

Abstract
The early landmark discoveries in cancer metabolism research have uncovered metabolic processes that support rapid proliferation, such as aerobic glycolysis (Warburg effect), glutaminolysis, and increased nucleotide biosynthesis. However, there are limitations to the effectiveness of specifically targeting the metabolic processes which support rapid proliferation. First, as other normal proliferative tissues also share similar metabolic features, they may also be affected by such treatments. Secondly, targeting proliferative metabolism may only target the highly proliferating “bulk tumor” cells and not the slower-growing, clinically relevant cancer stem cell subpopulations which may be required for an effective cure. An emerging body of research indicates that altered metabolism plays key roles in supporting proliferation-independent functions of cancer such as cell survival within the ischemic and acidic tumor microenvironment, immune system evasion, and maintenance of the cancer stem cell state. As these aspects of cancer cell metabolism are critical for tumor maintenance yet are less likely to be relevant in normal cells, they represent attractive targets for cancer therapy.
Keywords: cancer, immune evasion, metabolism, metabolites, tumor microenvironment
INTRODUCTION
The class of drugs known as the antifolates was amongst the first successful chemotherapeutics beginning with aminopterin developed in the 1940s (Farber and Diamond, 1948), and antifolates such as methotrexate and pemetrexed are still widely used in cancer therapy (McGuire, 2003; Visentin et al., 2012). These drugs competitively inhibit dihydrofolate reductase and block the synthesis of tetrahydrofolate, a cofactor required in purine biosynthesis and other metabolic functions. Antifolates can thus target cancer cells based on their highly proliferative nature and requirement for nucleic acids.
Antifolates and other strategies that also exploit the rapid proliferation of cells, for example DNA- damaging agents such as the platins, have proven to be effective tools against cancer. Furthermore, numerous metabolic pathways, such as glycolysis and one carbon metabolism, have been shown to be altered in cancer cells in a manner that supports rapid proliferation, and these remain attractive targets for therapy.
However, highly proliferating normal cells, such as intestinal cells or cells during embryonic development, feature similar proliferative metabolism, such as pyruvate kinase M2 isoform (PKM2) expression or high folate demand (Barker, 2014; Di Francesco et al., 2016; Kim et al., 2015b). Such cells are impacted by these chemotherapeutic strategies, and this becomes the basis for side effects, dose limiting toxicities, and chemotherapy contraindication in pregnancy. An additional limitation is that there are subsets of cancer cells that do not display high proliferation rates, and these have been postulated to be the chemotherapy resistant, tumor-repopulating “cancer stem cells” that confound successful therapy (Moore and Lyle, 2011; Vinogradov and Wei, 2012).
On the other hand, when considering the cancer cell within its context i.e. the intratumoral environment, it is evident that there are specific conditions and demands placed on a cancer cell that distinguish it from any normal cell, that may provide even more attractive avenues for metabolism-based therapy (Fig. 1). First, in the solid tumor environment, due to irregular vasculature and rapid growth that outpaces vessel formation, many regions lack proper blood flow leading to oxygen and nutrient deficiency. Secondly, in addition to stromal involvement, there is critical immune cell involvement that both acts against or in some cases supports tumor progression. Finally, in secondary sites of tumor formation or following therapeutic intervention, the maintenance of a cancer “stem cell” population may play a key role in repopulating the tumor. Recent research suggests that in cancer cells, metabolic activities are altered in ways that allow them to overcome these challenges.
Fig. 1.
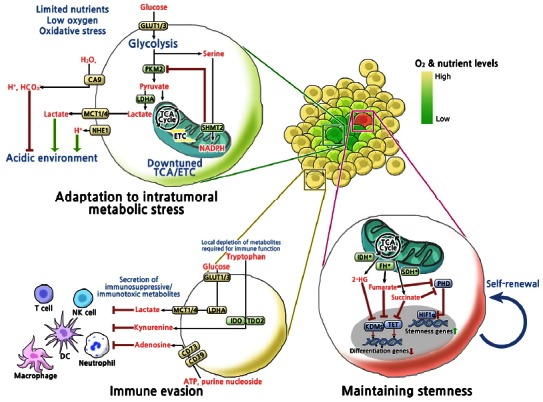
Overview of metabolic mechanisms involved in adaptation to intratumoral metabolic stress, maintaining stemness and immune evasion. Metabolic reprogramming of cancer cells enhances fitness toward intratumoral metabolic stress and immune surveillance, and contributes to maintaining stemness. GLUT1/3, Glucose transporter 1/3; PKM2, M2 isoform of pyruvate kinase; SHMT2, Serine hydroxymethyltransferase; LDHA, Lactate dehydrogenase A; NHE1, Na+/H+-exchanger; MCT1/4, Monocarboxylate transporters; CA9, Carbonic anhydrase 9; IDH*, Gain-of-function mutation in isocitrate dehydrogenase1/2; FH*, Loss-of-function mutation in fumarate hydratase; SDH*, Loss-of-function mutation in succinate dehydrogenase; PHD, Prolyl hydroxylases; HIF1α, Hypoxia-inducible factor 1α; KDMs, Histone lysine demethylases; TET, Ten eleven translocation family of 5-methylcytosine (3mC) hydroxylases; IDO1, Indoleamine-2, 3-dioxygenase 1; TDO2, tryptophan-2, 3-dioxygenase 2; CD73, Ecto-5′-nucleotidases; CD39, Ectonucleoside triphosphate diphosphohydrolase 1.
ADAPTATION TO INTRATUMORAL METABOLIC STRESS
The combination of limited perfusion to and from the tumor, and the metabolic activities of the cancer cells themselves, contributes to a harsh and toxic environment. The growth of a tumor can outpace blood vessel formation; also, tumor vasculature is irregular and prone to disruption and collapse (Siemann, 2011). Thus, tumor cells, particularly those that are located distally from functioning vasculature, are subjected to an environment with low levels of oxygen, glucose, amino acids, and other nutrients. Many of the metabolic alterations found in cancer cells appear to aid their survival within such conditions. In particular, a central cancer cell metabolism that aids their survival within such an environment is a feature referred to as aerobic glycolysis: regardless of oxygen tension, cancer cells have a high rate of glycolysis and lactate production, with low oxygen consumption. This feature was perhaps the earliest defined biochemical characteristic of cancer cells (Warburg, 1956), and supports rapid proliferation by diverting glycolytic carbons to biosynthetic processes such as the pentose phosphate pathway and serine biosynthesis (Hsu and Sabatini, 2008; Vander Heiden et al. 2009).
