

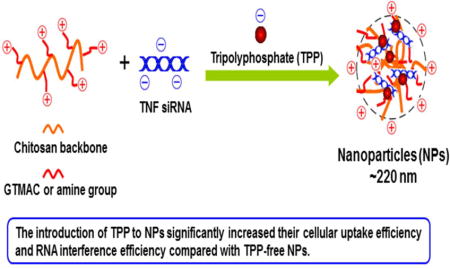
Abstract
Tumor necrosis factor-α (TNF-α) is a major pro-inflammatory cytokine that is mainly secreted by macrophages during inflammation. Here, we synthesized a series of N-(2-hydroxy)propyl-3-trimethyl ammonium chitosan chlorides (HTCCs), and then used a complex coacervation technique or tripolyphosphate (TPP)-assisted ionotropic gelation strategy to complex the HTCCs with TNF-α siRNA (siTNF) to form nanoparticles (NPs). The resultant NPs had a desirable particle size (210–279 nm), a slightly positive zeta potential (14–22 mV), and negligible cytotoxicity against Raw 264.7 macrophages and colon-26 cells. Subsequent cellular uptake tests demonstrated that the introduction of TPP to the NPs markedly increased their cellular uptake efficiency (to nearly 100%) compared with TPP-free NPs, and yielded a correspondingly higher intracellular concentration of siRNA. Critically, in vitro gene silencing experiments revealed that all of the TPP-containing NPs showed excellent efficiency in inhibiting the mRNA expression level of TNF-α (by approximately 85–92%, which was much higher than that obtained using Oligofectamine/siTNF complexes). Collectively, these results obviously suggest that our non-toxic TPP-containing chitosan-based NPs can be exploited as efficient siTNF carriers for the treatment of inflammatory diseases.
Keywords: Chitosan, N-(2-hydroxy)propyl-3-trimethyl ammonium chitosan chloride, Tripolyphosphate, Nanoparticle, RNA interference, Macrophage
Graphical abstract
1. Introduction
Tumor necrosis factor-α (TNF-α) is mainly secreted by macrophages and plays a central role in the pathogenesis of various inflammatory disorders, such as inflammatory bowel diseases, acute hepatic injury and rheumatoid arthritis [1–3]. Thus, TNF-α has become an important therapeutic target. Currently, biological strategies against TNF-α (e.g., infliximab and adalimumab) have been shown to successfully relieve inflammatory diseases in multiple clinical trials [4, 5]. However, serious infections and side effects have also been reported, including infusion reactions and auto-immunity to antibodies [6].
RNAi interference (RNAi) mediated by small interfering RNAs (siRNAs) of 19–23 base pairs is a powerful tool for post-transcriptional silencing gene expression. It has been recognized as an efficient approach for downregulating TNF-α expression in macrophages [7, 8], but the therapeutic efficacy of naked siRNA is limited by its rapid enzymatic degradation and poor internalization into cells, which reflects its low stability, hydrophilicity and negative charges [9, 10]. To overcome these obstacles, various carriers have been used to deliver siRNA into cells. They can be divided into two main categories: viral and non-viral carriers [11]. Though viral carriers have the advantage of high transfection efficiency, they have been associated with immunogenicity and oncogenic effects [12, 13]. Therefore, a wide range of non-viral delivery systems (e.g., lipids, dendrimers and polymers) have been proposed as alternatives for viral carriers, due to their minimal host immune responses, ease of synthesis/chemical modification and relative stability in storage [14–16].
Recently, chitosan-based carriers have been widely applied in siRNA delivery, as they offer the benefits of biocompatibility, biodegradability and good siRNA-binding ability [17–19]. Chitosan is a linear, natural cationic copolymer of glucosamine and N-acetyl-glucosamine [20]. It is a weak base with a pKa value of approximately 6.5, and can be soluble in acidic medium due to the protonation of its amine groups [21]. However, its amino groups are only partially protonated in neutral and physiological environments, thus limiting their interactions with siRNA [22]. In addition, chitosan can precipitate from solution under the latter conditions, which has limited its application as a suitable siRNA carrier [23, 24]. Many techniques have been developed to overcome this restriction, including quaternization or guanidination of the NH2 groups, or conjugation of dextran and/or polyethylene glycol to the chitosan backbone [25–28]. Quaternary chitosan has attracted a great deal of interest based on its numerous merits, including its well-defined structure, enhanced positive charge and improved solubility [24]. The modification of the primary amino groups of chitosan with glycidyltrimethylammonium chloride (GTMAC) appears to be a promising approach for synthesizing cationic quaternary chitosan; the resulting polymer is called N-(2-hydroxy)propyl-3-trimethyl ammonium chitosan chloride (HTCC). Quaternization has been shown to improve the nucleic acid binding capability of chitosan and enhance its cellular uptake efficiency by improving the electrostatic affinity between quaternized chitosan and cell membrane [28, 29].
Tripolyphosphate (TPP) is a soluble, non-toxic and very common polyanionic crosslinker that can interact with the positively charged groups in chitosan or its derivatives [30]. Our group and others have shown that TPP can assist the formation of stable nanoparticles (NPs) between polycation and siRNA under neutral and physiological conditions [15, 31, 32]. Katas et al. [33] fabricated chitosan/siRNA NPs based on three methods (simple complexation, TPP-assisted ionotropic gelation and surface adsorption). They found that chitosan/siRNA NPs formed via TPP-assisted ionotropic gelation technique exhibited a much higher biological activity compared to the other two types of chitosan/siRNA NPs, suggesting that TPP-chitosan/siRNA NPs could be ideal vectors for siRNA delivery. To the best of our knowledge, however, no attempts have been made to examine the effect of TPP on the HTCC-facilitated delivery of siRNA, and no previous study has used this formulation to deliver TNF-α siRNA (siTNF) to macrophages for anti-inflammation.
Here, we described the first fabrication of TPP-HTCC/siRNA NPs, the characterization of their physicochemical properties (siRNA complexation capability, hydrodynamic particle size and zeta-potential), and the assessment of their cytotoxicity and TNF-α knockdown profiles in macrophages.
