

Abstract
Pericytes, resident fibroblasts, and mesenchymal stem cells are poorly described cell populations. They have recently been characterized in much greater detail in rodent lungs and have been shown to play important roles in development, homeostasis, response to injury and pathogens, as well as recovery from damage. These closely related mesenchymal cell populations form extensive connections to the lung's internal structure, as well as its internal and external surfaces. They generate and remodel extracellular matrix, coregulate the vasculature, help maintain and restore the epithelium, and act as sentries for the immune system. In this review, we revisit these functions in light of significant advances in characterizing and tracking lung fibroblast populations in rodents. Lineage tracing experiments have mapped the heritage, identified functions that discriminate lung pericytes from resident fibroblasts, identified a subset of mesenchymal stem cells, and shown these populations to be the predominant progenitors of pathological fibroblasts and myofibroblasts in lung diseases. These findings point to the importance of resident lung mesenchymal populations as therapeutic targets in acute lung injury as well as fibrotic and degenerative diseases. Far from being passive and quiescent, pericytes and resident fibroblasts are busily sensing and responding, through diverse mechanisms, to changes in lung health and function.
The architecture of the lungs juxtaposes a large external surface area with an extensive vascular bed for efficient gas exchange. In between these epithelial and endothelial layers lies an elastic matrix, lymphatics, smooth muscle, resident and migratory leukocytes, as well as a poorly characterized population of mesenchymal stromal cells.1 Many of these cells have been described to contribute to normal lung function, as well as loss of function, in disease states. This review focuses on the pleiotropic roles played by two distinct lung mesenchymal populations, known as pericytes and resident fibroblasts, that until now have been challenging to identify. We will describe their roles during development, homeostasis, and fibrotic diseases. New discoveries made by lineage tracing, conditional ablation, and targeted gene deletion of mesenchymal cell subpopulations, in rodents, have expanded on and reinforced the conclusion that mesenchymal fibroblasts are crucial for forming and maintaining vascular networks, sensing damage, recruiting inflammatory cells, and remodeling the extracellular matrix of the lung and other organs. These actions are beneficial when maintaining or returning a tissue to homeostasis, but can become pathological when prolonged, excessive, or recurrent. Injuries, infections, and cellular damage provoke differentiation of resident mesenchymal cells into activated or pathological fibroblasts that drive inflammation, deposit new extracellular matrix, and withdraw their support from endothelial cells. When pathological fibroblasts additionally express the contractile protein α-smooth muscle actin (α-SMA) they are known as myofibroblasts. Persistent myofibroblast activation can cumulate in fibrosis with progressive scarring, loss of lung function, morbidity, and mortality.
Characteristics of Lung Pericytes and Resident Fibroblasts
Pericytes are mesenchymal cells closely related to vascular smooth muscle cells (VSMCs) that underlie and envelop capillaries, forming focal contacts with adjacent endothelial cells. Pericytes may be strictly defined anatomically by the presence of processes within capillary basement membrane (Figure 1). They may also be distinguished by molecular criteria, including the lack of leukocyte, endothelial, and parenchymal hallmarks, and the presence of markers, including platelet-derived growth factor (PDGF) receptors, and, often, proteoglycan neural/glial antigen 2 (NG2; alias chondroitin sulfate proteoglycan-4).2, 3, 4, 5 Pericytes embed themselves within the capillary basement membrane and may extend peg-socket contacts with the endothelium ending in adherence, gap, and tight junctions between each pericyte and one or more endothelial cells.2, 3, 4, 5, 6 The extent of pericyte vascular coverage varies by organ and anatomy and is relatively high in the lung, correlating with a stronger barrier and lower turnover of endothelial cells.3
Figure 1.

Tissue architecture and mesenchymal lineages in the lung. A: Electron microscopy photomicrographs of normal mouse lung showing fibroblast processes abutting epithelium and pericyte processes attached to endothelium and within a loose capillary basement membrane. Fluorescence images of inflated lung (B) and scheme (C) showing FoxD1 lineage pericytes (red) and cells producing collagen (Col) 1 (green). Note extensive mesenchymal cell populations in normal lung. In disease, both pericyte-derived cells and Col1+, platelet-derived growth factor receptor-α+ resident fibroblast–derived cells contribute to Col1 production in the foci of fibrosis. Scale bars: 1 μm (A); 50 μm (B and C). a, alveolus; c, capillary; cbm, capillary basement membrane; ec, endothelial cell; epi, epithelial cell; f, fibroblast; p, pericyte; rbc, red blood corpuscle.
The lung also contains a second resident mesenchymal population with similar morphology and shared expression of key markers. These resident fibroblasts descend from different precursors and position themselves beneath epithelial cells or are scattered through the interstitium between the epithelial and endothelial layers, but without directly contacting the vasculature.7, 8, 9, 10, 11 Together, pericytes and other fibroblasts constitute 10% to 20% of all lung cells, and both populations differentiate into matrix-generating activated myofibroblasts.9, 10 However, they are also heterogeneous and plastic populations, which has complicated their study and contributed to contrasting interpretations of their role in fibrotic lung diseases.2, 3, 6, 9, 10, 12, 13, 14, 15, 16 Such broad adaptability is proposed to enable pericytes and resident fibroblasts to repeatedly adjust to support growing, injured, or regenerating vascular or airway structures.2, 6, 13
Extensive investigation into the origins of pathogenic myofibroblasts identified three other candidates, hematopoietic fibrocytes and reprogrammed epithelial or endothelial cells.2, 13 Although these types of cells can assume a myofibroblast identity, key lineage-tracing experiments demonstrate that the pericytes and resident fibroblasts are the predominant source of myofibroblasts in the lung during development, in its mature healthy state, and after injury.2, 9, 10, 13
Pericytes and resident lung fibroblasts can be identified independently of their location, with appropriate criteria for excluding other cell types, by their expression of markers, including PDGF receptor (PDGFR) β and α, CD146, NG2, α-SMA, collagen I(α)1 (Col1), vimentin, and desmin (Table 1).2, 3, 9, 10, 17
Table 1.
Expression Patterns of Markers for Pericytes and Resident Fibroblasts in Normal Adult Lung Tissue (Mainly from Rodent Studies)
| Marker | Pericytes | Resident fibroblasts | Other cell types | References |
|---|---|---|---|---|
| PDGFR-β | + | − | Vascular smooth muscle | 2, 3, 5, 9, 10, 18, 19, 20 |
| PDGFR-α | + | + | Vascular, bronchiolar smooth muscle | 9, 10, 11, 21, 22 |
| NG2 | +/− | − | Macrophages | 2, 3, 5, 9, 10, 17, 20, 23 |
| α-SMA | +/− | − | Vascular, bronchiolar smooth muscle | 5, 9, 10, 19, 22, 24 |
| Col1 | −/+ | + | Fibrocytes | 3, 10, 24, 25, 26, 27, 28, 29, 30 |
| S100a4 | − | − | Leukocytes | 9, 31, 32 |
| Vimentin | + | + | Macrophages | 3, 9, 33 |
| Desmin | − | + | Vascular, bronchiolar smooth muscle | 3, 9, 34 |
Col, collagen; NG, neural/glial antigen; PDGFR, platelet-derived growth factor receptor; α-SMA, α-smooth muscle actin; +, present; –, absent.
However, the fidelity of these markers varies by context. They differ between species and organs; can be present on other cell types; are altered by stage of organ development, activation signals, or injury; and can undergo either rapid or gradual change during cell culture.2, 3, 9, 10 The most durable marker of pericytes in healthy lung is PDGFR-β.
Tracing the Origins and Subsets of Lung Pericytes and Resident Fibroblasts
Fate mapping studies in transgenic mice have made crucial advances toward identifying the ontogenies and identities of pericytes, resident fibroblasts, and disease-associated fibroblasts and myofibroblasts (Figure 2 and Table 2).
Figure 2.

Model of mesenchymal lineages in development of the lung. Solid lines show the relationships between cell types identified by lineage tracing studies in the embryonic, healthy adult, and injured lung (Table 2). Dashed lines indicate additional hypotheses for the origins of α-smooth muscle actin (α-SMA)− pathogenic fibroblasts in injured lung. The earliest embryonic expression of genes used for tracing are depicted on the left. Note that the timing of inducible tracing also determines which populations will be marked. Col, collagen; E, embryonic day; NG, neural/glial antigen; PDGFR, platelet-derived growth factor receptor.
Table 2.
Phenotypes of Lineage-Traced Lung Pericytes, Resident Fibroblasts, and Precursors in Rodent Models
| Lineage | Locations |
Phenotypic markers |
Negative markers | References | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Perivascular | Interstitial | Subepithelial | PDGFR-β | PDGFR-α | NG2 | α-SMA | Col1 | |||
| Embryonic | ||||||||||
| Tbx4enh | + | + | − | + | − | CD31, cytokeratin | 8, 35 | |||
| − | − | + | − | CD31, cytokeratin | ||||||
| Healthy adult | ||||||||||
| Gli1 | + | − | + | + | + | − | − | CD31, CD45 | 20 | |
| Tbx4enh | + | − | + | ± | ± | CD31, T1α | 8 | |||
| PDGFR-α | + | − | + | + | + | − | ± | CD31 | 9 | |
| − | + | + | − | + | − | − | CD31 | |||
| FoxD1 (negative) | − | + | + | − | + | − | − | + | CD31, CD34, CD45, EpCAM, aquaporin V | |
| FoxD1 | + | − | − | + | + | + | − | + | CD31, CD34, CD45, EpCAM, aquaporin V | 10 |
| + | − | − | + | − | + | − | − | CD31, CD34, CD45, EpCAM, aquaporin V | ||
| FoxJ1 | + | − | − | + | − | 9 | ||||
| NG2 | + | − | − | + | + | − | CD31 | |||
| ABCG2 | + | − | + | + | − | − | − | CD31, CD45, CD133, CD144, CD146 | 17, 36 | |
| Bleomycin-injured adult | ||||||||||
| Gli1 | Fibrotic foci | + | + | − | + | CD31, CD45 | 20 | |||
| FoxD1+ | + | + | 10 | |||||||
| FoxJ1 | + | − | 9 | |||||||
| NG2 | + | + | − | CD31 | ||||||
| ABCG2 | + | − | − | + | CD31, CD45, CD133, CD144, CD146 | 17, 36 | ||||
Col, collagen; NG, neural/glial antigen; PDGFR, platelet-derived growth factor receptor; α-SMA, α-smooth muscle actin; +, present; –, absent.