Key changes in metabolic gene expression appear to underlie aerobic glycolysis. In contrast to most normal cells, cancer cells predominantly express the M2 isoform of pyruvate kinase (PKM2), which limits phosphoenolpyruvate (PEP) to pyruvate conversion and thus overall glucose carbon flux into the TCA cycle (Christofk et al., 2008). Furthermore, increased expression of pyruvate dehydrogenase kinase 1 (PDK1), which phosphorylates and inhibits pyruvate dehydrogenase, also acts to limit glucose carbon flux into TCA cycle (Kim et al., 2006; Papandreou et al., 2006). As the TCA cycle is coupled to oxidative phosphorylation, these changes result in decreased oxygen consumption at the tumor and individual cell level, and improve cancer cell survival under oxygen limiting conditions (Kim et al., 2006; Papandreou et al., 2006).
An additional consequence of the hypoxic environment is electron transport chain failure and increased production of reactive oxygen species (ROS) within cancer cells (Kim et al., 2006; Klimova and Chandel, 2008). Furthermore, outside of the cancer cell, cancer-associated fibroblast or cancer-associated macrophages or neutrophils contribute establishment of a prooxidant environment (Condeelis and Pollard, 2006; Giannoni et al., 2011). Therapeutic strategies such as ionizing radiation can further contribute to a prooxidant environment. Decreasing carbon flux into TCA can decrease ROS production, and increase cancer cell survival in experimental conditions of hypoxia and/or chemically induced ROS (Anastasiou et al., 2011; Kim et al., 2006).
The Warburg phenotype appears to be triggered in large part by the tumor microenvironment itself. Hypoxia-inducible factor 1 (HIF1) is stabilized in hypoxic conditions and induces the expression of a number of metabolic enzymes and transporters involved in central carbon metabolism, including PKM2, PDK1, glucose transporters 1 and 3 (GLUT1/3), and lactate dehydrogenase A (LDHA), all of which contribute to the aerobic glycolysis phenotype (Luo et al., 2011; Semenza, 2012). In addition, the tetramerization and activity of PKM2 is induced by levels of metabolites such as fructose bisphosphate and serine (Chaneton et al., 2012; Christofk et al., 2008), and thus a nutrient limited tumor environment, such as those lacking glucose and serine, may limit PKM2 activity, reinforcing the bottleneck of carbons into the TCA cycle and the Warburg phenotype.
As a result of their glycolytic metabolism, cancer cells produce and excrete high levels of lactate; lactate secretion and proton efflux from cancer cells, exacerbated by poor perfusion and a hypercellular environment, result in an acidic environment (pH 5.6–6.8) (Chiche et al., 2010), which poses an additional challenge for cancer cells. It appears that cancer cells are adapted towards this challenge; solid tumors highly express carbonic anhydrase IX (CA9), another HIF-1 target, which catalyzes the conversion of CO2 and H2O to HCO3−(bicarbonate), which can neutralize low pH (Parks et al., 2011). Furthermore, cancer cells overexpress proton-exporting transporters such as monocarboxylate transporters (MCTs) and Na+/H+-exchanger (NHE1), which allow them to form a pH gradient in which their intracellular pH is high relative to their environment (Chiche et al., 2010; Webb et al., 2011).
Elevated expression and functional relevance of serine/glycine metabolism enzymes have recently been reported in a variety of cancers (Jain et al., 2012; Kim et al., 2015a; Possemato et al., 2011; Yang and Vousden, 2016; Ye et al., 2014; Zhang et al., 2012), and appear to play key roles in aiding cell survival in the tumor environment. Overexpression of mitochondrial serine hydroxymethyltransferase (SHMT2) promotes survival in hypoxic and ischemic environments (Kim et al., 2015a; Ye et al., 2014). This effect of SHMT2 occurs by limiting PKM2 activity and thus oxygen consumption (Kim et al., 2015a), and through increased one carbon metabolism leading to NADPH production and redox control (Fan et al., 2014; Ye et al., 2014).
IMMUNE EVASION
The immune system plays a critical role in preventing cancer, and avoiding immune surveillance is a hallmark of cancer (Hanahan and Weinberg, 2011). Cells of both the innate and adaptive immune systems are found in tumors, and how the tumor either evades or tolerates this immune component is a critical determinant of tumor growth and clinical outcome (Gajewski et al., 2013; Galon et al., 2006; Vesely et al., 2011). While cancer cells have multiple layers of adaptations that allow immune evasion and tolerance, such as downregulation of tumor antigens and resistance against immune cell cytotoxicity, recent studies show that cancer cells have diverse metabolic mechanisms of evading the immune system.
The aforementioned Warburg metabolism of cancer cells, aside from promoting survival within the tumor metabolic environment, also contributes to immune evasion. First, the high expression of glucose transporters and glucose utilization by cancer cells can competitively deplete the glucose required by activated T cells in the highly proliferative state, which are also glucose dependent (Fox et al., 2005; Pearce and Pearce, 2013). It was recently shown that the glycolytic intermediate PEP is necessary to sustain T-Cell receptor-mediated Ca2+-NFAT signaling and thus acts as a checkpoint for T-cell activity; thus local glucose depletion by tumor cells can not only impair T cell proliferation, but T cell activity itself (Ho et al., 2015).
In addition to the effects of local depletion of glucose, the high production of lactate from tumor cell metabolism also acts to suppress immune surveillance. LDHA-associated lactic acid production and acidification suppresses T and NK cell activation (Brand et al., 2016; Fischer et al., 2007). An abundance of lactic acid in the cancer microenvironment inhibits lactic acid export in T cells, thus disrupting their metabolism and function (Fischer et al., 2007). Lactate also inhibits monocyte migration and cytokine release (Goetze et al., 2011), and dendritic cell activation (Gottfried et al., 2006). Furthermore, survival of tumor-associated macrophages (TAM) is impaired by lactic acid accumulation in the tumor microenvironment (Carmona-Fontaine et al., 2013). Lactate promotes the polarization of TAM to a M2 state, which has a role in wound healing and immunosuppression (Colegio et al., 2014). In this case, however, polarization of TAM has an effect of promoting, rather than suppressing, tumor growth.
