling analyses at the Genetic Resources Core Facility (Johns Hopkins University, Baltimore, MD). Additional clinical parameters, such as the TNM classification, perineural and vascular invasion are depicted in Supplemental Figure 1. A.
Formalin fixed cells were characterized by incubation with (all antibodies from Abcam, Cambridge, MA) vimentin (1:200) or a pancytokeratin cocktail (AE1/AE3 + 5D3, 1:100,) antibodies, followed by incubation with FITC or Texas Red conjugated secondary antibodies (Abcam) with 4’,6’-Diaminidino-2-phenylindole dihydrochloride (DAPI) nuclear counterstaining. Images were obtained by using an Olympus BX51 microscope (Olympus, Japan), NikonDS-Fi1 digital camera (Nikon, Japan) and ImagePro 6.0 (Media-Cybernetics, Bethesda, MD). Chemopreventives [4-HPR (Cedarburg Pharmaceuticals, Grafton, WI), 2-ME (Sigma-Aldrich, St. Louis, MO) and tocilizumab (Ohio State University James Cancer Hospital Pharmacy)] treatment doses were derived from concurrent cell proliferation (BrdU) and viability (WST) assays with optimal doses defined as retention of comparable cell viability as control cultures that suppressed proliferation. Double and triple agent treatments reduced proliferation to a greater extent than monotherapy, yet cell viabilities remained comparable (data not shown). The highly tumorigenic ATTC CRL-2095 human tongue OSCC cell line (2095sc), which has been well characterized by our lab [18, 25], was also evaluated and used for in vitro and in vivo studies.
Cell line matched OSCC tumor, peritumor tissues and normal human oral mucosa pSTAT3 and pEGFR characterization
Formalin fixed (24-48 h) OSCC tumor tissues corresponding to central tumor, tumor free margins, and metastatic lymph nodes (for JSCC 1, 2 and 3), healthy oral mucosa and ulcerated, non-neoplastic oral mucosal tissues (obtained with Ohio State University IRB approval) were stained with hematoxylin and eosin in addition to signaling-relevant immunohistochemical stains: phospho-STAT3 rabbit monoclonal antibody (1:25, Cell Signaling Tec., Danvers, MA), phospho-EGF receptor rabbit monoclonal antibody (1:200, Cell Signaling Tec., Danvers, MA) or rabbit IgG isotype control (negative control) using standard preparation and incubation conditions, followed by biotinylated secondary antibodies incubation and Vectastain ABC reagent (Vector Laboratories, Burlingame, CA). IHC images were captured via an Olympus BX51 microscope (Olympus, Japan) and Nikon DS-Fi1 digital camera (Nikon, Japan).
Effect of receptor targeted inhibitors on OSCC signaling
OSCC cell lines were pretreated for 1 hour with 0.01% DMSO (vehicle control), 100nM afatinib (Selleckchem, Houston, TX) 100nM Vargatef (Selleckchem), or 100nM afatinib + 100nM Vargatef. Dosing levels were determined by concurrent proliferation and viability studies in conjunction with literature values [26]. The cells in every treatment group were then stimulated for 20 minutes with: vehicle (1μl ddH2O), 50ng/ml EGF, 50ng/ml VEGF, or 50ng/ml EGF + 50ng/ml VEGF, followed by standard immunoblotting and data normalization relative to GAPDH. Additional experiments investigated the effects of 5 μM 4-HPR and 2.5 μM 2-ME treatment on phosphorylation and nuclear translocation of constitutively active STAT3 (JSCC1 and JSCC2) and stimulated (JSCC3) cell lines. Immunoblot images were captured (Li-Cor Odyssey imager) and analyzed (Li-Cor Image Studio, Version 4.0) to depict effects on treatment on phosphorylation relative to respective levels of GAPDH.
Determination of OSCC Cultured Cells’ Endogenous Cytokine Secretion
Conditioned media from 24-hour sera-deprived JSCC-1, JSCC-2 and JSCC3 cells were analyzed using the Proteome Profiler Human Cytokine XL Array (R&D Systems, Minneapolis, MN), with image capture and analyses via the Li-Cor Odyssey imager and Image Studio software (Li-Cor Biosciences, Lincoln, NE). Sera-free conditioned media for IL-6, VEGF, TGF-α and EGF were analyzed by ELISAs (R&D Systems, Minneapolis, MN), with data expressed as pg/106 cells.
Molecular modeling studies to assess 4-HPR-STAT3 and related kinases, c-Src and c-Abl interactions
Molecular modeling studies to evaluate 4-HPR's interactions and potential binding to STAT3's, c-Src's and c-Abl's associated tyrosine kinases and other sites were conducted using AutoDock Vina software [27] with protein structures obtained from the Protein databank [28]. STAT3, c-Src and c-Abl structures were optimized using Yasara and the default minimization algorithm. All ligands were constructed in Spartan10 and minimized using Merck molecular force field. The optimized protein structures and ligands were docked using AutoDock Vina using an exhaustiveness of 100. Each calculation was repeated three times to ensure a thorough exploration of the binding site. Calculated binding free energies were used to determine a binding affinity (Ka) and dissociation constant (Kd) to compare to experimental data. ΔG = RT ln(Ka) or Ka = e−(ΔG/RT) and Kd = 1/Ka. As c-Src can adopt one of two distinct conformations i.e. inactive (closed confirmation) and active (open), 4-HPR interactions were evaluated using both conformations. Similarly, c-Abl also has two formations i.e. inactive DFG-in and active DFG-out and both conformations were analyzed with the same common set of ligands at the ATP binding site.
4-HPR's effects on tumorsphere formation and retention of proliferative capacity
5×105 JSCC1 cells (tumorsphere-formation competent unlike JSCC2 and JSCC3 cells) were plated in complete medium with either vehicle control (0.01% DMSO) or 5μM 4-HPR in Corning Ultra-Low attachment tissue culture flasks (Sigma-Aldrich, St. Louis, MO.) Suspension cultures received fresh medium and treatment q d for 7 days, with daily images capture via the Nikon DS-Ri + NIS Element. After 7 days, cells and medium were harvested, centrifuged, and cells replated in standard culture flasks containing complete medium. Images were obtained daily and cells harvested after 7 days in culture. Cell number (hemocytometer counts) and viability (trypan blue exclusion) were obtained.
Single and combined treatment effects on signaling, transcription factor activation and DNA binding, and cytokine release
Twenty four hour sera-deprived cells were treated for an additional 24 hours in sera free media with the following: 1) 0.01% DMSO (vehicle control), 2) 5 μM 4-HPR, 3) 2.5 μM 2-ME, 4) 1 μg/ml tocilizumab (TOC, ~2.55 μM), 5) 5 μM 4-HPR+ 2.5 μM 2-ME, 6) 5 μM 4-HPR+ 1 μg/ml TOC, 7) 2.5 μM 2-ME+1 μg/ml TOC, 8) 5 μM 4-HPR+2.5 μM 2-ME+1μg/ml TOC. Following treatment, conditioned media were collected (ELISAs for IL-6, sIL-6R, TGF-α, VEGF, EGF, R&D Systems, Minneapolis, MN) and nuclear and cytosolic lysates (Nuclear Extract Kit, Active Motif, Carlsbad, CA) isolated for EMSAs (STAT3, NFκB p50 and p65, TransAM Transcription Factor ELISAs, Active Motif) and cytosolic and nuclear extracts for Westerns (NE-PER Nuclear and Cytoplasmic Extraction Reagents, ThermoFisher Scientific, Waltham MA). Western blot analyses used the following antibodies and dilutions: p-EGFR rabbit monoclonal antibody (1:1000 Cell Signaling Technologies), EGFR rabbit monoclonal antibody (1:2000 Cell Signaling Technologies), p-STAT3 rabbit monoclonal antibody (1:1000 Cell Signaling Technologies), STAT3 rabbit monoclonal antibody (1:2000 Cell Signaling Technologies), Erk1/2 mouse monoclonal antibody (1:2000, Cell Signaling Tec.), and phospho-Erk1/2 rabbit polyclonal antibody (1:1000, Cell Signaling Tec.), NF-κB p65 (rabbit monoclonal, 1:1000, Cell Signaling) and NF-κB p50 (rabbit 1:1000, Cell Signaling), β-actin mouse monoclonal antibody (1:10000 Santa Cruz Biotechnology), BCR-ABL (7C6, 1:1,000, Thermo Fisher Scientific, Rockford, IL), GAPDH rabbit monoclonal antibody (1:10000 Cell Signaling Technologies), histone H3 and YY1 were used as the nuclear loading controls based on targeted protein molecular weight (Nuclear Loading Control and ChIP Grade monoclonal antibodies, Abcam, Cambridge MA). Densitometric analyses used Kodak 1D3 image analysis software, with data normalized to GAPDH (cytosolic) or histone H3 or YY1 (nuclear). For select experiments, cells were treated with the STAT3 inhibitor LY5 (0.5 μM) [29].
