

Abstract
The most important goals for the field of immuno-oncology are to improve the response rate and increase the number of tumor indications that respond to immunotherapy, without increasing adverse side effects. One approach to achieve these goals is to use tumor-directed immunotherapy, i.e., to focus the immune activation to the most relevant part of the immune system. This may improve anti-tumor efficacy as well as reduce immune-related adverse events. Tumor-directed immune activation can be achieved by local injections of immune modulators in the tumor area or by directing the immune modulator to the tumor using bispecific antibodies. In this review, we focus on therapies targeting checkpoint inhibitors and co-stimulatory receptors that can generate tumor-specific T cell responses through localized immune activation.
Keywords: Immunotherapy, Tumor-directed immunotherapy, Cancer, Intratumoral, Bispecific antibody, Immuno-oncology
Introduction
The groundbreaking results with CTLA-4 and PD-1/PD-L1 checkpoint blocking antibodies provide a solid foundation for the field of cancer immunotherapy to build on. The field is now geared toward identifying drug candidates that act complementary or synergistically with checkpoint inhibitors to enhance the response rates [1]. At the same time, treatments need to be safer in order to allow a broader use of cancer immunotherapy.
Tumor-directed immunotherapy is an approach to focus the immune activation to the most relevant part of the immune system (Fig. 1). This concept has also been termed in situ vaccination [2, 3]. The aim of tumor-directed immunotherapy is to activate immune cells that have already homed to the tumor/local lymph node where tumor antigens are present, while minimizing irrelevant activation of the rest of the immune system. Preclinical data suggest that this can reduce immune-related adverse events (irAE). A critical aspect of tumor-directed immunotherapy is that it must be able to generate a systemic anti-tumor response that eradicates distant metastases and induces long-term tumor immunity.
Fig. 1.
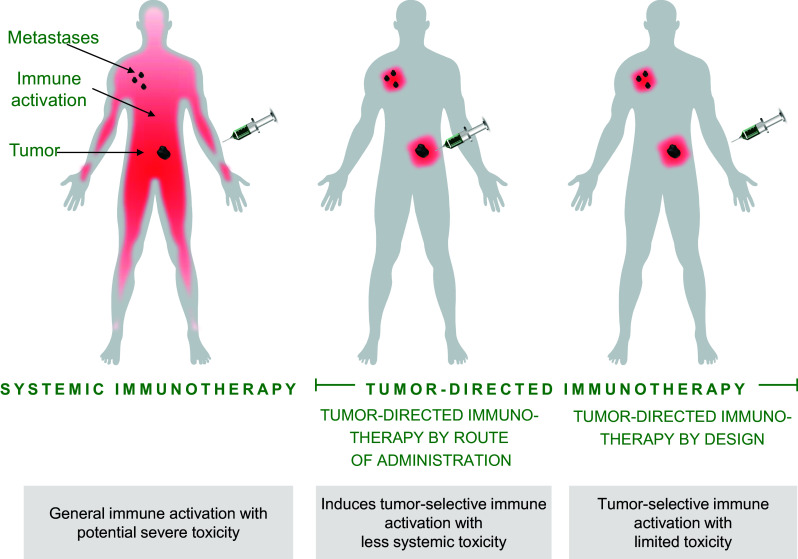
Illustration of tumor-directed immunotherapy (also termed in situ vaccination) compared to systemic immunotherapy. Intravenous administration of agonistic or checkpoint blocking antibodies activates tumor-directed T cells generating an anti-tumor response. However, these treatments can also induce cytokine release, cause liver problems, and activate autoreactive T cells, resulting in immune-related adverse events. Tumor-directed immunotherapy aims to direct immune activation to the tumor and tumor-draining lymph node axis. Activated tumor-directed T cells have the potential to migrate to distant tumors, eradicating also metastatic lesions. In contrast to systemic immunotherapy, the impact on immune cells irrelevant for the anti-tumor response is reduced. There are two approaches to tumor-directed immunotherapy: tumor-directed immunotherapy by administration route and tumor-directed immunotherapy by design. Tumor-directed immunotherapy by administration route is achieved by administering the immunomodulatory antibody directly into the tumor, into tumor-draining lymph nodes, or by a slow-release combination close to the tumor site. The immune stimulation is thereby focusing on the tumor area, minimizing systemic exposure and thus reducing systemic side effects. Tumor-directed immunotherapy by design can be achieved using bispecific cross-linking-dependent agonistic TNFR antibodies where a tumor-binding part mediates the cross-linking, replacing the need for FcγR-mediated cross-linking. In the absence of tumor cells, these types of bispecific antibodies will not be active, minimizing systemic immune activation and reducing systemic side effects
Tumor-directed immunotherapy would allow the use of highly potent immune modulating therapies and combinations without increasing the risk for the patients. In addition to decreasing the risk for inducing toxicity, tumor-directed immunotherapy may reduce secondary systemic anti-inflammatory feedback responses that dampen the anti-tumor immune response. In the case of monoclonal antibodies, tumor-directed immune activation can be achieved by local injection into the tumor area or by targeting the tumor using bispecific antibodies.