Enhanced tryptophan catabolism is another immune evading mechanism employed by tumors. Many types of cancers over-express tryptophan-2, 3-dioxygenase 2 (TDO2) and indoleamine-2, 3-dioxygenases 1 (IDO1), which convert the tryptophan into kynurenine (Munn and Mellor, 2007). The high utilization of extracellular tryptophan by cancer cells can limit utilization of tryptophan by T cells, resulting in amino acid deprivation-associated apoptosis of effector T cells (Fallarino et al., 2002). Kynurenine and its downstream metabolites, 3-hydroxykynurenine and 3-hydroxyanthranilic acid, induce cell cycle arrest and apoptosis of T-cells (Fallarino et al., 2003). Kynurenine also suppresses antitumor immune responses by acting as a natural ligand for aryl hydrocarbon receptor (Pilotte et al., 2012). However, the mechanisms which underlie the immunosuppressive effects of tryptophan catabolism pathway metabolites in immune cells are still largely unknown.
Another metabolic mechanism by which cancer cells achieve immune escape is through inducing the extracellular accumulation of the purine nucleoside adenosine. Adenosine accumulates in the tumor microenvironment, mainly initiating from a mechanism in which ATP is released from cells either through injury/necrosis or through altered nucleotide metabolism and transport (Antonioli et al., 2013; Stagg and Smyth, 2010). ATP is sequentially degraded by the cell surface enzymes CD39 and CD73 to yield adenosine. Several tumors overexpress CD39/CD73, leading to overproduction of adenosine. Additionally, some tumors downregulate nucleotide transporters and the cell surface adenosine deaminase CD26, both of which normally decrease adenosine levels. Interestingly, the extracellular level of adenosine is dramatically increased in hypoxic tissues (Takahashi et al., 2010; Vaupel and Mayer, 2016), via induction of CD39/CD73 by hypoxia, which may play a role in hypoxic tumor tissues. In turn, extracellular adenosine acts on a variety of adenosine receptors (A1, A2A, A2B, A3) on B, T, NK and NKT cells, dendritic cells, macrophages, neutrophils, and mast cells, leading to downstream signal transduction and suppression of immune functions such as NK cell cytotoxicity, dendritic cell maturation, and pro-inflammatory cytokine production (Bouma et al., 1997; Eppell et al., 1989; Hasko et al., 1996; 2008; Hausler et al., 2011; Ryzhov et al., 2011). Thus, adenosine is a physiologically important immune signaling metabolite that acts on a variety of immune cells, and this mechanism is ‘highjacked’ by the altered metabolism of cancer cells.
STEMNESS
Cancer stem cell (CSC) is a term that is used to describe cancer cells that possess self-renewal and tumor-seeding properties (Chen et al., 2013). The specific targeting of CSCs is thought to be an important objective in cancer therapy because of their observed drug-resistant and tumorigenic properties (Chen et al., 2013). Examinations of CSC metabolism have reported metabolic characteristics such as a glycolytic phenotype and dependence on one carbon metabolism, features that are undoubtedly important to CSCs, but do not clearly distinguish them from cancer cells in general (Jang et al., 2015; Sancho et al., 2016). However, recent evidence indicates that metabolic parameters can modulate the stemness of cancer cells, suggesting new avenues for therapeutic strategies.
The metabolite 2-hydroxyglutarate (2HG), referred to as an ‘oncometabolite’ due to its cancer-promoting properties, regulates the stemness of cancer cells. Wild-type isocitrate dehydrogenase 1/2 (IDH1/2) catalyzes reversible conversion of isocitrate to α-ketoglutarate (α-KG) as part of the TCA cycle. However, gain-of-function IDH1/2 mutations, found in gliomas and myeloid malignancies, cause it to catalyze the conversion of α-KG to D-2HG (Ward et al., 2010). At high levels, D-2HG, due to its structural similarity to α-KG, competitively binds and α-KG-dependent dioxygenases such as the ten eleven translocation family of 5-methylcytosine (3mC) hydroxylases (TETs) and the Jumonji C domain-containing histone lysine demethylases (KDMs), promoting hypermethylation (Chowdhury et al., 2011; Xu et al., 2011), which results in suppression of genes involved in cell differentiation (Losman et al., 2013; Lu et al., 2012). Indeed, targeting mutant IDH1 or IDH2 promotes cellular differentiation and can delay tumor growth (Rohle et al., 2013; Wang et al., 2013). Recently, L-2HG, an enantiomer of D-2HG, was reported to accumulate in hypoxia via LDHA and malate dehydrogenase 1 and 2 (MDH1/2) activity (Intlekofer et al., 2015). Though D-2HG and L-2HG can exert disparate roles in prolyl hydroxylases (PDHs), respectively (Koivunen et al., 2012; Terunuma et al., 2014), both D-2HG and L-HG act as a suppressor of histone demethylases.
Loss of function mutations in two other TCA cycle enzymes, succinate dehydrogenase (SDH) and fumarate hydratase (FH) promotes accumulation of succinate and fumarate, respectively (Yang et al., 2013). Succinate and fumarate are also known as oncometabolites, and similarly to D-2HG promote epigenetic alterations by inhibiting the aforementioned group of α-KG-dependent enzymes resulting in global histone and DNA hypermethylation (Xiao et al., 2012). In paraganglioma, a neuroendocrine tumor, SDH mutation is frequently observed in malignant tumors (Gimenez-Roqueplo et al., 2003; Neumann et al., 2004). SDH mutation-related tumors display hypermethylation and epigenetic silencing of crucial genes implicated in cell differentiation and epithelial-mesenchymal transition (EMT) (Letouze et al., 2013). EMT is a developmental process in which epithelial cells lose their epithelial characteristics to become motile mesenchymal stem cells, and cancer cells with epithelial properties can also undergo EMT to become cells with mesenchymal characteristics (Kalluri and Weinberg, 2009). This is a significant event as in addition to developing motile and invasive properties, cancer cells which undergo EMT are thought to obtain stem-cell like properties. In FH-deficient mouse and human cells, accumulation of fumarate induces EMT by suppression of TET-mediated demethylation of the antimetastatic miRNA cluster, which result in expression of EMT-related genes (Sciacovelli et al., 2016).