Formulation of controlled-release polylactide-co-glycolide (PLGA) 4-HPR implants for xenograft studies
PLGA millicylinders, which consisted of 60% 50:50 acid end capped PLGA (24-38 kDa), were prepared by solvent extrusion method by dissolve 60% 50:50 acid encapped PLGA (24-38 kDa) in acetone, followed by addition of 4-HPR and excipients [MgCO3 and sodium deoxycholate (NaDC)], polymer drying and removal of the encasing silicon tubing. Optimization studies were conducted to enhance 4-HPR loading (up to 30%), release kinetics (20% NaDC), pore formation (MgCO3) and drug solubilization/prevention of crystallization (B-cyclodextrin, hydroxypropyl methyl cellulose (HPMC) K4M, and polyvinyl pyrollidone (PVP K30).
Determination of 4-HPR tumor levels entailed recording tumor wet mass, addition of internal standard (acitretin) and RIPA lysis buffer, followed by homogenization, addition of acetonitrile and centrifugation. Acetretin percent recovery was compared to a RIPA buffer/acetonitrile extraction control solution spiked with equivalent amount of acetretin and 4-HPR. Sera preparation entailed addition of acetretin and acetonitrile, sonication, followed by centrifugation. UPLC/UV analyses showed no evidence of the oxidized metabolite 4-oxo-4-HPR when compared to the calibration standard in either the tumor or sera samples.
Evaluation of therapeutic efficacy using OSCC tumor xenografts
SCC2095sc cells [106 cells suspended in 100 μl Matrigel (Corning Life Sciences, Corning, NY)] were subcutaneously injected in the flanks of 6 week old male nude mice (n=6 per treatment group). Tumor measurements were recorded daily with calipers (greatest length and greatest width) and final tumor volumes calculated via tumor volume V= (length × width2) × ½. Treatment groups consisted of: 1) control (PBS injections) + blank (no drug) PLGA implants, 2) 2-ME (1μg/100μl PBS, b.i.d.) + blank PLGA implant, 3) 4-HPR releasing PLGA implant + PBS injections, 4) TOC q.d. (0.3μg/100μl PBS) + PLGA blank implant, 5) 2-ME (1μg/100μl PBS, b.i.d.) + 4-HPR releasing PLGA implant, 6) 2-ME (1μg/100μl PBS, b.i.d.) + TOC q.d. (0.3μg/100μl PBS) + blank PLGA implant, 7) TOC q.d. (0.3μg/100μl PBS) + 4-HPR releasing PLGA implant, 8) 2-ME (1μg/100μl PBS, b.i.d.) + TOC q.d. (0.3μg/100μl PBS) + 4-HPR releasing PLGA implant. By day 14 post injection, all mice had measurable tumors and treatment began on day 15. Attempts to achieve uniform tumor size distribution among groups by animal transfer resulted in aggression toward the new cage mates. Pretreatment mean tumor volumes therefore varied among treatment groups. PLGA implants (via trocar) and injections were placed in the center of the tumor. At day 28 post OSCC xenograft placement final tumor measurements were obtained, the mice were euthanized and OSCC tumors along with lung, liver and sera were harvested for pharmacokinetic, histologic and IHC analyses (Ki-67, cleaved caspase-3 and involucrin). Image analyses of the nuclear stains (Ki-67 and cleaved caspase 3) were conducted using Image-Pro Plus 6.2 software (Media Cybernetics, Rockville, MD.).
Statistical analyses
Data normality (Shapiro Wilks normality test) determined whether parametric or nonparametric analyses were employed. The Wilcoxon matched pairs signed rank test was used to assess the effects of 4-HPR treatment on STAT3 activation. Effects of combination treatments on STAT3-DNA binding and cytokine release as well as image analyzed IHC tumor data were evaluated by the Kruskal Wallis ANOVA followed by the Dunn's Multiple Comparison post-hoc test (individual cell lines, IHC data) or One Way ANOVA, followed by Tukey's multiple comparison test (combined cell line data). A paired t test (pre versus post treatment individual tumor measurement) was used to assess the effects of treatment on tumor volume.
Results
JSCC cells retain features of their corresponding tumor tissues
All JSCC lines contained dual cytokeratin and vimentin staining cells. (Supplemental Figure 1.A.) Normal oral epithelia showed sparse pSTAT3 and no pEGFR nuclear staining in basal cells with modest increases noted following ulceration (Supplemental Figure 1.B.). Cell-matched tumor tissues, however, showed membrane-associated pEGFR with highest expression in the JSCC2 tumor tissue. pSTAT3 nuclear staining was highest in the JSCC1 and JSCC2 tumors; similar to corresponding cells lines’ constitutive STAT3 phosphorylation. All three JSCC lines also demonstrated constitutive EGFR and/or STAT3 signaling similar to their corresponding tumors’ high in vivo expression.
Afatinib and Vargatef's effects are cell line specific
JSCC2 cells demonstrated high constitutive activation of pSTAT3 and pERK1/2 and modest levels of pEGFR. The other lines showed modest (JSCC3-pEGFR and ERK1/2) and modest (JSCC1-pSTAT3) constitutive activation, respectively. EGF stimulation increased phosphorylation of EGFR (all lines) and ERK1/2 phosphorylation (JSCC1 and 2) whereas VEGF elicited more modest responses in the JSCC1 and 2 lines, primarily increasing pSTAT3 levels (Figure 1.A.). Responses to the EGFR tyrosine kinase inhibitor (Afatinib) and the triple angiogenic (VEGF, bFGF, PDGF) tyrosine kinase inhibitor (Vargatef) were also cell-line dependent. Although Afatanib and Vargatef treatment reduced phosphorylation in JSCC3 cells at the EGFR, ERK1/2, and STAT3 levels, high to moderate levels of phosphorylated STAT3 persisted in JSCC1 and JSCC2 cells despite treatment(s).
Figure 1.
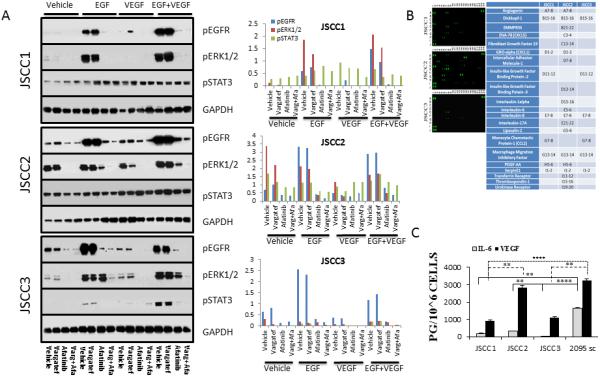
A. Effects of Afatanib and Vargatef on OSCC intracellular signaling. Cells were cultured for 24 h in sera free base medium, followed by a 1 hour pretreatment with Afatanib (100 nM) and/or Vargatef (100 nM) followed by a 20 minute stimulation with EGF (50 ng/ml) and/or VEGF (50 ng/ml) prior to harvesting. The extent of constitutive signaling (vehicle lane) was cell line dependent. JSCC2 cells showed the highest baseline signaling (constitutive pERK1/2 and pSTAT3), while JSCC1 and JSCC3 cells showed modest constitutive pSTAT3 and pERK1/2 activation, respectively. Growth factor challenge (50 ng/ml both EGF and VEGF singularly and in combination) increased receptor and downstream signaling activity with VEGF inducing some crossover EGFR activation. While Afatinib and Vargetef blocked STAT3 phosphorylation in the JSCC3 cells, pSTAT3 persisted in the JSCC1 and JSCC2 cell lines regardless of treatment. The accompanying histogram depicts levels of phosphorylated ERKR, ERK1/2 and STAT3 relative to GAPDH.
Figure 1.B. Proteome profiles reveal cell line specific cytokine release. Conditioned media from 24 hour sera-deprived JSCC cell lines were analyzed to determine extent of autologous cytokine release. Cytokines uniformly released by all cell lines were: Dickkofp-1, IL-8, and macrophage inhibitory factor. Only the Afatnaib and Vargetef refractory JSCC1 and JSCC2 cell lines released the proangiogenic proteins angiogenin, CXCL1, and PDGF-AA. Notably, Vargetef does not block PDGF-AA signaling.
