

Abstract
Inhaled diacetyl vapors are associated with flavorings-related lung disease, a potentially fatal airway disease. The reactive α-dicarbonyl group in diacetyl causes protein damage in vitro. Dicarbonyl/l-xylulose reductase (DCXR) metabolizes diacetyl into acetoin, which lacks this α-dicarbonyl group. To investigate the hypothesis that flavorings-related lung disease is caused by in vivo protein damage, we correlated diacetyl-induced airway damage in mice with immunofluorescence for markers of protein turnover and autophagy. Western immunoblots identified shifts in ubiquitin pools. Diacetyl inhalation caused dose-dependent increases in bronchial epithelial cells with puncta of both total ubiquitin and K63-ubiquitin, central mediators of protein turnover. This response was greater in Dcxr-knockout mice than in wild-type controls inhaling 200 ppm diacetyl, further implicating the α-dicarbonyl group in protein damage. Western immunoblots demonstrated decreased free ubiquitin in airway-enriched fractions. Transmission electron microscopy and colocalization of ubiquitin-positive puncta with lysosomal-associated membrane proteins 1 and 2 and with the multifunctional scaffolding protein sequestosome-1 (SQSTM1/p62) confirmed autophagy. Surprisingly, immunoreactive SQSTM1 also accumulated in the olfactory bulb of the brain. Olfactory bulb SQSTM1 often congregated in activated microglial cells that also contained olfactory marker protein, indicating neuronophagia within the olfactory bulb. This suggests the possibility that SQSTM1 or damaged proteins may be transported from the nose to the brain. Together, these findings strongly implicate widespread protein damage in the etiology of flavorings-related lung disease.
In 2000, a group of eight workers at a microwave popcorn manufacturing plant were diagnosed with clinical bronchiolitis obliterans, with severe fixed airways obstruction.1 Workers at the plant were found subsequently to have increased rates of lung disease and airways obstruction relative to the general population. The altered lung function of those workers correlated with exposure to diacetyl (2,3-butanedione), a flavoring present in natural butter and a common component of butter flavoring.1 Studies in rats and mice demonstrated remarkable airway epithelial cytotoxicity after diacetyl inhalation.2, 3, 4 In workers, high-resolution computed tomography scans demonstrated bronchial wall thickening in eight popcorn manufacturing workers, and two workers had biopsy findings consistent with constrictive bronchiolitis.5 Additional cases of fixed airways obstruction were subsequently identified in workers exposed to diacetyl vapors in other workplaces; those workplaces involved microwave popcorn, coffee, chemical, and flavorings production.6, 7, 8, 9 Recently, workers in flavoring production were found to have an increased prevalence of restrictive lung disease, suggesting that the full spectrum of pulmonary function abnormalities associated with inhaling flavoring vapors may include restrictive as well as obstructive lung disease.10 The occupational disease in workers is known as popcorn workers' lung or flavorings-related lung disease. Additional high-concentration diacetyl inhalation exposures occur in traditional cigarette smokers and e-cigarette vapers.11, 12
Diacetyl is a four-carbon compound with adjacent carbonyl carbons, putting it into a class of compounds known as α-dicarbonyl compounds. The adjacent carbonyl groups tend to increase the chemical reactivity of α-dicarbonyl compounds.13, 14 The importance of these adjacent carbonyl groups was recently underlined by the discovery that another α-dicarbonyl butter flavoring, 2,3-pentanedione, caused airway epithelial injury and airway fibrosis in rats, which was similar to diacetyl-induced airway injury.15, 16 Protein damage, particularly at arginine residues, can be caused by chemical interactions between proteins and diacetyl or the structurally related 3-carbon α-dicarbonyl compound, methylglyoxal.17, 18 Although most studies of α-dicarbonyl compounds are conducted in vitro, a recent study demonstrated arginine adducts in hemoglobin and albumin of rats and mice after intratracheal instillation or oropharyngeal aspiration of [14C]diacetyl in vivo.19 This suggests that protein damage occurs in vivo and could play a major role in the development of flavorings-related lung disease. However, neither protein damage nor biological responses to excessive protein damage have been identified in the airway epithelium after diacetyl inhalation.
Damaged proteins can fail to form the desired three-dimensional shape, resulting in unfolded or misfolded proteins. Unfolded and misfolded proteins are often cytotoxic.20, 21 Misfolded proteins tend to form aggregates. Aggregated proteins are extraordinarily toxic to cells, generally inaccessible to the proteasome, and sometimes form recognizable ubiquitin-containing inclusions or puncta within cells.22 Biological adaptation to misfolded proteins commonly involves the ubiquitin system.20 Ubiquitin is a 76–amino acid protein that conjugates with misfolded proteins, enabling their degradation into small peptides within the proteasome.20, 23, 24, 25 Ubiquitin conjugation most frequently occurs at one of the seven lysine residues present in ubiquitin. These seven sites are K6, K11, K27, K29, K33, K48, and K63. Conjugation at the different lysine residues of ubiquitin can have different consequences for proteins, a situation that is best described for K48- and K63-linked ubiquitination. Ubiquitin-mediated protein degradation in the proteasome principally involves K48-linked polyubiquitin chains and is generally regarded as a process that protects the cell from damaged proteins and preserves amino acids. By contrast, proteins linked by K63 ubiqutination generally do not undergo proteasomal degradation; in fact, K63-linked ubiquitination is enhanced by inhibition of the proteasome.26 K63-linked ubiquitination can affect cellular signaling, DNA repair, endosomal trafficking, and clearance of aggregated proteins through autophagy.27, 28, 29 Ubiquitin-mediated autophagy in the lysosome is particularly useful in eliminating aggregated proteins.21, 30 However, excessive autophagy of proteins can lead to cell death and potentially causes ubiquitin and amino acid depletion. This is important because ubiquitin plays an essential role in both protein quality control and regulating proteins important in signaling cascades.20, 23, 24, 25, 31, 32 In addition, several important human diseases involve intracellular accumulations of ubiquitin and protein aggregates.33, 34, 35 Thus, it is clear that the appearance of increased numbers of ubiquitin-positive puncta in conjunction with autophagy is a well-described response to altered protein homeostasis. However, it is not known if diacetyl inhalation alters these processes in vivo in the intrapulmonary airway epithelial cells, which are the target tissue in flavorings-related lung disease.
Diacetyl can be metabolized by the enzyme, dicarbonyl/l-xylulose reductase (DCXR).36 The product of DCXR-mediated diacetyl metabolism is acetoin, which contains an α-hydroxyketone in place of the reactive α-dicarbonyl group. In an acute inhalation toxicity mixed exposure study using ratios similar to those seen in workplaces, acetoin did not significantly alter diacetyl-induced changes in airway reactivity.37 This provides additional support for the role of the reactive α-dicarbonyl group in diacetyl-induced toxicity and suggests the possibility that DCXR may provide a degree of protection from diacetyl-induced airway injury by removing the α-dicarbonyl group implicated in causing protein damage.
In this study, we investigated the hypothesis that flavorings-related lung disease is caused by in vivo protein damage. Using immunofluorescence microscopy, morphometry, confocal microscopy, and transmission electron microscopy, we provide strong evidence that diacetyl disrupts protein homeostasis in the lung, and this is localized to the airway epithelium, the target tissue in flavorings-related lung disease. Using Dcxr knockout mice, we found limited evidence that DCXR provides a degree of protection from diacetyl-induced changes in protein homeostasis, suggesting that the reactive α-dicarbonyl group plays a role in diacetyl-induced protein damage. Surprisingly, we also found evidence that puncta of the autophagy-associated multifunctional scaffolding protein, sequestosome-1 (SQSTM1; p62) reach the olfactory bulb of the brain and can accumulate in microglia. Together, these data are consistent with protein damage as the fundamental mechanism for flavorings-related lung disease and suggest that SQSTM1, a mediator of ubiquitinated protein aggregation, may be transported from the diacetyl-exposed nose to the brain.
Materials and Methods
Animals
Dcxr knockout mice were produced to evaluate the role of DCXR-mediated diacetyl metabolism in protecting airways from diacetyl-induced damage. These mice were generated as part of the trans-NIH Knock-Out Mouse Project (KOMP, Davis, CA).38 Embryonic stem cells from the Dcxr knockout cell line 10365B-E12 (University of California, Davis, KOMP Repository, Davis, CA) were used to generate chimeric Dcxr knockout mice at the University of California, Davis. The embryonic stem cells were generated as a result of NIH grants to Velocigene (U01HG004085) at Regeneron Inc. (Tarrytown, NY) and the CSD Consortium (U01HG004080). C57BL6-Dcxr tm1 (KOMP)Vlcg/N knockout mice (Dcxr ko1 mice) were generated from the chimeras at the National Institute for Occupational Safety and Health (NIOSH) by crossing the chimeras with C57BL6/N mice (Taconic, Hudson, NY) and were then bred to homozygosity and maintained by brother-sister matings. Because the selection cassettes used to generate knockout mice have been reported to alter gene expression in nearby genes,39 the cassettes were removed in C57BL6-Dcxrtm1.1 (KOMP)Vlcg/N knockout mice (Dcxr ko2 mice) produced at the NIOSH. Dcxr ko2 mice were generated by crossing the Dcxr ko1 mice with germline Cre recombinase expressing C57BL/6N-Hprttm1(CMV-cre)Wtsi/Mmucd mice (Mutant Mouse Resource Center, Davis, CA) and then using PCR to identify the offspring in which the selection cassette had been deleted. The resulting mice were then crossed with C57BL6/N mice, and offspring without either Dcxr or Cre were selected for further breeding. The resulting Dcxr knockout mice without selection cassettes were bred to homozygosity and maintained by brother-sister matings at the NIOSH.
The housing of the mice was in the specific pathogen-free NIOSH barrier animal facility, which is fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International. The mice lived in polycarbonate individually ventilated cages on autoclaved α-Dri virgin cellulose chips and hardwood β-chips with enrichment until exposure. The mice received a commercial irradiated mouse diet (Harlan Teklad diet 7913; Harlan Teklad, Indianapolis, IN) and water ad libitum. The mice were monitored and found free of viral pathogens, internal and external parasites, Mycoplasma, Helicobacter, and cilia-associated respiratory bacillus. To comply with the principles of replacement, reduction, and refinement of animal use, the number of mice used in each experiment was the minimum number needed for statistical significance.40 The NIOSH Institutional Animal Care and Use Committee reviewed, approved, and provided oversight for the study and all animal procedures.
The flavoring dose-response experiment in wild-type and Dcxr ko1 mice used young adult mice between 9 and 12 weeks of age. Mice were randomly assigned to exposure groups. The average age range was comparable in the groups (an average of 62.2 to 68.3 days of age in each group).
A follow-up experiment was designed to identify the molecular pathology of diacetyl-induced airway epithelial changes in wild-type, Dcxr ko1, and Dcxr ko2 mice. This experiment used young adult mice between 7 and 12 weeks of age for fluorescence microscopy, confocal microscopy, real-time PCR, and Western blots. Mice for transmission electron microscopy were between 15.5 and 17 weeks. These mice were also randomly assigned to exposure groups. The average age range was comparable in the groups compared statistically.
Experimental Design
The flavoring dose-response experiment was designed to identify the effect of DCXR on the acute toxicity of inhaled diacetyl at three exposure concentrations. All exposures were for 6 hours, and mice were sacrificed at 18 to 22 hours after exposure. The number of animals was six in each sex/genotype/exposure group. The experimental design for this experiment is detailed in Table 1.
Table 1.
Experimental Design for the Dose-Response Experiment
| Exposure | Target concentration (ppm) | Genotype | End point∗ |
|---|---|---|---|
| Air | NA | Wild type and Dcxr ko1† | Lung and nose histopathology; olfactory bulb immunofluorescence |
| Diacetyl | 100 | Wild type and Dcxr ko1† | Lung and nose histopathology |
| Diacetyl | 200 | Wild type and Dcxr ko1† | Lung and nose histopathology; olfactory bulb immunofluorescence |
| Diacetyl | 300 | Wild type and Dcxr ko1† | Lung and nose histopathology |
NA, not applicable.
Separate mice were used for histopathology and mRNA end points. For histopathology exposure groups, both males and females were included. Six animals were in each sex/genotype/exposure group for histopathology.
Dcxr-ko1 mice are C57BL6-Dcxr tm1 (KOMP)Vlcg/N mice, which have the Dcxr gene deleted and replaced by a selection cassette that contains an initial LoxP site, followed by a human ubiquitin promoter driving a neomycin response element and a second LoxP site.
The molecular pathogenesis experiment was designed to further evaluate molecular changes identified in the dose-response study and to ensure that changes associated with the Dcxr knockout status were not because of the insertion of the selection cassette into the mouse genome.39 This experiment used wild-type, Dcxr ko1, and Dcxr ko2 mice exposed to either air or target concentrations of 200 ppm diacetyl for 6 hours. Mice were euthanized by an overdose of pentobarbital (Sleepaway; Fort Dodge Animal Health, Fort Dodge, IA) at 18 to 22 hours after exposure. Details of the experimental design are in Table 2.
Table 2.
Experimental Design for the Molecular Pathogenesis Experiment
| Exposure | Target concentration (ppm) | Genotype | End point∗ |
|---|---|---|---|
| Air | NA | Wild type, Dcxr ko1,†Dcxr ko2‡ | Lung and nose histopathology; lung and olfactory bulb mRNA; lung airway enriched fraction Western immunoblot; bronchial transmission electron microscopy |
| Diacetyl | 200 ppm | Wild type, Dcxr ko1,†Dcxr ko2‡ | Lung and nose histopathology; lung and olfactory bulb mRNA; lung airway enriched fraction Western immunoblot; bronchial transmission electron microscopy |
Wild-type mice and two different Dcxr-knockout mice were exposed to air or 200 ppm diacetyl.
NA, not applicable.
The left lung lobe was used for histopathology, and the right lung lobes were used for mRNA end points. The olfactory bulb was used for mRNA. To reduce animal use, immunofluorescence of olfactory bulb used tissues from wild type and Dcxr ko1 in the 200 ppm group from the dose-response study. Western immunoblot and electron microscopy samples were from air or 200 ppm diacetyl-exposed Dcxr-ko2 mice.
Dcxr-ko1 mice are C57BL6-Dcxr tm1 (KOMP)Vlcg/N mice, which have the Dcxr gene deleted and replaced by a selection cassette that contains an initial LoxP site, followed by a human ubiquitin promoter driving a neomycin response element and a second LoxP site.
Dcxr-ko2 mice are C57BL6-Dcxr tm1.1 (KOMP)Vlcg/N that have both the Dcxr gene and the selection cassette deleted from the mouse genome.