The impairment of α-KG-dependent dioxygenases by 2-HG, succinate, and fumarate may also have an additional route in which stemness is enhanced: the stabilization of HIF. The activation of a HIF response is observed in SDH- or FD-deficient cancer cells, which could be explained by the allosteric suppression of PHDs by accumulated succinate or fumarate (Isaacs et al., 2005; Selak et al., 2005). The activated HIF is known to induce the expression of stemness and EMT-related genes such as Oct-3/4, Nanog, Sox-2, snail, twist and LOX (Mimeault and Batra, 2013). Thus, 2-HG, succinate and fumarate might actively regulate stemness of cancer cells by activating the HIF response.
Additional metabolic pathways are emerging as regulators cancer cell differentiation. Dihydropyrimidine accumulation by dihydropyrimidine dehydrogenase, which catalyzes the rate-limiting step in pyrimidine degradation, is required in EMT, and it was noted that a number of other metabolic enzymes show similar mesenchymal-cell-enriched expression patterns that indicate they may also be involved in EMT (Shaul et al., 2014; 2016). Dendrogenin A (DDA), an enzymatic product of the conjugation of 5,6a-epoxy-cholesterol and histamine, promotes cancer differentiation, which results in suppression of tumor growth (de Medina et al., 2013). The underlying mechanisms for these functions are not yet well understood.
CONCLUSION
As overviewed in Fig. 1, the cancer cell faces a set of road-blocks including a harsh metabolic environment, surveillance and attack from the immune system, and the requirement for self-renewal - it is not surprising that their metabolic activities are altered in ways that enhance fitness toward these challenges. These adaptations, in contrast to cell proliferation, can be considered highly specific to cancer cells, and thus hold promise for future therapeutic strategies. The metabolic enzymes described here that support these adaptations (Table 1) are catalysts and thus are inherently druggable, interesting targets for therapy. As an example, recent clinical trials and FDA approval of drugs which trigger immune responses against cancers (Farkona et al., 2016) have shown great promise, and a strategy for blocking the metabolic mechanisms that cancer cells use for immune evasion in combination with these drugs may be particularly effective. Along these lines, further efforts are required to more fully understand how the various metabolic features of cancer cells confound their detection and elimination by the immune system. As another strategy, based on the unique set of challenges that cancer cells face, combinations of drugs to hinder multiple aspects of the metabolic adaptations described here may be a means to achieve additional selectivity towards cancer cells.
Table 1.
Working mechanisms of metabolites involved in adaptation to intratumoral metabolic stress, maintaining stemness and immune evasion
| Working enzymes in cancer | Metabolites | Working mechanism | References | |
|---|---|---|---|---|
| Adaptation to intratumoral metabolic stress | Pyruvate kinase isozymes M2, PKM2 | Decrease in downstream glycolytic products | Decreased carbon flux into TCA cycle | Christofk et al., 2008 |
|
| ||||
| Serine hydroxymethyltransferase, SHMT2 (Serine→Glycine) | Excessive Glycine and NADPH | Limiting PKM2 activity and thus oxygen consumption decrease Redox control via production of NADPH |
Fan et al., 2014; Kim et al., 2015a; Ye et al., 2014 | |
|
| ||||
| Carbonic anhydrase 9, CA9 (CO2, H2O→Bicarbonate, H+) | Excessive Bicarbonate | Binding with hydrogen forming H2CO3 | Parks et al., 2011 | |
|
| ||||
| Immune evasion | Lactate dehydrogenase A, LDHA (Glucose→Lactate) | Limited glucose | Limit utility of glucose by T cells→ limiting for immune response | Fox et al., 2005 |
|
| ||||
| Excessive lactate | Disturbing T cell metabolism and function by inhibition of lactic acid export of T cells | Fischer et al., 2007 | ||
|
| ||||
| Suppression of T and NK cell activation | Brand et al., 2016; Fischer et al., 2007 | |||
|
| ||||
| Inhibition of monocyte migration and cytokine release | Goetze et al., 2011 | |||
|
| ||||
| Inhibition of dendritic cell activation | Gottfried et al., 2006 | |||
|
| ||||
| Inhibition of TAM survival | Carmona-Fontaine et al., 2013 | |||
|
| ||||
| Polarization of TAM to a M2 state, (M2 state TAM has a role in immunosuppression) | Colegio et al., 2014 | |||
|
| ||||
| Indoleamine2,3-Dioxygenase1/Tryptophan 2,3-dioxygenase2, IDO1/TDO2 (Tryptophan→Kynurenine) | Limited tryptophan | Limit utility of tryptophan by T cells → amino acid deprivation-associated apoptosis of effector T cells | Fallarino et al., 2002 | |
|
| ||||
| Excessive kynurenine | Cell cycle arrest and apoptosis of T-cell by kynurenine | Fallarino et al., 2003 | ||
|
| ||||
| Kynurenine acting as a ligand for aryl hydrocarbon receptor | Pilotte et al., 2012 | |||
|
| ||||
| Ecto-5′-nucleotidases/Ectonucleoside triphosphate diphosphohydrolase1, CD39/CD73 (Nucleotide→Adenosine) | Excessive adenosine | Inhibition of T cell response and NK cell cytotoxicity | Hasko et al., 2008; Hausler et al., 2011 | |
|
| ||||
| Suppression of dendritic cell maturation and its pro-inflammatory cytokines production | Ryzhov et al., 2011 | |||
|
| ||||
| Activation of immunosuppressive functions of MDSCs | Ryzhov et al., 2011 | |||
|
| ||||
| Inhibition of phagocytic activity and nitric oxide and superoxide production in macrophage | Eppell et al., 1989; Hasko et al., 1996 | |||
|
| ||||
| Inhibition of neutrophil degranulation | Bouma et al., 1997 | |||
|
|
||||
| Maintaining Stemness | gain-of-function mutation in isocitrate dehydrogenase1/2, IDH1/2 (αKG→D-2HG) | Excessive D-2HG | Suppression of gene involved in cell differentiation | Losman et al., 2013; Lu et al., 2012 |
|
| ||||
| Lactate dehydrogenase A, Malate dehydrogenase, LDHA MDH1/2 (αKG→L-2HG) | Excessive L-2HG | |||