Mesenchymal Progenitors Traced from Development
Gli1+ Wnt2+ Isl1+ progenitors from the developing heart produce the lung's mesodermal lineages, including its developing vascular and airway muscle, proximal vessel endothelium, pericytes, and fibroblasts.37 In the mature lung, as well as kidney, liver, and heart, Gli1 expression is sustained within a small mesenchymal stem cell (MSC) subset of PDGFR-β+ perivascular and subepithelial cells that could differentiate into α-SMA+ myofibroblasts after lung injury.20 Lineage tracing showed that these Gli1+ MSCs do not express NG2 in healthy tissue nor do fibroblasts derived from them in the injured lung, yet >60% of pericytes in healthy lung express NG2.10, 20
Separate studies identified mesenchymal progenitors expressing the transcription factor FoxD1 transiently in posterior lung buds during early development. Lineage tracing of these FoxD1 progenitors, combined with a Col1 reporter system, identified four distinct mesenchymal populations in the healthy adult lung (Figure 2 and Table 2): FoxD1 progenitor-derived Col1−, FoxD1 progenitor-derived Col1+, FoxD1-independent Col1+, and VSMC (referred to as FoxD1+/Col1−, FoxD1+/Col1+, FoxD1−/Col1+, and VSMC).10, 38 These mesenchymal cell types accounted for approximately 20% of all lung cells. FoxD1 expression was only detected between days 11 and 13 of embryogenesis in the lung buds, and the descendants of the transiently FoxD1-expressing population are crucial for the developing lung to mature. Targeted deletion of the enzyme Dicer1 in FoxD1+ cells, which disrupts normal cell function, was neonatally lethal because of severe alveolar branching and septation defects.39
A subset of FoxD1+ progenitors differentiates into α-SMA+ VSMCs sheathing arterioles, reflecting a common mesenchymal origin and late divergence in ontogeny with pericytes.2, 3, 18 VSMCs excluded, approximately 80% of the FoxD1-traced cells did not express Col1 or α-SMA but fit the definition of pericytes: stellate shaped with long cell processes, attached to endothelial cells, separated from the airways, exclusively PDGFR-β+, 60% NG2+, and composing 5% to 15% of lung's cells. The similarly abundant FoxD1−/Col1+ cells were not pericytes in adults. Instead, they frequently underlay the epithelium, their cell bodies abutting type II airway epithelial cells. They lacked α-SMA, PDGFR-β, and NG2, but expressed PDGFR-α, and are likely the equivalent of lipofibroblasts described below.21, 40 The smallest, FoxD1+/Col1+, population made up 2% to 5% of the lung and were also α-SMA− perivascular cells that co-expressed PDGFR-α and PDGFR-β and expressed NG2+. On the basis of this study, normal mature lung contains two types of pericytes with close ancestry, distinguished in adults by collagen I and PDGFR-α/β, and derived from FoxD1 progenitors. It also contains a more distantly related resident fibroblast population that exclusively expresses PDGFR-α (Figure 2 and Table 2).10
Observations based on FoxD1 lineage connect well with an inducible progenitor tracing system using a lung-specific enhancer of Tbx4, a T-box transcription factor expressed at all stages of lung development by various mesenchymal cell types (Table 2).8, 35 Among the populations traced by Tbx4, by either labeling throughout embryogenesis or only after embryonic day 15.5, were NG2+ α-SMA− pericytes and NG2− α-SMA+ perialveolar adipophilin-stained lipofibroblasts. Thus, embryonic Tbx4 enhancer activity and adult Col1 transcription appear to match in spatially separated mesenchymal populations with the features of pericytes and resident fibroblasts.8, 10, 21 The pattern of PDGFR-α expression also matches this connection. A large population of PDGFR-α+, α-SMA+ myofibroblasts position themselves adjacent to PDGF-A+ epithelial cells and play a crucial role in generating the septae between alveoli before embryonic day 18.5.11 As this process is completed, the epithelium ceases to produce PDGF-A, and PDGFR-α expression shifts to cells scattered between the epithelium and endothelium or in blood vessel walls. Moreover, in adult lung PDGFR-α, lineage marking included lipid-bearing lipofibroblasts able to support the growth and differentiation of type II airway epithelial cells.21, 40 Thus, both Tbx4− and PDGFR-α tracing correspond with the identification of PDGFR-α+ Col1+ interstitial fibroblasts and pericytes in adult lung.10
Mesenchymal Cells Traced in Adult Lung
An earlier study of adult lung cell lineages reached notably similar conclusions (Table 2).9 A PDGFR-β+ NG2+ α-SMA− perivascular population arose from NG2-traced cells. FoxJ1-expressing progenitors also labeled NG2+ α-SMA− pericytes. But, PDGFR-α tracing found three groups of descendants. Like the FoxD1−/Col1+ subset, one of these PDGFR-α+ populations did not express PDGFR-β, NG2, or α-SMA and was likewise widely distributed through the lung interstitium, with some cells lying beneath the epithelium but not endothelium. The other two PDGFR-α–traced populations co-expressed PDGFR-β, stained variably for α-SMA but not NG2, and were found in either perivascular or peribronchiolar locations.
Mesenchymal Stem Cells
A population of resident MSCs in contact with both endothelium and epithelium also resides in adult human and mouse lung and has been distinguished from pericytes and resident fibroblasts by its expression of the ABCG2 transporter.3, 5, 17, 20, 36, 41 ABCG2-expressing, or lineage-marked, NG2− MSCs express mostly the same genes at similar levels as NG2+ cells, but lower levels of extracellular matrix components and their interacting receptors than the remaining ABCG2− NG2− lung mesenchymal fibroblasts (Table 2). In vitro ABCG2+ cells can acquire the phenotype of pericytes, myofibroblasts, or endothelial cells. But, in healthy adult lung, ABCG2-traced descendants were barely detectable in any of these populations within 3 weeks, suggesting that once differentiated, these populations either replenish only themselves or turn over slowly in normal tissue.36 Nonetheless, ABCG2-marked cells either directly or indirectly differentiated into myofibroblasts at fibrotic foci after lung injury.17 Whether these MSCs are the same as Gli1+ MSCs remains to be established.
Mesothelial Cells
Finally, after covering the developing mesenchymal surface, several studies have shown that mesothelial cells potentially produce multiple lung mesenchymal lineages, including fibroblasts. Both Tbx4− and Wt1-based experimental systems traced the progeny of mesothelial cells to smooth muscle and endothelial populations, as well as fibroblasts identified by desmin and/or the absence of other lineage markers.34, 35, 42 These results also indicate that mesothelium is not the main source of pericytes or fibroblasts in the adult lung, but may instead contribute to rare fibroblast subgroups whose distinguishing features and ancestry remain ambiguous.
Together, these largely concordant findings demonstrate both the close relationships between pericytes and resident fibroblasts and the heterogeneity among these populations (Figure 2 and Table 2). Col1 expression in healthy lung tissue insightfully divided their phenotypes (Figure 3).10 The predominant features of Col1+ cells, both inherently and by comparison to FoxD1+/Col1− pericytes, were pathways associated with extracellular matrix synthesis and remodeling, adhesion, and lung development (Figure 3). These pathways represent the most widely acknowledged functions of fibroblasts, yet the Col1− express higher levels of immune sensing and response genes as well as a range of chemokines, suggesting they are poised to recognize and rapidly respond to injury or infection and may also enable surveillance of the lung by migrating leukocytes. A similar study dividing cells by ABCG2 and NG2 expression instead associated immune functions with NG2− fibroblasts but presumably combined Col1− and Col1+ pericytes as a single, NG2+ group.36
Figure 3.