2. Materials and Methods
2.1 Materials
Chitosan, GTMAC (purity ≥ 90%), TPP (purity ≥ 98%), sodium nitrite (purity ≥ 97%), lipopolysaccharides (LPS) from Salmonella enteric serotype typhimurium (purity ≥ 99%) were purchased from Sigma-Aldrich (St. Louis, USA). Chitosan was purified before its subsequent application, and its molecular weight was tailored by depolymerization using sodium nitrite following a reported method [34]. Viscosity-average molecular weight of the resultant chitosan was measured as 1.8×104 Da using a 0.5 M CH3COOH/0.2 M CH3COONa solvent system [35]. Oligofectamine (OF), 4′,6-diamidino-2-phenyl-indole dihydrochloride (DAPI), FITC fluorescently tagged siRNA (FITC-siRNA) and Vybrant® MTT cell proliferation assay kit (MTT) were obtained from Invitrogen (Eugene, USA). GelRed was from Biotium (Hayward, USA). siTNF was purchased from Santa Cruz Biotechnology (Santa Cruz, CA), and its sequences were CGUCGUAGCAAACCACCAATT (sense strand) and UUGGUGGUUUGCUACGACGTG (anti-sense strand). All commercial products were used without further purification unless otherwise stated. Deionized water was obtained using a Milli-Q water purification system.
2.2 Synthesis of HTCC
HTCC was prepared according to our previous method [20, 24]. Briefly, chitosan powder was suspended in deionized water. The mixture was stirred for 30 min prior to dropwise addition of GTMAC with continuous stirring. The weight ratio of GTMAC to chitosan changed from 0.5:1 to 1.5:1 to produce HTCC samples. The reaction mixture was stirred at 85 °C for 6 h. After being precipitated and washed by hot alcohol, the product was collected by filtration. The collected polymer was dissolved in distilled water and dialyzed (MWCO=3500) against NaCl solution for 5 days and lyophilized. The final products were named as HTCC-1, HTCC-2 and HTCC-3, respectively, and the degrees of quaternization (DQs) of quaternary ammonium groups were denoted by the numbers followed HTCC.
2.3 Characterization
NMR spectrum was recorded on a Bruker Avance spectrometer (INOVA-400 NMR). 70 mg of chitosan and HTCC-1 was dissolved in respective DCl (1%, v/v) solution and D2O for 1H NMR measurement.
DQs of HTCC were measured by conductometric titration of chloride ions. HTCC samples were dissolved in 0.1 M HAc solutions and the resultant solutions were titrated with an aqueous AgNO3 solution to measure the amount of Cl− ions which were proportional to the DQs of HTCC samples. The reaction conditions and the DQs of quaternized chitosan are summarized in Table 1.
Table 1.
Basic parameters of different polymers.
| Samples | Feed ratio of GTMAC to chitosan (wt./wt.) | Reaction time (h) | DD (%)a | DQ (%)b |
|---|---|---|---|---|
| Chitosan | — | — | 86.4 | — |
| HTCC-1 | 0.5:1 | 6 | — | 10.9 ± 1.5 |
| HTCC-2 | 1.0:1 | 6 | — | 23.4 ± 1.4 |
| HTCC-3 | 1.5:1 | 6 | — | 33.7 ± 2.1 |
DD is the degree of deacetylation; DQ is the degree of quaternization.
DD of chitosan was determined by 1H NMR.
DQs of HTCC were measured by conductometric titration and data are quoted as mean ± S.E.M.. (n = 3).
2.4 Preparation of NPs
NPs were fabricated by a complex coacervation or TPP-assisted ionotropic gelation technique. NPs were fabricated based on different polymers to siRNA weight ratios and the weight ratio of polymer to TPP was set to 4:1. Initially, polymers and TPP were dissolved in 0.1 M sodium acetate/0.1 M acetic acid buffers (pH 5.5) to obtain solutions with the concentration of 4 mg/mL and 1 mg/mL, respectively. The siRNA stock solution was prepared in RNase-free water with the concentration of 0.1 mg/mL. Equivalent volumes of siRNA solutions were subsequently mixed with or without TPP solution, followed by the addition of different amount of polymer solutions, and vortexed for 10 s. The resulting NPs were further allowed to incubate for 30 min at room temperature for complete NPs formation. All the NPs were prepared immediately prior to the experiments.
2.5 Agarose gel electrophoresis assay
The siRNA condensing capacities of polymer/siRNA NPs and TPP-polymer/siRNA NPs were evaluated by agarose gel electrophoresis. The NPs were fabricated at various weight ratios of polymer/siRNA ranging from 10:1 to 100:1. Agarose gel (4%, W/V) containing GelRed solution (0.5 μg/mL) was prepared in Tris-Acetate-EDTA buffer. After 30 min of incubation at room temperature, the samples were electrophoresed at 100 V for 20 min. The resulting siRNA migration patterns were viewed under UV transilluminator.
2.6 Particle size, zeta-potential and morphology measurement
Particle sizes (nm) and zeta potential (mV) of NPs were measured by dynamic light scattering (DLS) using 90 Plus/BI-MAS (Multi-angle particle sizing) or DLS after applying an electric field using a ZetaPlus (Zeta potential analyzer, Brookhaven Instruments Corporation). The average of the diameters or zeta potential was calculated using 3 runs. Each run is an average of 10 measurements.
For morphology test, a drop of suspension of TPP-HTCC-1/siRNA with the weight ratio of 60:1 was mounted on a freshly cleaved glass slide using carbon adhesive tape and sputter-coated with a mixture of gold and palladium (60:40) in an argon atmosphere under low pressure. The image was measured by scanning electron microscopy (SEM, LEO 1450VP, Zeiss, Germany).
2.7 Cytotoxicity assay
For MTT assay, Raw 264.7 macrophages and colon-26 cells were seeded at a respective density of 8×103 and 2×104 cells/well in 96-well plates and incubated overnight. The cells were subsequently incubated with freshly prepared NPs suspensions for 4 h or 24 h, and the siRNA concentration in the medium is set as 100 nM. Cells were then incubated with MTT (0.5 mg/mL in supplemented 100 μL of serum free DMEM) at 37 °C for 4 h. Thereafter, the media were discarded and 50 μL dimethyl sulfoxide (DMSO) was added to each well prior to spectrophotometric measurements at 490 nm. Untreated cells were used as negative references, whereas cells were treated with 0.5% Triton X-100 as positive controls.