Inhalation Exposures
For the flavorings dose-response experiment, the inhalation exposures were divided into two separate exposure blocks. Target diacetyl concentrations for the dose-response experiment were 100, 200, and 300 ppm (Figure 1A). Three additional exposure blocks were used for the follow-up molecular pathology experiment, and for each of those blocks, the target diacetyl concentration was 200 ppm (Figure 1B). Mice were exposed in a glass and stainless steel whole-body inhalation exposure chamber, as previously described for diacetyl and 2,3-pentanedione exposures in rats but modified for exposing mice.3, 16 In brief, a computer-controlled syringe pump injected diacetyl into stainless steel tubing, which was heated to 150°C. Diluent air flowed through the tubing so that the air in the tubing was at 70°C when the diacetyl was vaporized and then flowed into the inhalation chamber, where the concentration was monitored using a volatile organic compound meter (PGM-760; RAE Systems, San Jose, CA). The temperature in the chamber was maintained between 27.0°C and 28.0°C. Each mouse was housed individually in a wire cage during the exposures. Each exposure lasted 6 hours after the initial increase in diacetyl concentration (Figure 1), and mice were removed from the exposure chamber when all diacetyl was exhausted from the chamber. Exposure data were acquired during both the dose-response (Figure 1A) and the molecular pathogenesis (Figure 1B) experiments. In the dose-response study exposures, measured diacetyl concentrations (means ± SEM) were 101.0 ± 0.0 and 98.0 ± 0.1 ppm for the two 100-ppm exposures; 200.3 ± 0.2 and 205.4 ± 0.0 ppm for the two 200-ppm exposures; and 300.3 ± 0.2 and 298.7 ± 0.19 ppm for the two 300-ppm exposures. The follow-up 200-ppm exposures for molecular pathogenesis study had measured diacetyl concentrations (means ± SEM) of 207.2 ± 1.1, 211.1 ± 0.9, and 188.9 ± 0.9 ppm during the 6-hour exposure period. For simplicity, the target concentrations are used to identify dose groups. Control mice were exposed to filtered room air that did not contain diacetyl in wire cages within a separate inhalation exposure chamber.
Figure 1.

Exposure concentrations as monitored by the volatile organic compound meter. A: Inhalation exposure concentrations for diacetyl during the dose-response study were designed to produce an average chamber concentration of 100 ppm (green), 200 ppm (blue), or 300 ppm (red) of diacetyl. B: Inhalation exposure concentrations for diacetyl during the molecular pathogenesis experiments.
Airway Nomenclature
In humans, bronchi are distinguished from bronchioles by the presence of cartilage in the walls of bronchi. However, in the mouse, cartilage is usually only present in the proximal portion of the mainstem bronchus and is absent from other intrapulmonary airways. For the purposes of this study, because of size comparability with the smallest bronchi of humans, the two largest intrapulmonary airways in the sagittal section of the mouse left lung lobe were designated as bronchi.41 Terminal bronchioles were identified by the adjacent alveolar ducts.
Histopathology
Lungs were inflation fixed by airway perfusion with 10% neutral-buffered formalin and were processed within 24 hours of necropsy and embedded in paraffin. In the dose-response study, the entire brain was removed from the calvarium. The brain and the nose were immersion fixed in 10% neutral-buffered formalin. The nose was decalcified in 13% formic acid. Nose sections T1, T2, and T3 were taken at three different levels, as previously described.42 The olfactory bulb of the brain was embedded en bloc. Lung, olfactory bulb, and nose sections were stained with hematoxylin and eosin. Slides were evaluated by a board-certified veterinary pathologist (A.F.H.), according to best practices guidelines for histopathology evaluation in toxicologic pathology studies.43 Histopathological changes in lung and nose sections were assigned semiquantitative pathology scores, as previously described.3 The pathology score is the sum of the severity and distribution (extent) scores.
Fluorescence Assays
Immunohistochemical detection of ubiquitinated protein aggregates is used to identify protein misfolding, which exceeds the proteostasis capacity of cells.44 Immunohistochemical detection of ubiquitinated protein puncta, SQSTM1, and lysosomal markers lysosomal-associated membrane protein (LAMP) 1 and LAMP2 was used as established method to identify in vivo autophagy. By using immunofluorescence (fluorescent immunohistochemistry), we were able to colocalize ubiquitinated proteins to SQSTM1, the autophagy-associated scaffolding/adaptor protein that sequesters ubiquitinated proteins in the cell. By using lysosomal markers, we were able to further localize the ubiquitinated proteins to the lysosomes. An additional antibody was used to identify cytokeratins, which are cytoskeletal components of airway epithelial cells, to visualize the cytoskeleton and localize ubiquitin within the cytoskeleton.45
Primary antibodies (Table 345) were commercially available and tested/approved for immunohistochemistry by the manufacturer. Three different antibodies were used to detect antigens important in autophagy of misfolded proteins. The antibody used for morphometric quantification of total ubiquitin (mouse monoclonal antibody Ubi-1) had supplier-listed specificity for conjugated and unconjugated ubiquitin (76 amino acids, 8.5 kDa). Testis and sections of lung or nose previously identified to have ubiquitin-positive puncta were positive control tissues for ubiquitin immunostaining. The antibody used for morphometric quantification of K63-ubiquitin (rabbit monoclonal antibody EPR5890-448) was stated by the supplier to be specific for K63-ubiquitin and to have been tested for specificity by Western blot. The antibody used for quantification of SQSTM1 (immunoaffinity purified rabbit polyclonal antibody LS-B4617) had been identified by the supplier as specific for SQSTM1. Product details are listed in Table 3.45 Remaining antibodies were used to localize total ubiquitin, K63-ubiquitin, and SQSTM1 to specific cellular and subcellular sites. In addition, we correlated their presence with diacetyl-induced morphological changes or changes in RNA expression in the lung, nose, and brain. This design provided converging lines of evidence to ensure validity and reproducibility.
Table 3.
Primary Antibodies Used for Immunofluorescence Assays
| Antigen | Host/clone | Company | Catalog no. | Dilution |
|---|---|---|---|---|
| Catenin β-1 (CTNNB1) | Rabbit/E247 | Abcam (Cambridge, MA) | ab32572 | 1:100 |
| Keratin, type II cytoskeletal 8 (KRT8)∗ | Rabbit/EP1628Y | Abcam (Cambridge, MA) | ab53280 | 1:200 |
| Cadherin-1 (CDH1) | Mouse/36 | BD Biosciences (San Jose, CA) | 610181 | 1:50 |
| Olfactory marker protein (OMP) | Goat (polyclonal) | Wako (Richmond, VA) | 544-10001 | 1:100 |
| Ubiquitin, conjugated and unconjugated | Mouse/Ubi-1 | Millipore (Billerica, MA) | MAB1510 | 1:400 |
| Ubiquitin (linkage-specific, K63) | Rabbit/EPR8590-448 | Abcam (Cambridge, MA) | ab179434 | 1:100 |
| Allograft inflammatory factor 1 (AIF1) | Chicken (polyclonal) | Abcam (Cambridge, MA) | Ab139590 | 1:200 |
| Mouse lysosomal membrane glycoprotein 1 (LAMP1) | Rat/monoclonal† | Developmental Studies Hybridoma Bank (Iowa City, IA) | 1D4B | 1:50 |
| Mouse lysosomal membrane glycoprotein 2 (LAMP2) | Rat/monoclonal† | Developmental Studies Hybridoma Bank (Iowa City, IA) | ABL-93 | 1:50 |
| SQSTM 1/p62 | Rabbit (polyclonal) | Lifespan Biosciences, Inc. (Seattle, WA) | LS-B4617 | 1:200 |
This antibody also recognizes keratin, type I cytoskeletal 18 (KRT18). Both KRT8 and KRT18 are cytoskeletal components of airway epithelial cells.45
The monoclonal antibody developed by J. Thomas August was obtained from the Developmental Studies Hybridoma Bank, generated by the National Institute of Child Health and Human Development of the NIH, and maintained at the Department of Biology, The University of Iowa (Iowa City).
Fluorochrome-conjugated secondary antibodies and antibody fragments were used at dilutions of 1:200 and obtained from Jackson ImmunoResearch (West Grove, PA). These products were as follows: i) Alexa Fluor 594 AffiniPure F(ab')2 fragment donkey anti-mouse IgG (H + L), ii) Alexa Fluor 594 AffiniPure F(ab')2 fragment donkey anti-rabbit IgG, iii) Alexa Fluor 647 AffiniPure F(ab')2 fragment donkey anti-chicken IgY (IgG) (H + L), iv) Alexa Fluor 488 AffiniPure F(ab')2 donkey anti-rat IgG (H + L), v) Alexa Fluor 488 AffiniPure donkey anti-rabbit IgG (H + L), vi) Alexa Fluor 488 AffiniPure donkey anti-mouse IgG (H + L), and vii) Alexa Fluor 488 AffiniPure donkey anti-mouse IgG (H + L).
Paraffin-embedded tissue sections of lung and nose were prepared for fluorescence assays by deparaffinizing in xylene and alcohols. For the immunofluorescence assays, antigenicity was retrieved with 1 mmol/L EDTA, as previously described.16 Slides were then rinsed in distilled water and blocked for 2 hours at room temperature using 10% donkey serum (Jackson ImmunoResearch). Primary antibodies were applied using capillary action with pairs of ProbeOn Plus slides (Fisher Scientific, Pittsburgh, PA), which were incubated overnight at 4°C. Nonimmune serum or immunoglobulin was used in place of the primary antibodies in the negative control slides. Negative control and positive control slides were included in each staining run. The following day, slides were rinsed and washed three times in 1× phosphate-buffered saline (Sigma-Aldrich, St. Louis, MO) and then blocked in 10% donkey serum, followed by a 2-hour incubation in the dark with the secondary antibodies (Jackson ImmunoResearch). Slides were then rinsed three times in 1× phosphate-buffered saline, and nuclei were stained using DAPI FlouroPure (Molecular Probes, Eugene, OR) at a concentration of 300 nmol/L in phosphate-buffered saline. After three additional phosphate-buffered saline rinses, the slides were coverslipped using ProLong Gold Antifade Reagent (Molecular Probes) and stored at 4°C until imaged. Apoptosis was quantified using a terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling assay kit (Promega, Madison, WI), followed by morphometry.
Immunofluorescence staining in paraffin-embedded, formalin-fixed olfactory bulb from wild-type and Dcxr ko1 mice in the 200-ppm group of the dose-response study was performed as described above with an additional blocking step. Before the 10% donkey serum was added, a 1-hour block with 200 mmol/L glycine in Tris buffer at 4°C was performed. This step was beneficial in reducing the amount of red background staining.
Morphometry
Immunofluorescence was visualized using an Olympus AX70 fluorescent photomicroscope (Olympus, Center Valley, PA) and quantified by morphometry using ImageJ software version 1.47 (NIH, Bethesda, MD; https://imagej.nih.gov).46 Photomicrographs for morphometry were taken with a SPOT Slider model 17.4 4 Mp camera (SPOT Imaging Solutions, Sterling Heights, MI). Morphometry provided quantitative data on molecular changes in protein quality control systems. The terminal bronchioles, intrapulmonary bronchi, and olfactory bulb were selected for morphometric evaluation because we had no evidence of significant necrosis in these tissues.
Confocal Microscopy
Confocal microscopy was used as an established procedure for obtaining spatial localization of autophagic processes.47 The three-dimensional localization of immunofluorescence within airway epithelial cells was evaluated using an LSM 510 laser scanning confocal microscope (Zeiss, Thornwood, NY). The three-dimensional localization of immunofluorescence within olfactory neurons and microglia was evaluated using an LSM 780 laser scanning confocal microscope (Zeiss).
Transmission Electron Microscopy
At 18 to 22 hours after exposure, Dcxr ko2 mice exposed to air or 200-ppm diacetyl for 6 hours were perfused with heparinized sterile USP normal saline and then perfused with 2% glutaraldehyde in 0.84 mol/L sodium cacodylate buffer (pH 7.4) via the right ventricle of the heart. The left and right mainstem bronchus and the pulmonary parenchyma adjacent to the left mainstem bronchus were trimmed the following day. The airways were post-fixed with osmium tetroxide and subsequently mordanted in tannic acid, stained with uranyl acetate, alcohol dehydrated, embedded in Epon, and then stained with uranyl acetate and lead citrate. Ultrastructural changes were evaluated using a JEOL JEM-1400Plus transmission electron microscope (JEOL, Peabody, MA).
Western Immunoblots
Lung airway-enriched fraction (LAEF) was dissected free hand by a veterinary pathologist (A.F.H.) from the mainstem bronchus, its bifurcation, and immediately adjacent hilar region of the lung from eight air and eight diacetyl-exposed animals. LAEFs from two animals each were pooled to obtain sufficient tissue for preparation of protein homogenates, giving a total n of 4 per experimental group.
LAEF tissues were homogenized in tissue protein extraction reagent (Pierce Biotechnologies, Inc., Rockford, IL) containing protease inhibitors. The homogenates were centrifuged, and the supernatant was carefully collected (soluble protein fraction). The pellet was rehomogenized in 2% SDS solution and centrifuged, and the supernatant was collected (insoluble protein fraction). Total protein was determined according to the micro-bicinchoninic acid method (Pierce Biotechnologies, Inc.) using bovine serum albumin as a standard. Protein extracts were stored at −75°C until use.
Aliquots of the soluble protein homogenates (10 μg total protein) were diluted in Laemmli sample buffer, boiled, and loaded onto 10% SDS-polyacrylamide gels. Proteins then were electrophoretically resolved and transferred to 0.45-μm Immobilon-FL PVDF Membrane (Millipore, Billerica, MA). After transfer, immunoblot analysis was performed as previously described.48 The primary antibodies used were as follows: i) anti-ubiquitin antibody (rabbit polyclonal, Millipore, catalog 07-375, lot 2682376; used at a concentration of 2.5 μg/mL), ii) anti-ubiquitin antibody (mouse monoclonal, Millipore, catalog MAB1510, clone Ubi-1, lot 2681282; used at a concentration of 17.5 μg/mL), iii) anti-ubiquitin linkage-specific K63 antibody (Ub-K63, rabbit monoclonal, Abcam, catalog Ab179434, clone EPR8590-448; used at a concentration of 5.4 μg/mL), or iv) β-actin (mouse monoclonal, Santa Cruz Biotechnology, Inc., catalog sc47778, clone C4, lot J2313; used at a concentration of 130 ng/mL). Similarly, the insoluble protein homogenates were blotted with anti-ubiquitin or β-actin antibodies. After primary antibody incubations, the membranes were incubated with appropriate IRDye 680 or 800 Secondary Antibodies (LI-COR Biosciences, Lincoln, NE), and near-infrared fluorescence detection was performed on the Odyssey Imaging System (LI-COR Biosciences). The fluorescent signal intensities (k counts) of the individual bands were determined and normalized to the endogenous control, β-actin.