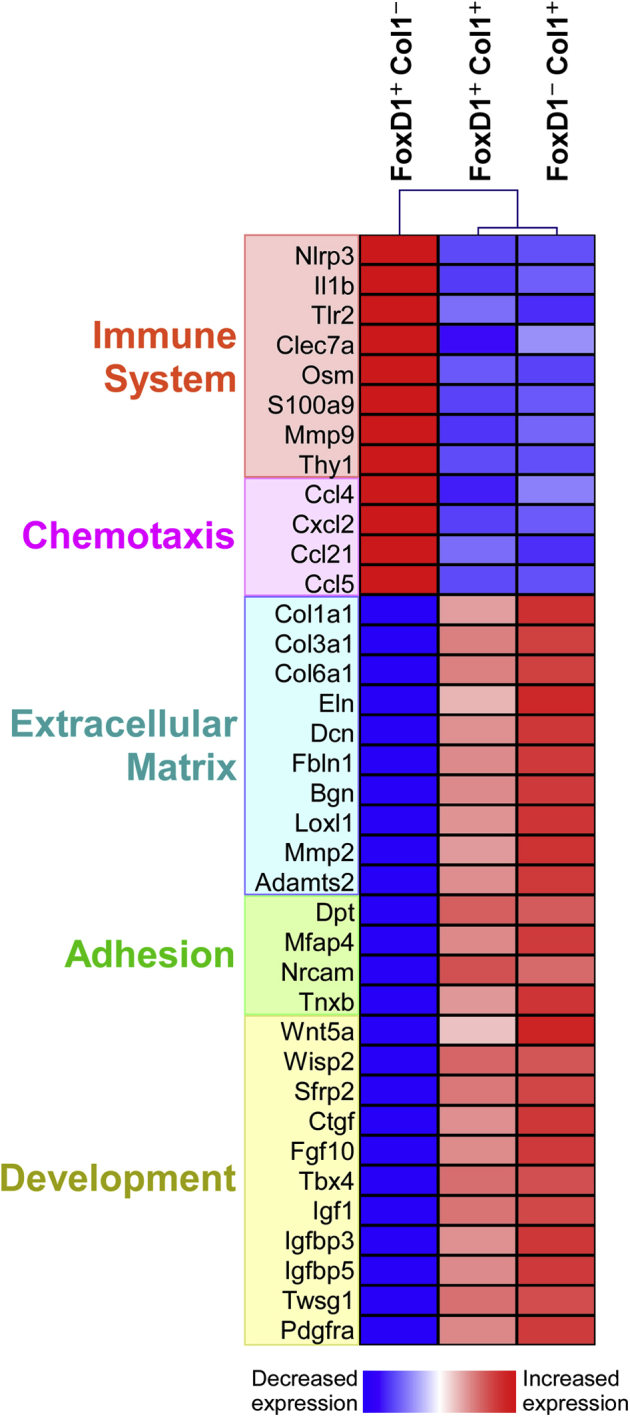
Collagen (Col) 1 expression distinguishes between immune and matrix-oriented lung fibroblasts. Hung et al10 characterized the most enriched Kyoto Encyclopaedia of Genes and Genomes gene ontology pathways (www.genome.jp/kegg, last accessed June 2016) in the three FoxD1/Col1 fibroblast subsets in healthy lung. Immune and chemotaxis pathways are more enriched in FoxD1+/Col1− cells, whereas matrix, adhesion, and developmental pathways are more strongly engaged in Col1+ cells. Top differentially expressed genes playing important roles in inflammation, tissue remodeling, and lung genesis, or degeneration were selected from the complete data set10 for discussion.
Pericytes and Endothelial Cells Maintain the Vascular System
Pericytes and endothelial cells regulate one another during embryonic development, adult homeostasis, and in response to injury. The PDGF, angiopoietin, transforming growth factor (TGF)-β, notch, epidermal growth factor, hedgehog, ephrin, S1P, and stromal derived factor-1 pathways all engage pericytes with endothelial cells and pattern the vasculature during development by controlling their numbers (proliferation), differentiation, position, and stabilization.1, 2, 3, 5, 18, 19, 43, 44, 45, 46, 47 Pericytes and endothelial cells cooperatively deposit and assemble vascular basement membrane, and pericyte detachment from the endothelium and differentiation into myofibroblasts contributes to loss of vascular homeostasis.2, 3, 5, 10, 18, 19, 43, 44, 46, 48 For example, pericytes have been reported to down-regulate tissue inhibitor of metalloproteinases-3 and up-regulate the ADAM metalloproteinase with thombospondin motifs-1 (ADAMTS1) when differentiating into myofibroblasts, tissue inhibitor of metalloproteinases-3 inhibits proteolysis by ADAMTS1, and ADAMTS1 destabilizes capillary networks in vitro.45 But, compared to the well-established general models for pericyte-endothelial coordination during embryogenesis, far less is understood of how lung pericytes act on the vasculature during homeostasis, degeneration, or regeneration.5, 6, 18
The receptor, PDGFR-β, marks pericytes and establishes their interactions with endothelial cells. Endothelial cells produce its ligand PDGF-B when sprouting, to which the pericytes respond by proliferating and migrating to attach to and cover the growing vessel. The primary consequence of PDGF pathway defects is vascular dysfunction, with endothelial hyperplasia, poor vessel integrity, microcapillary aneurysms, and loss of pericyte coverage in the lung and other organs.2, 3, 5, 19, 44 Furthermore, the endothelial-pericyte dependence on PDGF-B to prevent vascular leakage increases during sustained lung inflammation and is required for angiopoietin-1 (Ang-1) to help stabilize the endothelium.49
Paracrine signaling by Ang-1 from pericytes through its receptor Tie-2 on endothelial cells generates a reciprocal interaction to the PDGF pathway. Ang-1 matures and stabilizes the endothelium, increasing its integrity, and also enables new blood vessels to sprout and grow without leaking.3, 5, 18, 46 Unlike the PDGF pathway, disconnecting Ang-1 from Tie-2 does not interfere with pericyte positioning or reduce their vascular coverage, although it does alter the number and proportions of vessels during embryonic development.3, 5, 43 Instead, targeted Ang-1 deficiency exacerbated fibrosis in response to physical damage or vascular stress, implying that the Ang-1/Tie2 connection between pericytes and endothelial cells regulates their coordinated response to injury.3, 43
TGF-β plays complicated and interdependent important roles for pericytes, endothelial cells, and their precursors beginning early in development.3, 5, 18, 50, 51, 52 Both partners produce TGF-β and its receptors, and TGF-β pathway deficiencies cause vascular defects, including loss of pericyte coverage. However, the TGF-β pathway is so broadly engaged in development and pathogenesis in all organs, including its potential to stimulate opposing effects in angiogenesis, that without data from inducible, cell type–specific inhibition experiments, it remains problematic to assign discrete, TGF-β–dependent roles to lung pericytes in vascular biology.3, 5, 18, 53
Pericytes sense vasoactive molecules and neurotransmitters, and one of their first proposed functions was to control blood flow by regulating the diameter of capillary vessels.3 Ang-1 overexpression did increase capillary diameter, although whether periyctes could exert sufficient mechanical force to squeeze or stretch a capillary structure has been debated.3, 5, 46 In the brain and kidney, pericytes directly regulate blood flow in vivo and cause vasoconstriction after ischemic brain injury.54, 55 Moreover, in sophisticated in vitro microfluidics experiments, primary human lung pericytes also caused vasoconstriction.56 It is therefore likely that pericytes directly constrict or relax a subset of blood vessels as well as more broadly coregulating the lung vascular system.
Pericytes Act as the Immune System's Sentries
Leukocytes maintain immune surveillance of the lung's enormous mucosal surface by migrating through the tissue.57, 58 However, the lung's microvascular architecture, including capillaries narrower than rounded leukocytes, fluid dynamics, and expression pattern of adhesion molecules do not conform to the classic tether-roll-adhere-transmigrate paradigm of migration across the endothelium.59 Instead, neutrophils mobilized into the lung at sites of dynamic vascular leakage and migrated in a pattern suggesting rapid chemotaxis up highly localized and short-lived chemokine gradients.60, 61 Pericytes can be crucial for recruiting neutrophils into nonlymphoid organs. After passing between endothelial cells, neutrophils were observed adhering to and crawling along pericytes, then migrating through the gaps between pericytes into the underlying tissue.62 Furthermore, locally administering either tumor necrosis factor-α or IL-1β caused pericytes to change shape, transiently increasing the spacing between themselves, and with coincident timing, for neutrophils to migrate across the pericyte vascular sheath. Another investigation found that factors from NG2+ pericytes attracted monocytes as well as neutrophils, and also altered the subsequent migration and responsiveness of these passing inflammatory leukocytes after they moved into the interstitial tissue.23
Expression profiling revealed that the chemotaxis pathway was more active in FoxD1+/Col1− pericytes than in other lung fibroblasts, including higher levels of Ccl4 (macrophage inflammatory protein-1β), Cxcl2 (macrophage inflammatory protein-2), Ccl21 (secondary lymphoid-tissue chemokine), and Ccl5 (regulated on activation normal T cell expressed and secreted), which combined could attract neutrophils, monocytes, macrophages, dendritic cells, NK cells, and other classes of innate lymphocytes, and T cells (Figure 3).10, 63 The Col1− pericyte subset also preferentially expressed a variety of immune sensing and response genes. Activation of the NLRP3 inflammasome to produce IL-1β is one of the strongest defenses against infections, but can also severely damage tissue and plays a highly pathogenic role in lung fibrosis.53, 64 Toll-like receptor-2 and Clec7a (Dectin-1) detect microbial molecular signatures, but also may react to host-derived ligands generated by cellular damage.65 Oncostatin-M (OSM), an IL-6 family cytokine, has been strongly implicated in allergic and fibrotic lung diseases and stimulates endothelial, smooth muscle, fibroblast, leukocyte, and epithelial cells from the lung in synergy with other inflammatory cytokines.66 S100A9 is a pleiotropic intracellular and extracellular factor for immune activation, playing roles in signal transduction, oxidation, cytoskeletal and extracellular matrix structure, and as an alarmin.67
Pericytes expressing immune surveillance and effector genes may therefore function in roles traditionally envisioned for macrophages.6, 53, 58, 64, 68 Indeed, most of the immunological genes and functions identified in pericytes were long since recognized and still most commonly studied in monocytes and macrophages.10, 23, 53, 58, 62 Distinctions between resident tissue macrophages and macrophages differentiating from immigrating monocytes have recently become far better appreciated. Resident tissue macrophages have unexpectedly but commonly been found to react with low sensitivity to signals from microbes or damage, and to respond without producing high levels of proinflammatory and profibrotic cytokines.53, 57, 58, 68, 69 One potential explanation is that because alveolar macrophages and similar resident cells are highly exposed to and must cope with foreign stimuli they are tuned to low reactivity, and therefore cannot be deployed as sentries. By contrast, positioning cells with immune sensory and response capabilities beneath endothelial and epithelial layers might better enable a sensitive reaction to distress, invasion, or breach of these barriers.
