

Abstract
The treatment of advanced renal cell carcinoma has posed a challenge for decades, in part because of common themes related to intrinsic resistance to cytotoxic chemotherapy, and the obscure biology of these cancer types. Forward movement in the treatment of the renal cell carcinomas can thus be approached in two ways: splitting the tumor types along histologic and molecular features, in the hopes of coupling highly precision-focused therapy on a subset of disease with most potential for benefit, or lumping the various biologies and histologies together, in order to carry the rarer renal cell carcinoma types with more common disease. The former strategy satisfies our desire for customized precision in treatment delivery, while the latter strategy allows us to offer a wider therapeutic menu in a set of diseases we are continuing to learn about on a physiologic and molecular level.
Keywords: Renal cell carcinoma, clear cell, papillary, molecular subtypes
2. Introduction
The study of biology has been propelled forward over the years by field-shifting technological advances that bring scientific details into greater resolution. These advances have revolutionized multiple fields, including cancer biology. Often, the perspectives that these technologies provide results in the division of a seemingly homogeneous cohort into multiple further carefully defined subtypes. A prime example is lung cancer. Classically, lung cancer was subdivided into small cell and non-small cell types based on histologic appearance. Subsequently, non-small cell lung cancer was further divided into distinct histologic subtypes including adenocarcinoma, squamous, and large cell, although treatment with cisplatin-based doublets was long held as the standard of care for all of them1. Adenocarcinoma of the lung is now further subdivided based on the presence or absence of driver mutations. Given the development of potent inhibitors targeting the protein products of these driver mutations, these molecularly-defined subtypes of adenocarcinoma now split the disease along mutation lines to guide therapeutic decisions. For example, tumors harboring EGFR mutations are particularly sensitive to EGFR inhibitors such as erlotinib2. Thus, the technology-driven subtyping or “splitting” of cancers has directly impacted treatment paradigms.
The EGFR lung adenocarcinoma breakthrough is but one of several such examples in oncology where molecular features of a patient’s tumor directly dictates therapeutic decisions. The success of these select examples, coupled with the momentum stemming from advanced technologies in molecular biology, has fueled enthusiasm for precision medicine. Precision, or “personalized” cancer medicine, as defined by the National Cancer Institute, is the use of “specific information about a person’s tumor to help diagnose, plan treatment, find out how well treatment is working, or make a prognosis”. Thus, if cancer care is to pursue this model of medicine, one could envision a path towards further and further “splitting” of tumors. In fact, an extreme interpretation of personalized medicine is that no two patients and thus no two tumors are identical. Furthermore, we now know that individual tumors are heterogeneous and constantly evolving3. Thus, “splitting” to the extreme will result in the identification of genomically and phenotypically distinct subregions within an individual patient’s tumor.
Identifying individual tumors in individual patients and designing individualized treatment plans based on this data is exciting and appealing. However, practically, such pursuits make the development of novel therapeutics, provider education, and regulation significantly more difficult. In addition, given the finite size of the human genome, the number of ways in which a cancer cell can program itself is immense and diverse but perhaps not infinite. Thus, it is not surprising that the pursuit of “personalized cancer medicine” has seen examples of the pendulum swinging away from ever-more subtypes or “splitting” of tumors and towards a system in which tumors that share a certain molecular feature are “lumped” together for study purposes, including drug development. For example, it was discovered that a subgroup of esophagogastric adenocarcinoma overexpressed the receptor tyrosine kinase HER2. HER2 is also overexpressed in breast cancer where multiple agents that target this specific protein have been tested and shown to be effective. Indeed, when treated with targeted drugs, the metastatic HER2-overexpressing esophagogastric adenocarcinomas demonstrated a response4. Thus, in this example, HER2-overexpressing tumors can potentially be “lumped” together with use of similar drugs. This system of “lumping” of otherwise very dissimilar tumors, in this case breast and gastric adenocarcinomas, with distinct sites of origin, epidemiology, and pathogenesis can be viewed as the antithesis to the parsing of similar tumors into further and further subdivisions. In this review, we seek to describe these seemingly antagonistic trends in renal cell carcinoma specifically and explore how these trends may impact patient care in the near future.
3. The Evolution of Renal Cell Carcinoma Sub-classification: A Path of “Splitting”
3.1. Introduction to Renal Cell Carcinoma Histologic Subtypes
It is now well accepted across the renal cell carcinoma (RCC) community that kidney tumors that look very different when examined under magnification are distinct diseases with divergent biology. This simple conclusion, which may seem self-evident to the modern physician and investigator, actually took decades of analysis and the molecular biology revolution to become realized. For decades, most or all kidney tumors were lumped under a single title5. While other early pathologists described kidney tumors with papillary features6, not until the 1970s was “papillary” renal cell carcinoma studied in a detailed manner with clinical and pathologic correlation7. These and other studies marked a transition in which investigators began to increasingly appreciate and describe distinct histologic subtypes of kidney cancer. Thus, what was once collectively known as “hypernephromas” would eventually be “split” into at least 16 distinct subtypes through the application of various histologic and molecular criteria (Table 1)8. Enthusiasm for this system of splitting renal cell carcinoma into many subtypes was fueled by recognition that core biology, clinical features, and even response to treatments can be very different among the subtypes.
Table 1. 2016 WHO Classification of Kidney Tumors.