In this review, we focus on therapies targeting checkpoint inhibitors and co-stimulatory receptors that facilitate tumor-specific T cell responses through localized immune activation. Cancer vaccines, oncolytic viruses, local injections of cytokines, and Toll-like receptor (TLR) agonists are covered elsewhere [3, 4].
Cancer immunotherapy results in activation or reactivation of tumor-specific T cells
The ultimate goal of cancer immunotherapy is to generate a strong tumor-specific T cell response enabling effector T cells to find and kill tumor cells, irrespective of localization or number of tumor lesions. Immune checkpoint therapy based on either anti-CTLA-4 or anti-PD-1/PD-L1 blocking antibodies inactivates the brakes on T cells, allowing broad activation of T cells, including tumor-specific T cells. In addition, experimental model systems have revealed that antibodies targeting CTLA-4 deplete Treg in the tumor microenvironment [5]. Although there are currently no clinical data confirming this, ex vivo studies support this proposed mode of action of ipilimumab [6].
CTLA-4 or PD-1/PD-L1 blockade is associated with increased survival in melanoma, renal cell cancer, non-small cell lung cancer, bladder cancer, and Hodgkins lymphoma [7]. In fact, the combination of anti-CTLA-4 and anti-PD-1 therapy appears to be even more effective, albeit at the cost of a higher frequency of irAE. Nevertheless, this has sparked considerable optimism in the cancer immunotherapy field. Antibodies blocking additional checkpoint inhibitors such as LAG-3, TIM-3, and VISTA are currently in early clinical trials.
Strategies targeting checkpoint inhibitors have proven particularly successful in T-cell-infiltrated immunogenic tumors. However, turning non-immunogenic tumors into immunogenic tumors remains a challenge. Co-stimulatory agonistic antibodies may prove to be valuable to this end. Currently, agonist antibodies targeting the co-stimulatory receptors CD40, OX40, ICOS, CD27, GITR, and CD137 are evaluated in the clinic [8–10]. Most of these co-stimulatory receptors are expressed on T cells. In contrast, CD40 is mainly expressed on antigen-presenting cells, such as dendritic cells (DC). Activation of CD40 on DC improves their cross-presentation of tumor antigens and release of IL-12, thereby boosting the number of activated tumor-directed T effector cells.
Tumor-directed immunotherapy can be achieved by local administration of immune modulating drugs
Use of T cell co-stimulators is often associated with cytokine release. When these agents are combined with checkpoint inhibitors, there is a risk for aggregated toxicity. Tumor-directed immunotherapy may be a way to allow for such combinations while avoiding an increase in frequency and severity of irAE. There are two conceptually different ways to generate a tumor-specific T cell response through localized co-stimulation, i.e., either by local administration of the co-stimulator in the tumor area, or by developing co-stimulators designed to exert their effect predominantly in the tumor microenvironment (Fig. 1).
In the clinical setting, local administration of antibodies targeting checkpoint inhibitors and co-stimulatory receptors in the tumor area has been proposed primarily for the treatment of unresectable tumors or as pre- or postsurgical adjuvant therapy to prevent local recurrence. However, a growing number of studies have suggested that intratumoral injections of antibodies targeting checkpoint inhibitors and co-stimulatory receptors can generate a systemic anti-tumor response and immunity, eradicating also metastases distant to the injection site. Compared to intravenous administration, the intratumoral route may reduce acute as well as overall systemic exposure, and hence reduce the risk of acute reactions such as cytokine release syndrome and late-onset irAE.
Preclinical studies using tumor-directed immunotherapy
Several studies in animal models have shown that local co-stimulation using agonistic antibodies can drive systemic anti-tumor effects and induce T-cell-dependent anti-tumor immunological memory [11–15]. The most widely studied co-stimulatory target using local drug administration is CD40 where the concept has been demonstrated in multiple models. Such models include virally transduced tumors [12, 13], multiple myeloma, lung cancer [12] as well as bladder tumors [11, 15]. Injection in or near the tumor is critical for generating the systemic tumor effect. This was demonstrated using experimental tumor models with multiple tumors, where the anti-tumor effect was severely impaired when CD40 activating therapy was administered at a site distant from the tumor [11, 13].