Unfortunately, no means of selectively identifying, tracing, or targeting the subepithelial population of lung fibroblasts independently of their positioning has yet been discovered to test this possibility (Table 2). Moreover, the failed PANTHER idiopathic pulmonary fibrosis (IPF) trial testing immunosuppression by prednisone, azathioprine, and N-acetylcysteine was designed to inhibit adaptive immunity; these drugs do not effectively block innate immune pathways.70 The growing understanding of how pericyte-endothelial interactions control the integrity and reactivity of the vasculature prompts the important question of whether epithelial cells and subepithelial fibroblasts likewise cooperatively control the airway barrier, initiate immune responses, and guide inflammatory leukocytes.
Homeostatic Lung Remodeling by Interstitial and Perivascular Fibroblasts
Although extracellular matrix structures can be extremely stable in vivo, even in normal adult lung, homeostatic turnover of collagens is estimated to proceed at approximately 5% of the total tissue content each week.27 Extensive cross-linking reduces susceptibility to digestion, so it is believed that segregated pools of collagens are replaced at different rates with stability proportional to the extent collagens become deeply embedded within the lung extracellular matrix.27, 47 The low frequency of pericytes producing collagen suggests that there is little replacement of the extracellular matrix supporting small vascular structures in the normal mature lung.9, 10, 47 Because approximately 80% of the collagen-producing cells are separated from the vasculature, either positioned in the lung interstium or underlying the epithelium,10 most collagens that are homeostatically broken down and replaced may be anatomically as well as biochemically compartmentalized to the vicinity of these cells.
The Col1+ homeostatic lung populations selectively transcribed a diverse suite of extracellular matrix component genes, including collagens type I, III, and VI, elastin, decorin, and biglycan (Figure 3). They were also a major source of factors that cross-link or degrade matrix proteins for remodeling, including Lxl1 and most differentially expressed metallopeptidases. One exception, matrix metalloproteinase-9 (Mmp9), is predominantly expressed by FoxD1+/Col1− pericytes, but it is functionally associated with immune pathways and activated macrophages.53, 64 Genes involved in adhesion, particularly between cells and matrix, are likewise overrepresented in Col1+ cells. FoxD1+/Col1− pericytes transcribe type IV, V, and VIII collagens but at 5- to 20-fold lower levels and likewise express only approximately 5% of the genes in the Kyoto Encyclopaedia of Genes and Genomes (KEGG) extracellular membrane pathway at >50% the levels detected in FoxD1−/Col1+ counterparts (data not shown).
Expression profiling suggests that many genes important in lung development are also functioning homeostatically in Col1+ cells (Figure 2). An active Wingless/INT-1 (WNT) pathway is a prominent feature of both FoxD1-derived and FoxD1-independent Col1+ cells. Lung morphogenesis depends on both canonical and noncanonical WNT signaling, including Wnt5a, which, in turn, regulates fibroblast growth factor-10 (Fgf10) and adjusts the proliferation, patterning, and interactions between epithelial and mesenchymal cells.1, 71, 72, 73, 74, 75 Col1+ cells express higher levels of T-box-4 (Tbx4) as well, which is engaged in the early stages of lung formation by transcribing Fgf10.76 Insulin-like growth factor-1 (Igf1), a crucial stimulus for lung development, is likewise still relatively highly expressed by Col1+ cells. Moreover, IGF binding proteins and connective tissue growth factor are important modulators of key pathways for lung development, including WNT, PDGF, and TGF-β.12, 30, 77 Thus, the gene expression profiles imply that in the mature organ these Col1+ populations are still operating much of the developmental program used to generate the lung, most plausibly in maintenance and repair.
Myofibroblasts in Lung Fibrosis and Degeneration
Infection, injury, cellular distress, and inflammation elicit a wound healing process that ideally restores the affected tissue to its functional state and then ceases. Persistent or repeated insults drive excessive production of growth and angiogenic factors, and inflammatory and fibrogenic cytokines. Such dysregulated and nonresolving wound healing responses cause fibrosis, the pathological loss of organ function as scarring replaces normal tissue.15, 53, 64 Fibrotic lung diseases are a growing and serious cause of morbidity and mortality with scant therapeutic options.13, 78, 79
Pollutants, such as tobacco smoke or asbestos, autoimmune diseases, and inflammatory disorders, directly cause lung fibrosis. When injured, lung epithelial cells release a diverse and potent assortment of factors that can activate fibroblasts, including TGF-β, connective tissue growth factor, prostaglandin E2 (PGE2), sonic hedgehog (SHH), WNT1 inducible signaling pathway protein 1 (WISP1), and IL-1α.15, 80 For idiopathic lung fibrosis, association studies identified the genes for surfactant proteins C and A2, the lipid transporter ABCA3, telomerase components, and immune response genes.81 The functions of these genes and the effects of their risk-associated polymorphisms have drawn attention to the endoplasmic reticulum stress response, resilience, survival, and replenishment of lung epithelial cells.15, 75 Moreover, these connections between epithelial cell death and lung fibrosis help justify the continued and widespread use of lung epithelial injury by bleomcyin to cause fibrosis, despite this model's acknowledged flaws.82 A strengthening hypothesis to generally explain fibrotic lung diseases is that injury or infection overcomes an already stressed epithelium's capacity for recuperation and engages innate immune pathways that may circumstantially promote or protect from harm.13, 15, 53, 64, 75, 78, 81
Myofibroblasts are the key effector cell type in fibrotic disease because they are dominant builders and remodelers of extracellular matrix that also exert strong force on their surroundings.9, 10, 12, 13, 14, 27, 30, 38, 53, 64, 78, 83 Myofibroblasts may be distinguished by expressing α-SMA and similar proteins in common with muscle, forming stress fibers, and by contractility.14 Because these cells are rare in healthy organs, abundant in actively scarring lesions, and the primary therapeutic target for fibrotic diseases, the origin of myofibroblasts has been intensely studied.
Cultured cells with these features can be generated from pericytes, interstitial α-SMA− fibroblasts, epithelial cells, endothelial cells, or hematopoietic fibrocytes.13, 14, 53 The interpretations of whether each of these cell types become a significant part of the myofibroblast populations in animal models or human fibrotic lung disease have conflicted, due in large part to markers open to multiple interpretations and differences between the fidelities of the lineage tracing methods.9, 10, 25, 29, 32, 84, 85, 86, 87, 88 In two independent experiments, a substantial population of myofibroblasts reported transgenic expression from an incomplete section of the human surfactant protein C promoter after lung injury.32, 86 Because transcription of endogenous surfactant protein C is specific for type II airway epithelial cells, these results support the epithelial-to-mesenchymal transition hypothesis that pathogenic myofibroblasts originate from dedifferentiated epithelial cells.79
However, a different reporter system inserted into the endogenous mouse Sftpc locus found epithelial cells did not become myofibroblasts in the bleomycin lung injury model.9 In the same study, lineage tracing by either NG2 or FoxJ1 showed that activated lung pericytes proliferated in fibrotic regions, although these cells did not have highly up-regulated α-SMA. FoxD1 lineage tracing found a stronger connection between pericytes and myofibroblasts, with at least half the α-SMA+ and Col1+ cells derived from FoxD1+ precursors, and FoxD1-independent PDGFR-α+ interstitial fibroblasts proposed to account for the others.10 Perivascular Gli1+ or ABCG2+ MSCs also differentiate into myofibroblasts, although they might transition to become pericytes first.17, 20, 41 Caveats apply to these data. Lineage tracing systems are limited by specificity, efficiency, and sensitivity. The source of myofibroblasts may proportionately vary between etiologies of fibrotic diseases (eg, by epithelial versus vascular injury or dependence on inflammation and the involvement of leukocytes). Nevertheless, these findings best support the conclusion that the lung myofibroblast population originates from pericytes, interstitial fibroblasts, and MSCs, but without a major contribution from epithelial, endothelial, or hematopoietic cells.13, 53
Most potential therapeutic agents, including the first two approved to treat IPF, have targets important to myofibroblasts.64, 89, 90 The most direct approach for drug development has been to inhibit key growth and developmental factors or their receptors, later joined by strategies focused on extracellular matrix structure, stress, and metabolism. Immunosuppression might be considered an indirect approach and potential risk, especially in diseases where leukocytes do not seem a primary concern.13, 70 However, because evidence is building to support the importance of active immunological pathways in epithelial cells and fibroblasts, leads developed for autoimmune diseases, inflammatory pathologies, and the stromal aspects of immuno-oncology may also affect myofibroblasts.53, 89
Pericytes and Interstitial Fibroblasts in Lung Regeneration
One proposed etiology for IPF is that lung developmental programs aberrantly restart.79 But, the pathogenesis of fibrotic and degenerative lung diseases may also be considered developmental programs that fail to stop within the limits of homeostasis.53, 64, 75 The programing to regenerate lung tissue clearly can operate as demonstrated (eg, by the similar pathways and epithelial-fibroblast communication activated to septate the alveoli during development and reactivated to recreate alveoli after pneumectomy).1, 22, 75 The scientific and therapeutic challenges are to stimulate or limit these processes for beneficial changes.