Figure 1.C. Inter-cell line heterogeneity extends to OSCC-relevant cytokine release. To provide more quantitative assessments, ELISA analyses were conducted to assess autologous production and release of three OSCC-relevant cytokines i.e. IL-6, VEGF, EGF and TNFα from 24 hour sera-deprived cells (n=9 for every cell line, mean±s.e.m. pg/106 cells). None of the lines released detectable levels of epidermal growth factor (assay level of detection 3.9 pg/ml), with only released low levels of TGFα. Inter-cell line comparisons of IL-6 and VEGF release revealed significant differences (Kruskal Wallis followed by a Dunn's Multiple Comparison post hoc test, **=p<0.01, ****=p<0.0001).
Cell lines release unique cytokine profiles
Proteome profiling showed JSCC2 cells released the most cytokines (19) relative to 9 and 6 for the JSCC1 and JSCC3 cells respectively (Figure 1.B.). All three cell lines released Dickkopf-1, Interleukin-8, and Macrophage migration inhibitory factor. The constitutive pSTAT3 and tyrosine kinase blockade resistant JSCC1 and JSCC2 cells exclusively released Angiogenin, CXCL1 and PDGF-AA. Quantification of the STAT3 activating cytokines (IL-6, EGF, and TGF-α) by ELISAs revealed high levels of IL-6 release in JSCC2 and the highly tumorigenic 2095sc cells, followed by moderate and negligible IL-6 release in JSCC1 and JSCC3 cells respectively (Figure 1.C.). None of the cell lines released EGF or TGF-α. 4-HPR inclusion (5 μM, fresh treatment q.d., base medium) increased IL-6 release, which was greatest at the 48h time point with increases of~50% and ~20% in the JSCC1 and JSCC2 cells respectively (data not shown).
4-HPR (5μM) singularly and in combination with 2-ME (2.5μM) and TOC (1 ug/ml) inhibits STAT3 cytosolic phosphorylation and nuclear translocation
Cytosolic and nuclear extract immunoblots demonstrated that 5 μM 4-HPR suppressed STAT3 phosphorylation and nuclear translocation in the STAT3 constitutively phosphorylated cell lines (Figure 2.A.). Also, 4-HPR was appreciably more effectively at suppressing STAT3 activation and nuclear translocation in JSCC1 cells relative to the STAT3 inhibitor LY5 [29].
Figure 2. Treatment effects of fenretinide (4-HPR, 5μM), 2-methoxyestradiol (2-ME, 2.5 μM) and the STAT3 inhibitor LY5 (0.5 μM) [30] on STAT3 phosphorylation and nuclear translocation.
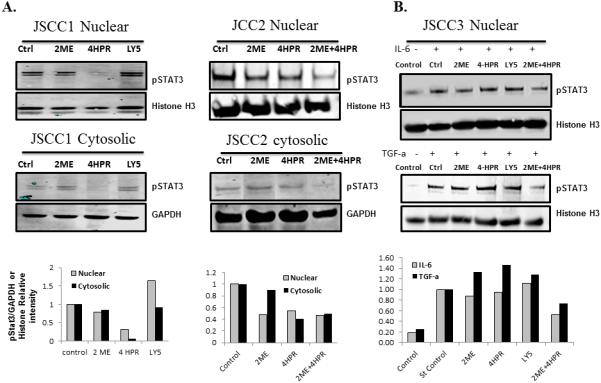
Figure 2.A. (JSCC1 cells), B. (JSCC2 cells) and C. (JSCC3 cells). Cytosolic and nuclear proteins were harvested from sera deprived JSCC1 and JSCC2 cell lines following 24 h of treatment (5 μM 4-HPR, 2.5 μM 2-ME, or 0.5 μM LY5) or vehicle (0.1% DMSO) control. Multiple experiments confirmed 5 μM 4-HPR treatment significantly reduced constitutive STAT3 phosphorylation and pSTAT3 nuclear translocation in the STAT3 constitutively active JSCC1 and JSCC2 cells (p< 0.05, Wilcoxon matched pairs signed rank test, n=7). C. As the JSCC3 cells don't constitutively express pSTAT3, these cells underwent 24h stimulation in base medium supplemented with 10 ng/mL of IL-6 or 5 ng/mL of TGF-α, with or without 4-HPE, 2-ME or LY5 treatment, followed by harvest. The combination treatment of 2ME and 4-HPR significantly inhibited IL-6 or TGF-α induced pSTAT3 nuclear translocation in JSCC3 cells. (n=3, p<0.05, Wilcoxon matched pairs signed rank test). Figure 2.D. Effect of single and combination treatments of fenretinide (4-HPR, 5μM), 2-methoxyestradiol (2-ME, 2.5 μM) and the IL-6 receptor inhibitor TOC (1 ug/ml) on STAT3 and pSTAT3 levels. Figure D, E. All cell lines with constitutive pSTAT3 expression (2095sc, JSCC1 and JSCC2) were sera deprived for 24h, followed by an additional 24 hour of treatment in sera-free medium that contained: control (0.1% DMSO), 5μM 4-HPR, 2.5 μM 2-ME, 1 μg/ml TOC (initial agent concentrations same for single and combinations). As the JSCC3 cells don't constitutively express pSTAT3, these cells underwent 24h stimulation in base medium supplemented with10 ng/mL of IL-6 and 5 ng/mL of TGF-α, with or without 4-HPR, 2-ME and TOC followed by harvest. Selective treatments induced reduction of both STAT3 and pSTAT3 levels in2095sc cells, and to a lesser extent JSCC1 and JSCC3 cells. The treatment combinations of TOC + 4-HPR and TOC+4-HPR+2-ME induced significant inhibition of STAT3 phosphorylation [p<0.05, n=12 total with n=3 for every individual cell line (including JSCC3)], Kruskal Wallis followed by Dunn's Multiple Comparison post hoc test).
Selected combination treatments induced reduction of STAT3 and pSTAT3 levels in 2095sc cells and to a lesser extent the JSCC1 and 2 lines (Figure 2. C). In addition the treatment combinations of TOC + 4-HPR and TOC+4-HPR+2-ME significantly inhibited STAT3 phosphorylation [p<0.05, n=12 total with n=3 for every individual cell line (Figure 2.D., including JSCC3)], Kruskal Wallis followed by Dunn's Multiple Comparison post hoc test).
Molecular modeling of 4-HPR interactions with STAT3 and additional nonreceptor kinases c-Abl and c-Src
Molecular modeling data revealed that 4-HPR and its oxidized metabolite 4-oxo-4HPR bind along the arm (right side of the figure above with the amino acid side chains displayed), while other STAT3 inhibitors bind in the small pocket with the beta-sheet near Tyr 640 and Lys 591 (lower left side of the amino acids with side chains showing, consistent with models for XZH-5, STA-21, pCinn-Nle-mPro-Gln-NHBn, LYS5 and others. (Figure 3.A.). Notably, 4-HPR also binds with extremely strong affinity (−14.2kcal/mol) at an additional binding site in the SH2 domain. At this energy level, 4-HPR should demonstrate efficacy at nanomolar levels, interfere with STAT3 dimerization and due to Tyr705's proximity to SH2, possibly perturb phosphorylation.
Figure 3.
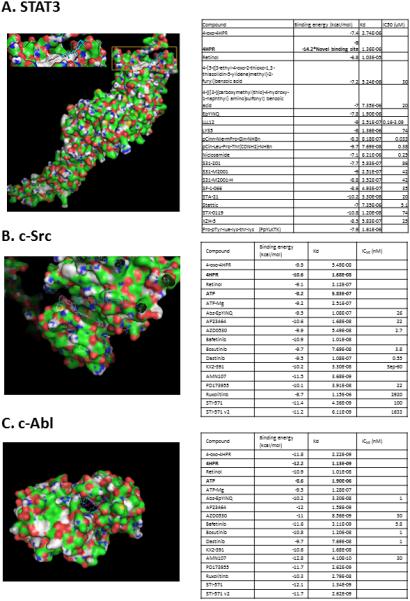
A. 4-HPR demonstrates both moderate and high-affinity binding at STAT3's SH2 dimerization site. The STAT3 protein structure was obtained from the Protein databank1 1BG11a. The STAT3 structure was optimized via Yasara3 using the default minimization algorithm. All ligands were constructed in Spartan102 and minimized using MMFF5. The optimized protein structure and ligands were docked using AutoDock Vina4 employing an exhaustiveness of 100. Each calculation was repeated three times to ensure a thorough exploration of the binding site. Previous results have shown that flexible amino acid side chains provide for better results6 therefore the side chains for amino acids: 591, 592, 595, 597, 609, 611-613, 620, 623, 635, 637, 638, 640, 657, 705-710 and 712 were made flexible to ensure a more realistic binding mode for all calculations. The binding energy (−14.2) observed for 4-HPR at the newer binding site imply nM levels of 4-HPR would impede STAT3 dimerization.