RNA Isolation, cDNA Synthesis, and Real-Time PCR
Lung and brain regions selected for real-time PCR were immediately stabilized in RNALater (Applied Biosystems, Foster City, CA), and stored at −75°C until RNA isolation. DNase-free RNA was isolated, cDNA was synthesized, and real-time PCR was performed as previously described.16 The following TaqMan gene expression assays were used: Dcxr (Mm01246774_g1), Cbr2 (Mm00483074_g1), Cxcl2 (Mm00436450_m1), Il1β (Mm00434228_m1), Il6 (Mm00446190_m1), Tnfα (Mm00443258_m1), Scgb1a1 (Mm00442046_m1), Ubc (Mm01201237_m1), Stam1 (Mm00488457_m1), Hgs (Mm00468635_m1), Lamp1 (Mm00495262_m1), Lamp2 (Mm00495267_m1), Sqstm1 (Mm00448091_m1), Omp (Mm00448081_s1), and the housekeeping gene Gapdh (Mm99999915_g1), which was used for normalization.
Statistical Analysis
Recognizing the complexity of analyzing scientific data,49, 50, 51 statistical analyses were performed by the co-authors who are statisticians (M.L.K. and J.C.H.). Consistent with recent recommendations from the American Statistical Association,50, 51 we used a spectrum of techniques (immunofluorescence, morphometry, confocal microscopy, transmission electron microscopy, real-time PCR, and Western immunoblots) to identify in vivo phenomena associated with protein damage in the airway epithelium of the lung and the neuroepithelium, transparently summarized those findings graphically and by photomicroscopy, and identified converging lines of evidence regarding in vivo protein damage and altered proteostasis in diacetyl-induced cytotoxicity. The α value for statistical significance was set at 0.05. Lower P values are not more significant, as it is inappropriate to interpret P values in this manner; however, they have been included for completeness. Data were evaluated for exposure effects, effects of exposure on each genotype, and effect of genotype on exposure. P values in figures represent comparisons between diacetyl- and air-exposed mice of the same genotype and comparisons between the knockout and wild type at the same exposure concentration. Morphometry and semiquantitative histopathology data were analyzed using Kruskal-Wallis nonparametric tests because the assumptions of an analysis of variance were not met. The Kruskal-Wallis test was followed by pairwise comparisons using the Wilcoxon rank-sum test. Real-time PCR mRNA expression data were converted to fold-change values relative to air-exposed genotype- and sex-matched controls and subjected to one-way analysis of variance tests. Western immunoblot band intensities were analyzed for decreases in free ubiquitin and increases in ubiquitinated proteins using a one-sided Wilcoxon two-sample test. Statistical analyses were conducted using SAS/STAT software version 9.1 of the SAS System for Windows (SAS Institute, Inc., Cary, NC).
Results
Dose-Response in Wild-Type and Dcxr ko1 Mice
Histopathology
In the transitional and respiratory epithelium of the nose, the principal diacetyl-associated histopathological changes were degeneration and necrosis (Figure 2, A–D, and Supplemental Table 1), which were similar to diacetyl-induced changes previously described in diacetyl-exposed mice.4 The respiratory and transitional epithelia of air-exposed mice were within normal histological limits (Figure 2A). In contrast, necrosis (Figure 2B) was significantly increased in the respiratory and transitional epithelia in section T1 in all diacetyl-exposed groups (Figure 2E and Supplemental Table 1). The pathology score for necrosis is the sum of the severity and distribution of necrosis, and the mean pathology score increased with increasing diacetyl exposure concentration (Figure 2E and Supplemental Table 1). The mean pathology score decreased as the distance from the nares increased (Figure 3 and Supplemental Table 1). This is consistent with absorption of some of the diacetyl from the airstream along the course of the nasal passageways.52, 53 Sex had no effect on diacetyl-induced necrosis pathology scores (Supplemental Table 1). The pathology score for knockout mice was significantly greater than for wild-type mice (P = 0.0224) at level T1 after the 300-ppm exposure (Figure 2E).
Figure 2.

Cellular degeneration and death in the epithelium lining nasal airways and bronchi with corresponding pathology scores. A: Maxilloturbinate from the first nasal section (T1) of a mouse 1 day after inhaling air in an inhalation exposure chamber. The maxilloturbinate is lined by normal transitional epithelium admixed with occasional ciliated epithelial cells. B: Maxilloturbinate from the first nasal section (T1) of a mouse 1 day after inhaling 300 ppm diacetyl. Necrotic cellular debris has replaced the normal transitional epithelium. C: Nasoturbinate from an air-exposed mouse. D: Degenerative changes in respiratory epithelium lining the nasoturbinate of a mouse exposed to 200 ppm diacetyl are characterized by enlarged epithelial cells with rarified cytoplasm and displaced nuclei. E: The pathology score (severity + distribution) for necrosis in the respiratory and transitional epithelium of the nose increased with exposure concentration and decreased as the sections moved away from the front of the nose (T1) sequentially toward the back of the nose (T3). Necrosis was not seen in the air controls (0 ppm). Necrosis was unaffected by sex. Necrosis pathology scores in knockout mice (closed bars) and wild-type mice (open bars) were similar except at 300 ppm at level T1. F: The pathology score (severity + distribution) for necrosis in the olfactory neuroepithelium of the nose increased with diacetyl exposure concentration. Necrosis pathology scores in knockout mice (closed bars) and wild-type mice (open bars) were not significantly different. G: Control bronchus from an air-exposed Dcxr knockout mouse (mouse P11-1126) shows no evidence of degeneration. H: Bronchus of a Dcxr knockout mouse exposed to diacetyl (P11-1129) with epithelial changes characterized by enlargement, cytoplasmic rarefaction, and nuclear displacement. I: Semiquantitative pathology scores from air-exposed control mice demonstrate no evidence of bronchial epithelial degeneration (mean pathology score of 0), whereas diacetyl-exposed mice had bronchial epithelial degeneration, which increased with increasing exposure concentration. Diacetyl significantly increased vacuolar degeneration at all exposure concentrations irrespective of genotype. There was no effect of sex on the diacetyl-induced vacuolar degeneration, and the sexes were combined for analysis of the diacetyl-induced changes. The pathology score for vacuolar degeneration was significantly greater in knockout mice (closed bars) compared to wild-type mice (open bars) at the 200 ppm exposure concentration. J: The number of apoptotic cells detected by the terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling assay in the epithelium of bronchi was increased in all diacetyl-exposed groups. Apoptosis was not significantly different in knockout (closed bars) compared with wild-type (open bars) mice. ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001 versus air-exposed controls; †P < 0.05 versus wild type. Scale bar = 20 μm (A–D and G–H).
Figure 3.

Ubiquitin is increased by diacetyl exposure and localized within intracytoplasmic accumulations in the neuroepithelium of the nose and in intrapulmonary bronchi. A: The olfactory neuroepithelium of a male wild-type mouse exposed to 300 ppm diacetyl, stained by immunofluorescence double labeling for total ubiquitin (red) and OMP (green), demonstrates large ubiquitin (conjugated and unconjugated) accumulations in the neuroepithelium. B: Lung stained by immunofluorescence double label for total ubiquitin (red) and catenin-β1 (green) demonstrates ubiquitin in the airway epithelium of a female wild-type mouse exposed to 300 ppm diacetyl; ubiquitin (conjugated and unconjugated) forms large puncta. C: In the bronchi, diacetyl increased the number of airway epithelial cells containing ubiquitin. The number of cells with ubiquitin puncta was significantly greater in knockout (closed bars) mice compared with wild-type (open bars) mice at the 200 ppm exposure concentration. ∗∗P < 0.01, ∗∗∗P < 0.001 compared with air controls of the same genotype; †††P < 0.001 compared with wild-type mice. Scale bar = 50 μm (A and B).
Vacuolar degeneration of the respiratory and transitional epithelia was often seen in conjunction with necrosis, as is anticipated with irreversible cell injury that is a precursor to cell death. However, the diacetyl-induced degeneration was somewhat unusual in being characterized by cell enlargement, with distortion of the apical cell membrane, cytoplasmic rarefaction, and frequent apical displacement of the nucleus (Figure 2D). This type of cell degeneration was most frequently seen in section T2 and was more prominent in mice inhaling 100 or 200 ppm than in mice inhaling 300 ppm diacetyl.
In the olfactory neuroepithelium, the principal diacetyl-associated histopathological change was necrosis. The pattern of olfactory neuroepithelial necrosis was characterized by regular foci of more severe necrosis, a pattern that has been previously described after acute 2,3-pentanedione exposures in the rat.16 The mean pathology score for necrosis in olfactory neuroepithelium increased with increasing diacetyl exposure concentration, but was unaffected by distance from the nares or genotype (Figure 2F and Supplemental Table 2).
In the lung, degeneration of airway epithelial cells was the principal histopathological change. Epithelial degeneration was associated with diacetyl inhalation but was not seen in air controls (Figure 2, G–I). Similar to the epithelial degeneration seen in the respiratory epithelium of the nose, the degeneration of bronchiolar epithelium was often characterized by cytoplasmic rarefaction, distortion of the apical cell membrane, and apical displacement of the nucleus. This vacuolar degeneration was not seen in terminal bronchioles, but was frequently observed in bronchi (Figure 2H and Supplemental Table 3). Diacetyl-induced vacuolar degeneration in the airway epithelium of bronchi was seen in both males and females and in both wild-type and knockout mice. Sex had no significant effect on diacetyl-induced vacuolar degeneration or necrosis, and the sexes were pooled for the statistical analysis of the diacetyl and genotype effects. At 200 ppm, the mean pathology score for degeneration was significantly greater in Dcxr knockout than in wild-type mice (Figure 2I).
By morphological evaluation, necrotic cell death was uncommon and not significantly increased in bronchi (Supplemental Table 3). Apoptosis, as detected by terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling assay, was also uncommon but was significantly increased in the bronchi of the lung, but was unaffected by exposure concentration, sex, or Dcxr genotype (Figure 2J). Because the intrapulmonary airway epithelium is the putative target for flavorings-related lung disease, had no significant necrosis, and limited apoptosis, these airways were optimal samples for investigating mechanisms of injury.
Ubiquitin-Positive Puncta in Nasal and Intrapulmonary Airways of Diacetyl-Exposed Mice
Because diacetyl causes protein damage and damaged proteins often form ubiquitin conjugates, we examined ubiquitin in the nose and the lung using dual-label immunofluorescence microscopy for total ubiquitin. In the nose, sections were stained for total ubiquitin and olfactory marker protein (OMP). Staining for both ubiquitin and OMP allowed ubiquitin accumulation to be identified within the olfactory neuroepithelium. The complex arrangement of cell types in the olfactory neuroepithelium and the presence of necrosis in diacetyl-exposed mice prevented an accurate count of the number of affected cells in the olfactory neuroepithelium. Nevertheless, large ubiquitin-positive foci were not observed in controls and were frequently observed in the neuroepithelium of mice inhaling diacetyl (Figure 3A).
In the lung, sections were stained for total ubiquitin and catenin-β1 (CTNNB1). The CTNNB1 staining allowed the identification of intercellular junctions of airway epithelial cells to identify the intrapulmonary sites of damage within the lung. This allowed accurate counts of the number of airway epithelial cells with ubiquitin-positive puncta and the area of immunoreactive ubiquitin/mm basement membrane (Figure 3, B and C).
After diacetyl exposure, the number of cells containing ubiquitin puncta was not significantly increased in the terminal bronchioles of diacetyl-exposed mice (data not shown). However, prominent intraepithelial ubiquitin puncta were observed in the bronchi of diacetyl-exposed mice (Figure 3B). In these larger airways, the number of cells containing ubiquitin puncta was significantly increased by diacetyl exposure in both wild-type and knockout mice (Figure 3C). In addition, knockout mice had significantly more airway epithelial ubiquitin puncta compared with wild-type mice at 200 ppm (Figure 3C). Male mice had significantly more cells with ubiquitin puncta than female mice after inhaling 200 ppm diacetyl (P = 0.0014) or 300 ppm diacetyl (P = 0.027). The three-way analysis of variance further indicated an interaction between genotype and diacetyl exposure (P = 0.0096) as well as an interaction between sex and diacetyl exposure (P = 0.038).
K63-Ubiquitin Puncta in the Airway Epithelium of Diacetyl-Exposed Mice
K63-ubiquitination is ubiquitination at lysine 63 of ubiquitin and is generally associated with nonproteasomal functions, including involvement in autophagy and modulation of signaling cascades.27 K63-ubiquitination was rarely observed in air-exposed mice. In diacetyl-exposed mice, K63-ubiquitination was localized to the airway epithelium (Figure 4A). Rare K63-ubiquitin–positive puncta were seen in the terminal bronchioles of diacetyl-exposed mice and, when the sexes were pooled for analysis, the number of airway epithelial cells/mm basement membrane containing K63-ubiquitin puncta was significantly increased in the terminal bronchioles of wild-type mice inhaling 100 ppm diacetyl (Figure 4B). The number of cells with K63-ubiquitin puncta was increased in bronchi at all exposure concentrations (Figure 4C). In the 200-ppm diacetyl exposure group, knockout mice had significantly more K63-ubiquitin puncta in bronchi than did the wild-type mice (Figure 4C).
Figure 4.

Dual immunofluorescence for K63-ubiquitin and CDH1. K63-ubiquitin is localized as intraepithelial accumulations in the airway epithelium. The accumulations are associated with diacetyl exposure and are more frequently identified in bronchi than terminal bronchioles. A: Photomicrograph of immunofluorescence double label for K63-ubiquitin (red) and CDH1 (green) in the airway epithelium of the lung of a male Dcxr knockout mouse exposed to 200 ppm diacetyl. K63-ubiquitin forms large puncta similar to those identified by staining for total ubiquitin. B: Morphometric quantification of K63-ubiquitin positive cells in the epithelium of terminal bronchioles of wild-type (open bars) and knockout (closed bars) mice. K63 ubiquitin accumulations were only significantly increased in terminal bronchioles of wild-type mice inhaling 100 ppm. C: Morphometric quantification of K63-ubiquitin positive cells in the epithelium of bronchi. K63-ubiquitin puncta were rare except in diacetyl-exposed mice, and cells containing puncta were significantly increased relative to controls at all exposure concentrations. Knockout mice (closed bars) were significantly more susceptible than wild-type mice (open bars) at 200 ppm when the sexes were pooled for the statistical analysis. ∗P < 0.05, ∗∗∗P < 0.001 compared with air controls of the same genotype; †P < 0.05 compared with wild-type mice. Scale bar = 20 μm (A).
Identification of the Molecular Pathology of Diacetyl-Induced Airway Epithelial Changes in Wild-Type, Dcxr ko1, and Dcxr ko2 Mice
Additional groups of male wild-type and Dcxr knockout mice were exposed to 200 ppm diacetyl for 6 hours and necropsied 18 to 20 hours after exposure i) to confirm the significantly greater susceptibility of Dcxr knockout mice to diacetyl-induced ubiquitin puncta, ii) to localize ubiquitin accumulation and investigate changes in associated proteins, iii) to identify corresponding changes in mRNA expression in the lung and olfactory bulb, and iv) to ensure that changes associated with the Dcxr knockout status were not because of the presence of the selection cassette in the Dcxr ko1 mice.