Pericytes include a subpopulation of MSCs that may aid reconstructing damaged lung tissue.3, 17, 20, 36, 41 In support of this prospect, bleomycin lung injury reduces the number of ABCG2+ MSCs, transferring exogenous MSCs ameliorates fibrosis, disabling the antioxidative functions of MSCs increases vulnerability to hypoxic stress, and fewer ABCG2+ cells were detected in IPF than control samples from human donors.17, 36, 41 However, MSCs also generate myofibroblasts and, although not tested in the lung, depletion of Gli1+ MSCs reduces kidney and heart fibrosis.20 Therefore, delivering or expanding MSCs may not be beneficial, or even detrimental, unless the signals stimulating their differentiation into myofibroblasts are first inhibited.79
The intimate relationship between pericytes and endothelial cells controls both vessel integrity and angiogenesis, enabling successful vascular regeneration. Similar interactions potentially governing airway-oriented maintenance and regeneration have likewise been discovered between lung epithelial cells and their underlying fibroblasts. For example, soluble factors from lipid-containing PDGFR-α+ lung fibroblasts permit type II airway epithelial cells to proliferate, differentiate, and form alveolospheres in co-culture.21 TGF-β, connective tissue growth factor, ANGII, SHH, WISP1, and reactive oxygen species all mediate interactions between lung epithelium and fibroblasts, but these have been predominantly investigated as excessively induced drivers of fibrosis.13, 15, 16, 53, 78 However, these factors are also part of beneficial and self-limited wound healing, and epithelial restoration after injury is widely proposed to be the key difference between maintaining or losing lung function.13, 75, 78, 82
PGE2 is a notable exception because it seems deficient in disease. Airway epithelial cells are capable of high PGE2 output; PGE2 suppresses fibroblast migration, proliferation, and myofibroblast differentiation; and PGE2 levels are lower in IPF patients.15, 24, 91, 92 However, fibroblasts from both bleomycin-injured and IPF lung become less responsive to this feedback from epithelial cells because promoter hypermethylation reduces expression of a key PGE2 receptor, EP2.26, 28, 93 In addition, myofibroblasts are a major source of plasminogen activator inhibitor 1; plasminogen activator inhibitor 1 inhibits the conversion of plasminogen to plasmin, and plasmin stimulates PGE2 synthesis by lung epithelial cells.15, 94, 95 Thus, the PGE2-dependent negative feedback loop may serve as an informative example of how regeneration occurs from a self-limited wound healing response, whereas an imbalance in feedback inhibition between epithelial cells and fibroblasts leads, instead, to fibrosis.
Fibroblasts as Therapeutic Targets
Although pericytes and resident fibroblasts perform many important functions in regeneration and homeostasis, the pathological fibroblasts and myofibroblasts that derive from them are strongly implicated in chronic disease progression. Novel therapies to push pathological cells back to their state in health are likely to benefit patients. Nintedanib, a recently approved therapy for pulmonary fibrosis, is a multikinase inhibitor, known to inhibit PDGFR-α/β, fibroblast growth factor receptor, and VEGFR2.64, 90, 96, 97 Pericytes and fibroblasts predominantly express the PDGFRs and may be the primary target for this inhibitor. PDGFR signaling is known to be involved in MSC and pathological fibroblast expansion, as well as matrix accumulation.11, 19, 20, 22, 44 Although ample evidence has accrued to strongly suggest that idiopathic pulmonary fibrosis originates with epithelial dysfunction, the pathological fibroblast from IPF patients has undergone durable reprogramming in the form of epigenetic modifications, as exemplified by the capacity of adoptively transplanted human IPF pathological fibroblasts to recapitulate lung disease in immunodeficient mice.98 It is imperative that future studies focus on the epigenetic changes that could be modified to reduce myofibroblast differentiation and overactivated cell survival pathways.
Recent clinical trials in IPF have attempted to block the fibroblast-specific collagen cross-linking enzyme, LOXL2.64, 90, 97 The trial did not show efficacy (NCT01769196). It is possible that the trial failed because other LOXLs are active, including LOXL1 (Figure 3), leading to redundancy or because the drug failed to penetrate dense and robust collagen matrix.
Studies are underway to block active TGF-β formation at the interface between alveolar epithelium and the pathological fibroblast using antibodies against the integrin αvβ6.64, 90, 97 TGF-β is reported to be a driver of pathological fibroblast persistence, and this approach may be a safer and more tractable way than targeting TGF-β itself to reduce fibroblast activation.
Given the potential role of pericytes in the pathogenesis of acute lung injury, future studies to block innate immune pathways, including Toll-like receptor signaling in pericytes, and enhance ANG1/TIE2 signaling between pericytes and endothelium may prove to be beneficial in the short-term management of lung injury.2, 18, 43, 46, 49
Conclusions and Future Directions
Pericytes and lung fibroblasts are necessary for patterning and cellular differentiation of lung during development. They actively maintain homeostasis in the adult. They contribute to pathological inflammation and tissue remodeling in response to injury or infection, but may also be capable of regenerating its architecture and restoring its function if pathogenic stimuli resolve or are blocked. These populations are heterogeneous and dynamic, but these diverse features, which have tremendously complicated their study, may be the consequence of a high requirement for plasticity to perform their diverse functions.
Advances in the identification and classification of pericytes and lung fibroblasts have begun to resolve long-standing debates over their origins, subsets, and roles in disease. Although all precursors of myofibroblasts are not yet exhaustively accounted for, it is now possible to interrogate the importance of leading candidate genes in lung homeostasis and pathogenesis with much improved specificity and selectivity. Deeper study of tissue recuperation is particularly needed, where questions include how the pericyte, resident fibroblast, and myofibroblast populations are maintained and replenished after injury, and whether and by what means the damaged lung can regain its functions. Once devised, experiments to ascertain the importance of the fibroblasts underlying the lung epithelium to airway barrier function, immunity, and airway inflammation are also of high priority.
The study of lung pericytes is in its infancy currently, but as cells that regulate vascular permeability and sense tissue injury or pathogens, pericytes may be important cellular targets for management of both acute lung injury and chronic lung degeneration. Blockade of injury responses in pericytes may ameliorate vascular leak and lung inflammation in response to injury. The apparent switch from pericyte to fibroblast functions suggests a subpopulation of pathological lung fibroblasts may be returned to their beneficial phenotype. The fact that a subpopulation of pericytes also has true MSC potential supports the notion that pushing MSCs toward a pericyte fate as opposed to differentiation into pathological fibroblasts may actively stimulate regeneration in the adult lung.
Acknowledgments
We thank Dario Lemos and Iván Gomez for helpful discussions.
Footnotes
Lung Ontogeny and Injury Theme Issue
Supported by Biogen and NIH grant DK093493 (J.S.D.).
This article is part of a review series on lung ontogeny and injury.
Disclosures: L.B. and J.S.D. are employees of Biogen and both have Biogen stock. J.S.D. has patents for methods to treat fibrosis and stimulate regeneration.