Figure 3.B. Kinase inhibitor site molecular docking data suggest 4-HPR has nanomolar level affinity for c-Src's ATP binding site. With the exception of 4HPR and KX2-391 all other evaluated ligands bind in an analogous fashion to ATP i.e. lying in a “groove” on the protein surface between a beta-sheet and random coil. In contrast, 4HPR and KX2-391 bind with an orientation more perpendicular to the protein surface (in the same groove) and penetrate much deeper into c-Src's interior. This unique binding conformation is only 0.5 kcal/mol lower in binding energy compared to parallel to the groove. Finally, as c-Src is self-inhibited in the closed confirmation, binding and affinity data reflect modeling data employing c-Src's active “open” confirmation.
Figure 3.C. 4-HPR also demonstrates nanomolar level affinity for c-Abl's kinase ATP binding site. As Abl also has active “open” and inactive “closed” conformations, modeling data depict interactions with the open conformation. Notably, the fusion of BCR sequences to ABL during the translocation associated with CML (“Philadelphia Chromosome”), increases the tyrosine kinase activity of c-Abl.
4-HPR and 4-oxo-4HPR also demonstrated nanomolar level binding affinities (−10.6 kcal/mol for both) for c-Src's ATP binding site in both the active (Figure 3.B.) and inactive configurations (Supplemental Figure 2.A.). These affinities are comparable to the c-Src selective inhibitors Dastinib, AP23464 and PD173955. With the exception of 4-HPR and KX2-391, all other c-Src inhibitors bind similarly i.e. the inhibitor lies in a “groove” on the protein surface between a beta-sheet and random coil. In contrast, 4-HPR and KX2-391 bind with a more perpendicular orientation relative to the protein surface yet within the same groove as the other inhibitors (Figure 3.B.). 4-HPR's unique binding orientation enables deeper penetration into c-Src's protein interior.
Similarly, 4-HPR and 4-oxo-4-HPR are also nanomolar level inhibitors of c-Abl's ATP binding site in both its active and inactive conformations (binding energies of-12.2 and -13.1 kcal/mol, respectively) (Figure 3.C.). Although 4-HPR and 4-oxo-4HPR bind in the same pocket as other inhibitors, 4-HPR and to a lesser extent 4-oxo-4HPR bind in an orientation that places them deeper into c-Abl interior analogous to bosutinib, dastinib and KX2-391 (Figure 3.C.). Notably, 4-HPR binds with appreciably higher affinity at the ATP binding sites relative to ATP (10 and 100 fold higher for c-Src active and inactive sites and 1000 fold greater for c-Abl active and inactive sites (Supplemental Figure 2.B..).
4-HPR inhibits tumorsphere formation and eliminates further growth
While 5×105 JSCC1 cells (and not JSCC2 or 3 cells) readily formed tumorspheres in low attachment flasks + complete medium, 5 μM 4-HPR inclusion inhibited tumorsphere formation (Figure 4A.) Following re-plating in complete medium in standard tissue culture flasks, control cells reattached, resumed proliferation and retained ≥95% viability after 7 days in culture. In contrast, 5μM 4-HPR treated cells were not viable 100% trypan blue uptake) and failed to reattach. (Figure 4.B.).
Figure 4.
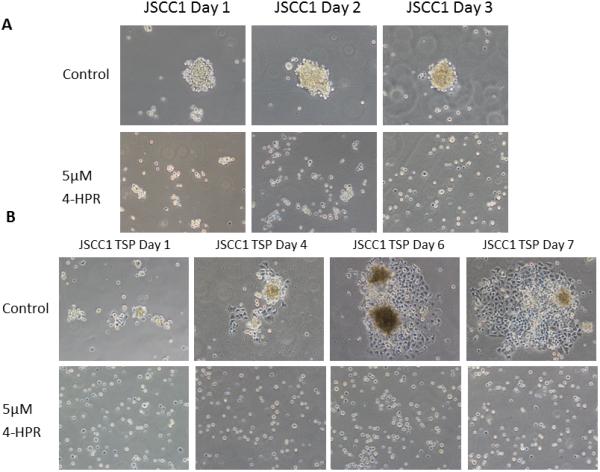
Figure 4.A. 4-HPR inhibits tumorsphere formation. JSCC1 cells were seeded at density of 5×105 cells in Advanced DMEM +5% FBS + 1x GlutaMAX Supplement in Ultra-Low Attachment surface T-25 flasks with DMSO control (0.05%) and 5μM 4-HPR treatment. Cell suspension cultures were treated for 3 days with fresh treatment every 24 h. While control cultures formed tumorspheres by 24 h, 4-HPR treated cellular interactions were limited to small cell aggregates. (Image scale at 100x.)
Figure 4.B. Cell attachment inhibition remains following 4-HPR removal. Following the treatment described in Figure 4.A. above, the JSCC1 control and 4-HPR treated cells were returned to standard tissue culture flasks for seven days in 4-HPR free complete medium (Advanced DMEM +5% FBS + 1x GlutaMAX Supplement). The control cultures readily reattached, formed cellular islands with readily apparent mitotic figures and possessed high viabilities (>95%, trypan blue exclusion). In contrast, the cells previously treated with 4-HPR failed to reattach and were uniformly nonvital. (Image scale 100x.)
Combination chemopreventive treatments inhibit STAT3-DNA binding
While monotherapy did not significantly inhibit STAT3-DNA binding, combination treatments that included 4-HPR [(4-HPR+2-ME, p<0.05), (4-HPR+TOC, p<0.01)] and all three agents (4-HPR+2-ME+tocilizumab, p<0.001) significantly inhibited STAT3-DNA binding (Figure 5.A.). Supplemental Figure 3.A. depicts individual cell line treatment effects.
Figure 5. Evaluation of the effects of single and combination treatment of fenretinide (4-HPR, 5μM), 2-methoxyestradiol (2-ME, 2.5 μM) and the IL-6R inhibitor TOC (1 ug/ml) on STAT3-DNA binding and on release of the stromal and tumor cell activating, trans-signaling molecule, sIL-6R.
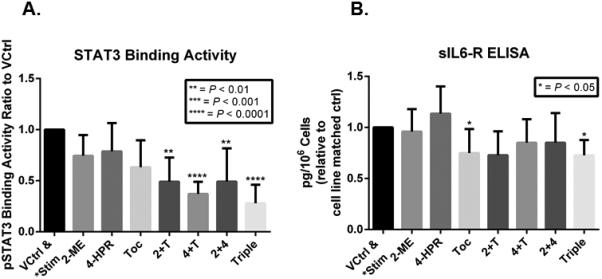
A. Treatment effects of STAT3-DNA binding. All cell lines with constitutive pSTAT3 expression (2095sc, JSCC1, JSCC2) were sera deprived for 24h, followed by an additional 24 hours of treatment in sera-free medium that contained: control (0.1% DMSO), 5μM 4-HPR, 2.5 μM 2-ME, 1 μg/ml TOC (initial agent concentrations same for single and combinations). As the JSCC3 cells don't constitutively express pSTAT3, these cells underwent 24h stimulation in base medium supplemented with10 ng/mL of IL-6 and 5 ng/mL of TGF-α, with or without 4-HPR, 2-ME and TOC followed by harvest. A. Collective cell line data (expressed as change relative to cell-line matched control) revealed dual and triple chemopreventive combinations significantly reduced STAT3 binding with its cognate DNA binding sites. (n=13, Kruskal Wallis ANOVA followed by Dunn's Multiple Comparison post hoc test). Corresponding analyses of single cell line data (n=3 for all cell lines with exception of n=4 for JSCC2) revealed triple treatment significantly (p<0.05) inhibited STAT3 binding in every individual cell line with constitutively active STAT3 (JSCC1, JSCC2 and SCC2095sc). B. Effects of treatment on cell line release of sIL-6R. All four cell lines released sIL-6R [levels ranges from ~750 fg/106 (JSCC2 cells baseline) to ~ 3,000 fg/106 cells (stimulated JSCC3 cells, baseline production JSCC3~2,000 fg/106 cells). The sIL-6R humanized monoclonal antibody (tocilizumab) when administered singularly and in a triple treatment combination significantly inhibited sIL-6 release (p<0.005, Kruskal Wallis followed by Dunns Multiple Comparison test). (Please see Supplemental Figure 3 to view individual cell line based sIL-6R production.)