Susceptibility of Dcxr Knockout Mice to Ubiquitin Accumulation
Inhaling 200 ppm diacetyl significantly increased the area of immunoreactive total ubiquitin in terminal bronchiolar airway epithelial cells in both wild-type (P = 0.003) and Dcxr knockout mice (P = 0.004 for Dcxr ko1 and Dcxr ko2). However, the area of immunoreactive ubiquitin was low in the terminal bronchioles of diacetyl-exposed mice (mean of 12 ± 2.9 μm2 for all diacetyl-exposed mice combined) compared with bronchi. In the bronchi, diacetyl inhalation significantly increased both the area of immunoreactive ubiquitin and the number of airway epithelial cells expressing ubiquitin. The area of immunoreactive ubiquitin in bronchial airway epithelial cells of diacetyl-exposed Dcxr ko1 (716 ± 82 μm2; mean ± SEM) was similar to the area of immunoreactive ubiquitin in Dcxr ko2 mice (679 ± 195 μm2; mean ± SEM). The area of immunoreactive ubiquitin in bronchial airway epithelial cells of wild-type mice (334 ± 96 μm2; mean ± SEM) was significantly less than in Dcxr ko1 mice (P = 0.016) but not Dcxr ko2 mice (Table 4).
Table 4.
Morphometric Analysis of Ubiquitin Area and Colocalization with Cytoplasmic Markers in the Intrapulmonary Bronchial Airway Epithelium of Air and Diacetyl-Exposed Mice
| Exposure (ppm) | Genotype | Area (μm2) of ubiquitin colocalized with KRT8 or KRT18/mm basement membrane∗ | Area of ubiquitin colocalized with LAMP1/mm basement membrane∗ | Area of ubiquitin colocalized with LAMP2/mm basement membrane∗ | Area of ubiquitin colocalized with SQSTM1/mm basement membrane∗ | Area of ubiquitin/mm basement membrane with CTNNB1 double label† | Mean ubiquitin area/mm basement membrane‡ |
|---|---|---|---|---|---|---|---|
| 0 | Wild type | 0.48 ± 0.48 | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0.02 ± 0.02 | 4.04 ± 3.13 |
| 0 | Dcxr ko1 | 0 ± 0 | 0.22 ± 0.22 | 0.71 ± 0.71 | 0 ± 0 | 0.25 ± 0.18 | 3.40 ± 3.12 |
| 0 | Dcxr ko2 | 0 ± 0 | 0.02 ± 0.02 | 7.3 ± 6.9 | 0 ± 0 | 0.32 ± 0.21 | 4.1 ± 2.7 |
| 0 | All genotypes (pooled data) | 0.16 ± 0.16 | 0.078 ± 0.07 | 2.7 ± 2.3 | 0 ± 0 | 0.20 ± 0.09 | 3.8 ± 1.6 |
| 200 | Wild type | 38 ± 27§ | 150 ± 88§ | 32 ± 8.3§ | 180 ± 90§ | 330 ± 96§ | 360 ± 140§ |
| 200 | Dcxr ko1 | 11 ± 2.8§ | 200 ± 85§ | 73 ± 17§ | 370 ± 110§ | 720 ± 82§¶ | 650 ± 97§ |
| 200 | Dcxr ko2 | 25 ± 19§ | 230 ± 98§ | 67 ± 33§ | 440 ± 130§ | 680 ± 200§ | 580 ± 130§ |
| 200 | All geno-types (pooled data) | 25 ± 9.3§ | 190 ± 50§ | 57 ± 13§ | 330 ± 280§ | 580 ± 110§ | 530 ± 72§ |
CTNNB1, catenin-β1; KRT, keratin; LAMP, lysosomal-associated membrane protein; SQSTM, sequestosome.
Area is expressed as the area in μm2/mm basement membrane of two bronchi in each of six mice per exposure and genotype group.
This is the area of ubiquitin/mm basement membrane measured in dual-label immunofluorescence for total ubiquitin and CTNNB1, which was performed as described in the dose-response study.
The mean ubiquitin area/mm basement membrane is the mean of the area of ubiquitin measured in dual-label immunofluorescence assays with five different second labels (CTNNB1, KRT8 and KRT 18, LAMP1, LAMP2, and SQSTM1) standardized per mm airway basement membrane.
Significantly different from air-exposed mice (P ≤ 0.05).
Significantly different from wild-type mice (P ≤ 0.05).
Subcellular Localization of Ubiquitin Accumulation
To identify the subcellular compartment(s) containing the ubiquitin puncta, lungs were stained by dual-label indirect immunofluorescence for the following: i) ubiquitin and LAMP1 (a marker of endosomes/lysosomes/exosomes), ii) ubiquitin and LAMP2, and iii) ubiquitin and cytoskeletal proteins of airway epithelium, keratins 8 and 18 (KRT8 and KRT18). Because damaged proteins are often sequestered within vesicles that also contain ubiquitin and SQSTM1,27 lungs were also stained by dual-label indirect immunofluorescence for ubiquitin and SQSTM1. The highest area of colocalization was between ubiquitin and SQSTM1, with a mean of 332.90 ± 278.60 μm2 of colocalized ubiquitin and SQSTM1/mm basement membrane in the epithelium lining the bronchial airways (Table 4). This demonstrated that most (64%) of the ubiquitin in bronchial airways of diacetyl-exposed mice was colocalized with SQSTM1 (Table 4). Consistent with a location in cytoplasmic vesicles, the ubiquitin puncta of diacetyl-exposed mice were also frequently colocalized with the endosomal/lysosomal markers LAMP1 and LAMP2 (Table 4). Of the cytoplasmic markers investigated, the smallest area of colocalization of airway ubiquitin was with cytoskeletal components (KRT8 and KRT18) (Table 4). However, the area of ubiquitin that was colocalized with each of the markers as well as total ubiquitin were significantly increased by diacetyl exposure. Neither genotype nor sex had a significant effect on ubiquitin colocalized with SQSTM1, LAMP1, LAMP2, or cytokeratins. Total ubiquitin measured as part of the dual-label immunofluorescence assays with five different second labels (CTNNB1, KRT8 and KRT18, LAMP1, LAMP2, and SQSTM1) was significantly increased by exposure, but was not significantly affected by genotype (Table 4). In addition, diacetyl exposure significantly increased airway epithelial SQSTM1/mm basement membrane in wild-type mice (P = 0.004), Dcxr ko1 mice (P = 0.004), and Dcxr ko2 mice (P = 0.004). Specifically, SQSTM1 increased from 0.03 ± 0.03 (means ± SEM) μm2/mm in control wild-type mice to 102.97 ± 120.94 μm2/mm in diacetyl-exposed wild-type mice; from 0.22 ± 0.20 μm2/mm in control Dcxr ko1 mice to 537.98 ± 129.18 μm2/mm in diacetyl-exposed Dcxr ko1; and from 0.11 ± 0.09 μm2/mm in control Dcxr ko2 mice to 627.86 ± 170.44 μm2/mm in diacetyl-exposed wild-type mice. Diacetyl-induced SQSTM1 was not significantly affected by genotype.
To further evaluate the intracytoplasmic location of ubiquitin accumulation in the airway epithelium of diacetyl-exposed mice, the dual-label immunofluorescence sections of intrapulmonary bronchi were evaluated by confocal microscopy. Ubiquitin puncta in diacetyl-exposed mice were often within spaces between cytokeratin intermediate filaments (Figure 5, A–C) and were located within intracytoplasmic vesicles, which also expressed LAMP1 (Figure 5, D–F) and LAMP2 (Figure 5, G–I), although LAMP2 staining of the cytoplasmic vesicles was not as intense as LAMP1 staining of the vesicles. Intraepithelial ubiquitin was usually associated with SQSTM1 (Figure 5, J–L). Together, these findings localized large quantities of ubiquitin to the endosomal/lysosomal compartment in conjunction with SQSTM1 and identified increased SQSTM1 in diacetyl exposed airways, indicating extensive autophagy.
Figure 5.

Ubiquitin puncta were localized to endosomal/lysosomal/exosomal vesicles and SQSTM1 puncta using confocal microscopy of immunofluorescence staining for ubiquitin (red), cytoskeletal components KRT8 and KRT18 (green, A–C), endosomal/lysosomal/exosomal marker LAMP1 (green, D–F), endosomal/lysosomal/exosomal marker LAMP2 (green, G–I), and the multifunctional scaffolding protein SQSTM1 (green, J–L) in the airway epithelium of diacetyl-exposed mice. A: In this merged image, ubiquitin appears red because it is in spaces between the cytoskeletal filaments (green). B: Ubiquitin in the section shown in A. C: The cytoskeletal intermediate filaments containing cytoskeletal components KRT8 or KRT18 (green) in A are distinct from the sites where ubiquitin is observed in B. D: In this merged image, the yellow demonstrates colocalization of ubiquitin (red) with the endosomal/lysosomal/exosomal marker LAMP1 (green). E: Ubiquitin (red) in the section shown in D. F: LAMP1 (green) in the section shown in D is present in the sites where uibiquitin is observed in E. G: In this merged image, the orange demonstrates colocalization of ubiquitin (red) with the endosomal/lysosomal/exosomal marker LAMP2 (green, G–I). H: Ubiquitin (red) in the section shown in G. I: LAMP2 (green) in the section shown in G is present in most areas where ubiquitin is present. J: In this merged image, ubiquitin (red) is associated with SQSTM1 (green, J–L), resulting in yellow within the sites containing the green puncta of the scaffolding protein, SQSTM1. K: Ubiquitin (red) in the section shown in J. L: SQSTM1 (green) in the section shown in J is present in the sites of cellular ubiquitin accumulation. Scale bar = 5 μm (A–L).
Transmission Electron Microscopy
Ultrastructural changes in the diacetyl-exposed airway epithelium confirmed the presence of diacetyl-induced autophagy. Air-exposed control mice had morphologically normal airway epithelium in ultrastructural sections (Figure 6A). Diacetyl-exposed airway epithelial cells sometimes showed degenerative changes, including dilation of endoplasmic reticulum, swelling of mitochondria, vacuolation, and autophagy. Autophagosomes were most frequently seen in the cytoplasm of club cells (Figure 6, B and C) but were observed in a few ciliated cells. Occasional diacetyl-exposed airway epithelial cells contained cytoplasmic aggregation of variably osmiophilic punctate structures, which were not surrounded by a membrane and which were morphologically consistent with aggresomes (Figure 6D).
Figure 6.

Ultrastructural confirmation of diacetyl-induced autophagy in airway epithelium. A: Normal ciliated respiratory epithelial cell (left) and club cell (right) in an air control mouse. B: Autophagy (arrows) in a club cell from a Dcxr ko2 mouse. The cytoplasm of the ciliated epithelial cell to the left of the club cell is vacuolated. C: A higher magnification of autophagy (arrows) in a club cell from a Dcxr ko2 mouse. The cytoplasm is vacuolated in the ciliated epithelial cell to the right. D: Aggregated electron-dense material is located within the cytoplasm without a surrounding membrane (arrow) and is morphologically consistent with an aggresome-like structure. Scale bar = 1 μm (A–D).
Western Immunoblotting
Neither ubiquitin nor β-actin bands were detected in the insoluble fraction of the protein homogenates, suggesting that there were no insoluble aggregated proteins in the diacetyl-exposed LAEF tissue samples. Subsequent immunoblot analyses were performed with the soluble protein fraction samples using two anti-ubiquitin antibodies. The anti-ubiquitin rabbit polyclonal antibody (07-375) detected various ubiquitinated protein bands (28 to 250 kDa), as well as the free ubiquitin band (approximately 9 kDa). The anti-ubiquitin mouse monoclonal antibody (MAB1510) did not detect free ubiquitin or high-molecular-weight (>150 kDa) ubiquitinated proteins, but detected ubiquitinated proteins in the range of 25 to 70 kDa. Inhalation of diacetyl (200 ppm) caused a significant reduction (59% decrease; P ≤ 0.05; nonparametric Wilcoxon two-sample test) in free ubiquitin levels in the LAEF, with concurrent increases in ubiquitinated proteins, particularly in the 50 kDa (approximately 53% increase; not statistically significant) and 70 kDa (approximately 87% increase; not statistically significant) range (Figure 7). The increases in the 50- and 70-kDa ubiquitinated bands were not statistically significant, which was expected because the decrease in free ubiquitin pools limited the amount of free ubiquitin available for ubiquitination of proteins at all molecular weights.54, 55 Fluorescence microscopy demonstrated that diacetyl-induced ubiquitin puncta were localized to the epithelium while the airway-enriched fraction contains the unaffected cells, such as the airway lamina propria, wall, and adjacent parenchyma. Immunoblot analysis with the anti-ubiquitin linkage-specific K63-ubiquitin antibody did not reveal any significant changes in K63 ubiquitination, a finding that is also consistent with localization of K63-ubiquitin puncta to the epithelial layer (data not shown).
Figure 7.

Free ubiquitin is depleted by diacetyl inhalation. A: Western immunoblot of ubiquitin and β-actin in protein from the airway-enriched fraction of air and diacetyl-exposed Dcxr ko2 mice. B: Bar graph of the band intensities in the Western immunoblot. ∗P < 0.05 when compared with air-exposed controls of the same genotype. MW, molecular weight.
mRNA Expression Changes in the Lungs of Diacetyl-Exposed Mice
Real-time PCR analysis of Dcxr mRNA expression in the lung was performed to confirm the deficiency of this gene in the knockout (Dcxr ko1 and Dcxr ko2) mice (Figure 8A). Exposure to 200 ppm diacetyl did not elicit robust inflammation in the lungs of either wild-type or Dcxr knockout mice (Figure 8A). Among the inflammatory mediators examined, only Il1β mRNA expression was induced (1.9-fold increase; P < 0.01) (Figure 8A) in the lungs of Dcxr ko2 mice, at 1 day after diacetyl exposure. The lung expression levels of other inflammatory mediators, including Il6 and Tnfα, were unaffected by either genotype or treatment (Figure 8A). In Dcxr ko2 mice, concurrent with the increase in Il1β expression, diacetyl exposure also caused a small decrease in the lung expression of Scgb1a1 (Ccsp; 30% decrease; P < 0.01) (Figure 8B), the gene coding for a club cell–specific 10-kD protein, which is thought to be associated with bronchiolitis obliterans. Diacetyl exposure did not alter the mRNA expression of the lysosomal markers, Lamp1 and Lamp2, or of markers associated with endosomal/lysosomal sorting and trafficking, like Stam1 and Hgs, components of the multi-ligand Escrt0 complex (Figure 8B).
Figure 8.