One of the critical issues when using local administration is the distribution of the antibody following injection. It has been demonstrated that injection in or near the tumor results in increased accumulation in the tumor-draining lymph nodes and that the systemic Cmax is reduced compared to systemic administration [11]. It has also been shown that increased affinity of the CD40 agonistic antibody results in increased accumulation of the antibody in the tumor area [15].
Moreover, a replication-deficient adenoviral vector expressing CD40 ligand (AdCD40L) has been studied in murine bladder cancer models. Weekly intra-/peritumoral injections of AdCD40L cured subcutaneous and orthotopic bladder tumors as well as distant tumors [16]. This was associated with increased T cell infiltration and generation of cytotoxic T cells [17].
Locally administered CTLA-4 blocking antibodies have also been assessed in several experimental tumor models, including transduced epithelial tumors [18], colon cancer [19], pancreatic cancer [20], and bladder cancer (unpublished data). CTLA-4 is of particular interest in this regard because of the high incidence of irAE associated with systemic CTLA-4 blockade [21].
Agonistic OX40 antibodies in combination with CTLA-4 antibodies and CpG have shown promising activity in experimental lymphoma and breast tumors upon local administration [22]. Further, Palazón et al. [23] showed that local stimulation of 4-1BB using agonistic antibodies may be a promising approach to treat colon tumors and avoid systemic side effects.
Clinical studies using tumor-directed immunotherapy
Preliminary data from studies of intratumoral ipilimumab in combination with IL-2 (NCT01672450) indicate good tolerability. Intratumoral administration of ipilimumab in combination with TLR9 agonists is also under investigation (NCT02254772), and local administration of TLR agonists has been studied in several clinical trials [24, 25], demonstrating both tolerability and clinical response. Further, a phase I/II study investigating intratumoral administration of tremelimumab in combination with TLR agonist (poly I:C) and systemic PD-L1 blockade has recently been initiated (NCT02643303). In addition, a first-in-man trial of intratumoral CD40 agonistic monoclonal antibody ADC-1013 was initiated in early 2015 (NCT02379741).
Several clinical studies have also been performed using adenoviral vectors expressing CD40 ligand (AdCD40L). A first-in-man phase I/IIa study was conducted with local instillation of AdCD40L in high-grade bladder cancer patients scheduled for cystectomy [26]. Patients received 3 weekly local instillations of the vector, followed by cystectomy. The treatments were well tolerated. Treated bladder tissue expressed CD40L as well as IFNγ and resulted in T cell infiltration and total or partial disappearance of malignant cells in 5/8 treated patients, thereby providing proof of concept [26]. This trial was followed by studies in dogs with spontaneous high-grade malignant melanoma [27]. In total, 19 dogs were treated, resulting in 5 complete and 8 partial responses, 4 stable disease and 2 progressive disease, including effects on distant metastases [28]. More recently, a phase I/IIa study of intratumor AdCD40L in patients with therapy-resistant metastatic malignant melanoma was initiated. Side effects were mild, and local and distant anti-tumor effects were observed in MR-PET imaging. Addition of low-dose i.v. cyclophosphamide resulted in prolonged survival compared to AdCD40L alone [29]. Further, replication-competent adenoviruses can be engineered to exhibit oncolytic, i.e., tumor cell selective killing and immunostimulatory, properties [30].
Technical aspects of intratumoral administration
Several challenges need to be addressed to bring this concept to the patients. Methods to prevent systemic leakage of the injected antibody using micro- or nanoparticles or emulsions have been described [31, 32]. Further, it is likely that immune cells both in the tumor and in tumor-draining lymph nodes are important, which may affect the requirement of appropriate exposure. The relative importance of the immune cell locations may vary depending on target and tumor type.
Intratumoral administration may be associated with adverse events such as local inflammation, pain, and bleeding. Local inflammation and associated pain could also be a sign of successful immune activation in the tumor area. The risk of bleeding after intratumoral injection is small (probably <1 % of injected tumors). Monitoring of patients for a few hours post-injection into deep tumors is however recommended. Another potential risk is that the immune activator may be mistakenly injected in a blood vessel during intratumoral administration, thus leading to rapid and complete uptake in the circulation with risk of acute cytokine release as a result. This risk is however considered small and can be further reduced by avoiding injections in well-vascularized tumors and by using an intratumoral injection guidance including aspiration prior to injection.
From a practical point a view, it is also important to consider factors such as injection volume, injection speed, diameter of needles, and size of tumors. When administrating intratumorally, the injection volume should be kept low (preferably below 500 µL) to minimize systemic leakage. For tumors larger than 3 cm in diameter, the dose may be administered close to the tumor margin in order to maximize the chance of exposing relevant immune cells. The size of the injected tumors may also affect the response. One hypothesis is that injection into larger tumors, e.g., larger than 5 cm in diameter, may lead to reduced efficacy due to large regions of central necrosis, while injection into tumors smaller than 1 cm may lead to increased leakage of the immune activator from the tumor. Further, the speed of injection may depend upon the density of the tumor tissue and the ease of distributing the study drug solution through the tumor while still allowing the retention of the study drug within the tumor.