2.8 Intracellular NPs uptake visualization
Raw 264.7 macrophages were seeded in four-chamber tissue culture glass slide (BD Falcon, Bedford, MA, USA) at a density of 1.0×104 cells/well and incubated overnight. The culture medium was exchanged to serum-free DMEM containing TPP-HTCC-1/FITC-siRNA NPs (weight ratio, 60:1). The FITC-siRNA concentration in the medium is set as 100 nM. After co-culture for various time points (1, 3 and 5 h), the cells were fixed in 4% paraformaldehyde for 20 min. To observe cellular uptake of NPs, DAPI was diluted 10,000 times and added to the wells for staining cells for 5 min. Images were acquired using an Olympus equipped with a Hamamatsu Digital Camera ORCA-03G.
2.9 Quantification of intracellular uptake
Raw 264.7 macrophages were seeded in 6-well plates at a density of 1×105 cells/well. After overnight incubation, medium was replaced with serum- and antibiotics-free medium and cells were treated with different NPs, at a final concentration of 100 nM FITC-siRNA. After 5 h of co-incubation, the cells were thoroughly rinsed with cold PBS to eliminate excess of NPs, which were not taken up by cells. Subsequently, the treated cells were harvested using trypsin, transferred to centrifuge tubes, and centrifuged at 1,500 rpm for 5 min. Upon removal of the supernatant, the cells were re-suspended in 0.5 mL of flow cytometry (FCM) buffer, transferred to round-bottom polystyrene test tubes (BD Falcon, 12 × 75 mm), and kept at 4 °C until analysis. Analytical FCM was performed using the FITC channel on the FCM Canto™ (BD Biosciences, San Jose, CA, USA). A total of 5,000 ungated cells were analyzed.
2.10 In vitro gene silencing efficiency test
Raw 264.7 macrophages were seeded in 6-well plates at a density of 1×105 cells/well and incubated overnight. Subsequently, various NPs were added into wells. The siTNF concentration in the medium is set as 100 nM. As controls, cells were transfected with OF/siTNF complexes. After co-culture for 5 h, the wells will be supplemented with DMEM medium containing 10% FBS and further incubated for 19 or 43 h. Cells were then stimulated with LPS (5 μg/mL) for 3 h. Total RNA was extracted using RNeasy Plus Mini Kit (Qiagen). The cDNA was generated from the total RNAs isolated above using the Maxima first strand cDNA synthesis kit (Fermentas) according to the manufacturer’s instruction. Levels of TNF-α mRNA expression were quantified by RT-PCR using Maxima® SYBR Green/ROX qPCR Master Mix (Fermentas). The data were normalized to the internal control: 36B4. Relative gene expression levels were calculated using the delta delta Ct (2−ΔΔCt) method. Sequences of all the primers used for RT-PCR are given in Supplementary Table 1.
2.11 Statistical analysis
Statistical analysis was performed using Student’s t-test. Data were expressed as mean ± standard error of mean (S.E.M.). Statistical significance was represented by *P<0.05 and **P<0.01.
3. Results and discussion
3.1 Preparation and characterization of HTCC
Quaternary chitosan with different DQs were prepared by using GTMAC as an etherification agent in deionized water. In an acidic or neutral environment, the epoxy groups of GTMAC prevalently react with the amine groups of chitosan, whereas under an alkaline condition, conjugations predominantly occur with the hydroxyl groups of chitosan [36–38]. In this study, GTMAC groups mainly conjugated to the amine groups of chitosan in water media. Under the utilized reaction conditions, when we fixed the reaction time as constant, the DQs of HTCC samples appeared to proportionally increase with the feeding ratio of GTMAC to chitosan, as shown in Table 1. This may suggest that the DQs of HTCCs can feasibly be controlled by adjusting the feeding ratio of these two reactants.
Fig. 1 presents the 1H NMR spectra of chitosan and HTCC-1. In comparison to chitosan, noticeable changes occur in the spectra of HTCC-1. A very strong peak at 3.14 ppm is observed in HTCC-1, indicating the presence of methyl groups in the quaternary ammonium side chains. The remnant peaks, which are assigned to protons in respective chitosan chains and those in quaternized side chains, are in good agreement with our published reports [20, 24]. Based on the NMR spectra of chitosan and HTCC-1, we conclude that quaternary ammonium side chains are successfully grafted onto the chitosan backbone.
Fig. 1.
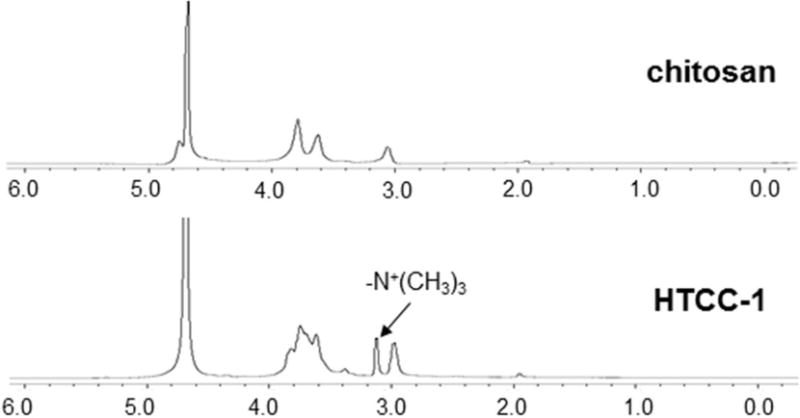
1H NMR spectrum of chitosan and N-(2-hydroxy)propyl-3-trimethyl ammonium chitosan chloride (DQ: 10.9%) dissolved in respective diluted DCl solution and D2O.
3.2 Agarose gel electrophoresis
Compared to plasmid DNA, siRNA has an extremely low charge density, high stiffness and small size (a double-stranded siRNA is only around 5.67 nm in length). These intrinsic properties largely limit its NP-forming ability [39]. To overcome these major obstacles, various covalent crosslinkers, such as glutaraldehyde and genipin, have been employed to assist in the formation of siRNA-loaded NPs. In the present study, we use the ionic crosslinker TPP, because whereas the residues of covalent crosslinkers can induce cytotoxicity, TPP is nontoxic and has been approved by FDA for application in the food industry [40]. TPP has a linear structure that carries five negative charges, and can thus crosslink protonated chitosan or HTCC polymers in an aqueous medium. Most importantly, TPP has been reported to enhance the siRNA-condensing capacity of polycations and increase the efficiency of NP-mediated gene silencing [15, 33].