Real-time PCR analysis of mRNA expression after diacetyl inhalation. Wild-type and Dcxr knockout mice were exposed to filtered air or diacetyl (200 ppm; 6 hours/day). A: At 1 day after exposure, the mRNA expression of dicarbonyl reductases and inflammatory mediators were analyzed in the lung. B: At 1 day after exposure, the mRNA expression levels of markers for lung club cells, as well as markers of lysosomal and endosomal protein trafficking, were analyzed in the lung. Normalized mRNA values are expressed as fold change. C: Real-time PCR analysis of mRNA expression in olfactory bulb of diacetyl-exposed mice. Wild-type and Dcxr knockout mice were exposed to filtered air or diacetyl (200 ppm; 6 hours/day). At 1 day after exposure, the mRNA expression levels of dicarbonyl reductases, inflammatory mediators, and marker of olfactory sensory neurons were analyzed in the olfactory bulb. Normalized mRNA values are expressed as fold change. ∗∗P < 0.01, ∗∗∗P < 0.001 versus air-exposed controls; †P < 0.05, ††P < 0.01 versus wild type. N, not detected.
mRNA Expression Changes in the Olfactory Bulb of Diacetyl-Exposed Mice
Similar to the observation in the lung, Dcxr mRNA expression was absent in the olfactory bulb of the knockout mice (Figure 8C). An interesting observation was the down-regulation/suppression of the mRNA for carbonyl reductase (Cbr2) that is also involved in metabolism of carbonyl compounds. Inflammation extended from the respiratory tract into the olfactory bulb, in mice inhaling 200 ppm diacetyl. The mRNA expression levels of inflammatory mediators, Il6 and tumor necrosis factor-α (Tnfα), were significantly elevated 1 day after diacetyl exposure. Although Il6 mRNA increased by 1.5- to 2.0-fold (P ≤ 0.001 for all genotypes combined) (Figure 8C), deficiency of Dcxr did not significantly influence or alter the expression of the Il6 mRNA. On the other hand, a significantly higher expression of Tnfα (approximately 12-fold; P ≤ 0.001 compared to air controls) (Figure 8C) was seen in the olfactory bulb of Dcxr-deficient mice when compared to the expression (fivefold; P ≤ 0.01 for each knockout) (Figure 8C) in wild-type mice, suggesting exacerbated inflammation. Diacetyl also caused a small reduction in the mRNA levels of the olfactory marker protein (Omp; 20% to 30% decrease; P ≤ 0.01), indicative of olfactory sensory neuron injury or loss (Figure 8C).
Diacetyl-Induced SQSTM1 Accumulation in the Olfactory Bulb
Neurons are able to transport nanoscale particulates.56, 57 We observed that mRNA levels for cytokines, particularly Tnfα, were increased in the olfactory bulb of the brain. Together, these observations suggested that diacetyl-induced misfolded proteins or SQSTM1 puncta could potentially be transported into the olfactory bulb and, thereby, activate signaling cascades. Consistent with this hypothesis, immunoreactive SQSTM1 was identified by immunofluorescence in the olfactory bulb of diacetyl-exposed mice. In the olfactory bulb, immunoreactive SQSTM1 had two patterns: punctate, scattered, nanoscale immunoreactivity and clustered accumulations of immunoreactive SQSTM1 (Figure 9). Quantitative morphometric analysis demonstrated statistically significant diacetyl-induced increases in the number (P ≤ 0.0001) and area (P ≤ 0.0001) of immunoreactive SQSTM1 (Figure 10). Neither sex nor genotype affected SQSTM1 accumulation in the olfactory bulb.
Figure 9.
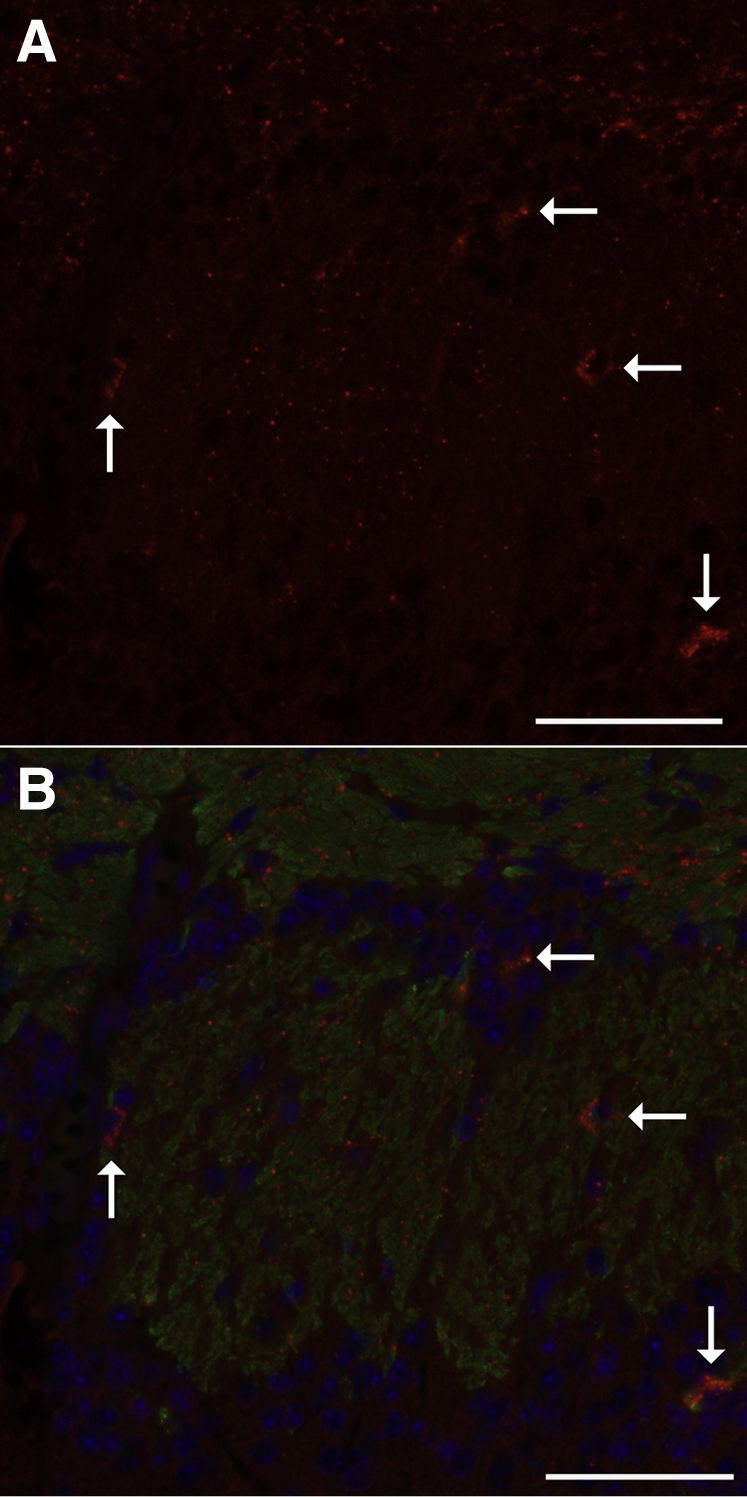
Low-magnification immunofluorescence localizing sites of SQSTM1 immunoreactivity within the olfactory bulb of the diacetyl-exposed mouse. A: Photomicrograph of immunofluorescence staining of SQSTM1 in red within the glomerular region of the mouse olfactory bulb, showing scattered submicrometer-sized immunoreactive SQSTM1 and larger accumulations (arrows) in specific foci. B: Photomicrograph of the same region shown in A, showing SQSTM1 in red, OMP in green, and DAPI in blue. The scattered SQSTM1 immunoreactivity localizes within or near OMP-positive axons. The clusters of SQSTM1-positive particles (arrows) appear to be within cells near the OMP-positive axons. Scale bar = 50 μm (A and B).
Figure 10.

Quantitative morphometric analysis of immunoreactive SQSTM1 demonstrates diacetyl-induced increases within the olfactory bulb of the diacetyl-exposed mouse. A: The number of SQSTM1-positive puncta was increased in the glomerular region at all exposure concentrations but unaffected by genotype. B: The area of SQSTM1-positive expression in the glomerular region was increased by diacetyl inhalation at all exposure concentrations but unaffected by genotype. ∗∗P < 0.01, ∗∗∗P < 0.001 compared with air controls of the same genotype.
Diacetyl-Induced Microglial Activation in the Olfactory Bulb
Triple-label immunofluorescence confocal microscopy for allograft inflammatory factor 1 (AIF1/IBA1), OMP, and SQSTM1 demonstrated AIF1-immunoreactive microglial cells within the glomerular region of the olfactory bulb of diacetyl-exposed mice. These AIF1-positive cells had an ameboid shape consistent with activated microglial cells (Figure 11A), were localized near OMP-positive axons (Figure 11B), and frequently contained intracytoplasmic SQSTM1 (Figure 11C) and OMP (Figure 11D), consistent with phagocytosis of olfactory neurons and SQSTM1.
Figure 11.

Representative immunofluorescence confocal photomicrographs demonstrating gliosis and localizing sites of SQSTM1 immunoreactivity within the glomerular region of the olfactory bulb in diacetyl-exposed mouse. A: SQSTM1 (red) puncta are within the cytoplasm of allograft inflammatory factor 1 (AIF1)–positive (white) reactive microglial cells (arrows) in a mouse inhaling 200 ppm diacetyl. B: In the merged image of OMP and AIF1 immunostaining, the AIF1-positive (white) microglial cells accumulating the SQSTM1 (red) and the scattered SQSTM1 particles are near neurons expressing OMP (green) in a mouse inhaling 200 ppm diacetyl. C: High-magnification image of an AIF1-positive (white) reactive microglial cell containing intracytoplasmic SQSTM1 (red) puncta in a mouse inhaling 200 ppm diacetyl. D: In the high-magnification merged image of OMP and AIF1 immunostaining, OMP (green) puncta are also present in AIF1-positive cells of the diacetyl-exposed mouse. Scale bars: 10 μm (A and B); 5 μm (C and D).
Diacetyl-Induced SQSTM1 Accumulation in the Neuroepithelium of the Nose
To identify whether SQSTM1 was potentially present in the olfactory neuroepithelium and could have been transported from there into the brain, we next stained nasal section T2 for immunoreactive SQSTM1 in mice inhaling 200 ppm diacetyl or air in the dose-response study. In both wild-type and Dcxr ko1 mice, SQSTM1 was abundant in the neuroepithelium of the diacetyl-exposed mouse, where it was principally located between OMP-positive olfactory neurons (Figure 12). However, in a few cells, SQSTM1 colocalized with OMP. This indicated that SQSTM1 was primarily, but not exclusively, in sustentacular cells. SQSTM1 was rare in the neuroepithelium of air-exposed mice (data not shown).
Figure 12.
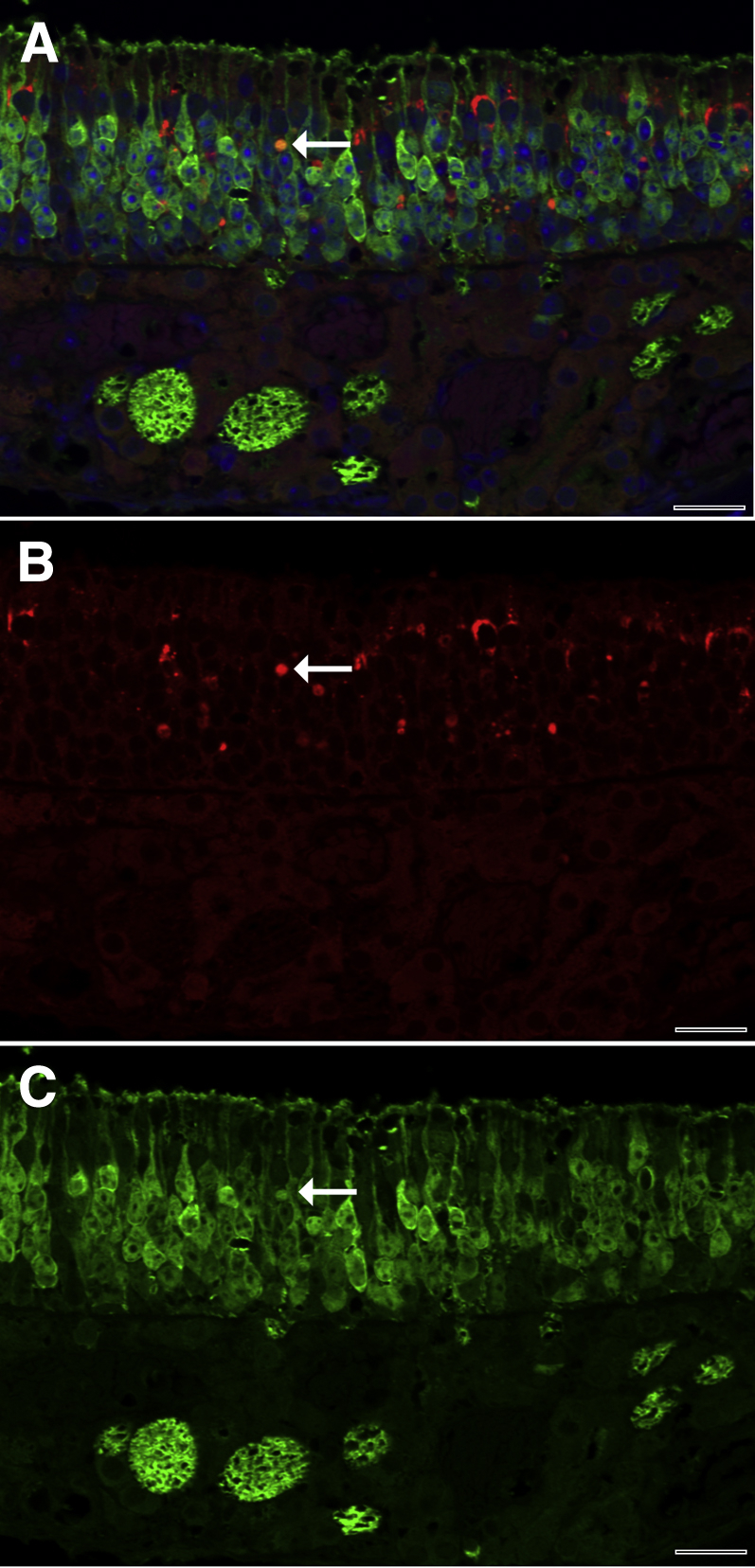
Representative immunofluorescence confocal photomicrograph of SQSTM1 in the neuroepithelium of the mouse nose. A: Merged double-label immunofluorescence demonstrates that SQSTM1 (red) is principally localized between olfactory neurons, which contain OMP (green). Rare cells expressed both SQSTM1 and OMP and were orange (arrow), consistent with neurons containing SQSTM1. B: Red fluorescence demonstrates that foci of SQSTM1 immunoreactivity are abundant and variably sized in the neuroepithelium. The neuron identified in the merged image shows strong SQSTM1 immunoreactivity (arrow). C: Green fluorescence demonstrates immunofluorescence for OMP. The neuron identified in the double-label image shows strong OMP immunoreactivity (arrow). Scale bar = 20 μm (A–C).