Selection of the tumor(s) to inject is primarily driven by feasibility. It could be argued that the primary tumor would offer the best opportunities to direct the response to “trunk” mutations, i.e., mutations that arise early in the tumorigenesis and are shared by the majority of the tumors in the patient, and may be associated with a better response than branch mutations that arise at later stages [33]. However, the quality and quantity of the immune infiltrate in different tumors in the same patient may also affect the choice of tumor to inject.
Tumor-directed immunotherapy can be achieved by designing drugs to act locally
Another approach to perform tumor-directed immunotherapy is to use immune-activating bispecific antibodies that incorporate tumor-targeting entities or monospecific antibodies that are preferentially active in the tumor milieu. Bispecific antibodies with a tumor-specific binding entity can be administered systemically, localize to the tumor area and thereby mediate tumor-directed immunotherapy.
Immunocytokines
Immunocytokines consist of a cytokine moiety fused to a monoclonal antibody or antibody fragment [34–39]. The antibody fragment binds to tumor-associated proteins, tumor vascular targets, or targets in the tumor stroma and can redirect the cytokine to the tumor area. The main purpose is to limit the systemic toxicity that is associated with cytokine treatments. Several studies have demonstrated that cytokines indeed can be redirected to the tumor area using this approach. Preclinical studies with immunocytokines using several different cytokines, e.g., TNFα, GM-CSF, IL-2, IL-12, IL-7, IL-15, IL-17, IL-18, IL-10, IFNγ, and IFNα, have generated impressive results with improved anti-tumor effects [34, 35]. However, most of the immunocytokines can be found outside the tumor area, and the increased half-life conferred by fusing an antibody to the cytokine can result in prolonged systemic exposure, which limits the therapeutic window. Intratumoral administration of immunocytokines may be an approach to address this [34, 35]. There are currently a number of ongoing clinical trials using immunocytokines using IL-2, IL-12, and TNF as the cytokine moiety [35]. However, the clinical progress has so far been modest.
Engineered monoclonal antibodies
Monoclonal antibodies can also be designed to preferentially accumulate in the tumor tissue by increasing the isoelectric point. The pH in tumors is significantly more acidic (6.6–7.0) than that of normal tissues (7.2–7.4). This acidity is primarily due to anaerobic glycolysis in tumor regions subjected to short-term or long-term hypoxia as a result of poorly organized neovasculature with diminished chaotic blood flow, and aerobic glycolysis (the Warburg effect) [36, 38]. Antibodies with higher isoelectric point may be retained in the acidic environment. Another promising approach would be to generate antibodies that preferentially bind their target at lower pH, designing the antibody binding site to depend on the protonated form of histidine residues [37, 39]. Further, a class of proteolytically triggered antibodies engineered to remain inert until enzymatically activated in the tumor microenvironment has been described [40].
Redirection of T cells using CD3-targeting bispecific antibodies
CD3-targeting bispecific antibodies can be utilized to redirect T cells toward tumor cells. This approach has proven successful, and blinatumomab, a bispecific T-cell-engaging antibody that binds CD19 on B cells and CD3 on T cells, was approved in 2014 for the treatment of relapsed/refractory B cell precursor acute lymphoblastic leukemia [41]. Several T cell redirecting therapies are currently in clinical development against solid tumors [42]. While CD3-targeting bispecific antibodies effectively kill tumor cells, they depend on polyclonal T cells for tumor killing, and they do not directly promote activation of T cells that specifically recognize tumor-associated antigens. This approach may therefore be less effective in inducing a long-term tumor immunity compared to immunotherapies targeting checkpoint molecules.
Bispecific antibodies selectively activated in the tumor microenvironment targeting TNFR-SF members
Another approach to achieve tumor-directed immune activation is to utilize bispecific antibodies that are activated upon binding to the tumor cell. To this end, cross-linking-dependent agonistic antibodies targeting the TNFR-super family may prove advantageous. Agonistic IgG1 antibodies targeting, e.g., CD137, CD40, or OX40, have limited agonistic effect without cross-linking provided by FcγR on adjacent cells [43–45]. Cross-linking may however be mediated by entities other than FcγR, such as a tumor-associated antigen. It is thus possible to generate bispecific anti-TNFR superfamily antibodies, where FcγR binding has been removed and replaced by tumor-associated antigen binding. Such compounds would only be active when cross-linked by the tumor-associated antigen in the tumor area. Stimulation of TNFR superfamily members is known to induce tumor-specific memory T cell responses [46, 47], and accordingly, this class of bispecific antibodies may have the potential to induce long-lasting tumor immunity.
