

Abstract
Cordyceps militaris is a fungus used for developing health food, but knowledge about its intraspecific differentiation is limited due to lack of efficient markers. Herein, we assembled the mitochondrial genomes of eight C. militaris strains and performed a comparative mitochondrial genomic analysis together with three previously reported mitochondrial genomes of the fungus. Sizes of the 11 mitochondrial genomes varied from 26.5 to 33.9 kb mainly due to variable intron contents (from two to eight introns per strain). Nucleotide variability varied according to different regions with non-coding regions showing higher variation frequency than coding regions. Recombination events were identified between some locus pairs but seemed not to contribute greatly to genetic variations of the fungus. Based on nucleotide diversity fluctuations across the alignment of all mitochondrial genomes, molecular markers with the potential to be used for future typing studies were determined.
Cordyceps militaris, which generally parasitizes larva or pupa of lepidopteran insects, is the type species of the genus Cordyceps (Hypocreales, Ascomycetes). This fungus is distributed worldwide from 0 to >2000 m above sea level1. Its fruiting body has now been mass-produced artificially and developed into health food (i.e. not just nutritious in an ordinary sense, but eaten specifically for its health-promoting properties), and the species is one of the most representative and widely-used species in Cordyceps sensu lato1. Biologically active compounds (e.g., cordycepin, polysaccharides, cordymin) isolated from the fungus exhibit a variety of pharmacological effects, including anti-cancer, antioxidant, anti-inflammatory, immune-enhancing, or antifungal activities2,3,4,5. In addition to the interest in artificial cultivation and pharmacological effects, researchers have studied the fungus broadly from the viewpoints of genomics6, transcriptomics7,8, methylome9, and proteomics7.
As a species with a worldwide distribution and a broad host range1, C. militaris serves as an ideal material to understand evolutionary biology of fungi. Our knowledge about the intraspecific genetic diversity of C. militaris, however, has been rather limited, and its population genetic structure is far from clear due to lack of highly efficient molecular markers. For example, the maximum kimura-2-parameter genetic distance based on nrDNA ITS sequences among C. militaris isolates from Britain, China, Japan, Korea, and Norway was less than 0.0110, lower than the value of 0.04 reported in Ophiocordyceps sinensis, an medicinally important fungus that parasitize on ghost moth insects endemic to the Tibetan Plateau11. Low genetic variations of the fungus were also observed in mating-type genes MAT1-1-1 and MAT1-2-112. Considering the vast host ranges and its worldwide distribution under various environmental conditions1, we expect a genetic diversity at least higher than what’s currently known based on nrDNA ITS. The limited variation of C. militaris on nrDNA ITS suggests that the nrDNA ITS region is not an appropriate marker for intraspecific genetic diversity study of C. militaris though proved useful in O. sinensis11. Population and diversity analyses of C. militaris require robust and reliable molecular markers.
Compared to nuclear DNA, mitochondrial DNA is much susceptible to damage and mutations mainly because of the presence of reactive oxygen species generated during ATP synthesis13. The high mutation number and the faster evolutionary rate of mitochondrial DNA, from 5 to 10 times higher than nuclear DNA14, make mitochondrial DNA suitable for discrimination of closely related organisms and elucidation of recent evolutionary events. To date, intraspecific comparative analyses of mitochondrial genome have been performed in some fungal species, including Candida albicans15, Lachancea kluyveri16, Lachancea thermotolerans17, Mycosphaerella graminicola18, Neurospora crassa19, Podospora anserina20, and Rhizophagus irregularis21. High variability of mitochondrial sequences and sizes were often observed in these fungal species.
Recently, the mitochondrial genome of C. militaris was reported22, and mitochondrial intron presence/absence dynamics (two to eight introns per strain) was documented for the fungus23, indicating a high variability of mitochondrial DNA in the fungus. The objectives for this work were 1) to investigate if there are unknown insertion sites for mitochondrial introns in C. militaris, 2) to compare nucleotide variations among different mitochondrial regions, 3) to identify mitochondrial regions with high variability and determine novel molecular markers suitable for typing studies. Therefore, we assembled the complete mitochondrial genomes of eight additional C. militaris strains and performed a comparative mitochondrial genomic analysis together with three previously published mitochondrial genomes of the fungus.