A previous report found that for the preparation of siRNA-loaded NPs, the optimum chitosan/TPP ratio is 4:1 [33]. Thus, this ratio was applied in the present study. Since cationic polymers are known to restrict negatively charged siRNA by forming NPs via electrostatic interactions, we used agarose gel electrophoresis to examine the siRNA-condensing ability of NPs generated with or without TPP. As shown in Fig. 2a, some siRNAs are released from TPP-free chitosan/siRNA NPs even at a weight ratio of 40:1. In contrast, HTCC polymers condense the siRNA completely at a weight ratio of 40:1 in the absence of TPP. This suggests that the introduction of GTMAC to the chitosan backbone critically improves the binding capacity of siRNA. The siRNA-condensing ability appears to increase with the increase of DQ, indicating that a higher positive charge density on the chitosan backbone supports the formation of more stable NPs. Furthermore, the chitosan and HTCCs appear to retard siRNA to a much higher degree in the presence of TPP in comparison to the results obtained without TPP. These results, which are consistent with previous findings [15, 33, 41], demonstrate that NPs formed by ionic gelation using TPP might have more charge interactions and should consequently be more stable. In addition, as shown in Fig. 2b, chitosan/siRNA NPs with weight ratios of over 60:1 are found to efficiently restrict the release of siRNA regardless of the presence of TPP. Therefore, to compare the biological activity of various NPs with the least polymer content, we utilize a weight ratio of 60:1 for the following experiments.
Fig. 2.
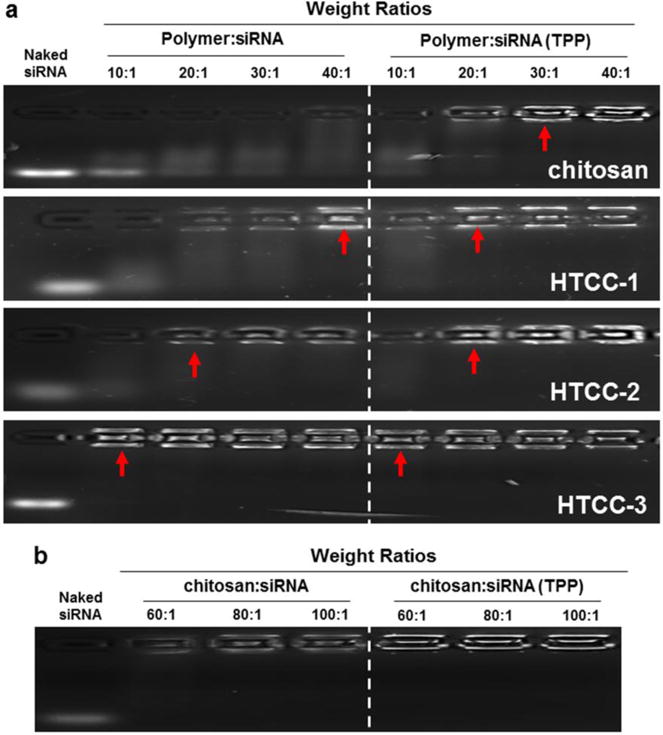
Agarose gel electrophoresis of various NPs. (a) chitosan/siRNA NPs and HTCC/siRNA NPs with different weight ratios (10:1, 20:1, 30:1 and 40:1) in the presence or absence of TPP. (b) chitosan/siRNA NPs with different weight ratios (60:1, 80:1 and 100:1) in the presence or absence of TPP.
3.3 Particle size, zeta potential and morphology of NPs
Particle size plays a key role in the cellular uptake of NPs, and is thus one of the most important parameters for cationic polymers intended for application as siRNA vectors. Several previous studies reported that cells typically take up NPs ranging from about 50 to several hundred nanometers [42, 43]. Fig. 3a shows the hydrodynamic diameter of NPs formed by siRNA plus chitosan or HTCCs in the presence or absence of TPP, and reveals that TPP clearly had a strong impact on NP size. The average sizes of the TPP-free polymer/siRNA NPs are in the range of 240.7–279.3 nm. The introduction of TPP significantly decreases the average sizes of NPs, with TPP-polymer/siRNA NPs falling in a range of 210.7–230.5 nm. The particle sizes of HTCC/siRNA NPs are smaller than those formed of chitosan/siRNA with or without TPP, due to the high siRNA complexation ability of HTCC. The sizes of the HTCC/siRNA NPs increase slightly as the DQ increased, perhaps because more free positive charges are contained within the high-DQ-HTCC-derived NPs compared to those formed with chitosan or lower-DQ derivatives. Their mutual repulsion and the permeation of water and hydrated counter-ions need to neutralize these charges would then cause swelling, thereby increasing the NP diameters. This could explain why chitosan derivatives with high degrees of positive charge cannot be facilitated to form small-size NPs. The zeta potentials of NPs determine their colloidal stability and influence the effectiveness of their interactions with negatively charged cell membranes. Therefore, the zeta potentials of NPs can strongly affect their transfection efficiencies [20]. The zeta potentials of the NPs generated in the present work are illustrated in Fig. 3b. All of the NPs show positive charges, and their zeta potential values are in the range of 14.1–22.0 mV. Moreover, their comparative positive values increase as the DQs of the chitosan derivatives increase (given a constant weight ratio). This reflects an increase in the number of positive groups in the HTCCs, as the amount of siRNA is fixed. Consistent with a previous report [33], the introduction of TPP into the NPs reduces their surface charge. A net positive charge is desirable for NPs, as it prevents their aggregation and promotes electrostatic interaction with the overall negatively charged cell membrane, yielding a high transfection efficiency. Thus, the NPs generated in the present work appear to have characteristics that would favor the siRNA delivery into cells.
Fig. 3.