Tumor-directed immunotherapy based on the above concept was first demonstrated using the natural ligand to CD137 (CD137L) fused to a tumor-targeting scFv [48, 49]. This concept may however be extended to any antibody activated after cross-linking, such as agonistic antibodies targeting TNFR superfamily members.
Concluding remarks
Tumor-directed immunotherapy, or in situ vaccination, is currently being evaluated in both preclinical studies and clinical trials. This approach has the potential to improve the response rate and increase the number of tumor indications that respond to immunotherapy, while limiting adverse side effects. Efforts are ongoing to generate next-generation immunotherapeutic drugs specifically designed for tumor-directed immunotherapy, and we predict that this field will grow substantially during the coming decades.
Abbreviations
- AdCD40L
Adenoviral vector expressing CD40 ligand
- Cmax
Peak plasma concentration of a drug after administration
- CTLA-4
Cytotoxic T-lymphocyte-associated protein 4
- FcɣR
Fc gamma receptor
- ICOS
Inducible T cell co-stimulator
- irAE
Immune-related adverse events
- LAG-3
Lymphocyte-activation gene 3
- PD-1
Programmed cell death protein 1
- PD-L1
Programmed death-ligand 1
- TIM-3
T cell immunoglobulin and mucin-domain containing-3
- Treg
Regulatory T cells
- VISTA
V-domain immunoglobulin (Ig)-containing suppressor of T cell activation
Compliance with ethical standards
Conflict of interest
Christina Furebring, Per Norlén, and Peter Ellmark are current employees of Alligator Bioscience AB. Thomas Tötterman has received research funding from and is a consultant to Alligator Bioscience AB. Sara Mangsbo has received research funding from, and is a consultant to Alligator Bioscience AB.
References
- 1.Ascierto PA. Immunotherapies and novel combinations: the focus of advances in the treatment of melanoma. Cancer Immunol Immunother. 2015;64:271–274. doi: 10.1007/s00262-014-1647-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hammerich L, Binder A, Brody JD. In situ vaccination: cancer immunotherapy both personalized and off-the-shelf. Mol Oncol. 2015;9:1966–1981. doi: 10.1016/j.molonc.2015.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pierce RH, Campbell JS, Pai SI, Brody JD, Kohrt HE. In-situ tumor vaccination: bringing the fight to the tumor. Hum Vaccin Immunother. 2015;11:1901–1909. doi: 10.1080/21645515.2015.1049779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wong KK, Li WA, Mooney DJ, Dranoff G. Advances in therapeutic cancer vaccines. Adv Immunol. 2016;130:191–249. doi: 10.1016/bs.ai.2015.12.001. [DOI] [PubMed] [Google Scholar]
- 5.Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, Roddie C, Henry JY, Yagita H, Wolchok JD, Peggs KS, Ravetch JV, Allison JP, Quezada SA. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J Exp Med. 2013;210:1695–1710. doi: 10.1084/jem.20130579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Romano E, Kusio-Kobialka M, Foukas PG, Baumgaertner P, Meyer C, Ballabeni P, Michielin O, Weide B, Romero P, Speiser DE. Ipilimumab-dependent cell-mediated cytotoxicity of regulatory T cells ex vivo by nonclassical monocytes in melanoma patients. Proc Natl Acad Sci. 2015;112:6140–6145. doi: 10.1073/pnas.1417320112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell. 2015;161:205–214. doi: 10.1016/j.cell.2015.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sanmamed MF, Pastor F, Rodriguez A, Perez-Gracia JL, Rodriguez-Ruiz ME, Jure-Kunkel M, Melero I. Agonists of co-stimulation in cancer immunotherapy directed against CD137, OX40, GITR, CD27, CD28, and ICOS. Semin Oncol. 2015;42:640–655. doi: 10.1053/j.seminoncol.2015.05.014. [DOI] [PubMed] [Google Scholar]