All cell lines produced sIL-6R. JSCC3 cells’ sIL-6R levels were the highest (albeit post 10 ng/mL of IL-6 and 5 ng/mL of TGF-α stimulation as opposed to other sera deprived lines), followed by constitutive production in the 2095sc, JSCC1 and JSCC2 cells respectively (Supplemental Figure 3.B.). Two treatments i.e. TOC and the 4-HPR, 2-ME and TOC triple treatment significantly reduced sIL-6R release (Figure 5.B.).
Studies to assess effects of treatment on binding of the NF-κB subunits p65 and p50 showed modest reduction only in the 2095sc (p65) and JSCC2 (both) cell lines (See Supplemental Figure 4 ).
In vivo studies show benefits of tumor-directed, multimodal therapy
All mice injected with Matrigel-2095sc cells developed OSCC tumors. Although all OSCC xenografts were well-vascularized, necrotic foci were observed in regions with high proliferation indices and adjacent to 4-HPR implants. Lymphovascular (several mice) and perineural (single mouse) invasion by tumor cells were observed in the 2-ME treatment group; the mouse with perineural invasion had lung metastases at sacrifice. Pretreatment tumor volumes varied appreciably within and among treatment groups (Figure 6), findings that may reflect variations in tumor growth capacity including angiogenesis. Only the TOC, TOC + 4-HPR implant, and TOC + 2-ME + 4-HPR implant treatments prevented a significant increase in tumor volume over 14 days (See Figure 6. A.). Notably, the TOC + 2-ME + 4-HPR implant group's mean pretreatment tumor volumes were nearly two fold higher than TOC group. The mean tumor fold size increases for the TOC and TOC + 2-ME + 4-HPR treatments were 2.13 and 1.72, respectively. 4-HPR implants released between 300-370 μg drug and achieved tissue levels of 331+/−134 μM. IHC image analysis of the OSCC tumors’ Ki-67 staining revealed that while all treatments decreased OSCC cell proliferation relative to control tumors, significant suppression was only observed in selected groups [2ME+TOC+4-HPR, TOC+4-HPR, 2-ME+TOC p≤0.001; 2-ME+4-HPR p≤ 0.05] (Figure 6). Finally, although treatments occurred just below the skin, no disruption, ulceration or histologic changes were observed in the overlying epidermis. Ki-67 staining was apparent in the basal layer keratinocytes consistent with normal proliferation. UPLC/UV analyses showed no evidence of the oxidized metabolite 4-oxo-4-HPR when compared to the calibration standard in either the tumor or sera samples. Furthermore, 4-HPR sera levels were below LLOQ (<50 ng/ml) in all mice that received PLGA implants.
Figure 6.
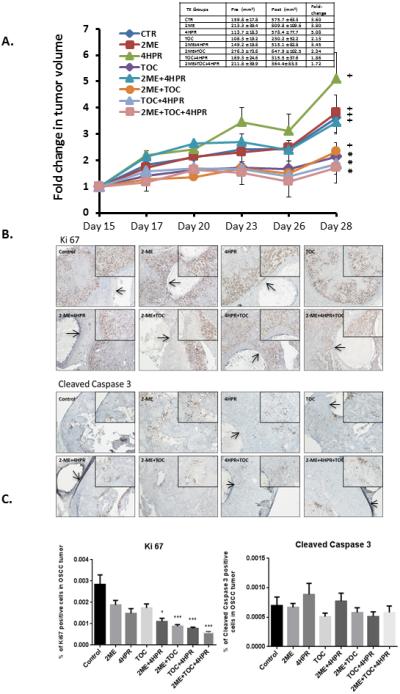
A. One million SCC2095sc cells (suspended in 100 μl Matrigel) were subcutaneously injected in in flanks of 6 to 7 week old male nude mice (n=6 per treatment group). By day 14 post injection, all mice had palpable and measurable tumors. Treatment began on day 15. Caliper-based tumor measurements were recorded daily and final tumor volumes calculated via tumor volume V= (length × width2) × ½. Treatment groups consisted of: 1) control (PBS injections) + blank (no drug) polylactide co-glycolide (PLGA) implants, 2) 2-methoxyestradiol (2-ME) (1μg/100μl PBS, b.i.d.) + blank PLGA implant, 3) fenretinide (4-HPR) releasing PLGA implant + PBS injections, 4) tocilizumab (TOC) q.d. (0.3μg/100μl PBS) + PLGA blank implant, 5) 2-ME (1μg/100μl PBS, b.i.d.) + 4-HPR releasing PLGA implant, 6) 2-ME (1μg/100μl PBS, b.i.d.) + TOC q.d. (0.3μg/100μl PBS) + blank PLGA implant, 7) TOC q.d. (0.3μg/100μl PBS) + 4-HPR releasing PLGA implant, 8) 2-ME (1μg/100μl PBS, b.i.d.) + TOC q.d. (0.3μg/100μl PBS) + 4-HPR releasing PLGA implant. In previous studies that employed younger mice tumors of varying sizes were disbursed among all treatment groups. Due to the older age of mice >6 weeks and the accompanying cage aggression tumor size distribution was not feasible in these studies. As a result, mean tumor volumes varied appreciably among treatment groups (See Table insert Figure 6). PLGA implants (trocar placement) and injections were placed in the center of the tumor. Fluid dispersion throughout tumor was noted during injections. At day 28 post OSCC xenograft placement the mice were sacrificed, OSCC tumors along with lung and liver tissues and sera were harvested (for PK and histologic analyses). Our data show three groups i.e. TOC, TOC + 4-HPR, and triple treatment (TOC+2-ME+4-HPR) were the only treatments that prevented a significant increase in tumor volume (paired t test). Provided the discrepancy in pretreatment tumor volumes (TOC the lowest, triple treatment second highest with its mean nearly two fold higher than TOC mean), the TOC + 2-ME + 4-HPR data are especially creditable.
Figure 6. B. Ki-67 and cleaved caspase 3 immunohistochemical stains were conducted to assess qualitative treatment effects on tumor cells’ proliferation and apoptosis. IHC images representative of all of the groups are presented. The PLGA implants are visible as a clear cylindrical object in every photomicrograph except the TOC and 2-ME + TOC treatment. To facilitate PLGA implant recognition, arrows have been placed within the implants. Both the Ki-67 and caspase 3 stains are located within the nucleus. Qualitative Ki67 staining assessment was most apparent at the periphery of the tumors in all groups. In contrast to the abundant Ki-67 staining, cleaved caspase 3 was not nearly as prominent.
Figure 6.C. Image analysis revealed that all treatments suppressed tumor cell proliferation relative to the untreated tumor. Furthermore, significant inhibition of proliferation (as assessed by % of tumor cells demonstrating Ki-67 labeling) was seen in these groups: 2-ME+4-HPR+TOC, TOC+4-HPR, 2-ME+TOC (all p<0.001) and 2-ME+4-HPR (p<0.05) Kruskal Wallis with Dunns’ Multiple Comparison post hoc test. Although cleaved caspase 3 nuclear staining was present, none of the treatments showed any significant effects relative to the matched control tumors. Image analysis did not reveal any inter-group differences with regard to caspase 3 labeling. (Image scale 4X and 10X for smaller and larger photomicrographs, respectively.).
Discussion
This study evaluated multifaceted chemopreventives’ abilities to suppress OSCC tumor-promoting pathways i.e. gratuitous signaling, constitutive transcription factor activation and DNA binding, anchorage independent growth and tumorigenesis [21-25]. Collectively, our results show combinations of agents with complementary mechanisms of action provided enhanced efficacy at both the in vitro and in vivo levels.