References
- 1.Morrisey E.E., Hogan B.L. Preparing for the first breath: genetic and cellular mechanisms in lung development. Dev Cell. 2010;18:8–23. doi: 10.1016/j.devcel.2009.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Armulik A., Abramsson A., Betsholtz C. Endothelial/pericyte interactions. Circ Res. 2005;97:512–523. doi: 10.1161/01.RES.0000182903.16652.d7. [DOI] [PubMed] [Google Scholar]
- 3.Armulik A., Genove G., Betsholtz C. Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell. 2011;21:193–215. doi: 10.1016/j.devcel.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 4.Weibel E.R. On pericytes, particularly their existence on lung capillaries. Microvasc Res. 1974;8:218–235. doi: 10.1016/0026-2862(74)90096-x. [DOI] [PubMed] [Google Scholar]
- 5.Kloc M., Kubiak J.Z., Li X.C., Ghobrial R.M. Pericytes, microvasular dysfunction, and chronic rejection. Transplantation. 2015;99:658–667. doi: 10.1097/TP.0000000000000648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rowley J.E., Johnson J.R. Pericytes in chronic lung disease. Int Arch Allergy Immunol. 2014;164:178–188. doi: 10.1159/000365051. [DOI] [PubMed] [Google Scholar]
- 7.Kapanci Y., Ribaux C., Chaponnier C., Gabbiani G. Cytoskeletal features of alveolar myofibroblasts and pericytes in normal human and rat lung. J Histochem Cytochem. 1992;40:1955–1963. doi: 10.1177/40.12.1333502. [DOI] [PubMed] [Google Scholar]
- 8.Zhang W., Menke D.B., Jiang M., Chen H., Warburton D., Turcatel G., Lu C.H., Xu W., Luo Y., Shi W. Spatial-temporal targeting of lung-specific mesenchyme by a Tbx4 enhancer. BMC Biol. 2013;11:111. doi: 10.1186/1741-7007-11-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rock J.R., Barkauskas C.E., Cronce M.J., Xue Y., Harris J.R., Liang J., Noble P.W., Hogan B.L. Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc Natl Acad Sci U S A. 2011;108:E1475–E1483. doi: 10.1073/pnas.1117988108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hung C., Linn G., Chow Y.H., Kobayashi A., Mittelsteadt K., Altemeier W.A., Gharib S.A., Schnapp L.M., Duffield J.S. Role of lung pericytes and resident fibroblasts in the pathogenesis of pulmonary fibrosis. Am J Respir Crit Care Med. 2013;188:820–830. doi: 10.1164/rccm.201212-2297OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bostrom H., Willetts K., Pekny M., Leveen P., Lindahl P., Hedstrand H., Pekna M., Hellstrom M., Gebre-Medhin S., Schalling M., Nilsson M., Kurland S., Tornell J., Heath J.K., Betsholtz C. PDGF-A signaling is a critical event in lung alveolar myofibroblast development and alveogenesis. Cell. 1996;85:863–873. doi: 10.1016/s0092-8674(00)81270-2. [DOI] [PubMed] [Google Scholar]
- 12.Kendall R.T., Feghali-Bostwick C.A. Fibroblasts in fibrosis: novel roles and mediators. Front Pharmacol. 2014;5:123. doi: 10.3389/fphar.2014.00123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Noble P.W., Barkauskas C.E., Jiang D. Pulmonary fibrosis: patterns and perpetrators. J Clin Invest. 2012;122:2756–2762. doi: 10.1172/JCI60323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hinz B., Phan S.H., Thannickal V.J., Prunotto M., Desmouliere A., Varga J., De Wever O., Mareel M., Gabbiani G. Recent developments in myofibroblast biology: paradigms for connective tissue remodeling. Am J Pathol. 2012;180:1340–1355. doi: 10.1016/j.ajpath.2012.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sakai N., Tager A.M. Fibrosis of two: epithelial cell-fibroblast interactions in pulmonary fibrosis. Biochim Biophys Acta. 2013;1832:911–921. doi: 10.1016/j.bbadis.2013.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Borok Z., Whitsett J.A., Bitterman P.B., Thannickal V.J., Kotton D.N., Reynolds S.D., Krasnow M.A., Bianchi D.W., Morrisey E.E., Hogan B.L., Kurie J.M., Walker D.C., Radisky D.C., Nishimura S.L., Violette S.M., Noble P.W., Shapiro S.D., Blaisdell C.J., Chapman H.A., Kiley J., Gail D., Hoshizaki D. Cell plasticity in lung injury and repair: report from an NHLBI workshop, April 19-20, 2010. Proc Am Thorac Soc. 2011;8:215–222. doi: 10.1513/pats.201012-067CB. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marriott S., Baskir R.S., Gaskill C., Menon S., Carrier E.J., Williams J., Talati M., Helm K., Alford C.E., Kropski J.A., Loyd J., Wheeler L., Johnson J., Austin E., Nozik-Grayck E., Meyrick B., West J.D., Klemm D.J., Majka S.M. ABCG2pos lung mesenchymal stem cells are a novel pericyte subpopulation that contributes to fibrotic remodeling. Am J Physiol Cell Physiol. 2014;307:C684–C698. doi: 10.1152/ajpcell.00114.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gaengel K., Genove G., Armulik A., Betsholtz C. Endothelial-mural cell signaling in vascular development and angiogenesis. Arterioscler Thromb Vasc Biol. 2009;29:630–638. doi: 10.1161/ATVBAHA.107.161521. [DOI] [PubMed] [Google Scholar]
- 19.Hellstrom M., Kalen M., Lindahl P., Abramsson A., Betsholtz C. Role of PDGF-B and PDGFR-beta in recruitment of vascular smooth muscle cells and pericytes during embryonic blood vessel formation in the mouse. Development. 1999;126:3047–3055. doi: 10.1242/dev.126.14.3047. [DOI] [PubMed] [Google Scholar]
- 20.Kramann R., Schneider R.K., DiRocco D.P., Machado F., Fleig S., Bondzie P.A., Henderson J.M., Ebert B.L., Humphreys B.D. Perivascular Gli1+ progenitors are key contributors to injury-induced organ fibrosis. Cell Stem Cell. 2015;16:51–66. doi: 10.1016/j.stem.2014.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barkauskas C.E., Cronce M.J., Rackley C.R., Bowie E.J., Keene D.R., Stripp B.R., Randell S.H., Noble P.W., Hogan B.L. Type 2 alveolar cells are stem cells in adult lung. J Clin Invest. 2013;123:3025–3036. doi: 10.1172/JCI68782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen L., Acciani T., Le Cras T., Lutzko C., Perl A.K. Dynamic regulation of platelet-derived growth factor receptor alpha expression in alveolar fibroblasts during realveolarization. Am J Respir Cell Mol Biol. 2012;47:517–527. doi: 10.1165/rcmb.2012-0030OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stark K., Eckart A., Haidari S., Tirniceriu A., Lorenz M., von Bruhl M.L., Gartner F., Khandoga A.G., Legate K.R., Pless R., Hepper I., Lauber K., Walzog B., Massberg S. Capillary and arteriolar pericytes attract innate leukocytes exiting through venules and “instruct” them with pattern-recognition and motility programs. Nat Immunol. 2013;14:41–51. doi: 10.1038/ni.2477. [DOI] [PubMed] [Google Scholar]
- 24.Kolodsick J.E., Peters-Golden M., Larios J., Toews G.B., Thannickal V.J., Moore B.B. Prostaglandin E2 inhibits fibroblast to myofibroblast transition via E. prostanoid receptor 2 signaling and cyclic adenosine monophosphate elevation. Am J Respir Cell Mol Biol. 2003;29:537–544. doi: 10.1165/rcmb.2002-0243OC. [DOI] [PubMed] [Google Scholar]
- 25.Hashimoto N., Jin H., Liu T., Chensue S.W., Phan S.H. Bone marrow-derived progenitor cells in pulmonary fibrosis. J Clin Invest. 2004;113:243–252. doi: 10.1172/JCI18847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang S.K., Wettlaufer S.H., Hogaboam C.M., Flaherty K.R., Martinez F.J., Myers J.L., Colby T.V., Travis W.D., Toews G.B., Peters-Golden M. Variable prostaglandin E2 resistance in fibroblasts from patients with usual interstitial pneumonia. Am J Respir Crit Care Med. 2008;177:66–74. doi: 10.1164/rccm.200706-963OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McKleroy W., Lee T.H., Atabai K. Always cleave up your mess: targeting collagen degradation to treat tissue fibrosis. Am J Physiol Lung Cell Mol Physiol. 2013;304:L709–L721. doi: 10.1152/ajplung.00418.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moore B.B., Ballinger M.N., White E.S., Green M.E., Herrygers A.B., Wilke C.A., Toews G.B., Peters-Golden M. Bleomycin-induced E prostanoid receptor changes alter fibroblast responses to prostaglandin E2. J Immunol. 2005;174:5644–5649. doi: 10.4049/jimmunol.174.9.5644. [DOI] [PubMed] [Google Scholar]
- 29.Phillips R.J., Burdick M.D., Hong K., Lutz M.A., Murray L.A., Xue Y.Y., Belperio J.A., Keane M.P., Strieter R.M. Circulating fibrocytes traffic to the lungs in response to CXCL12 and mediate fibrosis. J Clin Invest. 2004;114:438–446. doi: 10.1172/JCI20997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ponticos M., Holmes A.M., Shi-wen X., Leoni P., Khan K., Rajkumar V.S., Hoyles R.K., Bou-Gharios G., Black C.M., Denton C.P., Abraham D.J., Leask A., Lindahl G.E. Pivotal role of connective tissue growth factor in lung fibrosis: MAPK-dependent transcriptional activation of type I collagen. Arthritis Rheum. 2009;60:2142–2155. doi: 10.1002/art.24620. [DOI] [PubMed] [Google Scholar]
- 31.Lawson W.E., Polosukhin V.V., Zoia O., Stathopoulos G.T., Han W., Plieth D., Loyd J.E., Neilson E.G., Blackwell T.S. Characterization of fibroblast-specific protein 1 in pulmonary fibrosis. Am J Respir Crit Care Med. 2005;171:899–907. doi: 10.1164/rccm.200311-1535OC. [DOI] [PubMed] [Google Scholar]