Adapted from Moch et al.,8.
| Kidney Cancer Subtype | Description |
|---|---|
| Clear cell renal cell carcinoma | VHL mutated, most common adult kidney cancer |
| Multiloculated cystic clear cell renal cell neoplasm of low malignant potential | Low grade, little or no recurrence or metastatic potential |
| Papillary renal cell carcinoma | Second most common adult kidney cancer, subdivided into types 1 and 2 |
| Hereditary leiomyomatosis renal cell carcinoma syndrome-associated renal cell carcinoma | Familial kidney cancer syndrome with germline mutations in fumarate hydratase |
| Chromophobe renal cell carcinoma | Lower malignant potential, associated with Bitt-Hogg-Dubé syndrome |
| Collecting duct carcinoma | Rare, poor prognosis |
| Renal medullary carcinoma | Associated with sickle cell trait, young patients, poor prognosis |
| MiT family translocation renal cell carcinoma | Young patients, poor prognosis53, rare |
| Succinate-dehydrogenase deficient renal cell carcinoma | Young patients, germline SDH mutations, good prognosis54 |
| Mucinous tubular and spindle cell carcinoma | Rare, good prognosis, female predominance |
| Tubulocystic renal cell carcinoma | Rare, good prognosis |
| Acquired cystic disease associated renal cell carcinoma | Associated with end-stage renal disease, good prognosis |
| Clear cell papillary renal cell carcinoma | May occur in end-stage renal disease and VHL disease, good prognosis |
| Renal cell carcinoma, unclassified | |
| Papillary adenoma | ≤1.5 cm, low grade, unencapsulated, good prognosis |
| Oncocytoma | Benign, well-differentiated, mitochdrial accumulation55, 56 |
3.2. Histologic Subtypes of Renal Cell Carcinoma
3.2.1. Clear Cell Renal Cell Carcinoma
The most common histologic subtype of renal cell carcinoma is the clear cell variant, representing 70–75% of adult renal cell carcinomas. These tumors tend to be vascular and can have macroscopic evidence of hemorrhage and necrosis, which was astutely described by early pathologists5. Microscopically, these tumors are characterized by clear cytoplasm due to abundant accumulation of glycogen and lipids9. Certain molecular hallmarks have come to define clear cell RCC and are now thought to be critical to its pathogenesis. For example, loss of function of the von Hippel Lindau (VHL) protein or dysregulation of its downstream effectors is nearly ubiquitous in clear cell RCC. The VHL gene is a classic two-hit tumor suppressor, as demonstrated by the autosomal dominant germline hereditary cancer risk syndrome VHL disease10. One copy is either mutated or silenced in up to 90% of sporadic clear cell RCC tumors11 while the second copy is typically lost through chromosome 3p deletions12. Under physiologic and normoxic conditions, the VHL protein is an important component of an E3 ubiquitin ligase complex that targets the hypoxia-inducible factors (HIFs) for proteasome-mediated degradation. However, biallelic loss of VHL allows for the inappropriate stabilization of HIFs (irrespective of oxygen levels) which results in a pro-angiogenic gene expression signature critical to clear cell RCC tumorigenesis13. This “pseudohypoxic” response has come to define clear cell RCC biology and distinguishes it from other RCC histologies.
3.2.2. Papillary Renal Cell Carcinoma
Papillary RCC is the second most common histologic subtype of adult kidney cancer, representing about 15% of cases. As discussed earlier, this variant of kidney cancer with papillary or tubulopapillary architecture was recognized early6. However, its recognition as a distinct subtype of kidney cancer gained traction as it was recognized that these tumors had a distinct clinical course7 and molecular features relative to the more common clear cell variant. Specifically, while 3p loss is nearly universal in clear cell RCC, early kidney cancer cytogenetic studies demonstrated that this chromosomal abnormality was absent in most papillary RCC14 and thus further supported the concept that papillary RCC was biologically and clinically distinct from clear cell RCC. Later, as larger papillary RCC cohorts were studied, it became evident that this disease could be further subdivided on the basis of histology into two subtypes. Type 1 papillary RCC was characterized by papillary or tubular structures covered by small basophilic cells with clear cytoplasm with small nuclei and inconspicuous nucleoli whereas type 2 papillary RCC was characterized by larger cells with eosinophilic cytoplasm, pseudostratification of cells, and prominent nucleoli15. Moreover, familial studies reveal that germline mutations contribute specifically to inherited forms of both disease—hereditary papillary renal carcinoma syndrome with germline mutations in MET giving rise to type 1 tumors16, 17 and hereditary leiomyomatosis and renal cell carcinoma syndrome with germline mutations in fumarate hydratase giving rise to type 2 tumors. Large genomic studies have demonstrated strong molecular distinctions between sporadic papillary type 1 and 2 RCC. Type 1 disease is characterized by molecular events that result in amplification of signaling from the hepatocyte growth factor receptor MET, most commonly gain-of-function somatic mutations in the tyrosine kinase domain19. Meanwhile, type 2 disease itself is more heterogeneous with loss of CDKN2a function, activation of the NRF2 anti-oxidant response element pathway, and mutations in chromatin regulators19, allowing the type 2 papillary tumors to be further subdivided into at least three additional subclassifications. Thus, type 1 and 2 papillary RCC have become fully recognized as being not only distinct from clear cell RCC, but also distinct from each other.