|
| ||||
| Loss-of-function mutation in succinate dehydrogenase, SDH (Succinate→Fumarate) | Excessive succinate | Silencing of crucial genes implicated in cell differentiation and EMT | Letouze et al., 2013 | |
|
| ||||
| Activation of a HIF response by the allosteric suppression of PHDs | Isaacs et al., 2005; Selak et al., 2005 | |||
|
| ||||
| Loss-of-function mutation in fumarate hydratase, FH (Fumarate→Malate) | Excessive fumarate | Suppression of EMT-related genes | Sciacovelli et al., 2016 | |
|
| ||||
| Activation of a HIF response by the allosteric suppression of PHDs | Isaacs et al., 2005; Selak et al., 2005 | |||
|
| ||||
| Dihydropyrimidine dehydrogenase (5FU→FU-H2) | Excessive dihydropyrimidine | EMT activation | Shaul et al., 2014 | |
|
| ||||
| Unknown | Excessive dendrogenin | A Activation of cancer cell differentiation | de Medina et al., 2013 | |
While we have outlined some of the recent key discoveries into how metabolic activities support cancer activities beyond proliferation per se, our understanding of the many ways in which altered metabolic activities can benefit a cancer cell is likely far from complete. As metabolic activities are tightly intertwined with biological processes, unidentified metabolic activities in cancer cells are likely to support and even drive cancer functions not covered here, such as cell migration and evasion of apoptosis. Furthermore, there is a complex interplay between tumor environment, the immune component, and cancer cell “stemness” described here that requires further investigation and understanding. For example, immune cells themselves contribute significantly to the metabolic tumor environment; the metabolic environment exerts selective pressure towards cells with stem-like properties; and so forth. These complex interactions, as well as the metabolic heterogeneity of the tumor, will all have to be taken into consideration when considering therapeutic strategies. However, targeting cancer metabolism has shown great promise in recent years and it is hoped that efforts like those described here to understand the multitude of ways they can aid cancer cell fitness will continue to take us in the right direction.
ACKNOWLEDGMENTS
We thank Ohseop Kwon for designing the images in our figure sets. We apologize that we were not able to cite many key contributions to the cancer metabolism field due to space restrictions.
REFERENCES
- Anastasiou D., Poulogiannis G., Asara J.M., Boxer M.B., Jiang J.K., Shen M., Bellinger G., Sasaki A.T., Locasale J.W., Auld D.S., et al. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science. 2011;334:1278–1283. doi: 10.1126/science.1211485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonioli L., Blandizzi C., Pacher P., Hasko G. Immunity, inflammation and cancer: a leading role for adenosine. Nat Rev Cancer. 2013;13:842–857. doi: 10.1038/nrc3613. [DOI] [PubMed] [Google Scholar]
- Barker N. Adult intestinal stem cells: critical drivers of epithelial homeostasis and regeneration. Nat Rev Mol Cell Biol. 2014;15:19–33. doi: 10.1038/nrm3721. [DOI] [PubMed] [Google Scholar]
- Bouma M.G., Jeunhomme T.M., Boyle D.L., Dentener M.A., Voitenok N.N., van den Wildenberg F.A., Buurman W.A. Adenosine inhibits neutrophil degranulation in activated human whole blood: involvement of adenosine A2 and A3 receptors. J Immunol. 1997;158:5400–5408. [PubMed] [Google Scholar]
- Brand A., Singer K., Koehl G.E., Kolitzus M., Schoenhammer G., Thiel A., Matos C., Bruss C., Klobuch S., Peter K., et al. LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK cells. Cell Metab. 2016;24:657–671. doi: 10.1016/j.cmet.2016.08.011. [DOI] [PubMed] [Google Scholar]
- Carmona-Fontaine C., Bucci V., Akkari L., Deforet M., Joyce J.A., Xavier J.B. Emergence of spatial structure in the tumor microenvironment due to the Warburg effect. Proc Natl Acad Sci USA, 2013;110:19402–19407. doi: 10.1073/pnas.1311939110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaneton B., Hillmann P., Zheng L., Martin A.C., Maddocks O.D., Chokkathukalam A., Coyle J.E., Jankevics A., Holding F.P., Vousden K.H., et al. Serine is a natural ligand and allosteric activator of pyruvate kinase M2. Nature. 2012;491:458–462. doi: 10.1038/nature11540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K., Huang Y.H., Chen J.L. Understanding and targeting cancer stem cells: therapeutic implications and challenges. Acta Pharmacol Sin. 2013;34:732–740. doi: 10.1038/aps.2013.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiche J., Brahimi-Horn M.C., Pouyssegur J. Tumour hypoxia induces a metabolic shift causing acidosis: a common feature in cancer. J Cell Mol Med. 2010;14:771–794. doi: 10.1111/j.1582-4934.2009.00994.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury R., Yeoh K.K., Tian Y.M., Hillringhaus L., Bagg E.A., Rose N.R., Leung I.K., Li X.S., Woon E.C., Yang M., et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011;12:463–469. doi: 10.1038/embor.2011.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christofk H.R., Vander Heiden M.G., Harris M.H., Ramanathan A., Gerszten R.E., Wei R., Fleming M.D., Schreiber S.L., Cantley L.C. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008;452:230–233. doi: 10.1038/nature06734. [DOI] [PubMed] [Google Scholar]
- Christofk H.R., Vander Heiden M.G., Wu N., Asara J.M., Cantley L.C. Pyruvate kinase M2 is a phosphotyrosine-binding protein. Nature. 2008;452:181–186. doi: 10.1038/nature06667. [DOI] [PubMed] [Google Scholar]
- Colegio O.R., Chu N.Q., Szabo A.L., Chu T., Rhebergen A.M., Jairam V., Cyrus N., Brokowski C.E., Eisenbarth S.C., Phillips G.M., et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature. 2014;513:559–563. doi: 10.1038/nature13490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condeelis J., Pollard J.W. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell. 2006;124:263–266. doi: 10.1016/j.cell.2006.01.007. [DOI] [PubMed] [Google Scholar]