Results
Overview of different C. militaris mitochondrial genomes
The mitochondrial genomes of eight additional C. militaris strains were successfully assembled in this study. Together with three strains, CM01, V26–17, and EFCC-C2, whose mitochondrial genomes were reported previously22,23, we performed a comparative mitochondrial genomic analysis. Each mitochondrial genome consists of two ribosomal RNA genes (rnl and rns), 27 tRNA genes, and 14 standard protein-coding genes of the oxidative phosphorylation system. Sizes of these mitochondrial genomes, however, varied from 26,535 to 33,967 bp mainly due to different numbers of introns, two to eight depending on strains (Table 1). Compared to our previous publication23, no novel insertion sites of introns were found. That is, mitochondrial introns of C. militaris seemed to occur only at eight possible sites, four in the rnl gene and one each in cob, cox1, cox2, and cox3 (Fig. 1). The second rnl intron (i.e., rnl-i2) was present in all strains, while each of the remaining seven introns could be absent in one or many strains (Table 1). Every intron contains an ORF encoding for either GIY-YIG homing endonucleases (for cob-i1, cox2-i1, rnl-i1, and rnl-i2), the LAGLIDADG homing endonucleases (for cox1-i1, cox3-i1, and rnl-i3), or ribosomal protein S3 (for rnl-i4). Besides introns, the rns-cox3 intergenic region (IR) also contributed to mitochondrial genome size variations. This region was 650–661 bp in two strains (EFCC-C2 and CM06) but 1,343 bp in other nine strains. A 327-bp ORF coding for a hypothetical protein was present in the longer rns-cox3 IR, but absent in the shorter rns-cox3 IR. Finally, although trivial, various small insertion/deletion (indel) regions, as depicted in the following sections, also contributed to variations of mitochondrial genome sizes.
Table 1. C. militaris isolates used in this study and information on their mitochondrial genomes.
| Isolate | Source | GenBank accession no. | Size (bp) | Introna |
No. introns | rns-cox3 IRa | Cladeb | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| cob-i1 | cox1-i1 | cox2-i1 | cox3-i1 | rnl-i1 | rnl-i2 | rnl-i3 | rnl-i4 | |||||||
| V26-17 | Company | KP719097 | 29478 | + | + | + | + | 4 | + | A | ||||
| V40-4 | Company | KP722512 | 27235 | + | + | 2 | + | B | ||||||
| V40-5 | Company | KP722501 | 33967 | + | + | + | + | + | + | + | + | 8 | + | D |
| CM09-9-24 | Liaoning, China | KP722496 | 28285 | + | + | + | 3 | + | C | |||||
| CM09-31-28 | Liaoning, China | KP722513 | 27245 | + | + | 2 | + | B | ||||||
| CM552 | Liaoning, China | KP722502 | 30662 | + | + | + | + | + | 5 | + | E | |||
| CM01 | unknown | KP719096 | 31854 | + | + | + | + | + | + | 6 | + | F | ||
| CM06 | unknown | KP722500 | 26535 | + | + | 2 | B | |||||||
| F02 | unknown | KP722505 | 31854 | + | + | + | + | + | + | 6 | + | F | ||
| CMB | unknown | KP722507 | 30026 | + | + | + | + | + | 5 | + | G | |||
| EFCC-C2 | unknown | NC_022834 | 33277 | + | + | + | + | + | + | + | + | 8 | D | |
a“+” Indicates the presence of an intron or the large rns-cox3 intergenic region (IR).
bThe clade here referred to those classified according to intron presence/absence patterns as depicted in our previous paper (Zhang et al. 2015).
Figure 1. The circular map of the mitochondrial genome of C. militaris V40-5.

rRNA genes were shown in blue; the 14 standard protein-encoding genes were shown in dark violet; tRNA genes were shown in red or in cyan. Introns were shown in yellow. The rns-cox3 intergenic ORF was shown in black.
Nucleotide variations at genic regions
Alignment of the 11 C. militaris mitochondrial genomes generated 34,067 positions, where approximately 87% comprises genic regions. Nucleotide variations at genic regions were inspected according to exonic (58% of the alignment) and intronic (29%) regions. At exonic regions of the 14 standard protein-coding genes, no indels were detected, but SNPs, from one in nad3 to 24 in nad5, occurred at all genes with exception of atp8, atp9, and nad6 (Table 2). Most SNPs are synonymous, but some from four genes (cox3, nad2, nad4, and nad5) resulted in one to five amino acid changes. For tRNA genes, only one SNP (either unique to one strain or shared by more than one strain) was detected in each of seven tRNA genes (Table 2); no any variation was found in other tRNA genes. For the two rRNA genes, both SNPs and indels were detected (Table 2). Actually, at exonic regions of all mitochondrial genes, indels were detected just from rns and rnl, and each indel was unique to one strain.
Table 2. Variations at exonic regions of each mitochondrial gene among 11 C. militaris strains.