Afatinib and Vargatef signaling inhibition was cell-line dependent and negatively correlated with constitutive ERK1/2 and STAT3 signaling. There were also marked inter-line differences in both the numbers and levels of cytokines produced. Consistent with the STAT3-IL-6 feed forward loop, lines with constitutive STAT3 activation released higher IL-6 levels. IL-6 levels detected (range~100-1,600 pg/106 cells) were comparable to IL-6 release from other human cancer cells (ovarian 100-200 pg/106 cells, prostate 850-1250 pg/106 cells) [32, 33]. Similarly, VEGF release (range ~ 1,000-3,000 pg/106 cells) also compared favorably to previous OSCC cell data (1,500 pg/106 cells) [34]. Consistent with the presence of the tumor promoting Bcr-Abl oncogene, the 2095sc cells released the highest levels of IL-6 and VEGF among the OSCC cell lines.
Molecular modeling studies showed concordance between 4-HPR binding and other STAT3 inhibitors i.e. fairly weak binding (IC50 values in the μM range) at the “standard” SH2 STAT3 binding site. 4-HPR, however, also binds to a novel STAT3 SH2 site at a high affinity that conveys a nanomolar energy equivalency level. In addition, 4-HPR's binding orientation enables greater penetration relative to standard inhibitors into Src's and Abl's protein interiors at their ATP binding sites. This unique orientation results in nanomolar level affinities that are 10 fold (Src) and 100 fold (Abl) greater than the binding of the endogenous ligand, ATP. Notably, both Src and Abl kinases contribute to constitutive STAT3 activation [35] and Abl functions in cell transformation via a Src/Abl/Rac/JNK/STAT3 signaling cascade [36]. Thus, 4-HPR's enhanced STAT3 inhibition relative to standard inhibitors e.g. LY5 [29] likely reflects both its unique high affinity SH2 interaction plus its upstream Src and Abl inhibition. Although Bcr-Abl is generally associated with chronic myelogenous leukemia, two OSCC clinical trials evaluated systemically administered Bcr-Abl inhibitors [Saractinib (dual Src and Bcr-Abl inhibitor) and Dasatinib (multikinase inhibitor of PDGFR, Bcr-Abl, c-Kit)] in recurrent or metastatic OSCC patients [37, 38]. While unsuccessful, these studies confirmed a Bcr-Abl association with OSCC pathogenesis [37, 38]. As the Abl ATP binding site remains constant in the Bcr-Abl fusion protein [31], 4-HPR's ATP-blocking effects should extend to this oncoprotein. This current and a previous study from our lab [21] depict 4-HPR's capacity to perturb tyrosine kinases via binding at higher affinities than the natural ligand at functional sites. Although chemically derived from retinol i.e. retention of the trimethylcyclohexenyl group and polyene chain, 4-HPR replaces retinol's hydroxyl group with a stability enhancing amide and a redox-active phenol ring. These changes apparently provide 4-HPR with unique protein interactive and binding capacities. Structure function analyses to assess 4-HPR-retinol binding protein interactions suggested bound 4-HPR induced both steric hindrance and target protein conformational changes [39]. Our current and previous modeling-functional analyses [21] suggest 4-HPR's high affinity protein interactions extend to tyrosine kinases integral for carcinogenesis.
While 4-HPR suppressed STAT3 phosphorylation and nuclear translocation in a cell line dependent fashion, combining 4-HPR with TOC and 2-ME provided the most pervasive effects. Furthermore, 4-HPR, 2-ME and/or TOC combination treatments significantly inhibited pSTAT3 binding to its cognate DNA site. Numerous mechanisms may be attributable for these results including 4-HPR's inhibition of STAT3 phosphorylation, impaired nuclear translocation potentially attributable to 4-HPR-mediated steric hindrance, intranuclear dephosphorylation, and/or upregulation of SOCS-1 [40]. Of interest, unphosphorylated STAT3 can bind with unphosphorylated NF-κB resulting in complex-mediated activation of κB-dependent genes including IL-6, VEGF, COX-2 [41]. Studies to assess 4-HPR, 2-ME and TOC's effects on two primary NF- κB subunits (p50 and p65) at κB DNA binding sites revealed 4-HPR + TOC reduced DNA binding of the gene activating p65 subunit in the two high VEGF producing cell lines while binding of the apoptosis-associated p50 remained constant in the 2095sc line [42]. While previous studies demonstrated 2-ME disrupted NF-κB activation and DNA binding in neuroectodermal brain tumors [43], our data, which showed 2-ME didn't inhibit OSCC cells’ NF-κB activation (data not shown), imply tumor type specificity. As STAT3 retains p65 in the nucleus [13], our data may also reflect reduced nuclear STAT3 levels. Also, our results showed, for the first time, sIL-6R production by OSCC cells. Although OSCC sIL-6R levels released by 106 OSCC cells were negligible, the number of cells in an OSCC tumor could elevate production to tumor-enabling levels. As s-IL6R enables IL-6 signaling in cells without IL-6R, serves as an IL-6 carrier and extends IL-6 half-life, autologous OSCC sIL-6R release would enhance tumorigenesis. Not surprisingly, treatments that disrupted intracellular signaling concurrently inhibited sIL-6R release. These data suggest local implant-induced sIL-6R inhibition could also perturb sIL-6R release from high output cells like PMNs which would significantly impact the tumor milieu [44]. In addition, 4-HPR inclusion induced anoikis in otherwise tumorsphere-competent cells. These findings are consistent with STAT3's role in anoikis resistance, STAT3's and FAK's contributions to anchorage independent growth, and 4-HPR's interference with FAK and STAT3 function [21, 45, 46]. As tumorsphere formation is associated with adoption of cancer stem cell features including self-renewal and resistance to cytotoxic agents, 4-HPR's prevention of anchorage independent growth could be highly beneficial for chemoprevention.
Although the long range goal is to develop an effective locally delivered secondary OSCC chemopreventive strategy, in vivo studies assessed capacity to suppress growth in established OSCC tumors. Our rationale for selection of this in vivo model is as follows. As the OIN to OSCC transition can be extremely brief at former resection sites, formation of microtumor foci is a realistic clinical concern [2, 3]. Provided this scenario, secondary chemopreventives would be required to inhibit OSCC tumor development, albeit on a smaller scale than currently tested. From the experimental perspective, a tumor initiation xenograft model optimally includes a cell line that generates uniformly-sized OSCC tumors in 100% of injected mice. As our lab had not yet established such a line, we employed a model with a higher level of experimental control i.e. confirmation of tumor growth was confirmed prior to treatment and integration of pretreatment and posttreatment tumor size comparisons. Notably, the most aggressive OSCC cell line, characterized by high production of OSCC-relevant cytokines i.e. IL-6, VEGF and sIL-6R and presence of the Bcr-Abl oncoprotein was selected for implantation.
Previous studies demonstrated 2-ME liposomes (150 mg/kg, i.p.) significantly reduced HNSCC tumor formation and 2-ME + paclitaxel (20 mg/kg i.p.) suppressed growth of established tumors [47]. The lack of 2-ME efficacy to reduce tumor size in our studies likely reflects both drug instability (~4 h half-life) and use of appreciably (>2,000) lower doses. Additionally, tocilizumab (100 μg i.p.) significantly reduced HNSCC tumor initiation in scid mice; however this treatment didn't significantly inhibit established tumor growth [48]. In contrast, our data showed appreciably lower local tocilizumab doses (0.3 μg q.d.) significantly inhibited OSCC tumor growth. Further, as systemic tocilizumab can cause significant immune suppression, local delivery is preferable from both efficacy and safety perspectives. Despite achieving tumor 4-HPR levels that were appreciably greater than apoptosis-inducing 10 μM [21], 4-HPR implants only significantly inhibited tumor growth when used with other agents. These data suggest that released 4-HPR was largely inaccessible to the tumors, potentially due to high affinity binding to centrally located residual Matrigel and extravasted erythrocytes. Previous results from our pilot studies, which entailed placement of two 4-HPR releasing implants adjacent to OSCC tumors, demonstrated significant tumor inhibition and emphasize the importance of both implant location and number. Studies are ongoing to investigate the effects of both implant placement and 4-HPR tissue diffusion. Finally, the absence of any detectable levels of 4-HPR or its metabolites in the sera of any animals emphasizes the safety of PLGA local drug delivery
IHC studies provided further insights into treatment-tumor effects. Ki-67 labeling was most apparent at tumor peripheries; findings consistent with relatively hypoxic tumor cores. Quantitative Ki-67 analyses revealed that any treatment reduced tumor cell proliferation while significant reduction in tumor cell labeling was present in the triple treatment, TOC+4-HPR, 2-ME+TOC and 2ME+4-HPR groups (see Figure 6.B. and C.). As the tumors in the 4-HPR group were so large, part of the Ki-67 decrease may reflect necrotic tumor foci due to inadequate angiogenesis to support the tumor mass. Although two established apoptosis-inducing drugs (2-ME and 4-HPR) were employed, low levels of cleaved caspase 3 were detected in all groups (Figure 6. B. and C.). These data may reflect both a relatively short half-life (~ 8 hours for activated caspase 3 [49]) and additional forms of cancer cell death including necrosis, senescence and autophagy [50]. Treatment effects on differentiation were evaluated by involucrin staining. While all experimental groups contained involucrin, qualitatively the most intense expression was observed in the 2-ME and 4-HPR+TOC groups. Despite the challenges of daily injections of drugs with short or intermediate half-lives (2-ME~4 h, TOC~0.8 days), pH dependent binding (acidic pH decreases TOC binding), and limited diffusion capacity (4-HPR) our data show efficacy of the selected chemopreventives to suppress growth in established OSCC tumors. These data provide confidence regarding the chemopreventives’ potential to abort microtumor foci. Also, it is probable that inter-tumor differences will affect extent of agent responsiveness e.g. tumors with lower autologous VEGF production would likely be more susceptible to 2-ME's inhibition of HIF-1α signaling. Finally, the absence of deleterious effects to the overlying epidermis emphasizes the chemopreventives’ safety during local delivery.