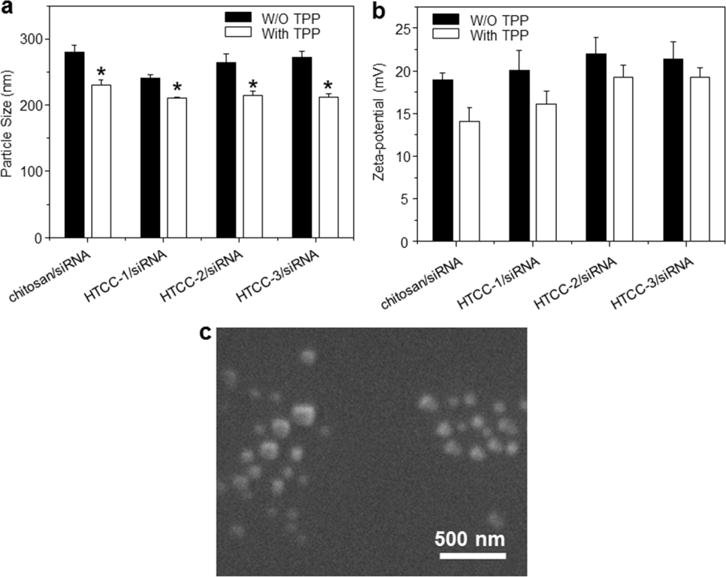
Particle size (nm), zeta potentials (mV) and morphological characterization of NPs. (a) Particle sizes and (b) zeta potentials of NPs generated using chitosan or HTCC with siRNA (weight ratio, 60:1) in the presence or absence of TPP. Each point represents the mean ± S.E.M. (n=3). Statistical significance was assessed using the Student’s t-test (*P<0.05 and **P<0.01). (c) Scanning electron microscopy (SEM) of TPP-HTCC-1/siRNA (weight ratio, 60:1).
Fig. 3c shows a representative SEM image of TPP-HTCC-1/siRNA NPs, which are largely spherical and have mean diameters ranging from 56.5 to 169.4 nm. The particle size measured by SEM is smaller than that determined by DLS, which is consistent with a previous finding [20]. This may reflect that fully hydrated (swollen) particles are measured during DLS, whereas SEM measurements are performed in a dry state. In addition, DLS calculates an intensity-average size, whereas SEM evaluates a number-average size, which is generally lower.
3.4 Cytotoxicity of HTCC
Cytotoxicity is a primary concern during the development of siRNA carriers. To evaluate the cytotoxicity of our developed NPs, we treated Raw 264.7 macrophages and colon-26 cells with various NPs, and examined their cell viability using MTT assays.
As presented in Fig. 4, polymer/siRNA NPs with or without TPP exhibit DQ-dependent cytotoxicity against Raw 264.7 macrophages after 4 h of treatment. Similar levels of cytotoxicity are obtained in colon-26 cells after 24 h of co-incubation. However, the viabilities of most treatment groups remain over 80% after these time points. The cytotoxicity of cationic polymers is known to depend on their surface charge: excess positive charges on the NP surface can interact with and functionally impair certain cellular components (e.g., cell membranes and intracellular enzymes) [44]. In the present study, the presence of TPP in the designed NPs should lower the surface positive charge, decreasing the interactions of these NPs with negatively charged cellular components.
Fig. 4.
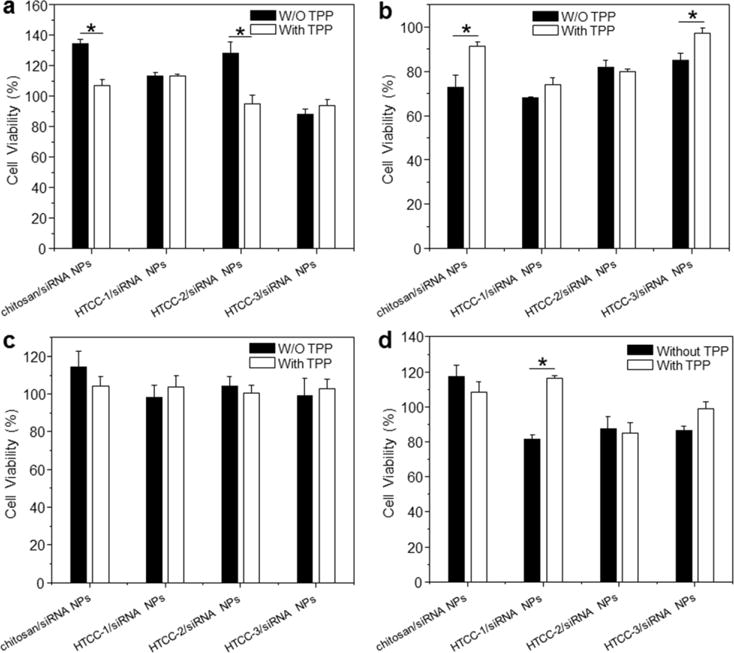
Cytotoxicity of chitosan/siRNA NPs (weight ratio, 60:1) and HTCC/siRNA NPs (weight ratio, 60:1) in the presence or absence of TPP against Raw 264.7 macrophages and colon-26 cells. (a) Raw 264.7 macrophages for 4 h, (b) Colon-26 cells for 4 h, (c) Raw 264.7 macrophages for 24 h, and (d) Colon-26 cells for 24 h. siRNA were used at a concentration of 100 nM. Toxicity is given as the percentage of viable cells remaining after treatment. Each point represents the mean ± S.E.M. (n=5). Statistical significance was assessed using the Student’s t-test (*P<0.05 and **P<0.01).
3.5 Cellular uptake of NPs by macrophages
Whether NPs can be internalized by macrophages is a vital concern for inflammatory disease therapy, since higher cellular uptake efficiency of NPs by macrophages should result in better therapeutic efficacy. To investigate the cellular uptake behavior of our NPs, we used fluorescence microscopy to monitor their time-dependent accumulation in Raw 264.7 macrophages.
Fig. 5 shows fluorescence images of cells treated with TPP-HTCC-1/FITC-siRNA NPs at different time points (0, 1, 3 and 5 h). These images show clearly time-dependent cellular uptake profiles of NPs (green fluorescence), whereas control cells are negative for this signal. An examination of individual cell images reveals the presence of weak intracellular fluorescence after 1 h of co-incubation, indicating that very few NPs are internalized into the cells during this short time period. However, cells incubated with NPs for 3 h or 5 h show bright and stable green fluorescence, suggesting that a relatively large amount of NPs has accumulated in cells. Green fluorescence is detected in almost all cells after 5 h of incubation. Furthermore, individual cell images reveal that the NPs are internalized into the cell cytoplasm rather than the nucleus. This likely reflects that the nuclear pore complex is typically 20–50 nm in size, and therefore limits the access of our NPs, which have a diameter greater than 200 nm.