- 9.Ellmark P, Mangsbo SM, Furebring C, Tötterman TH, Norlén P. Kick-starting the cancer-immunity cycle by targeting CD40. Oncoimmunology. 2015;4:e1011484-1-3. doi: 10.1080/2162402X.2015.1011484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vonderheide RH, Glennie MJ. Agonistic CD40 antibodies and cancer therapy. Clin Cancer Res. 2013;19:1035–1043. doi: 10.1158/1078-0432.CCR-12-2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sandin LC, Orlova A, Gustafsson E, Ellmark P, Tolmachev V, Tötterman TH, Mangsbo SM. Locally delivered CD40 agonist antibody accumulates in secondary lymphoid organs and eradicates experimental disseminated bladder cancer. Cancer Immunol Res. 2014;2:80–90. doi: 10.1158/2326-6066.CIR-13-0067. [DOI] [PubMed] [Google Scholar]
- 12.van Mierlo GJ, den Boer AT, Medema JP, van der Voort EI, Fransen MF, Offringa R, Melief CJ, Toes RE. CD40 stimulation leads to effective therapy of CD40− tumors through induction of strong systemic cytotoxic T lymphocyte immunity. Proc Natl Acad Sci. 2002;99:5561–5566. doi: 10.1073/pnas.082107699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fransen MF, Sluijter M, Morreau H, Arens R, Melief CJ. Local activation of CD8 T cells and systemic tumor eradication without toxicity via slow release and local delivery of agonistic CD40 antibody. Clin Cancer Res. 2011;17:2270–2280. doi: 10.1158/1078-0432.CCR-10-2888. [DOI] [PubMed] [Google Scholar]
- 14.Jackaman C, Lew AM, Zhan Y, Allan JE, Koloska B, Graham PT, Robinson BW, Nelson DJ. Deliberately provoking local inflammation drives tumors to become their own protective vaccine site. Int Immunol. 2008;20:1467–1479. doi: 10.1093/intimm/dxn104. [DOI] [PubMed] [Google Scholar]
- 15.Mangsbo SM, Broos S, Fletcher E, Veitonmäki N, Furebring C, Dahlén E, Norlén P, Lindstedt M, Tötterman TH, Ellmark P. The human agonistic CD40 antibody ADC-1013 eradicates bladder tumors and generates T-cell-dependent tumor immunity. Clin Cancer Res. 2015;21:1115–1126. doi: 10.1158/1078-0432.CCR-14-0913. [DOI] [PubMed] [Google Scholar]
- 16.Lindqvist C, Sandin LC, Fransson M, Loskog A. Local AdCD40L gene therapy is effective for disseminated murine experimental cancer by breaking T-cell tolerance and inducing tumor cell growth inhibition. J Immunother. 2009;32:785–792. doi: 10.1097/CJI.0b013e3181acea69. [DOI] [PubMed] [Google Scholar]
- 17.Loskog A, Björkland A, Brown MP, Korsgren O, Malmström PU, Tötterman TH. Potent antitumor effects of CD154 transduced tumor cells in experimental bladder cancer. J Urol. 2001;166:1093–1097. doi: 10.1016/S0022-5347(05)65928-9. [DOI] [PubMed] [Google Scholar]
- 18.Tuve S, Chen BM, Liu Y, Cheng TL, Touré P, Sow PS, Feng Q, Kiviat N, Strauss R, Ni S, Li ZY, Roffler SR. Lieber A (2007) Combination of tumor site-located CTL-associated antigen-4 blockade and systemic regulatory T-cell depletion induces tumor-destructive immune responses. Cancer Res. 2007;67:5929–5939. doi: 10.1158/0008-5472.CAN-06-4296. [DOI] [PubMed] [Google Scholar]
- 19.Fransen MF, van der Sluis TC, Ossendorp F, Arens R, Melief CJ. Controlled local delivery of CTLA-4 blocking antibody induces CD8+ T-cell-dependent tumor eradication and decreases risk of toxic side effects. Clin Cancer Res. 2013;19:5381–5389. doi: 10.1158/1078-0432.CCR-12-0781. [DOI] [PubMed] [Google Scholar]
- 20.Sandin LC, Eriksson F, Ellmark P, Loskog AS, Tötterman TH, Mangsbo SM. Local CTLA4 blockade effectively restrains experimental pancreatic adenocarcinoma growth in vivo. Oncoimmunology. 2014;3:e27614-1-8. doi: 10.4161/onci.27614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Michot JM, Bigenwald C, Champiat S, Collins M, Carbonnel F, Postel-Vinay S, Berdelou A, Varga A, Bahleda R, Hollebecque A, Massard C, Fuerea A, Ribrag V, Gazzah A, Armand JP, Amellal N, Angevin E, Noel N, Boutros C, Mateus C, Robert C, Soria JC, Marabelle A, Lambotte O. Immune-related adverse events with immune checkpoint blockade: a comprehensive review. Eur J Cancer. 2016;54:139–148. doi: 10.1016/j.ejca.2015.11.016. [DOI] [PubMed] [Google Scholar]