Risk reduction and primary chemoprevention clearly remain the optimal OSCC management approach. Provided the often fatal consequences of recurrent OSCCs and lack of effective intervention options to prevent tumor recurrence, development of a well-tolerated and effective secondary chemoprevention strategy is warranted. The potential benefits from optimized controlled-release local delivery implants that stabilize drugs, remove peak and valley drug levels, eliminate issues with systemic toxicities and patient compliance and facilitate drug diffusion throughout the previous surgical site to eradicate OSCC recurrence are readily apparent.
Supplementary Material
Acknowledgements
The authors would like to express their appreciation to Mary Lloyd and Mary Marin for their expert preparation of the histologic specimens and Mr. Matthew Chaney for his summer research efforts.
Grant Support. This study was supported by NIH grants R01 CA171329 (SR Mallery) and NIDCR T32DE014320 (JF Sheridan PI, B. Santiago Graduate Fellow).
Footnotes
Conflict of interest statement: The authors have no conflict of interest to disclose.
References
- 1.Huber MA, Tantiwongkosi B. Oral and Oropharyngeal Cancer. Med Clin N Am. 2014;98:1299–1321. doi: 10.1016/j.mcna.2014.08.005. [DOI] [PubMed] [Google Scholar]
- 2.Gleber-Netto FO, Braakhuis BJ, Triantafyllou A, Takes RP, Kelner N, Rodrigo JP, et al. Molecular events in relapsed oral squamous cell carcinoma: Recurrence vs secondary primary tumor. Oral Oncol. 2015;51:738–44. doi: 10.1016/j.oraloncology.2015.04.016. [DOI] [PubMed] [Google Scholar]
- 3.Brockstein BE. Management of Recurrent Head and Neck Cancer. Recent Progress and Future Directions. Drugs. 2011;71:1551–9. doi: 10.2165/11592540-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 4.Arora A, Scholar EM. Role of Tyrosine Kinase Inhibitors in Cancer Therapy. J Pharm Exp Therapeutics. 2005;315:971–9. doi: 10.1124/jpet.105.084145. [DOI] [PubMed] [Google Scholar]
- 5.Dziadziuszko R, Jassem J. Epidermal growth factor receptor (EGFR) inhibitors and derived treatments. Ann Oncology. 2012;23:193–6. doi: 10.1093/annonc/mds351. [DOI] [PubMed] [Google Scholar]
- 6.Molife LR, Omlin A, Jones RJ, Karavasilis V, Bloomfield D, Lumsden G, et al. Randomized Phase II trial of nintedanib, afatinib and sequential combination in castration-resistant prostate cancer. Future Oncol. 2014;10:219–31. doi: 10.2217/fon.13.250. [DOI] [PubMed] [Google Scholar]
- 7.Stanam A, Love-Homan L, Joseph TS, Espinosa-Cotton M, Simons AL. Upregulated interleukin-6 expression contributes to erlotinib resistance in head and neck squamous cell carcinoma. Mol Oncol. 2015;9:1371–83. doi: 10.1016/j.molonc.2015.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Specenier P, Vermorken JB. Cetuximab: its unique place in head and neck cancer treatment. Biologics. 2013;7:77–90. doi: 10.2147/BTT.S43628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sun C, Bernards R. Feedback and redundancy in receptor tyrosine kinase signaling: relevance to cancer therapies. Trends Biochem Sci. 2014;39:465–74. doi: 10.1016/j.tibs.2014.08.010. [DOI] [PubMed] [Google Scholar]
- 10.Bahleda R, Hollebecque A, Varga A, Gazzah A, Massard C, Deutsch E, et al. Phase I study of afatinib combined with nintedanib in patients with advanced solid tumours. Br J Cancer. 2015;113:1413–20. doi: 10.1038/bjc.2015.374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Furtek SL, Backos DS, Matheson CJ, Reigan P. Strategies and approaches of targeting STAT3 for cancer treatment. ACS Chem Biol. 2016;11:308–18. doi: 10.1021/acschembio.5b00945. [DOI] [PubMed] [Google Scholar]
- 12.Yu H, Lee H, Herrmann A, Buettner R, Jove R. Revisiting STAT3 signaling in cancer: new and unexpected biological functions. Nat Rev Cancer. 2014;14:736–46. doi: 10.1038/nrc3818. [DOI] [PubMed] [Google Scholar]
- 13.Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9:798–809. doi: 10.1038/nrc2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee H, Zhang P, Herrmann A, Yang C, Xin H, Wang Z, et al. Acetylated STAT3 is crucial for methylation of tumor-suppressor gene promoters and inhibition by resveratrol results in demethylation. Proc Natl Acad Sci USA. 2012;109:7765–69. doi: 10.1073/pnas.1205132109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jinno T, Kawano S, Maruse Y, Matsubara R, Goto Y, Sakamoto T, Hashiguchi Y, et al. Increased expression of interleukin-6 predicts poor response to chemoradiotherapy and unfavorable prognosis in oral squamous cell carcinoma. Oncol Rep. 2015;33:2161–68. doi: 10.3892/or.2015.3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chang PY, Kuo YB, Wu TL, Liao CT, Sun YC, Yen TC, et al. Association and prognostic value of serum inflammation markers in patients with leukoplakia and oral cavity cancer. Clin Chem Lab Med. 2013;51:1291–1300. doi: 10.1515/cclm-2012-0504. [DOI] [PubMed] [Google Scholar]
- 17.Ogura M, Uchida T, Terui Y, Hayakawa F, Kobayashi Y, Taniwaki M, et al. Phase I study of OPB-51602, an oral inhibitor of signal transducer and activator of transcription 3, in patients with relapsed/refractory hematological malignancies. Cancer Sci. 2015;106:896–901. doi: 10.1111/cas.12683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wong AL, Soo RA, Tan DS, Lee SC, Lim JS, Marban PC, et al. Phase I and biomarker study of OPB-51602, a novel signal transducer and activator of transcription (STAT) 3 inhibitor, in patients with refractory solid malignancies. Ann Oncol. 2015;26:998–1005. doi: 10.1093/annonc/mdv026. [DOI] [PubMed] [Google Scholar]
- 19.Logue JS, Morrison DK. Complexity in the signaling network: insights from the use of targeted inhibitors in cancer therapy. Genes Dev. 2012;26:641–50. doi: 10.1101/gad.186965.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Naithani R, Huma LC, Moriarty RM, McCormick DL, Mehta RG. Comprehensive review of cancer chemopreventive agents evaluated in experimental carcinogenesis models and clinical trials. Curr Med Chem. 2008;15:1044–71. doi: 10.2174/092986708784221403. [DOI] [PubMed] [Google Scholar]
- 21.Han BB, Li S, Tong M, Holpuch AS, Spinney R, Wang D, et al. Fenretinide perturbs focal adhesion kinase in premalignant and malignant human Oral Keratinocytes. Fenretinide's chemopreventive mechanisms include ECM interactions. Cancer Prev Res. 2015;8:419–30. doi: 10.1158/1940-6207.CAPR-14-0418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jones SA, Scheller J, Rose-John S. Therapeutic strategies for the clinical blockade of IL-6/gp130 signaling. J Clin Invest. 2011;121:3375–83. doi: 10.1172/JCI57158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mooberry SL. Mechanism of action of 2-methoxyestradiol: new developments. Drug Resist Updat. 2003;6:355–61. doi: 10.1016/j.drup.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 24.Takahashi N, Sausville EA, Breitman TR. N-(4-Hydroxyphenyl)retinamide (Fenretinide) in combination with retinoic acid enhances differentiation and retinoylation of proteins. Clin Cancer Res. 1995;1:637–42. [PubMed] [Google Scholar]