Discussion
In this study, we identified puncta of total ubiquitin, K63-ubiquitin, and SQSTM1 as a consistent finding in the damaged airway epithelium of mice after inhaling diacetyl. We were, furthermore, able to localize significant ubiquitin accumulation to the lysosome. This indicated that ubiquitinated cytoplasmic cargo reached the lysosome, a classic hallmark of autophagy.58 In addition, the airway epithelium had elevations in SQSTM1, a multifunction scaffolding protein that functions as an autophagy cargo receptor and accumulates in tissues during in vivo autophagy.59 Morphometric evaluation provided quantitative evidence that ubiquitin frequently colocalized with SQSTM1, LAMP1, and LAMP2 to confirm autophagy. We were, furthermore, able to use confocal microscopy to provide three-dimensional spatial confirmation of these autophagy-associated interactions. Consistent with the findings from immunofluorescence, morphometry, and confocal microscopy, transmission electron microscopy demonstrated autophagosomes within the in vivo diacetyl-exposed airway epithelium, the target for flavorings-related lung disease. Sequestration and selective autophagy are biological responses using SQSTM1 and ubiquitin, which are involved in the cellular response to cytotoxic protein aggregates.21, 22, 60, 61 K63-ubiquitination is particularly associated with the autophagic clearance of protein aggregates.27 These findings provide converging lines of evidence that are consistent with a striking up-regulation of protein sequestration and autophagy as an in vivo response of the respiratory epithelium to inhaled diacetyl vapors.21, 30, 62
Diacetyl is highly reactive and has a well-established ability to damage proteins.18 Diacetyl is particularly reactive with the amino acid arginine.18, 63 Diacetyl and related reactive α-dicarbonyl compounds directly damage proteins by interacting with the guanidinium group of arginine.18, 64 Recently, the relative in vitro chemical reactivity of three α-dicarbonyl flavorings (diacetyl, 2,3-pentanedione, and 2,3-hexanedione) was found to be similar to the pattern of relative toxicity reflected in bronchial fibrosis and pulmonary function changes after a 2-week inhalation exposure.63 In our study, the highly significant increases in ubiquitin and SQSTM1 puncta in airway epithelium indicate that inhaled diacetyl damages proteins in vivo in this target tissue for bronchiolitis obliterans.65
Normal protein homeostasis (alias proteostasis) is essential for cell survival.66 Because normal protein function is dependent on the correct three-dimensional structure, misfolded proteins generally lose their normal function. The lung is particularly dependent on normal proteostasis.66 In our study, the depletion of free ubiquitin in Western immunoblots provides further evidence of diacetyl-induced proteostasis imbalance in airways. Because ubiquitin is critical to many cellular functions and free ubiquitin is in limited supply, ubiquitin depletion is cytotoxic.54, 67, 68, 69 Thus, the diacetyl-induced depletion demonstrated in our study is biologically important. Similar to our findings, methylglyoxal, a three-carbon α-dicarbonyl compound, depletes free ubiquitin while increasing ubiquitin conjugates.70 Although polyubiquitination has long been known to efficiently target proteins for autophagy, mono-ubiquitination has recently been demonstrated to be sufficient for SQSTM1-mediated autophagy of proteins and organelles in mammalian cells.47 Indeed, in our study, polyubiquitin did not increase, but the lower-molecular-weight ubiquitinated proteins may be responsible for ubiquitin puncta, which reached the lysosome. None of the individual high-molecular-weight bands were significantly increased, but that was expected given depletion of the free ubiquitin pool, which prevented further ubiquitination, as well as the presence of unaffected cells in the airway wall and adjacent parenchyma in in vivo samples. Recently, proteostasis imbalance has been implicated in obstructive lung diseases.71 Similar to our findings in the diacetyl-exposed lung, ubiquitinated protein aggregates and the corresponding human protein, SQSTM1, accumulate in humans with chronic obstructive pulmonary disease.72, 73 Cigarette smoke is implicated in these aggregates in humans with chronic obstructive pulmonary disease.72 Because our study demonstrates that diacetyl alters proteostasis and cigarette smoke contains diacetyl,74, 75 the results suggest that diacetyl may play a role in chronic obstructive pulmonary disease in addition to its role in flavorings-related lung disease.11, 12 In addition, during autophagy, SQSTM1 is phosphorylated, a process that activates the Kelch-like ECH-associated protein 1 (Keap1)–nuclear factor E2–related factor 2 (Nrf2) pathway.62 Thus, our finding of SQSTM1 accumulation and autophagy may explain the recently described nuclear accumulation of NRF2 in the transitional and respiratory epithelium of the nose of diacetyl-exposed rats.76
More important, the autophagy of damaged proteins protects cells from aggregated proteins, but the amino acids, ubiquitin, and SQSTM1 are digested and potentially depleted in the lysosome.77 In addition, autophagy can induce apoptosis through caspase 8 activation.78 Thus, it seems likely that the protein damage, protein aggregates, ubiquitin depletion, and autophagy seen in our study explain the cytotoxicity in airway epithelium that was seen in this and in previous diacetyl inhalation studies.3, 4 However, we saw increased ubiquitin, SQSTM1, and autophagy in intrapulmonary airways with low levels of significant apoptosis and without significant necrosis, suggesting that ubiquitination and autophagy may have controlled the nature of the proteotoxic cell death. Indeed, autophagy is more frequently a protective biological response than a major contributor to a pathological response.21, 33, 79
Because the reactive α-dicarbonyl group is implicated in diacetyl-induced protein damage, we originally hypothesized that DCXR would provide protection from protein damage through metabolism of diacetyl to acetoin, which does not have the reactive α-dicarbonyl group.36 Consistent with this hypothesis, we did observe a significantly increased number of cells with ubiquitin puncta and K63-ubiquitin puncta in airway epithelium of DCXR knockout compared with wild-type mice in the 200 ppm exposure group in the dose-response experiment. There was a similar trend at this same exposure concentration in the molecular pathology experiment, although the effect did not consistently reach statistical significance, possibly as a result of the lower number of animals in that experiment. Although the magnitude of protection in the wild-type mouse relative to the knockout mouse was lower than anticipated, the findings add support to the importance of the reactive α-dicarbonyl group in the respiratory toxicity of diacetyl.80
At 300 ppm, ubiquitination in airway epithelium of wild-type and Dcxr knockout mice was not significantly different. A possible explanation for this could be the overwhelming of metabolism at this high exposure concentration. In light of the apparent protein damage observed in the diacetyl-exposed airway epithelium, another possible explanation could be damage to the DCXR protein itself, for which diacetyl is a substrate. This would render DCXR ineffective in the wild-type mouse at this high exposure concentration.
In the lung of diacetyl-exposed Dcxr knockout but not wild-type mice, there was significant down-regulation of mRNA for club cell secretory protein (SCGB1A1), the major product of the bronchiolar club cell. In humans, decreases in SCGB1A1 protein in bronchoalveolar lavage and serum are associated with the development of bronchiolitis obliterans.81, 82 A decrease in Scgb1a1 transcripts has also been described in rats during the development of bronchiolitis obliterans–like changes after intratracheal instillation of diacetyl.83 We believe that in our study, the decrease in Scgb1a1 mRNA results from a loss of club cells (previously known as Clara cells) in the airways of the diacetyl-exposed Dcxr ko2 mice. Consistent with that interpretation, ultrastructural documentation of autophagy was most prominent in club cells in our study.
Surprisingly, Tnfα mRNA was significantly up-regulated in the olfactory bulb, but not in the lung of wild-type and Dcxr ko mice inhaling 200 ppm diacetyl. Dcxr ko mice had greater elevation in Tnfα mRNA relative to air-exposed genotype controls when compared with the wild-type mice, suggesting that DCXR is protective. The up-regulation of olfactory bulb Tnfα was accompanied by a decrease in Omp mRNA, which suggests a loss of olfactory neurons. Because diacetyl is highly reactive and unlikely to diffuse into the brain, the selective increase in Tnfα in the olfactory bulb cannot be explained by diffusion of diacetyl. The increases in immunoreactive SQSTM1 observed within the olfactory bulbs of diacetyl-exposed mice suggest that one possible explanation could be that increases in SQSTM1 resulted in increased Tnfα mRNA expression, because it is believed to integrate multiple signaling cascades.84, 85, 86, 87
By confocal microscopy, we were able to localize SQSTM1 accumulation in the olfactory bulb of diacetyl-exposed mice to the glomerular region and immediately adjacent neuropil. Within that region, intracytoplasmic accumulations of SQSTM1 were localized within AIF1-positive activated microglial cells, which sometimes contained OMP-positive material. This phagocytosis of olfactory neurons and immunoreactive SQSTM1 within the olfactory bulb was consistent with the decreased Omp mRNA measured by real-time PCR. Together, these findings demonstrate olfactory neurotoxicity and microglial activation after inhaling 200 ppm diacetyl. It has recently been suggested that tumor necrosis factor-α stimulates neuronal phagocytosis by microglial cells.88 The concomitant observation of increased Tnfα mRNA expression and neuronal phagocytosis in the diacetyl-exposed mice is consistent with that hypothesis.
This is the first demonstration of SQSTM1 accumulation in the olfactory bulb of the brain after an inhalation exposure. SQSTM1 is important in a spectrum of cellular processes.62, 85, 86, 89 In addition, SQSTM1 and autophagy are increased in neuropathology associated with protein aggregates in diseases, such as Alzheimer, Parkinson, and Huntington.86, 90, 91 However, it is not known whether this association contributes to the pathogenesis of these diseases, is incidental, or is protective.91, 92 Because SQSTM1 was present in the olfactory neuroepithelium of diacetyl-exposed mice in our study and protein aggregates are easily transported by neurons,93, 94 neuronal transport of SQSTM1 or misfolded proteins from the nose to the brain could potentially explain the accumulation of SQSTM1 in the olfactory bulb. This is a concern because seeds of protein aggregates are believed to play a role in the spread of abnormal protein aggregates in the brain and SQSTM1 is a scaffold on which protein aggregates can form.85, 86, 94, 95, 96 Microglial activation and phagocytosis of olfactory nerves in diacetyl-exposed mice was also demonstrated. Changes in microglial populations and olfactory dysfunction are well described in neurodegenerative diseases,88, 97, 98, 99, 100, 101, 102 although their role in pathogenesis of neurodegenerative diseases remains controversial. Importantly, neurogenesis of olfactory neurons continues in adult life and can sometimes restore normal function.103 Further studies will be needed to investigate the potential importance of diacetyl-induced microglial activation, phagocytosis of olfactory neurons, enhanced Tnfα mRNA expression, and transport of immunoreactive SQSTM1 and/or altered proteins from the nose to the brain.
In summary, this study implicates in vivo protein damage of airway epithelial cells as the cause of flavorings-related lung disease. After diacetyl inhalation, we have demonstrated extensive accumulation of total ubiquitin, K63-ubiquitin, and SQSTM1 puncta in the respiratory epithelium, all hallmarks of excessive protein damage. The colocalization of ubiquitin to SQSTM1 and to the lysosome of airway epithelial cells indicates that diacetyl-induced respiratory toxicity involves widespread autophagy, further implicating protein damage in diacetyl-induced cytotoxicity. Autophagosomes were confirmed by transmission microscopy within club cells, the principal progenitor cells of the airway epithelium. Consistent with damage to these progenitor cells, club cell–specific Scgb1a1 was also down-regulated. The demonstration of free ubiquitin depletion is also consistent with the deduction that diacetyl-induced protein damage was excessive, because free ubiquitin pools are tightly regulated and depletion of free ubiquitin can mediate cytotoxicity.54, 55 The hypothesis that flavorings-related lung diseases is because of protein damage at sites of diacetyl absorption is further supported by the known ability of diacetyl and related α-dicarbonyl compounds to cause protein damage in cell-free systems and the somewhat greater susceptibility of Dcxr knockout mice to diacetyl toxicity. The conclusion that protein damage in airway epithelium is the likely mechanism of flavorings-related lung disease may help identify substitute flavorings predicted to cause similar airway injury.104 These findings also support the need for future investigations. These include investigating the potential importance of immunoreactive SQSTM1 in affecting extrapulmonary tissues and the potential ability of ubiquitin and SQSTM1 aggregates to serve as biomarkers of flavorings-related lung disease.
Acknowledgments
We thank Dr. Matthew W. Foster (Duke University) for helpful discussions; Patsy Willard (deceased) and Shannon Case for outstanding histotechnology throughout this study; the University of California, Davis, KOMP Repository, Velocigene, and the CSD Consortium for their role in generating Dcxr knockout mice.
Footnotes
Supported by National Institute for Occupational Safety and Health, Centers for Disease Control and Prevention, Intramural National Occupational Research Agenda Project 6927ZKQC, Causes of Flavorings-Related Lung Disease. M.P.G. was supported in part by a contract to West Virginia University under a National Institute for Occupational Safety and Health project, Pathophysiology of Popcorn Workers' Lung.
The findings and conclusions herein are those of the authors and do not necessarily represent the views of the National Institute for Occupational Safety and Health. Mention of brand name does not constitute product endorsement.
Disclosures: None declared.
Current address of J.L.B., Gryphon Scientific, Takoma Park, MD; of M.P.G., Covance, Madison, WI.
Supplemental material for this article can be found at http://dx.doi.org/10.1016/j.ajpath.2016.07.018.