| Gene | Exon |
Protein |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Length (bp) | Pi | S | Indel | Total | % | Length (aa) | Pi | S | Indel | Total | |
| atp6 | 783 | 1 | 3 | 0 | 4 | 0.5 | 260 | 0 | 0 | 0 | 0 |
| atp8 | 147 | 0 | 0 | 0 | 0 | 0.0 | 48 | 0 | 0 | 0 | 0 |
| atp9 | 225 | 0 | 0 | 0 | 0 | 0.0 | 74 | 0 | 0 | 0 | 0 |
| cob | 1161 | 6 | 6 | 0 | 12 | 1.0 | 386 | 0 | 0 | 0 | 0 |
| cox1 | 1593 | 4 | 7 | 0 | 11 | 0.7 | 530 | 0 | 0 | 0 | 0 |
| cox2 | 750 | 7 | 2 | 0 | 9 | 1.2 | 249 | 0 | 0 | 0 | 0 |
| cox3 | 810 | 5 | 7 | 0 | 12 | 1.5 | 269 | 1 | 0 | 0 | 1 |
| nad1 | 1104 | 3 | 4 | 0 | 7 | 0.6 | 367 | 0 | 0 | 0 | 0 |
| nad2 | 1686 | 4 | 8 | 0 | 12 | 0.7 | 561 | 1 | 4 | 0 | 5 |
| nad3 | 420 | 1 | 0 | 0 | 1 | 0.2 | 139 | 0 | 0 | 0 | 0 |
| nad4 | 1449 | 2 | 9 | 0 | 11 | 0.8 | 482 | 2 | 1 | 0 | 3 |
| nad4L | 270 | 3 | 0 | 0 | 3 | 1.1 | 89 | 0 | 0 | 0 | 0 |
| nad5 | 1995 | 12 | 12 | 0 | 24 | 1.2 | 664 | 1 | 1 | 0 | 2 |
| nad6 | 633 | 0 | 0 | 0 | 0 | 0.0 | 210 | 0 | 0 | 0 | 0 |
| rns | 1538–1558 | 3 | 1 | 23 | 27 | 1.7 | |||||
| rnl | 3120–3189 | 4 | 3 | 75 | 82 | 2.6 | |||||
| tRNA-Leu (TAA) | 82 | 1 | 0 | 0 | 1 | 1.2 | |||||
| tRNA-Phe (GAA) | 73 | 0 | 1 | 0 | 1 | 1.4 | |||||
| tRNA-Lys (TTT) | 73 | 0 | 1 | 0 | 1 | 1.4 | |||||
| tRNA-Gln (TTG) | 73 | 1 | 0 | 0 | 1 | 1.4 | |||||
| tRNA-His (GTG) | 73 | 0 | 1 | 0 | 1 | 1.4 | |||||
| tRNA-Arg (ACG) | 71 | 1 | 0 | 0 | 1 | 1.4 | |||||
| tRNA-Arg (CCT) | 71 | 0 | 1 | 0 | 1 | 1.4 | |||||
| Other 20 tRNAs | 1481 | 0 | 0 | 0 | 0 | 0.0 | |||||
| Total | 19681–19770 | 58 | 66 | 98 | 222 | 1.1 | 4328 | 5 | 6 | 0 | 11 |
Pi, parsimony informative sites; S, singleton sites; indel, insertion/deletion sites; %, the percentage of total variable sites (Pi, S, and indels) in each gene.
At intronic regions, no variation was found in cox3-i1, but SNPs and often indels were found in the remaining seven introns (Table 3). SNPs from introns of protein-coding genes occurred mostly at intronic ORF regions, while SNPs from introns of rnl occurred mostly at regions beyond intronic ORFs. Changes of amino acids were detected for all intronic proteins with exception of those present in cox3-i1 and rnl-i3, each of which was only possessed by two strains. Indels were found mainly at regions beyond intronic ORFs, but there were two exceptions. One is a 6-bp deletion found in the intronic ORF of rnl-i2 from CM09-9-24, which led to the deletion of two amino acids. The other is a truncated version of the intronic ORF of cox2-i1 from EFCC-C2, where a 62-bp sequence repeated twice in tandem and led to early translation stop. Unfortunately, mitochondrial sequences of EFCC-C2 reported by Sung (2015) could not be verified in this study.
Table 3. Variations at intronic regions of mitochondrial genes among 11 C. militaris strains.
| Intron | No. strains | Intron |
Intronic ORF |
Intronic protein |
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Length (bp) | Pi | S | Indel | Total | % | Length (bp) | Pi | S | Indel | Total | % | Length (aa) | Pi | S | Indel | Total | ||
| cob-i1 | 6 | 1256–1257 | 2 | 9 | 1 | 12 | 1.0 | 918 | 1 | 8 | 0 | 9 | 1.0 | 305 | 0 | 5 | 0 | 5 |
| cox1-i1 | 8 | 1051 | 2 | 4 | 0 | 6 | 0.6 | 945 | 2 | 3 | 0 | 5 | 0.5 | 314 | 0 | 1 | 0 | 1 |
| cox2-i1a | 6 | 1125–1187 | 1 | 5 | 62 (0) | 68 (6) | 5.7 (0.5) | 861 | 0 | 4 | 0 | 4 | 0.5 | 286 | 0 | 3 | 0 | 3 |
| cox3-i1 | 2 | 1213 | 0 | 0 | 0 | 0 | 0.0 | 933 | 0 | 0 | 0 | 0 | 0.0 | 310 | 0 | 0 | 0 | 0 |
| rnl-i1 | 6 | 1191–1192 | 2 | 7 | 1 | 10 | 0.8 | 738 | 1 | 3 | 0 | 4 | 0.5 | 245 | 1 | 1 | 0 | 2 |
| rnl-i2 | 11 | 1235–1241 | 1 | 2 | 6 | 9 | 0.7 | 756–762 | 1 | 1 | 6 | 8 | 1.0 | 251–253 | 1 | 1 | 2 | 4 |
| rnl-i3 | 2 | 893 | 0 | 2 | 0 | 2 | 0.2 | 624 | 0 | 1 | 0 | 1 | 0.2 | 207 | 0 | 0 | 0 | 0 |
| rnl-i4 | 10 | 1821–1828 | 1 | 11 | 7 | 19 | 1.0 | 1323 | 0 | 4 | 0 | 4 | 0.3 | 440 | 0 | 2 | 0 | 2 |
| Total | 3062–9861 | 9 | 40 | 77 (15) | 126 (64) | 7098–7104 | 5 | 24 | 6 | 35 | 0.5 | 2358–2360 | 2 | 13 | 2 | 17 | ||
aThe large number of indels present in cox2-i1 was due to an extra tandem repeat of a 62-bp fragment (at 200 bp 5′ normal stop codon), which was present merely in the strain EFCC-C2 and led to early translation stop of the intronic ORF. We could not confirm sequences from this strain, therefore, variations in cox2-i1 were also calculated by deleting this extra repeat and values after the repeat deletion were given within parenthesis. This repeat was also represented only once when calculating nucleotide variation indices for intronic ORF and intronic protein in this table.