- 25.Mueck AO, Seeger H. 2-Methoxyestradiol-Biology and mechanism of action. Steroids. 2010;75:625–31. doi: 10.1016/j.steroids.2010.02.016. [DOI] [PubMed] [Google Scholar]
- 26.Poindessous V, Ouaret D, El Ouadrani K, Battistella A, Megalophonos VF, Kamsu-Kom N, Petitprez A, Escargueil AE, Boudou P, Dumont S, et al. EGFR- and VEGF(R)-targeted small molecules show synergistic activity in colorectal cancer models refractory to combinations of monoclonal antibodies. Clin Cancer Res. 2011;17:6522–30. doi: 10.1158/1078-0432.CCR-11-1607. [DOI] [PubMed] [Google Scholar]
- 27.Trott O, Olson AJ. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455–61. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Berman H, Henrick K, Nakamura H. Announcing the worldwide Protein Data Bank. Nat Struct Biol. 2003;10:980. doi: 10.1038/nsb1203-980. [DOI] [PubMed] [Google Scholar]
- 29.Yu W, Xiao H, Lin J, Li C. Discovery of novel STAT3 small molecule inhibitors via in silico site-directed fragment-based drug design. J Med Chem. 2013;56:4402–12. doi: 10.1021/jm400080c. [DOI] [PubMed] [Google Scholar]
- 31.Hantschel O, Superti-Furga G. Regulation of the C-Abl and Bcr-Abl tyrosine kinases. Nat Rev. 2004;5:33–44. doi: 10.1038/nrm1280. [DOI] [PubMed] [Google Scholar]
- 32.Suh Y, Jo SY, Lee HY, Lee C. Inhibition of IL-6/STAT3 axis and targeting Axl and Tyro3 receptor tyrosine kinases by apigenin circumvent taxol resistance in ovarian cancer cells. Int J Oncol. 2015;46:1405–11. doi: 10.3892/ijo.2014.2808. [DOI] [PubMed] [Google Scholar]
- 33.Jemma AB, Sallami S, Ramarli D, Colombatti M, Oueslati R. The proinflammatory cytokine, IL-6, and its interference with bFGF signaling and PSMA in prostate cancer cells. Inflammation. 2013;36:643–50. doi: 10.1007/s10753-012-9586-7. [DOI] [PubMed] [Google Scholar]
- 34.Harada K, Ferdous T, Itashiki Y, Takii M, Mano T, Mori Y, et al. Cepharanthine inhibits angiogenesis and tumorigenicity of human oral squamous cell carcinoma cells by suppressing expression of vascular endothelial growth factor and interleukin-8. Int J Oncol. 2009;35:1025–35. doi: 10.3892/ijo_00000417. [DOI] [PubMed] [Google Scholar]
- 35.Garcia R, Bowman TL, Niu G, Yu H, Minton S, Muro-Cacho CA, et al. Constitutive activation of Stat3 by the Src and JAK tyrosine kinases participates in growth regulation of human breast carcinoma cells. Oncogene. 2001;20:2499–513. doi: 10.1038/sj.onc.1204349. [DOI] [PubMed] [Google Scholar]
- 36.Lin J, Arlinghaus R. Activated c-Abl tyrosine kinase in malignant solid tumors. Oncogene. 2008;27:4385–91. doi: 10.1038/onc.2008.86. [DOI] [PubMed] [Google Scholar]
- 37.Fury MG, Baxi S, Shen R, Kelly KW, Lipson BL, Carlson D, et al. Phase II study of saracatinib (AZD0530) for patients with recurrent or metastatic head and neck squamous cell carcinoma (HNSCC). Anticancer Res. 2011;31:249–53. [PMC free article] [PubMed] [Google Scholar]
- 38.Brooks HD, Glisson BS, Bekele BN, Johnson FM, Ginsberg LE, El-Naggar A, et al. Phase 2 study of dasatinib in the treatment of head and neck squamous cell carcinoma. Cancer. 2011;117:2112–9. doi: 10.1002/cncr.25769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Campos-Sandoval JA, Redondo C, Kinsella GK, Pal A, Jones G, Eyre GS, et al. Fenretinide derivatives act as disrupters of interactions of serum retinol binding protein (sRBP) with transthyretin and the sRBP receptor. J Med Chem. 2011;54:4378–87. doi: 10.1021/jm200256g. [DOI] [PubMed] [Google Scholar]
- 40.Lee TL, Yeh J, Van Waes C, Chen Z. Epigenetic modification of SOCS-1 differentially regulates STAT3 activation in response to interleukin-6 receptor and epidermal growth factor receptor signaling through JAK and/or MEK in head and neck squamous cell carcinomas. Mol Cancer Ther. 2006;5:8–19. doi: 10.1158/1535-7163.MCT-05-0069. [DOI] [PubMed] [Google Scholar]
- 41.Yang J, Liao X, Agarwal MK, Barnes L, Auron PE, Stark GR. Unphosphorylated STAT3 accumulates in response to IL-6 and activates transcription by binding to NFkappaB. Genes Dev. 2007;21:1396–1408. doi: 10.1101/gad.1553707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yu Y, Wan Y, Huang C. The biological functions of NF-κB1 (p50) and its potential as an anti-cancer target. Curr Cancer Drug Targets. 2009;9:566–71. doi: 10.2174/156800909788486759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kumar AP, Garcia GE, Orsborn J, Levin VA, Slaga TJ. 2-Methoxyestradiol interferes with NFκB transcriptional activity in primitive neuroectodermal brain tumors: implications for management. Carcinogenesis. 2003;24:209–16. doi: 10.1093/carcin/24.2.209. [DOI] [PubMed] [Google Scholar]
- 44.Chalaris A, Garbers C, Rabe B, Rose-John S, Scheller J. The soluble Interleukin 6 receptor: Generation and role in inflammation and cancer. Eur J Cell Biol. 2011;90:484–94. doi: 10.1016/j.ejcb.2010.10.007. [DOI] [PubMed] [Google Scholar]
- 45.Fofaria MN, Srivastava SK. STAT3 induces anoikis resistance, promotes cell invasion and metastatic potential in pancreatic cancer cells. Carcinogenesis. 2015;36:142–50. doi: 10.1093/carcin/bgu233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ward KK, Tancioni I, Lawson C, Miller NL, Jean C, Chen XL, et al. Inhibition of focal adhesion kinase (FAK) activity prevents anchorage-independent ovarian carcinoma cell growth and tumor progression. Clin Exp Metastasis. 2013;30:579–94. doi: 10.1007/s10585-012-9562-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee CH, Yu CC, Wang BY, Chang WW. Tumorsphere as an effective in vitro platform for screening anticancer stem cell drugs. Oncotarget. 2016;7:1215–26. doi: 10.18632/oncotarget.6261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ricker JL, Chen Z, Yang XP, Pribluda VS, Swartz GM, Van Waes C. 2-Methoxyestradiol inhibits Hypoxia-Inducible Factor 1α, tumor growth, and angiogenesis and augments Paclitaxel efficacy in Head and Neck Squamous Cell Carcinoma. Clin Cancer Res. 2004;10:8665–73. doi: 10.1158/1078-0432.CCR-04-1393. [DOI] [PubMed] [Google Scholar]
- 48.Shinriki S, Jono H, Ota K, Ueda M, Kudo M, Ota T, et al. Humanized anti-Interleukin-6 Receptor antibody suppresses tumor angiogenesis and in vivo growth of human Oral Squamous Cell Carcinoma. Clin Cancer Res. 2009;15:5426–34. doi: 10.1158/1078-0432.CCR-09-0287. [DOI] [PubMed] [Google Scholar]
- 49.Walsh JC, Logue SE, Luthi AU, Martin SJ. Caspase-1 promiscuity is counterbalanced by rapid inactivation of processed enzyme. J Biol Chem. 2011;286:32513–24. doi: 10.1074/jbc.M111.225862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brown JM, Attardi LD. The role of apoptosis in cancer development and treatment response. Nat Rev Cancer. 2005;5:231–7. doi: 10.1038/nrc1560. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.