- de Medina P., Paillasse M.R., Segala G., Voisin M., Mhamdi L., Dalenc F., Lacroix-Triki M., Filleron T., Pont F., Saati T.A., et al. Dendrogenin A arises from cholesterol and histamine metabolism and shows cell differentiation and anti-tumour properties. Nat Commun. 2013;4:1840. doi: 10.1038/ncomms2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Francesco A.M., Toesca A., Cenciarelli C., Giordano A., Gasbarrini A., Puglisi M.A. Metabolic modification in gastrointestinal cancer stem cells: characteristics and therapeutic approaches. J Cell Physiol. 2016;231:2081–2087. doi: 10.1002/jcp.25318. [DOI] [PubMed] [Google Scholar]
- Eppell B.A., Newell A.M., Brown E.J. Adenosine receptors are expressed during differentiation of monocytes to macrophages in vitro. Implications for regulation of phagocytosis. J Immunol. 1989;143:4141–4145. [PubMed] [Google Scholar]
- Fallarino F., Grohmann U., Vacca C., Bianchi R., Orabona C., Spreca A., Fioretti M.C., Puccetti P. T cell apoptosis by tryptophan catabolism. Cell Death Differ. 2002;9:1069–1077. doi: 10.1038/sj.cdd.4401073. [DOI] [PubMed] [Google Scholar]
- Fallarino F., Grohmann U., Vacca C., Orabona C., Spreca A., Fioretti M.C., Puccetti P. T cell apoptosis by kynurenines. Adv Exp Med Biol. 2003;527:183–190. doi: 10.1007/978-1-4615-0135-0_21. [DOI] [PubMed] [Google Scholar]
- Fan J., Ye J., Kamphorst J.J., Shlomi T., Thompson C.B., Rabinowitz J.D. Quantitative flux analysis reveals folate-dependent NADPH production. Nature. 2014;510:298–302. doi: 10.1038/nature13236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farber S., Diamond L.K. Temporary remissions in acute leukemia in children produced by folic acid antagonist, 4-aminopteroyl-glutamic acid. N Engl J Med. 1948;238:787–793. doi: 10.1056/NEJM194806032382301. [DOI] [PubMed] [Google Scholar]
- Farkona S., Diamandis E.P., Blasutig I.M. Cancer immunotherapy: the beginning of the end of cancer? BMC Med. 2016;14:73. doi: 10.1186/s12916-016-0623-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer K., Hoffmann P., Voelkl S., Meidenbauer N., Ammer J., Edinger M., Gottfried E., Schwarz S., Rothe G., Hoves S., et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood. 2007;109:3812–3819. doi: 10.1182/blood-2006-07-035972. [DOI] [PubMed] [Google Scholar]
- Fox C.J., Hammerman P.S., Thompson C.B. Fuel feeds function: energy metabolism and the T-cell response. Nat Rev Immunol. 2005;5:844–852. doi: 10.1038/nri1710. [DOI] [PubMed] [Google Scholar]
- Gajewski T.F., Schreiber H., Fu Y.X. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. 2013;14:1014–1022. doi: 10.1038/ni.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galon J., Costes A., Sanchez-Cabo F., Kirilovsky A., Mlecnik B., Lagorce-Pages C., Tosolini M., Camus M., Berger A., Wind P., et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313:1960–1964. doi: 10.1126/science.1129139. [DOI] [PubMed] [Google Scholar]
- Giannoni E., Bianchini F., Calorini L., Chiarugi P. Cancer associated fibroblasts exploit reactive oxygen species through a proinflammatory signature leading to epithelial mesenchymal transition and stemness. Antioxid Redox Signal. 2011;14:2361–2371. doi: 10.1089/ars.2010.3727. [DOI] [PubMed] [Google Scholar]
- Gimenez-Roqueplo A.P., Favier J., Rustin P., Rieubland C., Crespin M., Nau V., Khau Van Kien P., Corvol P., Plouin P.F., Jeunemaitre X., et al. Mutations in the SDHB gene are associated with extra-adrenal and/or malignant phaeochromocytomas. Cancer Res. 2003;63:5615–5621. [PubMed] [Google Scholar]
- Goetze K., Walenta S., Ksiazkiewicz M., Kunz-Schughart L.A., Mueller-Klieser W. Lactate enhances motility of tumor cells and inhibits monocyte migration and cytokine release. Int J Oncol. 2011;39:453–463. doi: 10.3892/ijo.2011.1055. [DOI] [PubMed] [Google Scholar]
- Gottfried E., Kunz-Schughart L.A., Ebner S., Mueller-Klieser W., Hoves S., Andreesen R., Mackensen A., Kreutz M. Tumor-derived lactic acid modulates dendritic cell activation and antigen expression. Blood. 2006;107:2013–2021. doi: 10.1182/blood-2005-05-1795. [DOI] [PubMed] [Google Scholar]
- Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Hasko G., Szabo C., Nemeth Z.H., Kvetan V., Pastores S.M., Vizi E.S. Adenosine receptor agonists differentially regulate IL-10, TNF-alpha, and nitric oxide production in RAW 264.7 macrophages and in endotoxemic mice. J Immunol. 1996;157:4634–4640. [PubMed] [Google Scholar]
- Hasko G., Linden J., Cronstein B., Pacher P. Adenosine receptors: therapeutic aspects for inflammatory and immune diseases. Nat Rev Drug Discov. 2008;7:759–770. doi: 10.1038/nrd2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausler S.F., Montalban del Barrio I., Strohschein J., Chandran P.A., Engel J.B., Honig A., Ossadnik M., Horn E., Fischer B., Krockenberger M., et al. Ectonucleotidases CD39 and CD73 on OvCA cells are potent adenosine-generating enzymes responsible for adenosine receptor 2A-dependent suppression of T cell function and NK cell cytotoxicity. Cancer Immunol Immunother. 2011;60:1405–1418. doi: 10.1007/s00262-011-1040-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho P.C., Bihuniak J.D., Macintyre A.N., Staron M., Liu X., Amezquita R., Tsui Y.C., Cui G., Micevic G., Perales J.C., et al. Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor T cell responses. Cell. 2015;162:1217–1228. doi: 10.1016/j.cell.2015.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu P.P., Sabatini D.M. Cancer cell metabolism: Warburg and beyond. Cell. 2008;134:703–707. doi: 10.1016/j.cell.2008.08.021. [DOI] [PubMed] [Google Scholar]