When we compared the divergence of exons and introns, exons were more divergent than introns in protein-encoding genes, while introns were more divergent than exons in rnl. When looking at all intron-containing genes, variability at intronic regions was similar to that at exonic regions (Tables 2 and 3).
Nucleotide variations at intergenic regions
Intergenic regions only accounted for approximately 13% (5,713–6,415 bp) of C. militaris mitochondrial genome, but both SNPs and indels were frequently detected (Table 4). Some indels (e.g., those found at nad3-atp9, cox1-nad1, and cox3-nad6 IRs) were shared by more than one strain. For the rns-cox3 IR, 9 of the 11 strains possessed a large region with an additional ORF, and two strains possessed a short region lacking the ORF. Nucleotide variability at the short rns-cox3 IR, however, was higher than that at the large rns-cox3 IR. When comparing the nucleotide variation frequencies, we found that intergenic regions were more variable than genic regions (Tables 2, 3, 4, ).
Table 4. Variations at intergenic regions of mitochondrial genes among 11 C. militaris strains.
| Intergenic regiona | No. strains | Actual length (bp) | Remaining length (bp)c | Pi | S | Indel | Total | %c | Note |
|---|---|---|---|---|---|---|---|---|---|
| rnl-nad2 | 11 | 1056–1065 | 166–175 | 4 | 0 | 9 | 13 | 7.6 | 12 tRNAs excluded |
| nad3-atp9 | 11 | 278–286 | 278–286 | 3 | 3 | 8 | 14 | 4.9 | |
| atp9-cox2 | 11 | 176–186 | 176–186 | 7 | 2 | 10 | 19 | 10.5 | |
| cox2-nad4L | 11 | 200–201 | 129–130 | 1 | 1 | 1 | 3 | 2.3 | 1 tRNA excluded |
| nad5-cob | 11 | 183 | 183 | 2 | 3 | 0 | 5 | 2.7 | |
| cob-cox1 | 11 | 529–536 | 457–464 | 1 | 4 | 9 | 14 | 3.0 | 1 tRNA excluded |
| cox1-nad1 | 11 | 866–879 | 795–808 | 3 | 6 | 21 | 30 | 3.7 | 1 tRNA excluded |
| nad1-nad4 | 11 | 242 | 242 | 0 | 5 | 0 | 5 | 2.1 | |
| nad4-atp8 | 11 | 67 | 67 | 0 | 0 | 0 | 0 | 0.0 | |
| atp8-atp6 | 11 | 80 | 80 | 0 | 0 | 0 | 0 | 0.0 | |
| atp6-rns | 11 | 356–358 | 356–358 | 0 | 0 | 3 | 3 | 0.8 | |
| large rns-cox3 IRb | 9 | 1343 | 963 | 0 | 1 | 0 | 1 | 0.1 | 5 tRNAs excluded |
| small rns-cox3 IRb | 2 | 650–661 | 270–281 | 0 | 5 | 11 | 16 | 5.8 | 5 tRNAs excluded |
| cox3-nad6 | 11 | 436–457 | 293–314 | 5 | 6 | 21 | 32 | 10.5 | 2 tRNAs excluded |
| nad6-rnl | 11 | 551 | 178 | 0 | 5 | 0 | 5 | 2.8 | 5 tRNAs excluded |
| Total | 5713–6415 | 3713–4415 | 26 | 41 | 93 | 160 | 3.6–4.3 | 27 tRNAs excluded |
aIntergenic regions are named by adjacent protein-encoding genes and/or ribosomal genes. tRNAs present in some of these fragments were excluded during calculation as noted in this table.
bFor the large rns-cox3 IR and the small rns-cox3 IR, identical five tRNAs were excluded. Therefore, the total number of tRNAs was still 27.
c“Remaining length” represents the length after excluding tRNAs. % was calculated based on remaining length.
Recombination among different mitochondrial regions
The overall index of association for exonic, intronic, and intergenic loci all rejected panmictic recombination (Table S1). Few phylogenetically incompatible locus pairs, however, were found because the proportion of compatible locus pairs was lower than 100%. Manual inspections found allelic combinations showing evidence of recombination between rnl-E1 and rnl-E2/nad3/cox2-E1, between rnl-E2 and nad3/cox2-E1/nad4L/nad3-atp9 IR/atp9-cox2 IR/cox2-nad4L IR/nad5-cob IR (Table S2).