- 22.Marabelle A, Kohrt H, Sagiv-Barfi I, Ajami B, Axtell RC, Zhou G, Rajapaksa R, Green MR, Torchia J, Brody J, Luong R, Rosenblum MD, Steinman L, Levitsky HI, Tse V, Levy R. Depleting tumor-specific Tregs at a single site eradicates disseminated tumors. J Clin Invest. 2013;123:2447–2463. doi: 10.1172/JCI64859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Palazón A, Martínez-Forero I, Teijeira A, Morales-Kastresana A, Alfaro C, Sanmamed MF, Perez-Gracia JL, Peñuelas I, Hervás-Stubbs S, Rouzaut A, de Landázuri MO, Jure-Kunkel M, Aragonés J, Melero I. The HIF-1α hypoxia response in tumor-infiltrating T lymphocytes induces functional CD137 (4-1BB) for immunotherapy. Cancer Discov. 2012;2:608–623. doi: 10.1158/2159-8290.CD-11-0314. [DOI] [PubMed] [Google Scholar]
- 24.Kim YH, Gratzinger D, Harrison C, Brody JD, Czerwinski DK, Ai WZ, Morales A, Abdulla F, Xing L, Navi D, Tibshirani RJ, Advani RH, Lingala B, Shah S, Hoppe RT, Levy R. In situ vaccination against mycosis fungoides by intratumoral injection of a TLR9 agonist combined with radiation: a phase 1/2 study. Blood. 2012;119:355–363. doi: 10.1182/blood-2011-05-355222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goldstein MJ, Varghese B, Brody JD, Rajapaksa R, Kohrt H, Czerwinski DK, Levy S, Levy RA. CpG-loaded tumor cell vaccine induces antitumor CD4+ T cells that are effective in adoptive therapy for large and established tumors. Blood. 2011;117:118–127. doi: 10.1182/blood-2010-06-288456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Malmström PU, Loskog AS, Lindqvist CA, Mangsbo SM, Fransson M, Wanders A, Gårdmark T, Tötterman TH. AdCD40L immunogene therapy for bladder carcinoma—the first phase I/IIa trial. Clin Cancer Res. 2010;16:3279–3287. doi: 10.1158/1078-0432.CCR-10-0385. [DOI] [PubMed] [Google Scholar]
- 27.von Euler H, Sadeghi A, Carlsson B, Rivera P, Loskog A, Segall T, Korsgren O, Tötterman TH. Efficient adenovector CD40 ligand immunotherapy of canine malignant melanoma. J Immunother. 2008;31:377–384. doi: 10.1097/CJI.0b013e31816a812d. [DOI] [PubMed] [Google Scholar]
- 28.Westberg S, Sadeghi A, Svensson E, Segall T, Dimopoulou M, Korsgren O, Hemminki A, Loskog AS, Tötterman TH, von Euler H. Treatment efficacy and immune stimulation by AdCD40L gene therapy of spontaneous canine malignant melanoma. J Immunother. 2013;36:350–358. doi: 10.1097/CJI.0b013e31829d8a1b. [DOI] [PubMed] [Google Scholar]
- 29.Loskog A, Maleka A, Mangsbo S, Svensson E, Lundberg C, Nilsson A, Krause J, Agnarsdóttir M, Sundin A, Ahlström H, Tötterman TH, Ullenhag G. Immunostimulatory AdCD40L gene therapy combined with low-dose cyclophosphamide in metastatic melanoma patients. Br J Cancer. 2016;114:872–880. doi: 10.1038/bjc.2016.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hemminki A. Oncolytic immunotherapy: where are we clinically? Scientifica (Cairo) 2014;2014:862925. doi: 10.1155/2014/862925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Broos S, Sandin LC, Apel J, Tötterman TH, Akagi T, Akashi M, Borrebaeck CA, Ellmark P, Lindstedt M. Synergistic augmentation of CD40-mediated activation of antigen-presenting cells by amphiphilic poly(γ-glutamic acid) nanoparticles. Biomaterials. 2012;33:6230–6239. doi: 10.1016/j.biomaterials.2012.05.011. [DOI] [PubMed] [Google Scholar]
- 32.Fransen MF, Cordfunke RA, Sluijter M, van Steenbergen MJ, Drijfhout JW, Ossendorp F, Hennink WE, Melief CJ. Effectiveness of slow-release systems in CD40 agonistic antibody immunotherapy of cancer. Vaccine. 2014;32:1654–1660. doi: 10.1016/j.vaccine.2014.01.056. [DOI] [PubMed] [Google Scholar]