3.2.3. Other Histologic Subtypes of Renal Cell Carcinoma
While clear cell and papillary RCC are the two most common subtypes of kidney cancer, representing 80–90% of cases, others have been described with increasingly robust molecular annotation. Chromophobe RCC represents ~5% of adult kidney cancer and microscopically features distinct cell borders and voluminous cytoplasm9. Two histologic subtypes have been described for even this rare tumor entity: classic and eosinophilic. Classic chromophobe has a pale cytoplasm and is more likely to demonstrate the characteristic monosomy chromosomal pattern that is often associated with chromophobe RCC, which includes loss of most or all of an entire copy of chromosomes 1, 2, 6, 10, 13, 17 and often 2120. In addition, the classic chromophobe subtype contains more somatic mutations, including mutations in TP53 and PTEN. Meanwhile, the eosinophilic subtype has dense pink cytoplasm due to mitochondrial accumulation. Interestingly, the eosinophilic subtype is enriched with mutations in mitochondrial genes, specifically those corresponding to complex 1 of the electron transport chain20. Thus, like other RCC histologies, variants of chromophobe RCC proved to have distinct molecular features and thus justifies their classification as distinct cancers.
Further to the splitters mindset, each of these subtypes can be further affected by the involvement of sarcomatoid histology. So-called sarcomatoid tumors sometimes have such overwhelming sarcomatoid features that the underlying primary tumor type is unidentifiable. Recent genetic analysis has demonstrated that a common profile of mutations can contribute to this aggressive histology21, and combination chemotherapy regimens incorporating targeted therapies can be used for this splinter group22.
Clear cell, papillary, and chromophobe RCC have a commonality in that they all are thought to originate in the renal cortex: clear cell and papillary RCC in the proximal convoluted tubule and chromophobe RCC in the distal convoluted tubule12, 19, 20. In contrast, other kidney tumors originate outside the renal cortex and include collecting duct (or Bellini’s tumors) and medullary carcinomas. Both are thought to arise from the renal medulla. Relatively less is known about these rare kidney cancer subtypes. Collecting duct carcinomas were recognized as a distinct clinical diagnosis in the 1980s and are characterized by a tubulopapillary pattern and desmoplastic stroma23 and in some cases are difficult to distinguish histologically from urothelial carcinomas. However, biologically they may be very different from urothelial carcinomas as the two cancers contain very different patterns of chromosomal gain and loss24. Medullary carcinomas most commonly arise in young patients with sickle cell trait25, 26. Even less is known about their molecular biology, although mutations in SMARCB1 appear to dominate their genetic profile27, rendering them further distinct from the other histologic subtypes described.
3.2.4. Clinical Significance of Histologic Subtypes of Renal Cell Carcinoma
While it is clear that several histologic subtypes exist within the RCC spectrum and detailed molecular studies have shed light on their divergent biology, enthusiasm for “splitting” this disease has been fueled by the clinical relevance of the subtypes. Specifically, the various histologic subtypes can vary greatly in prognosis. For example, initially it seemed that papillary RCC had a favorable prognosis relative to clear cell RCC7. However, with the emergence of histologic papillary RCC subtypes, the story became more complicated. It is now clear that the MET-driven papillary RCC type 1 tumors have a favorable prognosis with the majority of cases representing early stage disease while papillary RCC type 2 patients have a high rate metastasis and poor overall survival19. Distal tubule-derived chromophobe RCC rarely metastases and patients do well, while the tumors that arise from the renal medulla, namely collecting duct and medullary carcinomas, represent extremely aggressive disease with poor overall prognosis. In one series of renal medullary carcinoma, the mean survival was a mere 4 months26. Thus, precise histologic subclassification can provide a profound contribution to the understanding of an individual patient’s prognosis.
The clinical relevance of RCC histologic subtypes informs not only prognosis but, in some instances, can guide treatment decisions. For example, in the early studies of immunotherapy in RCC using high dose interleukin-2, it was observed that the response rate was superior for clear cell RCC relative to non-clear cell RCC subtypes for reasons that remain poorly understood28. These observations have been validated in more recent prospective analyses as well29.
However, a new class of medications would cause a dramatic shift in the design of prospective RCC trials. As discussed before, the molecular hallmark of clear cell RCC (but not other subtypes) is biallelic loss of VHL with resultant HIF stabilization and inappropriate hypoxia signaling, including profound increases in angiogenesis signaling13. This signaling includes increased production of vascular epithelial growth factor and subsequent signaling through the VEGF receptors (VEGFR), a class of receptor tyrosine kinases. Signaling through the VEGFRs, especially on endothelial cells, supports the tumor vasculature and growth. When a novel class of small molecule inhibitors were designed to target VEGFR, the large phase III trials typically included as an inclusion criteria a “clear cell component” in describing the histology30, 31. These trials established the VEGFR inhibitors as the new standard first-line therapy for metastatic clear cell RCC. The exclusion of non-clear cell RCC was justified because these VHL-intact patients were not predicted to have as much benefit from this class of drugs. Thus, for some time, it was unclear whether non-clear cell RCC patients benefited from VEGFR inhibitors. Attempts to design VEGFR inhibitors with an increased target spectrum tailored to papillary RCC biology has had mixed results. Specifically, foretinib has activity against both MET and VEGFR and was tested in a phase II trial for papillary RCC patients32. While foretinib had activity, the most robust responses were in patients with germline MET mutations (i.e. most likely a small subset of papillary type 1 patients). However, novel combinations such as bevacizumab and erlotinib that target angiogenesis and glucose transport, respectively, in the highly glycolytic FH mutant papillary type 2 group are being examined carefully33. Thus, in the anti-angiogenesis era, “splitting” RCC on the basis of histology has had a dramatic impact on clinical trial design.