Development of potential molecular markers
Nucleotide diversity across the alignment of 11 C. militaris mitochondrial genomes revealed some mutation hot regions (Figs 2 and 3), such as the rnl-nad2 intergenic region (named VG1), the region from 3′ nad3 to 5′ cox2 (VG2), the region from 3′ cox2 to 5′ cob (VG3), the region from 3′ cox1 to 5′ nad1 (VG4), the region from 3′ nad1 to 3′ nad4 (VG5), and the region from 3′ cox3 to 5′ nad6 (VG6). These fragments contained almost half of the total SNP sites detected among the 11 mitochondrial genomes (Fig. 3), showing the potential as novel molecular markers (Table 5).
Figure 2. Nucleotide diversity fluctuations across the alignment of 11 C. militaris mitochondrial genomes.
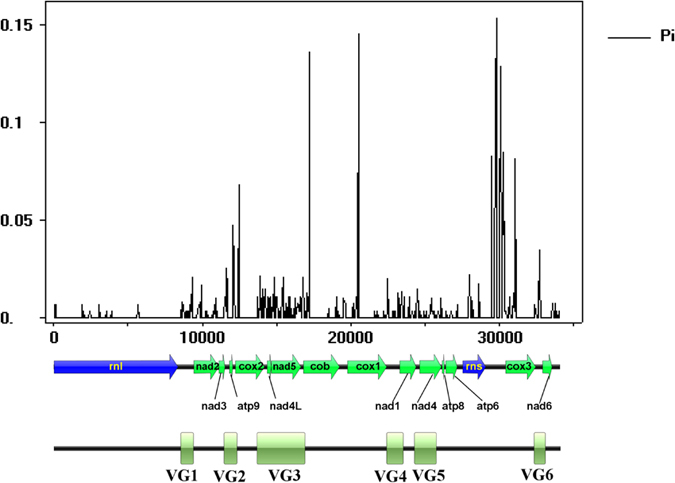
Gaps were considered during window sliding, but nucleotide variations at gap positions were not considered. The high nucleotide diversity at rns-cox3 intergenic region was due to ambiguous alignment as a result of presence of two different-length sequences among these genomes. The rns-cox3 intergenic region was not considered when selecting novel molecular markers. The relative positions of 14 standard protein-encoding genes, two ribosomal genes, and the six molecular markers were illustrated below the graph.
Figure 3. Variable sites across the alignment of 11 C. militaris mitochondrial genomes.

Digits above nucleotides indicate the nucleotide positions of each polymorphic site in the aligned sequences, reading from top to bottom. Dots mean identical nucleotides to the first sequence; dashes indicate indel. “***” represents an 864-bp sub-region from the rns-cox3 intergenic region; this sub-region was deleted due to ambiguous alignment. A total of 240 variable sites were detected with those present in indels considered. Relative positions of the six variable regions (VG1-6) were indicated in green boxes.
Table 5. Nucleotide variations and neutrality tests on the six fragments chosen as potential molecular markers.
| Locus | Actual length (bp) | Aligned length (bp) | Pi | S | Indel | Total | % | No. alleles | Nucleotide diversitya | Haplotype diversity a | Tajima’s D | Fu & Li’s D* | Fu & Li’s F* | P valueb |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| VG1 | 875–884 | 884 | 6 | 3 | 9 | 18 | 2.04 | 6 | 0.0035 ± 0.0007 | 0.800 ± 0.114 | 0.0248 | 0.1597 | 0.1420 | >0.1 |
| VG2 | 878–888 | 888 | 11 | 5 | 18 | 34 | 3.83 | 3 | 0.0079 ± 0.0013 | 0.618 ± 0.104 | 1.1208 | 0.2556 | 0.5389 | >0.1 |
| VG3 | 3230–3231 | 3231 | 25 | 17 | 1 | 43 | 1.33 | 7 | 0.0050 ± 0.0008 | 0.873 ± 0.089 | 0.6392 | −0.1260 | 0.0798 | >0.1 |
| VG3A | 1509–1510 | 1510 | 16 | 3 | 1 | 20 | 1.32 | 5 | 0.0059 ± 0.0008 | 0.782 ± 0.107 | ||||
| VG3B | 1850 | 1850 | 9 | 14 | 0 | 23 | 1.24 | 6 | 0.0039 ± 0.0009 | 0.836 ± 0.089 | ||||
| VG4 | 1119–1132 | 1132 | 4 | 7 | 21 | 32 | 2.83 | 6 | 0.0031 ± 0.0005 | 0.836 ± 0.089 | −0.6617 | −1.1442 | −1.1561 | >0.1 |
| VG5 | 1474 | 1474 | 1 | 15 | 0 | 16 | 1.09 | 5 | 0.0022 ± 0.0009 | 0.782 ± 0.093 | −1.8166 | −2.2777 | −2.4466 | <0.05 |
| VG6 | 750–771 | 771 | 6 | 7 | 21 | 34 | 4.41 | 4 | 0.0043 ± 0.0020 | 0.491 ± 0.175 | −1.1854 | −0.6463 | −0.8864 | >0.1 |
| Combined 6-locus dataset | 8348–8365 | 8380 | 53 | 54 | 70 | 177 | 2.11 | 8 | 0.0044 ± 0.0008 | 0.891 ± 0.092 | ||||
| Combined 5-locus dataset excluding VG5 | 6874–6891 | 6906 | 52 | 39 | 70 | 161 | 2.33 | 8 | 0.0048 ± 0.0008 | 0.891 ± 0.092 |
aNumbers after “±” were standard deviations.
bP values are those for denying neutrality.