Supplemental Data
References
- 1.Kreiss K., Gomaa A., Kullman G., Fedan K., Simoes E.J., Enright P.L. Clinical bronchiolitis obliterans in workers at a microwave-popcorn plant. N Engl J Med. 2002;347:330–338. doi: 10.1056/NEJMoa020300. [DOI] [PubMed] [Google Scholar]
- 2.Hubbs A.F., Battelli L.A., Goldsmith W.T., Porter D.W., Frazer D., Friend S., Schwegler-Berry D., Mercer R.R., Reynolds J.S., Grote A., Castranova V., Kullman G., Fedan J.S., Dowdy J., Jones W.G. Necrosis of nasal and airway epithelium in rats inhaling vapors of artificial butter flavoring. Toxicol Appl Pharmacol. 2002;185:128–135. doi: 10.1006/taap.2002.9525. [DOI] [PubMed] [Google Scholar]
- 3.Hubbs A.F., Goldsmith W.T., Kashon M.L., Frazer D., Mercer R.R., Battelli L.A., Kullman G.J., Schwegler-Berry D., Friend S., Castranova V. Respiratory toxicologic pathology of inhaled diacetyl in Sprague-Dawley rats. Toxicol Pathol. 2008;36:330–344. doi: 10.1177/0192623307312694. [DOI] [PubMed] [Google Scholar]
- 4.Morgan D.L., Flake G.P., Kirby P.J., Palmer S.M. Respiratory toxicity of diacetyl in C57BL/6 mice. Toxicol Sci. 2008;103:169–180. doi: 10.1093/toxsci/kfn016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Akpinar-Elci M., Travis W.D., Lynch D.A., Kreiss K. Bronchiolitis obliterans syndrome in popcorn production plant workers. Eur Respir J. 2004;24:298–302. doi: 10.1183/09031936.04.00013903. [DOI] [PubMed] [Google Scholar]
- 6.Kanwal R., Kullman G., Piacitelli C., Boylstein R., Sahakian N., Martin S., Fedan K., Kreiss K. Evaluation of flavorings-related lung disease risk at six microwave popcorn plants. J Occup Environ Med. 2006;48:149–157. doi: 10.1097/01.jom.0000194152.48728.fb. [DOI] [PubMed] [Google Scholar]
- 7.Kreiss K., Fedan K.B., Nasrullah M., Kim T.J., Materna B.L., Prudhomme J.C., Enright P.L. Longitudinal lung function declines among California flavoring manufacturing workers. Am J Ind Med. 2012;55:657–668. doi: 10.1002/ajim.21013. [DOI] [PubMed] [Google Scholar]
- 8.van Rooy F.G., Rooyackers J.M., Prokop M., Houba R., Smit L.A., Heederik D.J. Bronchiolitis obliterans syndrome in chemical workers producing diacetyl for food flavorings. Am J Respir Crit Care Med. 2007;176:498–504. doi: 10.1164/rccm.200611-1620OC. [DOI] [PubMed] [Google Scholar]
- 9.Centers for Disease Control and Prevention (CDC) Obliterative bronchiolitis in workers in a coffee-processing facility: Texas, 2008-2012. MMWR Morb Mortal Wkly Rep. 2013;62:305–307. [PMC free article] [PubMed] [Google Scholar]
- 10.Kreiss K. Work-related spirometric restriction in flavoring manufacturing workers. Am J Ind Med. 2014;57:129–137. doi: 10.1002/ajim.22282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Allen J.G., Flanigan S.S., LeBlanc M., Vallarino J., MacNaughton P., Stewart J.H., Christiani D.C. Flavoring chemicals in E-cigarettes: diacetyl, 2,3-pentanedione, and acetoin in a sample of 51 products, including fruit-, candy-, and cocktail-flavored E-cigarettes. Environ Health Perspect. 2016;124:733–739. doi: 10.1289/ehp.1510185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Farsalinos K.E., Kistler K.A., Gillman G., Voudris V. Evaluation of electronic cigarette liquids and aerosol for the presence of selected inhalation toxins. Nicotine Tob Res. 2015;17:168–174. doi: 10.1093/ntr/ntu176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miller A.G., Gerrard J.A. Assessment of protein function following cross-linking by alpha-dicarbonyls. Ann N Y Acad Sci. 2005;1043:195–200. doi: 10.1196/annals.1333.024. [DOI] [PubMed] [Google Scholar]
- 14.Rodriguez Mellado J.M., Ruiz Montoya M. Correlations between chemical reactivity and mutagenic activity against S. typhimurium TA100 for alpha-dicarbonyl compounds as a proof of the mutagenic mechanism. Mutat Res. 1994;304:261–264. doi: 10.1016/0027-5107(94)90218-6. [DOI] [PubMed] [Google Scholar]
- 15.Morgan D.L., Jokinen M.P., Price H.C., Gwinn W.M., Palmer S.M., Flake G.P. Bronchial and bronchiolar fibrosis in rats exposed to 2,3-pentanedione vapors: implications for bronchiolitis obliterans in humans. Toxicol Pathol. 2012;40:448–465. doi: 10.1177/0192623311431946. [DOI] [PubMed] [Google Scholar]
- 16.Hubbs A.F., Cumpston A.M., Goldsmith W.T., Battelli L.A., Kashon M.L., Jackson M.C., Frazer D.G., Fedan J.S., Goravanahally M.P., Castranova V., Kreiss K., Willard P.A., Friend S., Schwegler-Berry D., Fluharty K.L., Sriram K. Respiratory and olfactory cytotoxicity of inhaled 2,3-pentanedione in Sprague-Dawley rats. Am J Pathol. 2012;181:829–844. doi: 10.1016/j.ajpath.2012.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lo T.W., Westwood M.E., McLellan A.C., Selwood T., Thornalley P.J. Binding and modification of proteins by methylglyoxal under physiological conditions: a kinetic and mechanistic study with N alpha-acetylarginine, N alpha-acetylcysteine, and N alpha-acetyllysine, and bovine serum albumin, J Biol Chem. 1994;269:32299–32305. [PubMed] [Google Scholar]
- 18.Mathews J.M., Watson S.L., Snyder R.W., Burgess J.P., Morgan D.L. Reaction of the butter flavorant diacetyl (2,3-butanedione) with N-alpha-acetylarginine: a model for epitope formation with pulmonary proteins in the etiology of obliterative bronchiolitis. J Agric Food Chem. 2010;58:12761–12768. doi: 10.1021/jf103251w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fennell T.R., Morgan D.L., Watson S.L., Dhungana S., Waidyanatha S. Systemic uptake, albumin and hemoglobin binding of [(14)C]2,3-butanedione administered by intratracheal instillation in male Harlan Sprague Dawley rats and oropharyngeal aspiration in male B6C3F1/N mice. Chem Biol Interact. 2015;227:112–119. doi: 10.1016/j.cbi.2014.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goldberg A.L. Protein degradation and protection against misfolded or damaged proteins. Nature. 2003;426:895–899. doi: 10.1038/nature02263. [DOI] [PubMed] [Google Scholar]
- 21.Lim J., Lachenmayer M.L., Wu S., Liu W., Kundu M., Wang R., Komatsu M., Oh Y.J., Zhao Y., Yue Z. Proteotoxic stress induces phosphorylation of p62/SQSTM1 by ULK1 to regulate selective autophagic clearance of protein aggregates. PLoS Genet. 2015;11:e1004987. doi: 10.1371/journal.pgen.1004987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yao T.P. The role of ubiquitin in autophagy-dependent protein aggregate processing, Genes Cancer. 2010;1:779–786. doi: 10.1177/1947601910383277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hershko A. The ubiquitin system for protein degradation and some of its roles in the control of the cell-division cycle (Nobel lecture) Angew Chem. 2005;44:5932–5943. doi: 10.1002/anie.200501724. [DOI] [PubMed] [Google Scholar]
- 24.Ciechanover A. Intracellular protein degradation: from a vague idea, through the lysosome and the ubiquitin-proteasome system, and onto human diseases and drug targeting (Nobel lecture) Angew Chem. 2005;44:5944–5967. doi: 10.1002/anie.200501428. [DOI] [PubMed] [Google Scholar]
- 25.Rose I. Ubiquitin at Fox Chase (Nobel lecture) Angew Chem. 2005;44:5926–5931. doi: 10.1002/anie.200500995. [DOI] [PubMed] [Google Scholar]
- 26.Lim G.G., Chew K.C., Ng X.H., Henry-Basil A., Sim R.W., Tan J.M., Chai C., Lim K.L. Proteasome inhibition promotes Parkin-Ubc13 interaction and lysine 63-linked ubiquitination. PLoS One. 2013;8:e73235. doi: 10.1371/journal.pone.0073235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tan J.M., Wong E.S., Kirkpatrick D.S., Pletnikova O., Ko H.S., Tay S.P., Ho M.W., Troncoso J., Gygi S.P., Lee M.K., Dawson V.L., Dawson T.M., Lim K.L. Lysine 63-linked ubiquitination promotes the formation and autophagic clearance of protein inclusions associated with neurodegenerative diseases. Hum Mol Genet. 2008;17:431–439. doi: 10.1093/hmg/ddm320. [DOI] [PubMed] [Google Scholar]
- 28.Chen Z.J., Sun L.J. Nonproteolytic functions of ubiquitin in cell signaling. Mol Cell. 2009;33:275–286. doi: 10.1016/j.molcel.2009.01.014. [DOI] [PubMed] [Google Scholar]
- 29.Nathan J.A., Kim H.T., Ting L., Gygi S.P., Goldberg A.L. Why do cellular proteins linked to K63-polyubiquitin chains not associate with proteasomes? EMBO J. 2013;32:552–565. doi: 10.1038/emboj.2012.354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ding W.X., Yin X.M. Sorting, recognition and activation of the misfolded protein degradation pathways through macroautophagy and the proteasome. Autophagy. 2008;4:141–150. doi: 10.4161/auto.5190. [DOI] [PubMed] [Google Scholar]
- 31.Tauriello D.V., Maurice M.M. The various roles of ubiquitin in Wnt pathway regulation. Cell Cycle. 2010;9:3700–3709. doi: 10.4161/cc.9.18.13204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Iwai K. Diverse roles of the ubiquitin system in NF-kappaB activation. Biochim Biophys Acta. 2014;1843:129–136. doi: 10.1016/j.bbamcr.2013.03.011. [DOI] [PubMed] [Google Scholar]
- 33.Yamada T., Kawabata Y. Pneumocyte injury and ubiquitin-positive pneumocytes in interstitial lung diseases. Histopathology. 2015;66:161–172. doi: 10.1111/his.12528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dantuma N.P., Bott L.C. The ubiquitin-proteasome system in neurodegenerative diseases: precipitating factor, yet part of the solution. Front Mol Neurosci. 2014;7:70. doi: 10.3389/fnmol.2014.00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lowe J., Blanchard A., Morrell K., Lennox G., Reynolds L., Billett M., Landon M., Mayer R.J. Ubiquitin is a common factor in intermediate filament inclusion bodies of diverse type in man, including those of Parkinson's disease, Pick's disease, and Alzheimer's disease, as well as Rosenthal fibres in cerebellar astrocytomas, cytoplasmic bodies in muscle, and mallory bodies in alcoholic liver disease. J Pathol. 1988;155:9–15. doi: 10.1002/path.1711550105. [DOI] [PubMed] [Google Scholar]
- 36.Nakagawa J., Ishikura S., Asami J., Isaji T., Usami N., Hara A., Sakurai T., Tsuritani K., Oda K., Takahashi M., Yoshimoto M., Otsuka N., Kitamura K. Molecular characterization of mammalian dicarbonyl/L-xylulose reductase and its localization in kidney. J Biol Chem. 2002;277:17883–17891. doi: 10.1074/jbc.M110703200. [DOI] [PubMed] [Google Scholar]
- 37.Zaccone E.J., Thompson J.A., Ponnoth D.S., Cumpston A.M., Goldsmith W.T., Jackson M.C., Kashon M.L., Frazer D.G., Hubbs A.F., Shimko M.J., Fedan J.S. Popcorn flavoring effects on reactivity of rat airways in vivo and in vitro. J Toxicol Environ Health A. 2013;76:669–689. doi: 10.1080/15287394.2013.796302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lloyd K.C. A knockout mouse resource for the biomedical research community. Ann N Y Acad Sci. 2011;1245:24–26. doi: 10.1111/j.1749-6632.2011.06311.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meier I.D., Bernreuther C., Tilling T., Neidhardt J., Wong Y.W., Schulze C., Streichert T., Schachner M. Short DNA sequences inserted for gene targeting can accidentally interfere with off-target gene expression. FASEB J. 2010;24:1714–1724. doi: 10.1096/fj.09-140749. [DOI] [PubMed] [Google Scholar]
- 40.Flecknell P. Replacement, reduction and refinement. ALTEX. 2002;19:73–78. [PubMed] [Google Scholar]
- 41.Oldham M.J., Phalen R.F. Dosimetry implications of upper tracheobronchial airway anatomy in two mouse varieties. Anat Rec. 2002;268:59–65. doi: 10.1002/ar.10134. [DOI] [PubMed] [Google Scholar]
- 42.Herbert R., Leininger J.R. Nose, larynx and trachea. Pathology of the Mouse. In: Maronpot R.R., Boorman G.A., Gaul B.W., editors. Cache River Press; Vienna, IL: 1999. pp. 259–292. [Google Scholar]
- 43.Crissman J.W., Goodman D.G., Hildebrandt P.K., Maronpot R.R., Prater D.A., Riley J.H., Seaman W.J., Thake D.C. Best practices guideline: toxicologic histopathology. Toxicol Pathol. 2004;32:126–131. doi: 10.1080/01926230490268756. [DOI] [PubMed] [Google Scholar]
- 44.Kaniuk N.A., Brumell J.H. Examining ubiquitinated protein aggregates in tissue sections. Methods Mol Biol. 2010;648:175–182. doi: 10.1007/978-1-60761-756-3_11. [DOI] [PubMed] [Google Scholar]
- 45.Kasper M., Rudolf T., Verhofstad A.A., Schuh D., Muller M. Heterogeneity in the immunolocalization of cytokeratin-specific monoclonal antibodies in the rat lung: evaluation of three different alveolar epithelial cell types. Histochemistry. 1993;100:65–71. doi: 10.1007/BF00268879. [DOI] [PubMed] [Google Scholar]
- 46.Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim P.K., Hailey D.W., Mullen R.T., Lippincott-Schwartz J. Ubiquitin signals autophagic degradation of cytosolic proteins and peroxisomes. Proc Natl Acad Sci U S A. 2008;105:20567–20574. doi: 10.1073/pnas.0810611105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sriram K., Lin G.X., Jefferson A.M., Roberts J.R., Wirth O., Hayashi Y., Krajnak K.M., Soukup J.M., Ghio A.J., Reynolds S.H., Castranova V., Munson A.E., Antonini J.M. Mitochondrial dysfunction and loss of Parkinson's disease-linked proteins contribute to neurotoxicity of manganese-containing welding fumes. FASEB J. 2010;24:4989–5002. doi: 10.1096/fj.10-163964. [DOI] [PubMed] [Google Scholar]
- 49.Nuzzo R. Scientific method: statistical errors. Nature. 2014;506:150–152. doi: 10.1038/506150a. [DOI] [PubMed] [Google Scholar]
- 50.American Statistical Association Board of Directors ASA statement on statistical significance and P-values. Am Stat. 2016;70:131–133. doi: 10.1080/01621459.2017.1311265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wasserstein R.L., Lazar N.A. The ASA's statement on p-values: context, process and purpose. Am Stat. 2016;70:129–131. [Google Scholar]
- 52.Morris J.B., Hubbs A.F. Inhalation dosimetry of diacetyl and butyric acid, two components of butter flavoring vapors. Toxicol Sci. 2009;108:173–183. doi: 10.1093/toxsci/kfn222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gloede E., Cichocki J.A., Baldino J.B., Morris J.B. A validated hybrid computational fluid dynamics-physiologically based pharmacokinetic model for respiratory tract vapor absorption in the human and rat and its application to inhalation dosimetry of diacetyl. Toxicol Sci. 2011;123:231–246. doi: 10.1093/toxsci/kfr165. [DOI] [PubMed] [Google Scholar]