- Intlekofer A.M., Dematteo R.G., Venneti S., Finley L.W., Lu C., Judkins A.R., Rustenburg A.S., Grinaway P.B., Chodera J.D., Cross J.R., et al. Hypoxia induces production of L-2-hydroxyglutarate. Cell Metab. 2015;22:304–311. doi: 10.1016/j.cmet.2015.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacs J.S., Jung Y.J., Mole D.R., Lee S., Torres-Cabala C., Chung Y.L., Merino M., Trepel J., Zbar B., Toro J., et al. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: novel role of fumarate in regulation of HIF stability. Cancer Cell. 2005;8:143–153. doi: 10.1016/j.ccr.2005.06.017. [DOI] [PubMed] [Google Scholar]
- Jain M., Nilsson R., Sharma S., Madhusudhan N., Kitami T., Souza A.L., Kafri R., Kirschner M.W., Clish C.B., Mootha V.K. Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science. 2012;336:1040–1044. doi: 10.1126/science.1218595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang H., Yang J., Lee E., Cheong J.H. Metabolism in embryonic and cancer stemness. Arch Pharm Res. 2015;38:381–388. doi: 10.1007/s12272-015-0558-y. [DOI] [PubMed] [Google Scholar]
- Kalluri R., Weinberg R.A. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.W., Tchernyshyov I., Semenza G.L., Dang C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3:177–185. doi: 10.1016/j.cmet.2006.02.002. [DOI] [PubMed] [Google Scholar]
- Kim D., Fiske B.P., Birsoy K., Freinkman E., Kami K., Possemato R.L., Chudnovsky Y., Pacold M.E., Chen W.W., Cantor J.R., et al. SHMT2 drives glioma cell survival in ischaemia but imposes a dependence on glycine clearance. Nature. 2015a;520:363–367. doi: 10.1038/nature14363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H., Jang H., Kim T.W., Kang B.H., Lee S.E., Jeon Y.K., Chung D.H., Choi J., Shin J., Cho E.J., et al. Core pluripotency factors directly regulate metabolism in embryonic stem cell to maintain pluripotency. Stem Cells. 2015b;33:2699–2711. doi: 10.1002/stem.2073. [DOI] [PubMed] [Google Scholar]
- Klimova T., Chandel N.S. Mitochondrial complex III regulates hypoxic activation of HIF. Cell Death Differ. 2008;15:660–666. doi: 10.1038/sj.cdd.4402307. [DOI] [PubMed] [Google Scholar]
- Koivunen P., Lee S., Duncan C.G., Lopez G., Lu G., Ramkissoon S., Losman J.A., Joensuu P., Bergmann U., Gross S., et al. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature. 2012;483:484–488. doi: 10.1038/nature10898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letouze E., Martinelli C., Loriot C., Burnichon N., Abermil N., Ottolenghi C., Janin M., Menara M., Nguyen A.T., Benit P., et al. SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell. 2013;23:739–752. doi: 10.1016/j.ccr.2013.04.018. [DOI] [PubMed] [Google Scholar]
- Losman J.A., Looper R.E., Koivunen P., Lee S., Schneider R.K., McMahon C., Cowley G.S., Root D.E., Ebert B.L., Kaelin W.G., Jr (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science. 2013;339:1621–1625. doi: 10.1126/science.1231677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C., Ward P.S., Kapoor G.S., Rohle D., Turcan S., Abdel-Wahab O., Edwards C.R., Khanin R., Figueroa M.E., Melnick A., et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483:474–478. doi: 10.1038/nature10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo W., Hu H., Chang R., Zhong J., Knabel M., O’Meally R., Cole R.N., Pandey A., Semenza G.L. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell. 2011;145:732–744. doi: 10.1016/j.cell.2011.03.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuire J.J. Anticancer antifolates: current status and future directions. Curr Pharm Des. 2003;9:2593–2613. doi: 10.2174/1381612033453712. [DOI] [PubMed] [Google Scholar]
- Mimeault M., Batra S.K. Hypoxia-inducing factors as master regulators of stemness properties and altered metabolism of cancer- and metastasis-initiating cells. J Cell Mol Med. 2013;17:30–54. doi: 10.1111/jcmm.12004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore N., Lyle S. Quiescent, slow-cycling stem cell populations in cancer: a review of the evidence and discussion of significance. J Oncol. 2011;2011 doi: 10.1155/2011/396076. pii: 396076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munn D.H., Mellor A.L. Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J Clin Invest. 2007;117:1147–1154. doi: 10.1172/JCI31178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann H.P., Pawlu C., Peczkowska M., Bausch B., McWhinney S.R., Muresan M., Buchta M., Franke G., Klisch J., Bley T.A., et al. Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations. JAMA. 2004;292:943–951. doi: 10.1001/jama.292.8.943. [DOI] [PubMed] [Google Scholar]
- Papandreou I., Cairns R.A., Fontana L., Lim A.L., Denko N.C. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006;3:187–197. doi: 10.1016/j.cmet.2006.01.012. [DOI] [PubMed] [Google Scholar]
- Parks S.K., Chiche J., Pouyssegur J. pH control mechanisms of tumor survival and growth. J Cell Physiol. 2011;226:299–308. doi: 10.1002/jcp.22400. [DOI] [PubMed] [Google Scholar]
- Pearce E.L., Pearce E.J. Metabolic pathways in immune cell activation and quiescence. Immunity. 2013;38:633–643. doi: 10.1016/j.immuni.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilotte L., Larrieu P., Stroobant V., Colau D., Dolusic E., Frederick R., De Plaen E., Uyttenhove C., Wouters J., Masereel B., et al. Reversal of tumoral immune resistance by inhibition of tryptophan 2,3-dioxygenase. Proc Natl Acad Sci USA. 2012;109:2497–2502. doi: 10.1073/pnas.1113873109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Possemato R., Marks K.M., Shaul Y.D., Pacold M.E., Kim D., Birsoy K., Sethumadhavan S., Woo H.K., Jang H.G., Jha A.K., et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature. 2011;476:346–350. doi: 10.1038/nature10350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohle D., Popovici-Muller J., Palaskas N., Turcan S., Grommes C., Campos C., Tsoi J., Clark O., Oldrini B., Komisopoulou E., et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science. 2013;340:626–630. doi: 10.1126/science.1236062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryzhov S., Novitskiy S.V., Goldstein A.E., Biktasova A., Blackburn M.R., Biaggioni I., Dikov M.M., Feoktistov I. Adenosinergic regulation of the expansion and immunosuppressive activity of CD11b+Gr1+ cells. J Immunol. 