To determine whether the above six VGs were not under positive selection, we performed the Tajima’s D, and Fu and Li’s D* and F* tests of neutrality for each of them. The values obtained were not significantly different from zero with exception of VG5 (Table 5). Therefore, VGs1–4 and VG6 showed no deviation from the neutral model of evolution and are probably not under selective pressure. Mutations in VG5 are probably affected by natural selection, weakening its potential use in typing studies due to its constrained variability.
The combined sequence of VGs1–4 and 6 revealed eight haplotypes among the 11 strains (Table 5). When considering the complete mitochondrial genomes, the 11 strains also belonged to eight genotypes. Phylogeny constructed using the concatenated five-locus dataset was congruent to that constructed using the whole mitochondrial genome (Fig. 4). Using designed primers (Table S3), expected bands of the five fragments can be amplified from C. militaris strains (Fig. S1).
Figure 4. Tanglegram between phylogeny constructed using the whole mitochondrial genome (left) and that constructed using five loci (VGs 1-4, 6; right).
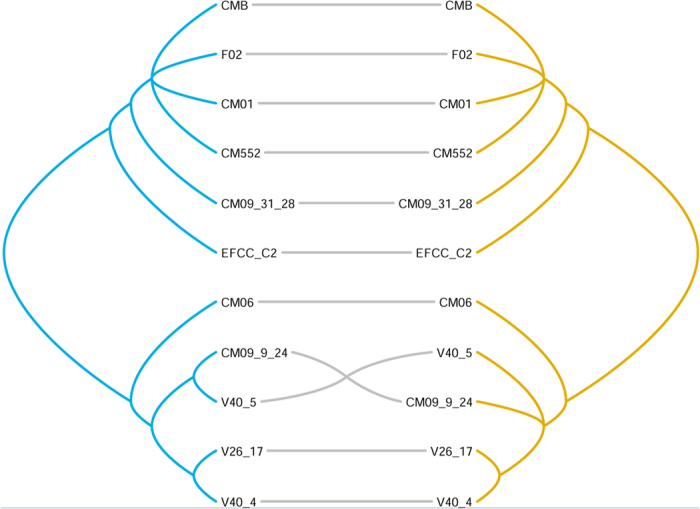
Please refer to Table 1 for the information of the 11 C. militaris strains used in this figure.
Discussion
In this study, we assembled successfully the complete mitochondrial genomes of eight C. militaris strains and performed a comparative mitochondrial genomic analysis together with three previously published mitochondrial genomes. All mitochondrial genomes were assembled based on Sanger sequencing results, and sequence errors were reduced to a minimum level as a result of the use of high-fidelity DNA polymerase and the manual check of sequencing chromatograms. The genome alignment of 34,067 positions revealed 286 polymorphic sites in genic regions (222 at exonic regions and >64 at intronic regions) and 160 polymorphic sites in intergenic regions among the 11 C. militaris strains (Tables 2, 3, 4, ). However, nucleotide variability at intergenic regions was higher than that at genic regions because intergenic regions (~13% of the whole mitochondrial genome) were much shorter than genic regions (~87%). With exception of one to five non-synonymous changes in cox3, nad2, nad4, and nad5, the majority of mutations in exonic regions of the 14 standard protein-coding genes were synonymous and located in the third codon positions (Table 2). In contrast, intronic ORFs were prone to amino acid changes (Table 3). Overall, variation levels are similar between intronic and exonic regions.
In addition to C. militaris, intraspecific comparisons of whole mitochondrial genomes have so far been performed in seven other fungal species15,16,17,18,19,20,21. These studies all revealed within-species variations of mitochondrial genomes. Intron presence/absence dynamics were clearly documented in Lachancea kluyveri16, Lachancea thermotolerans17, Podospora anserina20, and Rhizophagus irregularis21. Higher nucleotide variability at intergenic regions than at genic regions was also documented in Candida albicans15 and Lachancea kluyveri16.
Although we detected recombination events between rnl exons and some other coding or non-coding regions (Table S2), the overall evidence for panmictic recombination was insignificant (Table S1). Therefore, recombination seems not to contribute a lot to the above observed nucleotide variability. Because the mechanism for mitochondrial inheritance of C. militaris has not been investigated, how mitochondrial inheritance affects mitochondrial variations cannot be determined. However, as a heterothallic fungus6, there are chances for the generation of heteroplasmons when two C. militaris strains with opposite mating types fuse.