- 32.Tanjore H., Xu X.C., Polosukhin V.V., Degryse A.L., Li B., Han W., Sherrill T.P., Plieth D., Neilson E.G., Blackwell T.S., Lawson W.E. Contribution of epithelial-derived fibroblasts to bleomycin-induced lung fibrosis. Am J Respir Crit Care Med. 2009;180:657–665. doi: 10.1164/rccm.200903-0322OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mor-Vaknin N., Punturieri A., Sitwala K., Markovitz D.M. Vimentin is secreted by activated macrophages. Nat Cell Biol. 2003;5:59–63. doi: 10.1038/ncb898. [DOI] [PubMed] [Google Scholar]
- 34.Dixit R., Ai X., Fine A. Derivation of lung mesenchymal lineages from the fetal mesothelium requires hedgehog signaling for mesothelial cell entry. Development. 2013;140:4398–4406. doi: 10.1242/dev.098079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kumar M.E., Bogard P.E., Espinoza F.H., Menke D.B., Kingsley D.M., Krasnow M.A. Mesenchymal cells: defining a mesenchymal progenitor niche at single-cell resolution. Science. 2014;346:1258810. doi: 10.1126/science.1258810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chow K., Fessel J.P., Kaoriihida S., Schmidt E.P., Gaskill C., Alvarez D., Graham B., Harrison D.G., Wagner D.H., Jr., Nozik-Grayck E., West J.D., Klemm D.J., Majka S.M. Dysfunctional resident lung mesenchymal stem cells contribute to pulmonary microvascular remodeling. Pulm Circ. 2013;3:31–49. doi: 10.4103/2045-8932.109912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Peng T., Tian Y., Boogerd C.J., Lu M.M., Kadzik R.S., Stewart K.M., Evans S.M., Morrisey E.E. Coordination of heart and lung co-development by a multipotent cardiopulmonary progenitor. Nature. 2013;500:589–592. doi: 10.1038/nature12358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Humphreys B.D., Lin S.L., Kobayashi A., Hudson T.E., Nowlin B.T., Bonventre J.V., Valerius M.T., McMahon A.P., Duffield J.S. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am J Pathol. 2010;176:85–97. doi: 10.2353/ajpath.2010.090517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nakagawa N., Xin C., Roach A.M., Naiman N., Shankland S.J., Ligresti G., Ren S., Szak S., Gomez I.G., Duffield J.S. Dicer1 activity in the stromal compartment regulates nephron differentiation and vascular patterning during mammalian kidney organogenesis. Kidney Int. 2015;87:1125–1140. doi: 10.1038/ki.2014.406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ahlbrecht K., McGowan S.E. In search of the elusive lipofibroblast in human lungs. Am J Physiol Lung Cell Mol Physiol. 2014;307:L605–L608. doi: 10.1152/ajplung.00230.2014. [DOI] [PubMed] [Google Scholar]
- 41.Jun D., Garat C., West J., Thorn N., Chow K., Cleaver T., Sullivan T., Torchia E.C., Childs C., Shade T., Tadjali M., Lara A., Nozik-Grayck E., Malkoski S., Sorrentino B., Meyrick B., Klemm D., Rojas M., Wagner D.H., Jr., Majka S.M. The pathology of bleomycin-induced fibrosis is associated with loss of resident lung mesenchymal stem cells that regulate effector T-cell proliferation. Stem Cells. 2011;29:725–735. doi: 10.1002/stem.604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cano E., Carmona R., Munoz-Chapuli R. Wt1-expressing progenitors contribute to multiple tissues in the developing lung. Am J Physiol Lung Cell Mol Physiol. 2013;305:L322–L332. doi: 10.1152/ajplung.00424.2012. [DOI] [PubMed] [Google Scholar]
- 43.Jeansson M., Gawlik A., Anderson G., Li C., Kerjaschki D., Henkelman M., Quaggin S.E. Angiopoietin-1 is essential in mouse vasculature during development and in response to injury. J Clin Invest. 2011;121:2278–2289. doi: 10.1172/JCI46322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lindahl P., Johansson B.R., Leveen P., Betsholtz C. Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science. 1997;277:242–245. doi: 10.1126/science.277.5323.242. [DOI] [PubMed] [Google Scholar]
- 45.Schrimpf C., Xin C., Campanholle G., Gill S.E., Stallcup W., Lin S.L., Davis G.E., Gharib S.A., Humphreys B.D., Duffield J.S. Pericyte TIMP3 and ADAMTS1 modulate vascular stability after kidney injury. J Am Soc Nephrol. 2012;23:868–883. doi: 10.1681/ASN.2011080851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thurston G., Suri C., Smith K., McClain J., Sato T.N., Yancopoulos G.D., McDonald D.M. Leakage-resistant blood vessels in mice transgenically overexpressing angiopoietin-1. Science. 1999;286:2511–2514. doi: 10.1126/science.286.5449.2511. [DOI] [PubMed] [Google Scholar]
- 47.Townsley M.I. Structure and composition of pulmonary arteries, capillaries, and veins. Compr Physiol. 2012;2:675–709. doi: 10.1002/cphy.c100081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stratman A.N., Davis G.E. Endothelial cell-pericyte interactions stimulate basement membrane matrix assembly: influence on vascular tube remodeling, maturation, and stabilization. Microsc Microanal. 2012;18:68–80. doi: 10.1017/S1431927611012402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fuxe J., Tabruyn S., Colton K., Zaid H., Adams A., Baluk P., Lashnits E., Morisada T., Le T., O'Brien S., Epstein D.M., Koh G.Y., McDonald D.M. Pericyte requirement for anti-leak action of angiopoietin-1 and vascular remodeling in sustained inflammation. Am J Pathol. 2011;178:2897–2909. doi: 10.1016/j.ajpath.2011.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Urness L.D., Sorensen L.K., Li D.Y. Arteriovenous malformations in mice lacking activin receptor-like kinase-1. Nat Genet. 2000;26:328–331. doi: 10.1038/81634. [DOI] [PubMed] [Google Scholar]
- 51.Larsson J., Goumans M.J., Sjostrand L.J., van Rooijen M.A., Ward D., Leveen P., Xu X., ten Dijke P., Mummery C.L., Karlsson S. Abnormal angiogenesis but intact hematopoietic potential in TGF-beta type I receptor-deficient mice. EMBO J. 2001;20:1663–1673. doi: 10.1093/emboj/20.7.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lan Y., Liu B., Yao H., Li F., Weng T., Yang G., Li W., Cheng X., Mao N., Yang X. Essential role of endothelial Smad4 in vascular remodeling and integrity. Mol Cell Biol. 2007;27:7683–7692. doi: 10.1128/MCB.00577-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Duffield J.S., Lupher M., Thannickal V.J., Wynn T.A. Host responses in tissue repair and fibrosis. Annu Rev Pathol. 2013;8:241–276. doi: 10.1146/annurev-pathol-020712-163930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hall C.N., Reynell C., Gesslein B., Hamilton N.B., Mishra A., Sutherland B.A., O'Farrell F.M., Buchan A.M., Lauritzen M., Attwell D. Capillary pericytes regulate cerebral blood flow in health and disease. Nature. 2014;508:55–60. doi: 10.1038/nature13165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lemos D.R., Marsh G., Huang A., Campanholle G., Aburatani T., Dang L., Gomez I.G., Fisher K., Ligresti G., Peti-Peterdi J., Duffield J.S. Maintenance of vascular integrity by pericytes is essential for normal kidney function. Am J Physiol Renal Physiol. 2016 doi: 10.1152/ajprenal.00030.2016. [Epub ahead of print] doi: 10.1152/ajprenal.00030.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bichsel C.A., Hall S.R., Schmid R.A., Guenat O.T., Geiser T. Primary human lung pericytes support and stabilize in vitro perfusable microvessels. Tissue Eng Part A. 2015;21:2166–2176. doi: 10.1089/ten.TEA.2014.0545. [DOI] [PubMed] [Google Scholar]
- 57.Jakubzick C., Gautier E.L., Gibbings S.L., Sojka D.K., Schlitzer A., Johnson T.E., Ivanov S., Duan Q., Bala S., Condon T., van Rooijen N., Grainger J.R., Belkaid Y., Ma'ayan A., Riches D.W., Yokoyama W.M., Ginhoux F., Henson P.M., Randolph G.J. Minimal differentiation of classical monocytes as they survey steady-state tissues and transport antigen to lymph nodes. Immunity. 2013;39:599–610. doi: 10.1016/j.immuni.2013.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rodero M.P., Poupel L., Loyher P.L., Hamon P., Licata F., Pessel C., Hume D.A., Combadiere C., Boissonnas A. Immune surveillance of the lung by migrating tissue monocytes. Elife. 2015;4:e07847. doi: 10.7554/eLife.07847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hickey M.J., Westhorpe C.L. Imaging inflammatory leukocyte recruitment in kidney, lung and liver: challenges to the multi-step paradigm. Immunol Cell Biol. 2013;91:281–289. doi: 10.1038/icb.2012.83. [DOI] [PubMed] [Google Scholar]
- 60.Looney M.R., Thornton E.E., Sen D., Lamm W.J., Glenny R.W., Krummel M.F. Stabilized imaging of immune surveillance in the mouse lung. Nat Methods. 2011;8:91–96. doi: 10.1038/nmeth.1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kreisel D., Nava R.G., Li W., Zinselmeyer B.H., Wang B., Lai J., Pless R., Gelman A.E., Krupnick A.S., Miller M.J. In vivo two-photon imaging reveals monocyte-dependent neutrophil extravasation during pulmonary inflammation. Proc Natl Acad Sci U S A. 2010;107:18073–18078. doi: 10.1073/pnas.1008737107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Proebstl D., Voisin M.B., Woodfin A., Whiteford J., D'Acquisto F., Jones G.E., Rowe D., Nourshargh S. Pericytes support neutrophil subendothelial cell crawling and breaching of venular walls in vivo. J Exp Med. 2012;209:1219–1234. doi: 10.1084/jem.20111622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Griffith J.W., Sokol C.L., Luster A.D. Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu Rev Immunol. 2014;32:659–702. doi: 10.1146/annurev-immunol-032713-120145. [DOI] [PubMed] [Google Scholar]