3.3. Molecular Subtypes of Renal Cell Carcinoma
Given the importance of histologic sub-classification in prognosis and therapy, investigators have been aggressive in identifying molecular subtypes in an attempt to build upon this success. While some other non-clear cell RCC histologic subtypes, such as papillary and chromphobe RCC, can be further subdivided based on histology, clear cell RCC is relatively homogenous. Given that clear cell RCC represents the largest bulk of patient burden, there has been great interest in identifying molecular subtypes that can further inform prognosis, biology, and drug development. One such example emerged when investigators examined patterns of HIF staining in 160 sporadic human clear cell RCC tumors and split tumors into three groups: VHL wild-type, tumors positive for both HIF-1α and HIF-2α (H1H2), or tumors positive for only only HIF-2α (H2)34. The H1H2 tumors exhibited increased angiogenesis as well as increased AKT/mTOR and ERK/MAPK1 signaling while the H2 tumors demonstrated increased c-Myc activity and enhanced proliferation, features which could impact therapy effectiveness, but which remain untested. An alternative strategy to split clear cell RCC was developed from gene expression data. Using genome-wide mRNA microarray data, unsupervised clustering identified two dominant clear cell RCC subtypes called ccA and ccB35. Biologically, these subtypes are distinct with ccA tumors exhibiting increased angiogenesis while ccB tumors demonstrate increased TGF-beta and epithelial-to-mesenchymal signaling. Perhaps most importantly, this classification represents clinically distinct subtypes with ccB tumors demonstrating increased tumor size, grade, rate of metastasis as well as decreased recurrence-free and overall survival in multiple datasets12, 36, 37. In addition, these molecular subtypes feature distinct metabolic patterns with the aggressive ccB tumors demonstrating increased glucose uptake on imaging38. However, the other clear finding from this study was that individual tumors can harbor both ccA and ccB components, making this classification strategy difficult for informing therapeutic selection, although it has emerged as a strong prognostic indicator in numerous studies37, 39. Other investigators have successfully merged gene expression data with metabolomics profiling to parse clear cell RCC into multiple molecular subtypes, including an aggressive cohort with increased glutathione metabolism40. Finally, building off the results of large-scale genome-sequencing projects, another schema has emerged that focuses on two of the more commonly mutated genes in clear cell RCC: PBRM1 and BAP112. Both genes are involved in chromatin regulation and mutations in these genes are typically mutually exclusive. Using immunohistochemistry assays that evaluate protein expression and are known to reliably correlate with mutation status, large clinically-annotated tissue microarrays were utilized to segregate clear cell RCC tumors into 1 of 4 categories: tumors that express both PBRM1 and BAP1 (40%), BAP1-positive tumors (49%), PBRM1-positive tumors (9%), and tumors negative for both proteins (2%)41. The best prognosis was the group that expressed both PBRM1 and BAP1 (thus expected to lack mutations in these genes) whereas the worst prognosis was in the two groups that lacked expression of both proteins (therefore expected to be doubly mutated). Thus, using several powerful molecular platforms, several schemas to identify prognostically-significant molecular subtypes of clear cell RCC have emerged, which may indicate a future in which we deliver personalized therapy based on these features.
Until recently, molecular subtyping of non-clear cell RCC tumors was relatively unexplored. However, the Cancer Genome Atlas working group has used multiple platforms to molecularly annotate papillary RCC19. This integrated analysis identified 4 major molecular subtypes of papillary RCC. One subtype included predominantly papillary RCC type 1 tumors, including frequent activation of MET, whereas the other 3 subtypes included mostly papillary RCC type 2 tumors and thus reflects the molecular heterogeneity of this subtype. Among these 3 molecular subtypes of papillary RCC type 2 tumors was one notable for its extremely poor prognosis and young age of onset. Termed CpG island methylator phenotype or CIMP, these tumors were identified by their widespread pattern of genomic hypermethylation. These tumors also were frequently observed to have CDKN2a silencing and contained all the cases with germline mutations in fumarate hydratase. Thus, while papillary type 1 RCC was relatively homogeneous, papillary type 2 RCC displays far more molecular heterogeneity with multiple molecular subtypes identified.
4. The Case for Lumping
While it is relatively simpler (in the mind of an avowed splitter) to apply definitions with greater and greater specificity, there can be made a strong rational case for lumping strategies, particularly given the strong reinforcement that has come from decades of trials that effectively lump all of the renal cell carcinomas together. This strategy simplifies enrollment in studies, and provides treatment options for a greater range of patients. Although the non-clear cell histologies always make up a tiny minority of cases, clinical trials have not consistently shown inferiority for this practice42. Moreover, in spite of the extensive subclassification strategies available, histologically it remains not uncommon for tumors to be assigned a diagnosis of “unclassified” RCC, or renal epithelial tumor not otherwise specified. This is due to wide variation in histologic features even within subtypes, and a current lack of effective molecular tools to augment pathologic assignment. In fact, studies show activity of standard front line VEGF receptor targeted therapies in papillary RCC43 and even chromophobe RCC44. Even including the sarcomatoid variant classification, data suggesting VEGF receptor targeted therapies can be effective45 reinforces the lumping paradigm. Additionally, as these various histologies are treated with similar therapies, it is possible that convergent resistance mechanisms could emerge that likewise could be treated similarly.
The same was true for some pivotal, early trials with another class of medications: the mammaliam target of rapamycin or mTOR-inhibitors. Specifically, in a first-line trial comparing the mTOR-inhibitor temsirolimus to interferon-alpha or the combination in metastatic renal cell carcinoma, nearly 20% of enrollees had non-clear cell RCC histologies46. This trial was limited to patients with poor prognosis disease as defined by clinical criteria47. As temsirolimus proved to be superior to interferon-alpha and equivalent to the combination, it has emerged as an accepted therapy for poor-risk, first line RCC patients regardless of histologic subtype.