As a fungus with a worldwide distribution and a broad host range, C. militaris is expected to display a high intraspecific genetic diversity. Previous studies, however, failed to reveal a high intraspecific genetic differentiation based on nuclear DNA fragments10. The population genetic structure of C. militaris is far from clear due to lack of efficient molecular markers. By comparing mitochondrial genomes of different C. militaris strains, this study identified six mitochondrial DNA fragments (VG1–6) with potential as molecular markers. Among them, VG5 was later excluded due to its deviation from neutrality. The remaining five fragments (VG1–4 and VG6) showed the same number of haplotypes as the whole mitochondrial genome and were phylogenetically congruent to the whole mitochondrial genome. We, however, have to admit that C. militaris isolates employed in this study were all from China or Chinese companies, more SNPs might be detected when isolates from other countries were tested. Overall, these mitochondrial DNA fragments determined in this study are suitable to be used as molecular markers in future typing studies in C. militaris.
Materials and Methods
Fungal cultures
Based on our previous study23, eight additional C. militaris strains that showed multiple intron distribution patterns (Table 1) were chosen to assemble their complete mitochondrial genomes in this study. These isolates were cultivated at 25 °C for 10 days in potato dextrose agar media with a piece of cellophane paper covering the medium surface. Mycelia were collected and used for extracting genomic DNA using the cetyltrimethylammonium bromide method24.
Assembly of mitochondrial genomes and sequence annotations
Based on the published mitochondrial genome of C. militaris CM0123, multiple PCR primer pairs were designed to amplify mitochondrial fragments from strains used in this study. PCRs were performed using the DNA polymerase KOD FX (TOYOBO Bio-Technology Co. LTD, Japan), and sequences of amplicons were determined by Sanger sequencing at GENEWIZ Inc. (Suzhou, China). Different mitochondrial fragments of the same strain were assembled together under the aid of overlapping sequences. Annotation of mitochondrial genomes referred to those depicted previously23.
Sequence alignment and nucleotide variation analysis
Mitochondrial genome sequences from 11 C. militaris strains, eight from this study plus three from previous publications, were aligned by the online program MAFFT version 7 (http://mafft.cbrc.jp/alignment/server/). Individual exonic, intronic, and intergenic regions were extracted from the alignment using a perl script selectSites.pl written by Naoki Takebayashi (http://raven.iab.alaska.edu/~ntakebay/teaching/programming/perl-scripts/perl-scripts.html). Nucleotide variations, including parsimony informative sites, singleton sites, and indels, were summarized for each extracted region in MEGA 6.0625.
Recombination analysis
To know whether mitochondrial recombination might have contributed to the observed nucleotide variations, two complementary tests were conducted to examine evidence for recombination. In the first test, we examined allelic associations among alleles from different loci using the index of association (IA). In the second test, we calculated the phylogenetic incompatibility by looking for the proportion of loci with all possible recombinant genotypes. Both tests were performed using MultiLocus26 with 1,000 randomizations. Twenty-four exonic loci (five rnl exonic regions, two cob exonic regions, two cox1 exonic regions, two cox2 exonic regions, two cox3 exonic regions, atp6, atp8, atp9, nad1, nad2, nad3, nad4, nad4L, nad5, nad6, and rns), 8 intronic loci (rnl-i1, rnl-i2, rnl-i3, rnl-i4, cob-i1, cox1-i1, cox2-i1, and cox3-i1), and 15 intergenic loci (i.e., those among 14 standard protein-coding genes and ribosomal RNA genes) were examined.
Development of molecular markers
Nucleotide diversity across the alignment of 11 mitochondrial genomes was calculated in DnaSP 5.1027 using a sliding window of 50-bp window length and 50-bp step size with alignment gaps counted in the window length and slide. Regions with dense distribution of single nucleotide polymorphism (SNP) sites were investigated as potential molecular markers.
Neutrality test
For the above-selected markers, statistical tests of Tajima’s D and Fu and Li’s D* and F* for detection of deviation from the neutral model of evolution were performed using DnaSP 5.1027. Those without significant deviation from neutrality are suitable as molecular markers in future typing studies.
Phylogenetic tree construction and comparison
To know if the novel molecular markers are phylogenetically congruent to the complete mitochondrial genomes, we constructed phylogenetic trees using two datasets. One is the combined dataset of new markers chosen in the above section, and the other is the complete mitochondrial genomes but with partial rns-cox3 intergenic region excluded because of ambiguous alignment. Maximum parsimony trees were constructed using PAUP 4.0b10 (Sinauer Associates, Sunderland, MA, USA) with gaps being treated as missing data. A tanglegram was constructed from both trees using TREEMAP 3b124328.
Nucleotide accession numbers
Sequences of the eight newly assembled mitochondrial genomes of C. militaris were all submitted to GenBank under accession numbers as depicted in Table 1.