- 64.Friedman S.L., Sheppard D., Duffield J.S., Violette S. Therapy for fibrotic diseases: nearing the starting line. Sci Transl Med. 2013;5:167sr1. doi: 10.1126/scitranslmed.3004700. [DOI] [PubMed] [Google Scholar]
- 65.Brubaker S.W., Bonham K.S., Zanoni I., Kagan J.C. Innate immune pattern recognition: a cell biological perspective. Annu Rev Immunol. 2015;33:257–290. doi: 10.1146/annurev-immunol-032414-112240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Richards C.D. The enigmatic cytokine oncostatin m and roles in disease. ISRN Inflamm. 2013;2013:512103. doi: 10.1155/2013/512103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Markowitz J., Carson W.E., 3rd Review of S100A9 biology and its role in cancer. Biochim Biophys Acta. 2013;1835:100–109. doi: 10.1016/j.bbcan.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Okabe Y., Medzhitov R. Tissue biology perspective on macrophages. Nat Immunol. 2015;17:9–17. doi: 10.1038/ni.3320. [DOI] [PubMed] [Google Scholar]
- 69.Lin S.L., Castano A.P., Nowlin B.T., Lupher M.L., Jr., Duffield J.S. Bone marrow Ly6Chigh monocytes are selectively recruited to injured kidney and differentiate into functionally distinct populations. J Immunol. 2009;183:6733–6743. doi: 10.4049/jimmunol.0901473. [DOI] [PubMed] [Google Scholar]
- 70.Idiopathic Pulmonary Fibrosis Clinical Research Network. Raghu G., Anstrom K.J., King T.E., Jr., Lasky J.A., Martinez F.J. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med. 2012;366:1968–1977. doi: 10.1056/NEJMoa1113354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pongracz J.E., Stockley R.A. Wnt signalling in lung development and diseases. Respir Res. 2006;7:15. doi: 10.1186/1465-9921-7-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Li C., Xiao J., Hormi K., Borok Z., Minoo P. Wnt5a participates in distal lung morphogenesis. Dev Biol. 2002;248:68–81. doi: 10.1006/dbio.2002.0729. [DOI] [PubMed] [Google Scholar]
- 73.Li C., Hu L., Xiao J., Chen H., Li J.T., Bellusci S., Delanghe S., Minoo P. Wnt5a regulates Shh and Fgf10 signaling during lung development. Dev Biol. 2005;287:86–97. doi: 10.1016/j.ydbio.2005.08.035. [DOI] [PubMed] [Google Scholar]
- 74.Volckaert T., De Langhe S.P. Wnt and FGF mediated epithelial-mesenchymal crosstalk during lung development. Dev Dyn. 2015;244:342–366. doi: 10.1002/dvdy.24234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Beers M.F., Morrisey E.E. The three R's of lung health and disease: repair, remodeling, and regeneration. J Clin Invest. 2011;121:2065–2073. doi: 10.1172/JCI45961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sakiyama J., Yamagishi A., Kuroiwa A. Tbx4-Fgf10 system controls lung bud formation during chicken embryonic development. Development. 2003;130:1225–1234. doi: 10.1242/dev.00345. [DOI] [PubMed] [Google Scholar]
- 77.Veraldi K.L., Gibson B.T., Yasuoka H., Myerburg M.M., Kelly E.A., Balzar S., Jarjour N.N., Pilewski J.M., Wenzel S.E., Feghali-Bostwick C.A. Role of insulin-like growth factor binding protein-3 in allergic airway remodeling. Am J Respir Crit Care Med. 2009;180:611–617. doi: 10.1164/rccm.200810-1555OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Renzoni E., Srihari V., Sestini P. Pathogenesis of idiopathic pulmonary fibrosis: review of recent findings. F1000Prime Rep. 2014;6:69. doi: 10.12703/P6-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Selman M., Pardo A., Kaminski N. Idiopathic pulmonary fibrosis: aberrant recapitulation of developmental programs? PLoS Med. 2008;5:e62. doi: 10.1371/journal.pmed.0050062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Suwara M.I., Green N.J., Borthwick L.A., Mann J., Mayer-Barber K.D., Barron L., Corris P.A., Farrow S.N., Wynn T.A., Fisher A.J., Mann D.A. IL-1alpha released from damaged epithelial cells is sufficient and essential to trigger inflammatory responses in human lung fibroblasts. Mucosal Immunol. 2014;7:684–693. doi: 10.1038/mi.2013.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhou W., Wang Y. Candidate genes of idiopathic pulmonary fibrosis: current evidence and research. Appl Clin Genet. 2016;9:5–13. doi: 10.2147/TACG.S61999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Williamson J.D., Sadofsky L.R., Hart S.P. The pathogenesis of bleomycin-induced lung injury in animals and its applicability to human idiopathic pulmonary fibrosis. Exp Lung Res. 2015;41:57–73. doi: 10.3109/01902148.2014.979516. [DOI] [PubMed] [Google Scholar]
- 83.Lin S.L., Kisseleva T., Brenner D.A., Duffield J.S. Pericytes and perivascular fibroblasts are the primary source of collagen-producing cells in obstructive fibrosis of the kidney. Am J Pathol. 2008;173:1617–1627. doi: 10.2353/ajpath.2008.080433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Moore B.B., Kolodsick J.E., Thannickal V.J., Cooke K., Moore T.A., Hogaboam C., Wilke C.A., Toews G.B. CCR2-mediated recruitment of fibrocytes to the alveolar space after fibrotic injury. Am J Pathol. 2005;166:675–684. doi: 10.1016/S0002-9440(10)62289-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yamada M., Kuwano K., Maeyama T., Hamada N., Yoshimi M., Nakanishi Y., Kasper M. Dual-immunohistochemistry provides little evidence for epithelial-mesenchymal transition in pulmonary fibrosis. Histochem Cell Biol. 2008;129:453–462. doi: 10.1007/s00418-008-0388-9. [DOI] [PubMed] [Google Scholar]
- 86.Kim K.K., Wei Y., Szekeres C., Kugler M.C., Wolters P.J., Hill M.L., Frank J.A., Brumwell A.N., Wheeler S.E., Kreidberg J.A., Chapman H.A. Epithelial cell alpha3beta1 integrin links beta-catenin and Smad signaling to promote myofibroblast formation and pulmonary fibrosis. J Clin Invest. 2009;119:213–224. doi: 10.1172/JCI36940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Vyalov S.L., Gabbiani G., Kapanci Y. Rat alveolar myofibroblasts acquire alpha-smooth muscle actin expression during bleomycin-induced pulmonary fibrosis. Am J Pathol. 1993;143:1754–1765. [PMC free article] [PubMed] [Google Scholar]
- 88.Abe R., Donnelly S.C., Peng T., Bucala R., Metz C.N. Peripheral blood fibrocytes: differentiation pathway and migration to wound sites. J Immunol. 2001;166:7556–7562. doi: 10.4049/jimmunol.166.12.7556. [DOI] [PubMed] [Google Scholar]
- 89.Wynn T.A., Ramalingam T.R. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med. 2012;18:1028–1040. doi: 10.1038/nm.2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Datta A., Scotton C.J., Chambers R.C. Novel therapeutic approaches for pulmonary fibrosis. Br J Pharmacol. 2011;163:141–172. doi: 10.1111/j.1476-5381.2011.01247.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lama V., Moore B.B., Christensen P., Toews G.B., Peters-Golden M. Prostaglandin E2 synthesis and suppression of fibroblast proliferation by alveolar epithelial cells is cyclooxygenase-2-dependent. Am J Respir Cell Mol Biol. 2002;27:752–758. doi: 10.1165/rcmb.4857. [DOI] [PubMed] [Google Scholar]
- 92.Borok Z., Gillissen A., Buhl R., Hoyt R.F., Hubbard R.C., Ozaki T., Rennard S.I., Crystal R.G. Augmentation of functional prostaglandin E levels on the respiratory epithelial surface by aerosol administration of prostaglandin E. Am Rev Respir Dis. 1991;144:1080–1084. doi: 10.1164/ajrccm/144.5.1080. [DOI] [PubMed] [Google Scholar]
- 93.Huang S.K., Fisher A.S., Scruggs A.M., White E.S., Hogaboam C.M., Richardson B.C., Peters-Golden M. Hypermethylation of PTGER2 confers prostaglandin E2 resistance in fibrotic fibroblasts from humans and mice. Am J Pathol. 2010;177:2245–2255. doi: 10.2353/ajpath.2010.100446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ghosh A.K., Vaughan D.E. PAI-1 in tissue fibrosis. J Cell Physiol. 2012;227:493–507. doi: 10.1002/jcp.22783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bauman K.A., Wettlaufer S.H., Okunishi K., Vannella K.M., Stoolman J.S., Huang S.K., Courey A.J., White E.S., Hogaboam C.M., Simon R.H., Toews G.B., Sisson T.H., Moore B.B., Peters-Golden M. The antifibrotic effects of plasminogen activation occur via prostaglandin E2 synthesis in humans and mice. J Clin Invest. 2010;120:1950–1960. doi: 10.1172/JCI38369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Richeldi L., du Bois R.M., Raghu G., Azuma A., Brown K.K., Costabel U., Cottin V., Flaherty K.R., Hansell D.M., Inoue Y., Kim D.S., Kolb M., Nicholson A.G., Noble P.W., Selman M., Taniguchi H., Brun M., Le Maulf F., Girard M., Stowasser S., Schlenker-Herceg R., Disse B., Collard H.R., INPULSIS Trial Investigators Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2071–2082. doi: 10.1056/NEJMoa1402584. [DOI] [PubMed] [Google Scholar]
- 97.Woodcock H.V., Maher T.M. The treatment of idiopathic pulmonary fibrosis. F1000Prime Rep. 2014;6:16. doi: 10.12703/P6-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Trujillo G., Meneghin A., Flaherty K.R., Sholl L.M., Myers J.L., Kazerooni E.A., Gross B.H., Oak S.R., Coelho A.L., Evanoff H., Day E., Toews G.B., Joshi A.D., Schaller M.A., Waters B., Jarai G., Westwick J., Kunkel S.L., Martinez F.J., Hogaboam C.M. TLR9 differentiates rapidly from slowly progressing forms of idiopathic pulmonary fibrosis. Sci Transl Med. 2010;2:57ra82. doi: 10.1126/scitranslmed.3001510. [DOI] [PMC free article] [PubMed] [Google Scholar]