While many of the studies described thus far focused primarily on defining disease biology without much consideration to therapeutics, the biological underpinnings can in fact make the case strongly in favor of lumping—as in lumping tumors according to biological properties. For example, investigators identified 5 RCC patients with “exceptional” response to mTOR-inhibitors (median duration of response 28 months)48. Of note, 1 patient could not be histologically subtyped while the other 4 were clear cell RCC. Multiple specimens were obtained for each patient and genomic sequencing was performed. Interestingly, nearly all the samples identified activating alterations of the mTOR pathway. Thus, the use of genomic sequencing to identify a subgroup of patients more likely to respond to mTOR-inhibitors is an exciting approach to RCC therapeutics. This strategy is becoming increasingly popular, and the use of basket-style trials supports even broader lumping of disease types according to underlying genomic analysis rather than according to tissue of origin49.
Previously, the biological defining features of the major subtypes of renal cell carcinoma were highly distinct, as detailed above, which warranted the strong concern against lumping strategies. However, recent high throughput sequencing efforts have revealed new themes of somatic alterations that cross histologic boundaries. In addition to inactivating mutations in PTEN and related members of the TSC/mTOR signaling axis, mutations in chromatin modifiers such as SETD2 and PBRM1/BAF180 are also prevalent across the RCC spectrum12, 19, 20. As new therapeutics emerge that target cells with specific somatic dependencies, the options to lump tumors according to precision genomic strategies will only increase.
For better or worse, the newest therapies in this disease rely on targets for which we have very little knowledge to guide their utilization. The immunotherapies, formerly dominated by high dose interleukin-2 which was widely accepted to be exclusively active in clear cell RCC as well as melanoma, are now rapidly changing form with the advent of PD-1, PD-L1, and CTLA-4 inhibitory antibodies. These therapies hold great promise for deep, sustainable responses in a surprisingly broad array of tumor types. The pivotal, positive phase III trial of nivolumab versus everolimus in previously treated patients that resulted in the approval of nivolumab was limited to clear cell RCC patients50, although the indication was delivered broadly to patients with advanced previously treated RCC. In fact, programmed death ligand 1 (PDL1) has been shown to be expressed on both clear cell and non-clear cell RCC tumors51, 52. Despite this observation, many ongoing prospective trials including check-point inhibitors are following the pattern from the anti-angiogenesis era and limiting inclusion to those patients whose tumors contain a clear cell component (e.g. NCT01472081, NCT02575222). Thus, we may need to wait for retrospective analyses of off-study patients or novel trial design to address the question of the impact of histology on patient outcomes with check-point inhibitors. For now, however, many tumors are finding themselves lumped together as we search for clues to understand the arbiters of response to this new class of immune therapies.
5. Conclusion
Renal cell carcinoma is no longer recognized as a single disease but rather a collection of related, yet biologically distinct, cancers. Through the use of detailed molecular studies, we know that these histologically divergent tumors are also biologically divergent with unique mutational burdens, gene expression patterns, proteomic profiles, and metabolomic signatures. Even among populations of histologically indistinguishable tumors, molecular profiling has allowed us to parse out biologically unique subtypes. These molecular subtypes are clinically relevant as evidenced by their dissimilar recurrence and survival rates. This work has led to new knowledge and insight into the core biology of these tumors that is unparalleled in the history of the disease (Figure 1).
Figure 1.
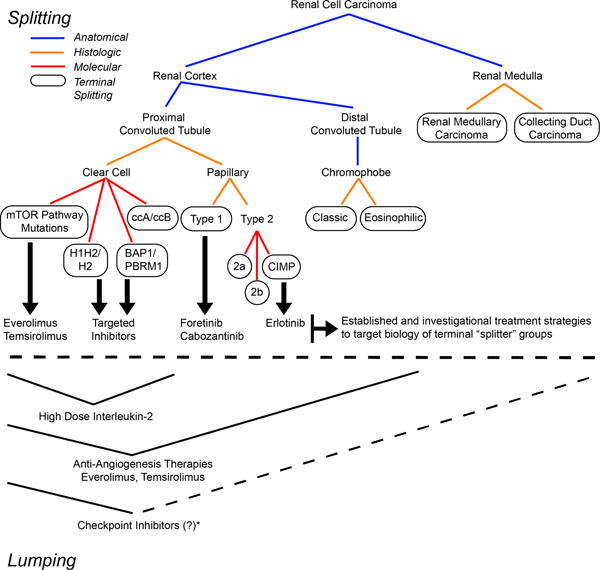
Representation of the two schools of thought: Splitting (top) demonstrating the narrower definitions obtained by applying increasingly strict criteria. Such strategies have been highly effective for application of specifically targeted therapy. Lumping (bottom) converges like groups according to similar features, or in this example, according to therapies that have demonstrated benefit to the group. *While PD-L1 staining has been observed in both clear cell and non-clear cell RCC, the activity of checkpoint inhibitors in non-clear cell RCC has not been well studied. However, PD-L1 staining is an important example of how biomarkers for specific classes of drugs can cross over previously defined “splitting schemas.