Fig. 5.
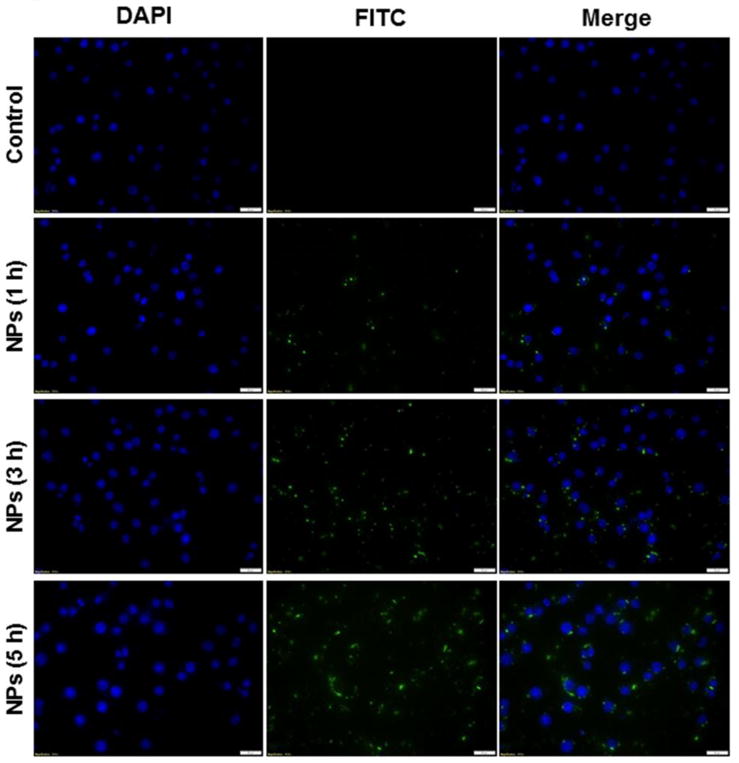
Cellular uptake profiles of TPP-HTCC-1/siRNA NPs (weight ratio, 60:1) in Raw 264.7 macrophages at different time points (1 h, 3 h and 5 h). Cells were treated with NPs loaded with FITC-siRNA (green) and processed for fluorescence staining. FITC-siRNA (100 nM) was used for the transfection. Fixed cells were stained with DAPI (purple) for visualization of nuclei. Scale bar represents 10 mm.
To quantitatively assess the cellular uptake efficiency of siRNA carried in different NPs, we treated Raw 264.7 macrophages with various NPs with or without TPP, and investigated their cellular uptake profiles after 5 h of co-incubation. In good agreement with our previous report [15], the cellular uptake of TPP-polymer/siRNA NPs is clearly higher than that of TPP-free polymer/siRNA NPs, while the cellular uptake of siRNA harbored in the latter increases slightly as the DQ increased (Fig. 6). These results indicate that the introduction of TPP to our NPs greatly improves the cellular uptake efficiency of siRNA by macrophages. Impressively, the percentage of green fluorescence-positive cells approaches 100%, which is consistent with the fluorescence imaging results shown in Fig. 5.
Fig. 6.
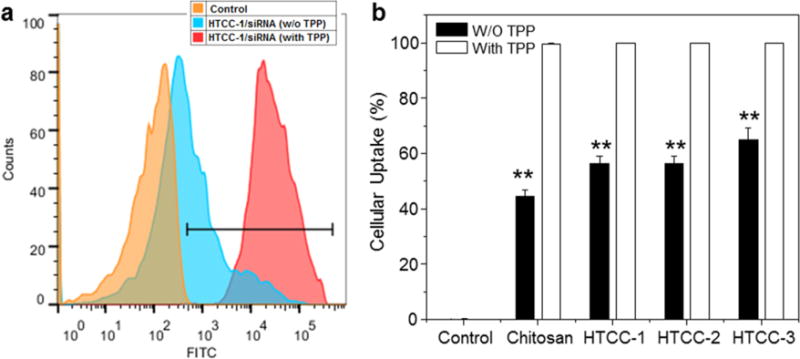
Quantification of cellular uptake of various NPs (weight ratio, 60:1) by Raw 264.7 macrophages. (a) Representative flow cytometry histograms of fluorescence intensity for cells treated with TPP-HTCC-1/siRNA NPs for 5 h. (b) Percentage of FITC fluorescence-positive cells after treatment with various NPs for 5 h. FITC-siRNA (100 nM) was used for the transfection. Each point represents the mean ± S.E.M. (n = 3; *P< 0.05 and **P< 0.01, Student’s t-test).
3.6 NP-mediated TNF-α silencing in vitro
To investigate the RNAi effectiveness of our NPs, we next tested whether siTNF-loaded NPs could be employed to knock down the expression of TNF-α in Raw 264.7 macrophages.
As demonstrated in Fig. 7a, in the context of NPs in the absence of TPP, relative to cells treated with LPS, only chitosan/siTNF NPs produce a marked decrease in TNF-α after 24 h of treatment. HTCC/siTNF NPs do not produce any obvious knockdown of TNF-α. This may reflect that HTCC samples have a stronger charge density that binds siRNA tightly and inhibits its release from the NPs, consequently restraining the functions of siTNF. A particularly large knockdown response is seen in cells treated with TPP-polymer/siTNF NPs, which decreases the expression of TNF-α to ~ 7.6–14.6% of the positive control level. In comparison, OF/siRNA complexes (positive control) decrease TNF-α expression to 26.5% of the control level, which was significantly higher than that of TPP-polymer/siTNF NPs-treated cells. Notably, the concentration of siTNF contained in our NPs is 2-fold less than that used in the OF/siTNF complexes. The similar trend of RNAi profiles has been observed after 48 h of NP treatment, as shown in Fig. 7b. In addition, free TPP exhibited negligible TNF-α mRNA downregulation effect on LPS-stimulated macrophages. The improved gene-silencing effects of our NPs could be attributed to their effective condensation of siRNA and increase intracellular internalization, both of which are improved by the introduction of TPP. The biological effects observed in the present study indicate that our TPP-containing NPs are taken up into the cells, and that they effectively release siTNF. This also indirectly demonstrates that siTNF could be efficiently released from endosome or lysosome to the cytoplasm.
Fig. 7.