- 54.Hanna J., Leggett D.S., Finley D. Ubiquitin depletion as a key mediator of toxicity by translational inhibitors. Mol Cell Biol. 2003;23:9251–9261. doi: 10.1128/MCB.23.24.9251-9261.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dantuma N.P., Groothuis T.A., Salomons F.A., Neefjes J. A dynamic ubiquitin equilibrium couples proteasomal activity to chromatin remodeling. J Cell Biol. 2006;173:19–26. doi: 10.1083/jcb.200510071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Goravanahally M.P., Hubbs A.F., Fedan J.S., Kashon M.L., Battelli L.A., Mercer R.R., Goldsmith W.T., Jackson M.C., Cumpston A., Frazer D.G., Dey R.D. Diacetyl increases sensory innervation and substance P production in rat trachea. Toxicol Pathol. 2014;42:582–590. doi: 10.1177/0192623313493689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Oberdorster G., Elder A., Rinderknecht A. Nanoparticles and the brain: cause for concern? J Nanosci Nanotechnol. 2009;9:4996–5007. doi: 10.1166/jnn.2009.gr02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kaur J., Debnath J. Autophagy at the crossroads of catabolism and anabolism. Nat Rev Mol Cell Biol. 2015;16:461–472. doi: 10.1038/nrm4024. [DOI] [PubMed] [Google Scholar]
- 59.Lee H.S., Daniels B.H., Salas E., Bollen A.W., Debnath J., Margeta M. Clinical utility of LC3 and p62 immunohistochemistry in diagnosis of drug-induced autophagic vacuolar myopathies: a case-control study. PLoS One. 2012;7:e36221. doi: 10.1371/journal.pone.0036221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kaganovich D., Kopito R., Frydman J. Misfolded proteins partition between two distinct quality control compartments. Nature. 2008;454:1088–1095. doi: 10.1038/nature07195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Miller S.B., Mogk A., Bukau B. Spatially organized aggregation of misfolded proteins as cellular stress defense strategy. J Mol Biol. 2015;427:1564–1574. doi: 10.1016/j.jmb.2015.02.006. [DOI] [PubMed] [Google Scholar]
- 62.Ichimura Y., Waguri S., Sou Y.S., Kageyama S., Hasegawa J., Ishimura R., Saito T., Yang Y., Kouno T., Fukutomi T., Hoshii T., Hirao A., Takagi K., Mizushima T., Motohashi H., Lee M.S., Yoshimori T., Tanaka K., Yamamoto M., Komatsu M. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol Cell. 2013;51:618–631. doi: 10.1016/j.molcel.2013.08.003. [DOI] [PubMed] [Google Scholar]
- 63.Morgan D.L., Jokinen M.P., Johnson C.L., Price H.C., Gwinn W.M., Bousquet R.W., Flake G.P. Chemical reactivity and respiratory toxicity of the alpha-diketone flavoring agents: 2,3-butanedione, 2,3-pentanedione, and 2,3-hexanedione. Toxicol Pathol. 2016;44:763–783. doi: 10.1177/0192623316638962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Toi K., Bynum E., Norris E., Itano H.A. Studies on the chemical modification of arginine, I: the reaction of 1,2-cyclohexanedione with arginine and arginyl residues of proteins. J Biol Chem. 1967;242:1036–1043. [PubMed] [Google Scholar]
- 65.King T.E., Jr. Bronchiolitis obliterans. Lung. 1989;167:69–93. doi: 10.1007/BF02714935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Balch W.E., Sznajder J.I., Budinger S., Finley D., Laposky A.D., Cuervo A.M., Benjamin I.J., Barreiro E., Morimoto R.I., Postow L., Weissman A.M., Gail D., Banks-Schlegel S., Croxton T., Gan W. Malfolded protein structure and proteostasis in lung diseases. Am J Respir Crit Care Med. 2014;189:96–103. doi: 10.1164/rccm.201306-1164WS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shabek N., Ciechanover A. Degradation of ubiquitin: the fate of the cellular reaper. Cell Cycle. 2010;9:523–530. doi: 10.4161/cc.9.3.11152. [DOI] [PubMed] [Google Scholar]
- 68.Groothuis T.A., Dantuma N.P., Neefjes J., Salomons F.A. Ubiquitin crosstalk connecting cellular processes. Cell Div. 2006;1:21. doi: 10.1186/1747-1028-1-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tan Z., Tu W., Schreiber S.S. Downregulation of free ubiquitin: a novel mechanism of p53 stabilization and neuronal cell death. Brain Res Mol Brain Res. 2001;91:179–188. doi: 10.1016/s0169-328x(01)00117-6. [DOI] [PubMed] [Google Scholar]
- 70.Bento C.F., Marques F., Fernandes R., Pereira P. Methylglyoxal alters the function and stability of critical components of the protein quality control. PLoS One. 2010;5:e13007. doi: 10.1371/journal.pone.0013007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cantin A.M., Richter M.V. Cigarette smoke-induced proteostasis imbalance in obstructive lung diseases. Curr Mol Med. 2012;12:836–849. doi: 10.2174/156652412801318746. [DOI] [PubMed] [Google Scholar]
- 72.Tran I., Ji C., Ni I., Min T., Tang D., Vij N. Role of cigarette smoke-induced aggresome-formation in COPD-emphysema pathogenesis. Am J Respir Cell Mol Biol. 2015;53:159–173. doi: 10.1165/rcmb.2014-0107OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Min T., Bodas M., Mazur S., Vij N. Critical role of proteostasis-imbalance in pathogenesis of COPD and severe emphysema. Curr Mol Med (Berl) 2011;89:577–593. doi: 10.1007/s00109-011-0732-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fujioka K., Shibamoto T. Determination of toxic carbonyl compounds in cigarette smoke. Environ Toxicol. 2006;21:47–54. doi: 10.1002/tox.20153. [DOI] [PubMed] [Google Scholar]
- 75.Pierce J.S., Abelmann A., Spicer L.J., Adams R.E., Finley B.L. Diacetyl and 2,3-pentanedione exposures associated with cigarette smoking: implications for risk assessment of food and flavoring workers. Crit Rev Toxicol. 2014;44:420–435. doi: 10.3109/10408444.2014.882292. [DOI] [PubMed] [Google Scholar]
- 76.Cichocki J.A., Smith G.J., Morris J.B. Tissue sensitivity of the rat upper and lower extrapulmonary airways to the inhaled electrophilic air pollutants diacetyl and acrolein. Toxicol Sci. 2014;142:126–136. doi: 10.1093/toxsci/kfu165. [DOI] [PubMed] [Google Scholar]
- 77.Komatsu M., Ichimura Y. Physiological significance of selective degradation of p62 by autophagy. FEBS Letters. 2010;584:1374–1378. doi: 10.1016/j.febslet.2010.02.017. [DOI] [PubMed] [Google Scholar]
- 78.Young M.M., Takahashi Y., Khan O., Park S., Hori T., Yun J., Sharma A.K., Amin S., Hu C.D., Zhang J., Kester M., Wang H.G. Autophagosomal membrane serves as platform for intracellular death-inducing signaling complex (iDISC)-mediated caspase-8 activation and apoptosis. J Biol Chem. 2012;287:12455–12468. doi: 10.1074/jbc.M111.309104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lopez-Alonso I., Aguirre A., Gonzalez-Lopez A., Fernandez A.F., Amado-Rodriguez L., Astudillo A., Batalla-Solis E., Albaiceta G.M. Impairment of autophagy decreases ventilator-induced lung injury by blockade of the NF-kappaB pathway. Am J Physiol Lung Cell Mol Physiol. 2013;304:L844–L852. doi: 10.1152/ajplung.00422.2012. [DOI] [PubMed] [Google Scholar]
- 80.NIOSH: Criteria for a Recommended Standard: Occupational Exposure to Diacetyl and 2,3-Pentanedione (draft). Edited by Department of Health and Human Services. Online external review document, 2011, Available at: http://www.cdc.gov/niosh/docket/archive/pdfs/NIOSH-245/0245-081211-draftdocument.pdf (accessed April 12, 2016).
- 81.Nord M., Schubert K., Cassel T.N., Andersson O., Riise G.C. Decreased serum and bronchoalveolar lavage levels of Clara cell secretory protein (CC16) is associated with bronchiolitis obliterans syndrome and airway neutrophilia in lung transplant recipients. Transplantation. 2002;73:1264–1269. doi: 10.1097/00007890-200204270-00013. [DOI] [PubMed] [Google Scholar]
- 82.Mattsson J., Remberger M., Andersson O., Sundberg B., Nord M. Decreased serum levels of clara cell secretory protein (CC16) are associated with bronchiolitis obliterans and may permit early diagnosis in patients after allogeneic stem-cell transplantation. Transplantation. 2005;79:1411–1416. doi: 10.1097/01.tp.0000158354.39635.ab. [DOI] [PubMed] [Google Scholar]
- 83.Palmer S.M., Flake G.P., Kelly F.L., Zhang H.L., Nugent J.L., Kirby P.J., Foley J.F., Gwinn W.M., Morgan D.L. Severe airway epithelial injury, aberrant repair and bronchiolitis obliterans develops after diacetyl instillation in rats. PLoS One. 2011;6:e17644. doi: 10.1371/journal.pone.0017644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wooten M.W., Geetha T., Seibenhener M.L., Babu J.R., Diaz-Meco M.T., Moscat J. The p62 scaffold regulates nerve growth factor-induced NF-kappaB activation by influencing TRAF6 polyubiquitination. J Biol Chem. 2005;280:35625–35629. doi: 10.1074/jbc.C500237200. [DOI] [PubMed] [Google Scholar]
- 85.Moscat J., Diaz-Meco M.T., Wooten M.W. Signal integration and diversification through the p62 scaffold protein. Trends Biochem Sci. 2007;32:95–100. doi: 10.1016/j.tibs.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 86.Geetha T., Vishwaprakash N., Sycheva M., Babu J.R. Sequestosome 1/p62: across diseases. Biomarkers. 2012;17:99–103. doi: 10.3109/1354750X.2011.653986. [DOI] [PubMed] [Google Scholar]
- 87.Park S., Ha S.D., Coleman M., Meshkibaf S., Kim S.O. p62/SQSTM1 enhances NOD2-mediated signaling and cytokine production through stabilizing NOD2 oligomerization. PLoS One. 2013;8:e57138. doi: 10.1371/journal.pone.0057138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Neniskyte U., Vilalta A., Brown G.C. Tumour necrosis factor alpha-induced neuronal loss is mediated by microglial phagocytosis. FEBS Letters. 2014;588:2952–2956. doi: 10.1016/j.febslet.2014.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bertrand M., Petit V., Jain A., Amsellem R., Johansen T., Larue L., Codogno P., Beau I. SQSTM1/p62 regulates the expression of junctional proteins through epithelial-mesenchymal transition factors. Cell Cycle. 2015;14:364–374. doi: 10.4161/15384101.2014.987619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zatloukal K., Stumptner C., Fuchsbichler A., Heid H., Schnoelzer M., Kenner L., Kleinert R., Prinz M., Aguzzi A., Denk H. p62 Is a common component of cytoplasmic inclusions in protein aggregation diseases. Am J Pathol. 2002;160:255–263. doi: 10.1016/S0002-9440(10)64369-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bjorkoy G., Lamark T., Brech A., Outzen H., Perander M., Overvatn A., Stenmark H., Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171:603–614. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ramesh Babu J., Lamar Seibenhener M., Peng J., Strom A.L., Kemppainen R., Cox N., Zhu H., Wooten M.C., Diaz-Meco M.T., Moscat J., Wooten M.W. Genetic inactivation of p62 leads to accumulation of hyperphosphorylated tau and neurodegeneration. J Neurochem. 2008;106:107–120. doi: 10.1111/j.1471-4159.2008.05340.x. [DOI] [PubMed] [Google Scholar]
- 93.Maday S., Twelvetrees A.E., Moughamian A.J., Holzbaur E.L. Axonal transport: cargo-specific mechanisms of motility and regulation. Neuron. 2014;84:292–309. doi: 10.1016/j.neuron.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kalani A., Tyagi A., Tyagi N. Exosomes: mediators of neurodegeneration, neuroprotection and therapeutics. Mol Neurobiol. 2014;49:590–600. doi: 10.1007/s12035-013-8544-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pegtel D.M., Peferoen L., Amor S. Extracellular vesicles as modulators of cell-to-cell communication in the healthy and diseased brain. Philosoph Trans R Soc Lon B Biol Sci. 2014;369:20130516. doi: 10.1098/rstb.2013.0516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lim J., Yue Z. Neuronal aggregates: formation, clearance, and spreading. Dev Cell. 2015;32:491–501. doi: 10.1016/j.devcel.2015.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kan M.J., Lee J.E., Wilson J.G., Everhart A.L., Brown C.M., Hoofnagle A.N., Jansen M., Vitek M.P., Gunn M.D., Colton C.A. Arginine deprivation and immune suppression in a mouse model of Alzheimer's disease. J Neurosci. 2015;35:5969–5982. doi: 10.1523/JNEUROSCI.4668-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Medeiros R., Kitazawa M., Passos G.F., Baglietto-Vargas D., Cheng D., Cribbs D.H., LaFerla F.M. Aspirin-triggered lipoxin A4 stimulates alternative activation of microglia and reduces Alzheimer disease-like pathology in mice. Am J Pathol. 2013;182:1780–1789. doi: 10.1016/j.ajpath.2013.01.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ruan Y., Zheng X.Y., Zhang H.L., Zhu W., Zhu J. Olfactory dysfunctions in neurodegenerative disorders. J Neurosci Res. 2012;90:1693–1700. doi: 10.1002/jnr.23054. [DOI] [PubMed] [Google Scholar]
- 100.Neher J.J., Neniskyte U., Zhao J.W., Bal-Price A., Tolkovsky A.M., Brown G.C. Inhibition of microglial phagocytosis is sufficient to prevent inflammatory neuronal death. J Immunol. 2011;186:4973–4983. doi: 10.4049/jimmunol.1003600. [DOI] [PubMed] [Google Scholar]
- 101.Brown G.C., Neher J.J. Microglial phagocytosis of live neurons. Nat Rev Neurosci. 2014;15:209–216. doi: 10.1038/nrn3710. [DOI] [PubMed] [Google Scholar]
- 102.Gorlovoy P., Larionov S., Pham T.T., Neumann H. Accumulation of tau induced in neurites by microglial proinflammatory mediators. FASEB J. 2009;23:2502–2513. doi: 10.1096/fj.08-123877. [DOI] [PubMed] [Google Scholar]
- 103.Lazarini F., Gabellec M.M., Moigneu C., de Chaumont F., Olivo-Marin J.C., Lledo P.M. Adult neurogenesis restores dopaminergic neuronal loss in the olfactory bulb. J Neurosci. 2014;34:14430–14442. doi: 10.1523/JNEUROSCI.5366-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zimmerman J.B., Anastas P.T. Chemistry: toward substitution with no regrets. Science. 2015;347:1198–1199. doi: 10.1126/science.aaa0812. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.