While our understanding of the biologic subtypes of RCC has progressed relatively quickly, the pace of clinical discovery to meet these diseases with precision approaches has lagged. There are several possible explanations why identification of more and more molecularly-distinct subtypes of RCC has not always resulted in immediate clinical advances. First, a lack of effective therapies is an obvious hindrance to improving RCC care. Regardless of how many times one “splits” RCC into biologically-defined subtypes, if all subtypes are resistant to available therapies, little clinical progress can be made. By the same token, if targeted therapies lack specificity for driver events, as in the case of the VEGF receptor targeted therapies which target tumor vasculature, then it is not surprising that these treatments can have broad activities in a variety of tumor types. However, as precision medicine tools, we have yet to secure a highly potent and durable targeted therapy approach that takes full advantage of the deviant biology of one or more of the renal tumor subtypes. These precise tools are on the horizon, and it will be in our best interest as a community to be ready to integrate such new drugs with accurate classification schemes and annotation strategies for renal cell carcinomas.
While a detailed understanding of RCC biologic subtypes has undoubtedly led to a deeper understanding of the diverse biology within this disease spectrum, and the development of matched therapies that target the driver biology is arguably the best approach to obtaining the ideals of precision medicine, difficulties ascertaining this ideal should not paralyze drug development. For example, the checkpoint inhibitors represent an exciting, novel therapeutic approach in RCC. As mentioned previously, this method of escaping immune surveillance, including expression of PLD1, is utilized by many RCC subtypes. As more is learned about this biology across the RCC spectrum, likely new “splitting” paradigms will emerge that incorporate this important biology with what has been previously described. However, in the present, it is practical and even essential that we “lump” tumors based on this biology for purposes of biomarker development and clinical trial design.
Acknowledgments
Funding to support this work was provided by the following: SMH is supported by Conquer Cancer Foundation Young Investigator Award program and American Urologic Association Research Scholar program. WKR is supported by the NIH K24CA172355.
Footnotes
SMH and WKR have no disclosures to report.
References
- 1.Chemotherapy in non-small cell lung cancer: a meta-analysis using updated data on individual patients from 52 randomised clinical trials. Non-small Cell Lung Cancer Collaborative Group. BMJ. 1995;311:899–909. [PMC free article] [PubMed] [Google Scholar]
- 2.Rosell R, Carcereny E, Gervais R, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012;13:239–246. doi: 10.1016/S1470-2045(11)70393-X. [DOI] [PubMed] [Google Scholar]
- 3.Gerlinger M, Rowan AJ, Horswell S, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–892. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bang YJ, Van Cutsem E, Feyereislova A, et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet. 2010;376:687–697. doi: 10.1016/S0140-6736(10)61121-X. [DOI] [PubMed] [Google Scholar]
- 5.Jellinek EO. Grawitz Tumor of Kidney. Cal State J Med. 1904;2:78–79. [PMC free article] [PubMed] [Google Scholar]
- 6.Kelly AOJ. On hypernephromas of the kidney. Philadelphia Med J. 1898;11:223–233. [Google Scholar]
- 7.Mancilla-Jimenez R, Stanley RJ, Blath RA. Papillary renal cell carcinoma: a clinical, radiologic, and pathologic study of 34 cases. Cancer. 1976;38:2469–2480. doi: 10.1002/1097-0142(197612)38:6<2469::aid-cncr2820380636>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 8.Moch H, Cubilla AL, Humphrey PA, Reuter VE, Ulbright TM. The 2016 WHO Classification of Tumours of the Urinary System and Male Genital Organs-Part A: Renal, Penile, and Testicular Tumours. Eur Urol. 2016 doi: 10.1016/j.eururo.2016.02.029. [DOI] [PubMed] [Google Scholar]
- 9.Shuch B, Amin A, Armstrong AJ, et al. Understanding pathologic variants of renal cell carcinoma: distilling therapeutic opportunities from biologic complexity. Eur Urol. 2015;67:85–97. doi: 10.1016/j.eururo.2014.04.029. [DOI] [PubMed] [Google Scholar]
- 10.Richards FM, Phipps ME, Latif F, et al. Mapping the Von Hippel-Lindau disease tumour suppressor gene: identification of germline deletions by pulsed field gel electrophoresis. Hum Mol Genet. 1993;2:879–882. doi: 10.1093/hmg/2.7.879. [DOI] [PubMed] [Google Scholar]
- 11.Nickerson ML, Jaeger E, Shi Y, et al. Improved identification of von Hippel-Lindau gene alterations in clear cell renal tumors. Clin Cancer Res. 2008;14:4726–4734. doi: 10.1158/1078-0432.CCR-07-4921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013;499:43–49. doi: 10.1038/nature12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shen C, Kaelin WG., Jr The VHL/HIF axis in clear cell renal carcinoma. Semin Cancer Biol. 2013;23:18–25. doi: 10.1016/j.semcancer.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kovacs G, Wilkens L, Papp T, de Riese W. Differentiation between papillary and nonpapillary renal cell carcinomas by DNA analysis. J Natl Cancer Inst. 1989;81:527–530. doi: 10.1093/jnci/81.7.527. [DOI] [PubMed] [Google Scholar]