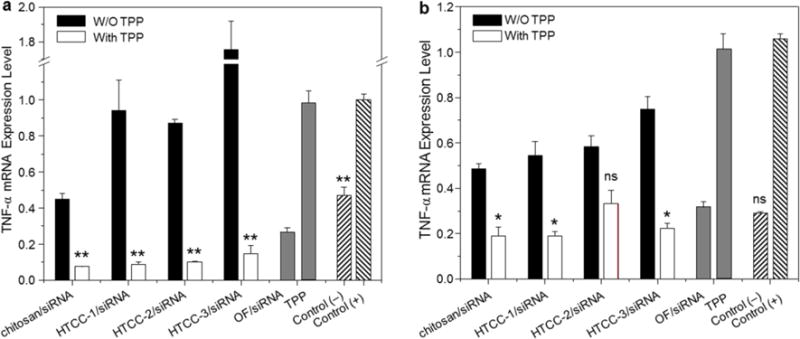
In vitro RNAi ability of various NPs (weight ratio, 60:1) against Raw 267.4 macrophages for 24 h (a) and 48 h (b). Cells were transfected by NPs (siTNF, 100 nM) for 24 h or 48 h and then treated with LPS (5 mg/mL) for 3 hours. OF was used according to the manufacturer’s standard protocols. Each point represents the mean ± S.E.M. (n = 3; *P< 0.05 and **P< 0.01, Student’s t-test).
4. Conclusion
Tumor necrosis factor-α (TNF-α), mainly secreted by macrophages, is an important therapeutic target for inflammatory diseases [2]. Previously, chitosan has been utilized as a siRNA delivery material in the presence or absence of tripolyphosphate (TPP) [23, 33]. In the present study, we successfully prepared N-(2-hydroxy)propyl-3-trimethyl ammonium chitosan chlorides (HTCCs) with different degrees of quaternization. For the first time, we fabricated TPP-containing HTCC/siTNF nanoparticles (NPs), and further investigated their physicochemical properties, cellular uptake profiles and RNAi efficiency in comparison to its counterparts (chitosan/siTNF NPs, TPP-chitosan/siTNF NPs and HTCC/siTNF NPs). Chitosan and HTCCs have much stronger siRNA condensation capacities in the presence of TPP compared with the results obtained without TPP. The TPP-containing NPs showed an enhanced siRNA condensation capacity, a desirable particles size (around 220 nm), and a slightly positive zeta potential. Importantly, the cellular uptake efficiencies of these NPs were close to 100%. Further RNA interference experiments demonstrated that TPP-containing NPs downregulated TNF-α mRNA expression level to ~7.6–14.6% of the positive control level, which was significantly lower than that of NPs without TPP and Oligofectamine/siTNF complexes. These results collectively indicate that TPP-assisted chitosan-based NPs could be exciting candidates as non-cytotoxic and efficient siTNF carriers for the treatment of inflammatory disease. Further studies are under progress in order to endow the TPP-containing chitosan-based NPs with macrophage-targeted property.
Supplementary Material
Acknowledgments
This work was supported by grants from the Department of Veterans Affairs (BX002526), the National Institutes of Health of Diabetes and Digestive and Kidney by the grant (RO1-DK-071594), the National Natural Science Foundation of China (51503172 and 81571807), the Fundamental Research Funds for the Central Universities (SWU114086 and XDJK2015C067) and the Scientific Research Foundation for the Returned Overseas Chinese Scholars (State Education Ministry). D.M. is a recipient of a Career Scientist Award from the Department of Veterans Affairs.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kriegel C, Amiji M. J Control Release. 2011;150:77–86. doi: 10.1016/j.jconrel.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wolf AM, Wolf D, Rumpold H, Ludwiczek S, Enrich B, Gastl G, Weiss G, Tilg H. Proc Natl Acad Sci U S A. 2005;102:13622–13627. doi: 10.1073/pnas.0501758102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Feldmann M. Nat Rev Immunol. 2002;2:364–371. doi: 10.1038/nri802. [DOI] [PubMed] [Google Scholar]
- 4.Hanauer SB, Feagan BG, Lichtenstein GR, Mayer LF, Schreiber S, Colombel JF, Rachmilewitz D, Wolf DC, Olson A, Bao W, Rutgeerts P, ACCENT I Study Group Lancet. 2002;359:1541–1549. doi: 10.1016/S0140-6736(02)08512-4. [DOI] [PubMed] [Google Scholar]
- 5.Cohen RD, Tsang JF, Hanauer SB. Am J Gastroenterol. 2000;95:3469–3477. doi: 10.1111/j.1572-0241.2000.03363.x. [DOI] [PubMed] [Google Scholar]
- 6.Sandborn WJ. Best Pract Res Clin Gastroenterol. 2003;17:105–117. doi: 10.1053/bega.2002.0345. [DOI] [PubMed] [Google Scholar]
- 7.Hannon GJ. Nature. 2002;418:244–251. doi: 10.1038/418244a. [DOI] [PubMed] [Google Scholar]
- 8.Kim SS, Ye C, Kumar P, Chiu I, Subramanya S, Wu H, Shankar P, Manjunath N. Mol Ther. 2010;18:993–1001. doi: 10.1038/mt.2010.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Whitehead KA, Langer R, Anderson DG. Nat Rev Drug Discov. 2009;8:129–138. doi: 10.1038/nrd2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Han L, Tang C, Yin CH. Biomaterials. 2013;34:5317–5327. doi: 10.1016/j.biomaterials.2013.03.060. [DOI] [PubMed] [Google Scholar]
- 11.Lu B, Xu XD, Zhang XZ, Cheng SX, Zhuo RX. Biomacromolecules. 2008;9:2594–2600. doi: 10.1021/bm8004676. [DOI] [PubMed] [Google Scholar]
- 12.Putnam D. Nat Mater. 2006;5:439–451. doi: 10.1038/nmat1645. [DOI] [PubMed] [Google Scholar]