- 33.McGranahan N, Furness AJ, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, Jamal-Hanjani M, Wilson GA, Birkbak NJ, Hiley CT, Watkins TB, Shafi S, Murugaesu N, Mitter R, Akarca AU, Linares J, Marafioti T, Henry JY, Van Allen EM, Miao D, Schilling B, Schadendorf D, Garraway LA, Makarov V, Rizvi NA, Snyder A, Hellmann MD, Merghoub T, Wolchok JD, Shukla SA, Wu CJ, Peggs KS, Chan TA, Hadrup SR, Quezada SA, Swanton C. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 2016;351:1463–1469. doi: 10.1126/science.aaf1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kiefer JD, Neri D. Immunocytokines and bispecific antibodies: two complementary strategies for the selective activation of immune cells at the tumor site. Immunol Rev. 2016;270:178–192. doi: 10.1111/imr.12391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Neri D, Sondel PM. Immunocytokines for cancer treatment: past, present and future. Curr Opin Immunol. 2016;40:96–102. doi: 10.1016/j.coi.2016.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sondel PM, Gillies SD. Current and potential uses of immunocytokines as cancer immunotherapy. Antibodies. 2012;1:149–171. doi: 10.3390/antib1020149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Danielli R, Patuzzo R, Ruffini PA, Maurichi A, Giovannoni L, Elia G, Neri D, Santinami M. Armed antibodies for cancer treatment: a promising tool in a changing era. Cancer Immunol Immunother. 2015;64:113–121. doi: 10.1007/s00262-014-1621-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yoshida GJ. Metabolic reprogramming: the emerging concept and associated therapeutic strategies. J Exp Clin Cancer Res. 2015;34:1–10. doi: 10.1186/s13046-015-0221-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vaughn DE, Bjorkman PJ. Structural basis of pH-dependent antibody binding by the neonatal Fc receptor. Structure. 1998;6:63–73. doi: 10.1016/S0969-2126(98)00008-2. [DOI] [PubMed] [Google Scholar]
- 40.Polu KR, Lowman HB. Probody therapeutics for targeting antibodies to diseased tissue. Expert Opin Biol Ther. 2014;14:1049–1053. doi: 10.1517/14712598.2014.920814. [DOI] [PubMed] [Google Scholar]
- 41.Sanford M. Blinatumomab: first global approval. Drugs. 2015;75:321–327. doi: 10.1007/s40265-015-0356-3. [DOI] [PubMed] [Google Scholar]
- 42.Zhukovsky EA, Morse RJ, Maus MV. Bispecific antibodies and CARs: generalized immunotherapeutics harnessing T cell redirection. Curr Opin Immunol. 2016;40:24–35. doi: 10.1016/j.coi.2016.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wilson NS, Yang B, Yang A, Loeser S, Marsters S, Lawrence D, Li Y, Pitti R, Totpal K, Yee S, Ross S, Vernes JM, Lu Y, Adams C, Offringa R, Kelley B, Hymowitz S, Daniel D, Meng G, Ashkenazi A. An Fcγ receptor-dependent mechanism drives antibody-mediated target-receptor signaling in cancer cells. Cancer Cell. 2011;19:101–113. doi: 10.1016/j.ccr.2010.11.012. [DOI] [PubMed] [Google Scholar]
- 44.Li F, Ravetch JV. Antitumor activities of agonistic anti-TNFR antibodies require differential FcγRIIB coengagement in vivo. Proc Natl Acad Sci. 2013;110:19501–19506. doi: 10.1073/pnas.1319502110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.White AL, Chan HT, Roghanian A, French RR, Mockridge CI, Tutt AL, Dixon SV, Ajona D, Verbeek JS, Al-Shamkhani A, Cragg MS, Beers SA, Glennie MJ. Interaction with FcγRIIB is critical for the agonistic activity of anti-CD40 monoclonal antibody. J Immunol. 2011;187:1754–1763. doi: 10.4049/jimmunol.1101135. [DOI] [PubMed] [Google Scholar]
- 46.French RR, Chan HT, Tutt AL, Glennie MJ. CD40 antibody evokes a cytotoxic T-cell response that eradicates lymphoma and bypasses T-cell help. Nat Med. 1999;5:548–553. doi: 10.1038/5505. [DOI] [PubMed] [Google Scholar]
- 47.Melero I, Shuford WW, Newby SA, Aruffo A, Ledbetter JA, Hellström KE, Mittler RS, Chen L. Monoclonal antibodies against the 4-1BB T-cell activation molecule eradicate established tumors. Nat Med. 1997;3:682–685. doi: 10.1038/nm0697-682. [DOI] [PubMed] [Google Scholar]
- 48.Müller D, Frey K, Kontermann RE. A novel antibody-4-1BBL fusion protein for targeted costimulation in cancer immunotherapy. J Immunother. 2008;31:714–722. doi: 10.1097/CJI.0b013e31818353e9. [DOI] [PubMed] [Google Scholar]
- 49.Hornig N, Kermer V, Frey K, Diebolder P, Kontermann RE, Müller D. Combination of a bispecific antibody and costimulatory antibody-ligand fusion proteins for targeted cancer immunotherapy. J Immunother. 2012;35:418–429. doi: 10.1097/CJI.0b013e3182594387. [DOI] [PubMed] [Google Scholar]