- 15.Delahunt B, Eble JN. Papillary renal cell carcinoma: a clinicopathologic and immunohistochemical study of 105 tumors. Mod Pathol. 1997;10:537–544. [PubMed] [Google Scholar]
- 16.Zhuang Z, Park WS, Pack S, et al. Trisomy 7-harbouring non-random duplication of the mutant MET allele in hereditary papillary renal carcinomas. Nat Genet. 1998;20:66–69. doi: 10.1038/1727. [DOI] [PubMed] [Google Scholar]
- 17.Lubensky IA, Schmidt L, Zhuang Z, et al. Hereditary and sporadic papillary renal carcinomas with c-met mutations share a distinct morphological phenotype. Am J Pathol. 1999;155:517–526. doi: 10.1016/S0002-9440(10)65147-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Toro JR, Nickerson ML, Wei MH, et al. Mutations in the fumarate hydratase gene cause hereditary leiomyomatosis and renal cell cancer in families in North America. Am J Hum Genet. 2003;73:95–106. doi: 10.1086/376435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Linehan WM, Spellman PT, Ricketts CJ, et al. Comprehensive Molecular Characterization of Papillary Renal-Cell Carcinoma. N Engl J Med. 2016;374:135–145. doi: 10.1056/NEJMoa1505917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Davis CF, Ricketts CJ, Wang M, et al. The somatic genomic landscape of chromophobe renal cell carcinoma. Cancer Cell. 2014;26:319–330. doi: 10.1016/j.ccr.2014.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Malouf GG, Ali SM, Wang K, et al. Genomic Characterization of Renal Cell Carcinoma with Sarcomatoid Dedifferentiation Pinpoints Recurrent Genomic Alterations. Eur Urol. 2016 doi: 10.1016/j.eururo.2016.01.051. [DOI] [PubMed] [Google Scholar]
- 22.Michaelson MD, McKay RR, Werner L, et al. Phase 2 trial of sunitinib and gemcitabine in patients with sarcomatoid and/or poor-risk metastatic renal cell carcinoma. Cancer. 2015;121:3435–3443. doi: 10.1002/cncr.29503. [DOI] [PubMed] [Google Scholar]
- 23.Fleming S, Lewi HJ. Collecting duct carcinoma of the kidney. Histopathology. 1986;10:1131–1141. doi: 10.1111/j.1365-2559.1986.tb02553.x. [DOI] [PubMed] [Google Scholar]
- 24.Becker F, Junker K, Parr M, et al. Collecting duct carcinomas represent a unique tumor entity based on genetic alterations. PLoS One. 2013;8:e78137. doi: 10.1371/journal.pone.0078137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Davis CJ, Jr, Mostofi FK, Sesterhenn IA. Renal medullary carcinoma. The seventh sickle cell nephropathy. Am J Surg Pathol. 1995;19:1–11. doi: 10.1097/00000478-199501000-00001. [DOI] [PubMed] [Google Scholar]
- 26.Swartz MA, Karth J, Schneider DT, Rodriguez R, Beckwith JB, Perlman EJ. Renal medullary carcinoma: clinical, pathologic, immunohistochemical, and genetic analysis with pathogenetic implications. Urology. 2002;60:1083–1089. doi: 10.1016/s0090-4295(02)02154-4. [DOI] [PubMed] [Google Scholar]
- 27.Calderaro J, Moroch J, Pierron G, et al. SMARCB1/INI1 inactivation in renal medullary carcinoma. Histopathology. 2012;61:428–435. doi: 10.1111/j.1365-2559.2012.04228.x. [DOI] [PubMed] [Google Scholar]
- 28.Upton MP, Parker RA, Youmans A, McDermott DF, Atkins MB. Histologic predictors of renal cell carcinoma response to interleukin-2-based therapy. J Immunother. 2005;28:488–495. doi: 10.1097/01.cji.0000170357.14962.9b. [DOI] [PubMed] [Google Scholar]
- 29.McDermott DF, Cheng SC, Signoretti S, et al. The high-dose aldesleukin “select” trial: a trial to prospectively validate predictive models of response to treatment in patients with metastatic renal cell carcinoma. Clin Cancer Res. 2015;21:561–568. doi: 10.1158/1078-0432.CCR-14-1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Motzer RJ, Hutson TE, Tomczak P, et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med. 2007;356:115–124. doi: 10.1056/NEJMoa065044. [DOI] [PubMed] [Google Scholar]
- 31.Sternberg CN, Davis ID, Mardiak J, et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: results of a randomized phase III trial. J Clin Oncol. 2010;28:1061–1068. doi: 10.1200/JCO.2009.23.9764. [DOI] [PubMed] [Google Scholar]
- 32.Choueiri TK, Vaishampayan U, Rosenberg JE, et al. Phase II and biomarker study of the dual MET/VEGFR2 inhibitor foretinib in patients with papillary renal cell carcinoma. J Clin Oncol. 2013;31:181–186. doi: 10.1200/JCO.2012.43.3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Linehan WM, Rouault TA. Molecular pathways: Fumarate hydratase-deficient kidney cancer–targeting the Warburg effect in cancer. Clin Cancer Res. 2013;19:3345–3352. doi: 10.1158/1078-0432.CCR-13-0304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gordan JD, Lal P, Dondeti VR, et al. HIF-alpha effects on c-Myc distinguish two subtypes of sporadic VHL-deficient clear cell renal carcinoma. Cancer Cell. 2008;14:435–446. doi: 10.1016/j.ccr.2008.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brannon AR, Reddy A, Seiler M, et al. Molecular Stratification of Clear Cell Renal Cell Carcinoma by Consensus Clustering Reveals Distinct Subtypes and Survival Patterns. Genes Cancer. 2010;1:152–163. doi: 10.1177/1947601909359929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brooks SA, Brannon AR, Parker JS, et al. ClearCode34: A prognostic risk predictor for localized clear cell renal cell carcinoma. Eur Urol. 2014;66:77–84. doi: 10.1016/j.eururo.2014.