2011;187:6120–6129. doi: 10.4049/jimmunol.1101225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancho P., Barneda D., Heeschen C. Hallmarks of cancer stem cell metabolism. Br J Cancer. 2016;114:1305–1312. doi: 10.1038/bjc.2016.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciacovelli M., Goncalves E., Johnson T.I., Zecchini V.R., da Costa A.S., Gaude E., Drubbel A.V., Theobald S.J., Abbo S.R., Tran M.G., et al. Fumarate is an epigenetic modifier that elicits epithelial-to-mesenchymal transition. Nature. 2016;537:544–547. doi: 10.1038/nature19353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selak M.A., Armour S.M., MacKenzie E.D., Boulahbel H., Watson D.G., Mansfield K.D., Pan Y., Simon M.C., Thompson C.B., Gottlieb E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell. 2005;7:77–85. doi: 10.1016/j.ccr.2004.11.022. [DOI] [PubMed] [Google Scholar]
- Semenza G.L. Hypoxia-inducible factors: mediators of cancer progression and targets for cancer therapy. Trends Pharmacol Sci. 2012;33:207–214. doi: 10.1016/j.tips.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaul Y.D., Freinkman E., Comb W.C., Cantor J.R., Tam W.L., Thiru P., Kim D2, Kanarek N., Pacold M.E., Chen W.W., et al. Dihydropyrimidine accumulation is required for the epithelial-mesenchymal transition. Cell. 2014;158:1094–1109. doi: 10.1016/j.cell.2014.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaul Y.D., Yuan B., Thiru P., Nutter-Upham A., McCallum S., Lanzkron C., Bell G.W., Sabatini D.M. MERAV: a tool for comparing gene expression across human tissues and cell types. Nucleic Acids Res. 2016;44:D560–566. doi: 10.1093/nar/gkv1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siemann D.W. The unique characteristics of tumor vasculature and preclinical evidence for its selective disruption by tumor-vascular disrupting agents. Cancer Treat Rev. 2011;37:63–74. doi: 10.1016/j.ctrv.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stagg J., Smyth M.J. Extracellular adenosine triphosphate and adenosine in cancer. Oncogene. 2010;29:5346–5358. doi: 10.1038/onc.2010.292. [DOI] [PubMed] [Google Scholar]
- Takahashi T., Otsuguro K., Ohta T., Ito S. Adenosine and inosine release during hypoxia in the isolated spinal cord of neonatal rats. Br J Pharmacol. 2010;161:1806–1816. doi: 10.1111/j.1476-5381.2010.01002.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terunuma A., Putluri N., Mishra P., Mathe E.A., Dorsey T.H., Yi M., Wallace T.A., Issaq H.J., Zhou M., Killian J.K., et al. MYC-driven accumulation of 2-hydroxyglutarate is associated with breast cancer prognosis. J Clin Invest, 2014;124:398–412. doi: 10.1172/JCI71180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden M.G., Cantley L.C., Thompson C.B. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaupel P., Mayer A. Hypoxia-driven adenosine accumulation: a crucial microenvironmental factor promoting tumor progression. Adv Exp Med Biol. 2016;876:177–183. doi: 10.1007/978-1-4939-3023-4_22. [DOI] [PubMed] [Google Scholar]
- Vesely M.D., Kershaw M.H., Schreiber R.D., Smyth M.J. Natural innate and adaptive immunity to cancer. Annu Rev Immunol. 2011;29:235–271. doi: 10.1146/annurev-immunol-031210-101324. [DOI] [PubMed] [Google Scholar]
- Vinogradov S., Wei X. Cancer stem cells and drug resistance: the potential of nanomedicine. Nanomedicine. 2012;7:597–615. doi: 10.2217/nnm.12.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visentin M., Zhao R., Goldman I.D. The antifolates. Hematol Oncol Clin North Am. 2012;26:629–648. ix. doi: 10.1016/j.hoc.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F., Travins J., DeLaBarre B., Penard-Lacronique V., Schalm S., Hansen E., Straley K., Kernytsky A., Liu W., Gliser C., et al. Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science. 2013;340:622–626. doi: 10.1126/science.1234769. [DOI] [PubMed] [Google Scholar]
- Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- Ward P.S., Patel J., Wise D.R., Abdel-Wahab O., Bennett B.D., Coller H.A., Fantin V.R., Hedvat C.V., Perl A.E., Rabinowitz J.D., et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 2010;17:225–234. doi: 10.1016/j.ccr.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb B.A., Chimenti M., Jacobson M.P., Barber D.L. Dysregulated pH: a perfect storm for cancer progression. Nat Rev Cancer. 2011;11:671–677. doi: 10.1038/nrc3110. [DOI] [PubMed] [Google Scholar]
- Xiao M., Yang H., Xu W., Ma S., Lin H., Zhu H., Liu L., Liu Y., Yang C., Xu Y., et al. Inhibition of alpha-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 2012;26:1326–1338. doi: 10.1101/gad.191056.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W., Yang H., Liu Y., Yang Y., Wang P., Kim S.H., Ito S., Yang C., Wang P., Xiao M.T., et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19:17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M., Soga T., Pollard P.J. Oncometabolites: linking altered metabolism with cancer. J Clin Invest. 2013;123:3652–3658. doi: 10.1172/JCI67228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M., Vousden K.H. Serine and one-carbon metabolism in cancer. Nat Rev Cancer. 2016;16:650–662. doi: 10.1038/nrc.2016.81. [DOI] [PubMed] [Google Scholar]
- Ye J., Fan J., Venneti S., Wan Y.W., Pawel B.R., Zhang J., Finley L.W., Lu C., Lindsten T., Cross J.R., et al. Serine catabolism regulates mitochondrial redox control during hypoxia. Cancer Discov. 2014;4:1406–1417. doi: 10.1158/2159-8290.CD-14-0250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W.C., Shyh-Chang N., Yang H., Rai A., Umashankar S., Ma S., Soh B.S., Sun L.L., Tai B.C., Nga M.E., et al. Glycine decarboxylase activity drives non-small cell lung cancer tumor-initiating cells and tumorigenesis. Cell. 2012;148:259–272. doi: 10.1016/j.cell.2011.11.050. [DOI] [PubMed] [Google Scholar]