- 13.Kim TH, Ihm JE, Choi YJ, Nah JW, Cho CS. J Control Release. 2003;93:389–402. doi: 10.1016/j.jconrel.2003.08.017. [DOI] [PubMed] [Google Scholar]
- 14.Kim TH, Ihm JE, Choi YJ, Nah JW, Cho CS. J Control Release. 2005;105:354–366. [Google Scholar]
- 15.Xiao B, Laroui H, Ayyadurai S, Viennois E, Charania MA, Zhang Y, Merlin D. Biomaterials. 2013;34:7471–7482. doi: 10.1016/j.biomaterials.2013.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tseng YC, Mozumdar S, Huang L. Adv Drug Deliv Rev. 2009;61:721–731. doi: 10.1016/j.addr.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang CX, Gao S, Kjems J. J Mater Chem B. 2014;2:8608–8615. doi: 10.1039/c4tb01374c. [DOI] [PubMed] [Google Scholar]
- 18.Song W, Zhao LZ, Fang KX, Chang B, Zhang YM. J Mater Chem B. 2015;3:8567–8576. doi: 10.1039/c5tb01062d. [DOI] [PubMed] [Google Scholar]
- 19.Choi B, Cui ZK, Kim S, Fan J, Wu BM, Lee M. J Mater Chem B. 2015;3:6448–6455. doi: 10.1039/C5TB00843C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xiao B, Wan Y, Wang X, Zha Q, Liu H, Qiu Z, Zhang S. Colloids Surf B. 2012;91:168–174. doi: 10.1016/j.colsurfb.2011.10.053. [DOI] [PubMed] [Google Scholar]
- 21.Dehousse V, Garbacki N, Colige A, Evrard B. Biomaterials. 2010;31:1839–1849. doi: 10.1016/j.biomaterials.2009.11.028. [DOI] [PubMed] [Google Scholar]
- 22.Dehousse V, Garbacki N, Jaspart S, Castagne D, Piel G, Colige A, Evrard B. Int J Biol Macromol. 2010;46:342–349. doi: 10.1016/j.ijbiomac.2010.01.010. [DOI] [PubMed] [Google Scholar]
- 23.Xiao B, Wan Y, Zhao MQ, Liu YQ, Zhang SM. Carbohydr Polym. 2011;83:144–150. [Google Scholar]
- 24.Xiao B, Wang X, Qiu Z, Ma J, Zhou L, Wan Y, Zhang SM. J Biomed Mater Res A. 2013;101:1888–1897. doi: 10.1002/jbm.a.34493. [DOI] [PubMed] [Google Scholar]
- 25.Gao Y, Xu Z, Chen S, Gu W, Chen L, Li Y. Int J Pharm. 2008;359:241–246. doi: 10.1016/j.ijpharm.2008.03.037. [DOI] [PubMed] [Google Scholar]
- 26.Janciauskaite U, Rakutyte V, Miskinis J, Makuska R. React Funct Polym. 2008;68:787–796. [Google Scholar]
- 27.Sugimoto M, Morimoto M, Sashiwa H, Saimoto H, Shigemasa Y. Carbohydr Polym. 1998;36:49–59. [Google Scholar]
- 28.Thanou M, Florea BI, Geldof M, Junginger HE, Borchard G. Biomaterials. 2002;23:153–159. doi: 10.1016/s0142-9612(01)00090-4. [DOI] [PubMed] [Google Scholar]
- 29.Mao Z, Ma L, Jiang Y, Yan M, Gao C, Shen J. Macromol Biosci. 2007;7:855. doi: 10.1002/mabi.200700015. [DOI] [PubMed] [Google Scholar]
- 30.Rudzinski WE, Aminabhavi TM. Int J Pharm. 2010;399:1–11. doi: 10.1016/j.ijpharm.2010.08.022. [DOI] [PubMed] [Google Scholar]
- 31.Csaba N, Koping-Hoggard M, Alonso MJ. Int J Pharm. 2009;382:205–214. doi: 10.1016/j.ijpharm.2009.07.028. [DOI] [PubMed] [Google Scholar]
- 32.Nasti A, Zaki NM, de Leonardis P, Ungphaiboon S, Sansongsak P, Rimoli MG, Tirelli N. Pharm Res. 2009;26:1918–1930. doi: 10.1007/s11095-009-9908-0. [DOI] [PubMed] [Google Scholar]
- 33.Katas H, Alpar HO. J Control Release. 2006;115:216–225. doi: 10.1016/j.jconrel.2006.07.021. [DOI] [PubMed] [Google Scholar]
- 34.Lavertu M, Méthot S, Tran-Khanh N, Buschmann MD. Biomaterials. 2006;27:4815–4824. doi: 10.1016/j.biomaterials.2006.04.029. [DOI] [PubMed] [Google Scholar]
- 35.Badawy MEI, Rabea EI. Postharvest Biol Technol. 2009;51:110–117. [Google Scholar]
- 36.Cho J, Grant J, Piquette-Miller M, Allen C. Biomacromolecules. 2006;7:2845–2855. doi: 10.1021/bm060436s. [DOI] [PubMed] [Google Scholar]
- 37.Lim SH, Hudson SM. Carbohydr Res. 2004;339:313–319. doi: 10.1016/j.carres.2003.10.024. [DOI] [PubMed] [Google Scholar]
- 38.Sun Y, Wan AJ. J Appl Polym Sci. 2007;105:552–561. [Google Scholar]
- 39.Mok H, Lee SH, Park JW, Park TG. Nat Mat. 2010;9:272–278. doi: 10.1038/nmat2626. [DOI] [PubMed] [Google Scholar]
- 40.Palmeira-de-Oliveira R, Palmeira-de-Oliveira A, Gaspar C, Silvestre S, Martinez-de-Oliveira J, Amaral MH, Breitenfeld L. Int J Pharm. 2011;421:130–134. doi: 10.1016/j.ijpharm.2011.09.030. [DOI] [PubMed] [Google Scholar]
- 41.Janes KA, Calvo P, Alonso MJ. Adv Drug Deliv Rev. 2001;47:83–97. doi: 10.1016/s0169-409x(00)00123-x. [DOI] [PubMed] [Google Scholar]
- 42.Song Y, Sun Y, Zhang X, Zhou J, Zhang L. Biomacromolecules. 2008;9:2259–2264. doi: 10.1021/bm800429a. [DOI] [PubMed] [Google Scholar]
- 43.Liu YM, Reineke TM. J Am Chem Soc. 2005;127:3004–3015. doi: 10.1021/ja0436446. [DOI] [PubMed] [Google Scholar]
- 44.Ryser HJ. Nature. 1967;215:934–936. doi: 10.1038/215934a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.