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Haake SM, Brooks SA, Welsh E, et al. Patients with ClearCode34-identified molecular subtypes of clear cell renal cell carcinoma represent unique populations with distinct comorbidities. Urol Oncol. 2015 doi: 10.1016/j.urolonc.2015.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brooks SA, Khandani AH, Fielding JR, et al. Alternate Metabolic Programs Define Regional Variation of Relevant Biological Features in Renal Cell Carcinoma Progression. Clin Cancer Res. 2016 doi: 10.1158/1078-0432.CCR-15-2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Buttner F, Winter S, Rausch S, et al. Survival Prediction of Clear Cell Renal Cell Carcinoma Based on Gene Expression Similarity to the Proximal Tubule of the Nephron. Eur Urol. 2015;68:1016–1020. doi: 10.1016/j.eururo.2015.05.045. [DOI] [PubMed] [Google Scholar]
- 40.Hakimi AA, Reznik E, Lee CH, et al. An Integrated Metabolic Atlas of Clear Cell Renal Cell Carcinoma. Cancer Cell. 2016;29:104–116. doi: 10.1016/j.ccell.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Joseph RW, Kapur P, Serie DJ, et al. Clear Cell Renal Cell Carcinoma Subtypes Identified by BAP1 and PBRM1 Expression. J Urol. 2016;195:180–187. doi: 10.1016/j.juro.2015.07.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Motzer RJ, Michaelson MD, Redman BG, et al. Activity of SU11248, a multitargeted inhibitor of vascular endothelial growth factor receptor and platelet-derived growth factor receptor, in patients with metastatic renal cell carcinoma. J Clin Oncol. 2006;24:16–24. doi: 10.1200/JCO.2005.02.2574. [DOI] [PubMed] [Google Scholar]
- 43.Ravaud A, Oudard S, De Fromont M, et al. First-line treatment with sunitinib for type 1 and type 2 locally advanced or metastatic papillary renal cell carcinoma: a phase II study (SUPAP) by the French Genitourinary Group (GETUG)dagger. Ann Oncol. 2015;26:1123–1128. doi: 10.1093/annonc/mdv149. [DOI] [PubMed] [Google Scholar]
- 44.Choueiri TK, Plantade A, Elson P, et al. Efficacy of sunitinib and sorafenib in metastatic papillary and chromophobe renal cell carcinoma. J Clin Oncol. 2008;26:127–131. doi: 10.1200/JCO.2007.13.3223. [DOI] [PubMed] [Google Scholar]
- 45.Tannir NM, Plimack E, Ng C, et al. A phase 2 trial of sunitinib in patients with advanced non-clear cell renal cell carcinoma. Eur Urol. 2012;62:1013–1019. doi: 10.1016/j.eururo.2012.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hudes G, Carducci M, Tomczak P, et al. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N Engl J Med. 2007;356:2271–2281. doi: 10.1056/NEJMoa066838. [DOI] [PubMed] [Google Scholar]
- 47.Motzer RJ, Mazumdar M, Bacik J, Berg W, Amsterdam A, Ferrara J. Survival and prognostic stratification of 670 patients with advanced renal cell carcinoma. J Clin Oncol. 1999;17:2530–2540. doi: 10.1200/JCO.1999.17.8.2530. [DOI] [PubMed] [Google Scholar]
- 48.Voss MH, Hakimi AA, Pham CG, et al. Tumor genetic analyses of patients with metastatic renal cell carcinoma and extended benefit from mTOR inhibitor therapy. Clin Cancer Res. 2014;20:1955–1964. doi: 10.1158/1078-0432.CCR-13-2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Redig AJ, Janne PA. Basket trials and the evolution of clinical trial design in an era of genomic medicine. J Clin Oncol. 2015;33:975–977. doi: 10.1200/JCO.2014.59.8433. [DOI] [PubMed] [Google Scholar]
- 50.Motzer RJ, Escudier B, McDermott DF, et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N Engl J Med. 2015;373:1803–1813. doi: 10.1056/NEJMoa1510665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Choueiri TK, Figueroa DJ, Fay AP, et al. Correlation of PD-L1 tumor expression and treatment outcomes in patients with renal cell carcinoma receiving sunitinib or pazopanib: results from COMPARZ, a randomized controlled trial. Clin Cancer Res. 2015;21:1071–1077. doi: 10.1158/1078-0432.CCR-14-1993. [DOI] [PubMed] [Google Scholar]
- 52.Choueiri TK, Fay AP, Gray KP, et al. PD-L1 expression in nonclear-cell renal cell carcinoma. Ann Oncol. 2014;25:2178–2184. doi: 10.1093/annonc/mdu445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Choueiri TK, Lim ZD, Hirsch MS, et al. Vascular endothelial growth factor-targeted therapy for the treatment of adult metastatic Xp11.2 translocation renal cell carcinoma. Cancer. 2010;116:5219–5225. doi: 10.1002/cncr.25512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shen SS, Ro JY, Tamboli P, et al. Mucinous tubular and spindle cell carcinoma of kidney is probably a variant of papillary renal cell carcinoma with spindle cell features. Ann Diagn Pathol. 2007;11:13–21. doi: 10.1016/j.anndiagpath.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 55.Dechet CB, Bostwick DG, Blute ML, Bryant SC, Zincke H. Renal oncocytoma: multifocality, bilateralism, metachronous tumor development and coexistent renal cell carcinoma. J Urol. 1999;162:40–42. doi: 10.1097/00005392-199907000-00010. [DOI] [PubMed] [Google Scholar]
- 56.Zerban H, Nogueira E, Riedasch G, Bannasch P. Renal oncocytoma: origin from the collecting duct. Virchows Arch B Cell Pathol Incl Mol Pathol. 1987;52:375–387. doi: 10.1007/BF02889979. [DOI] [PubMed] [Google Scholar]