Additional Information
How to cite this article: Zhang, S. et al. Comparative mitochondrial genomics toward exploring molecular markers in the medicinal fungus Cordyceps militaris. Sci. Rep. 7, 40219; doi: 10.1038/srep40219 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Material
Acknowledgments
This study was funded by the National Science Foundation of China (81102759), the Natural Science Foundation of Shanxi Province (2014021030-2, 201601D011065), the Fund Program for the Scientific Activities of Selected Returned Overseas Professionals in Shanxi Province, and the Special Fund for Large Scientific Instruments and Equipments in Shanxi Province.
Footnotes
Author Contributions Conceived and designed the experiments: S.Z. and Y.J.Z. Performed the experiments: A.J.H. and Y.X.Z. Analyzed the data: S.Z., X.Y.Z. and Y.J.Z. Wrote the paper: S.Z. and Y.J.Z.
References
- Shrestha B., Zhang W., Zhang Y. & Liu X. The medicinal fungus Cordyceps militaris: research and development. Mycological Progress 11, 599–614 (2012). [Google Scholar]
- Tuli H. S., Sharma A. K., Sandhu S. S. & Kashyap D. Cordycepin: A bioactive metabolite with therapeutic potential. Life Sciences 93, 863–869 (2013). [DOI] [PubMed] [Google Scholar]
- Wang M. et al. Cordyceps militaris polysaccharides can improve the immune efficacy of Newcastle disease vaccine in chicken. International Journal of Biological Macromolecules 59, 178–183 (2013). [DOI] [PubMed] [Google Scholar]
- Wong J. H. et al. Cordymin, an antifungal peptide from the medicinal fungus Cordyceps militaris. Phytomedicine 18, 387–392 (2011). [DOI] [PubMed] [Google Scholar]
- Das S. K., Masuda M., Sakurai A. & Sakakibara M. Medicinal uses of the mushroom Cordyceps militaris: Current state and prospects. Fitoterapia 81, 961–968 (2010). [DOI] [PubMed] [Google Scholar]
- Zheng P. et al. Genome sequence of the insect pathogenic fungus Cordyceps militaris, a valued traditional Chinese medicine. Genome Biology 12, R116 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Y. et al. Genome-wide transcriptome and proteome analysis on different developmental stages of Cordyceps militaris. Plos One 7, e51853 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong C., Xia Y., Zheng P., Shi S. & Wang C. Developmental stage-specific gene expression profiling for a medicinal fungus Cordyceps militaris. Mycology 1, 25–66 (2010). [Google Scholar]
- Wang Y.-l., Wang Z.-x., Liu C., Wang S.-b. & Huang B. Genome-wide analysis of DNA methylation in the sexual stage of the insect pathogenic fungus Cordyceps militaris. Fungal Biology 119, 1246–1254 (2015). [DOI] [PubMed] [Google Scholar]
- Wang L., Zhang W., Hu B., Chen Y. & Qu L. Genetic variation of Cordyceps militaris and its allies based on phylogenetic analysis of rDNA ITS sequence data. Fungal Diversity 31, 147–155 (2008). [Google Scholar]
- Zhang Y. et al. Genetic diversity of Ophiocordyceps sinensis, a medicinal fungus endemic to the Tibetan Plateau: Implications for its evolution and conservation. BMC Evolutionary Biology 9, 290 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Guo L., Zhang S. & Liu X. Determining mitochondrial molecular markers suitable for genetic diversity analysis of Cordyceps militaris. Acta Microbiologica Sinica 55, 826–833 (2015). [PubMed] [Google Scholar]
- Alexeyev M., Shokolenko I., Wilson G. & LeDoux S. The maintenance of mitochondrial DNA integrity–Critical analysis and update. Cold Spring Harbor perspectives in biology 5, a012641 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown W. M., George M. & Wilson A. C. Rapid evolution of animal mitochondrial DNA. Proceedings of the National Academy of Sciences 76, 1967–1971 (1979). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartelli T. F., Ferreira R. C., Colombo A. L. & Briones M. R. S. Intraspecific comparative genomics of Candida albicans mitochondria revealsnon-coding regions under neutral evolution. Infection, Genetics and Evolution 14, 302–312 (2013). [DOI] [PubMed] [Google Scholar]
- Jung P. P., Friedrich A., Reisser C., Hou J. & Schacherer J. Mitochondrial genome evolution in a single protoploid yeast species. G3-Genes Genomes Genetics 2, 1103–1111 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freel K. C., Friedrich A., Hou J. & Schacherer J. Population genomic analysis reveals highly conserved mitochondrial genomes in the yeast species Lachancea thermotolerans. Genome Biology and Evolution 6, 2586–2594 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torriani S. F. F., Goodwin S. B., Kema G. H. J., Pangilinan J. L. & McDonald B. A. Intraspecific comparison and annotation of two complete mitochondrial genome sequences from the plant pathogenic fungus Mycosphaerella graminicola. Fungal Genetics and Biology 45, 628–637 (2008). [DOI] [PubMed] [Google Scholar]
- McCluskey K. Variation in mitochondrial genome primary sequence among whole-genome-sequenced strains of Neurospora crassa. IMA Fungus 3, 93–98 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings D. J., McNally K. L., Domenico J. M. & Matsuura E. T. The complete DNA sequence of the mitochondrial genome of Podospora anserina. Current Genetics 17, 375–402 (1990). [DOI] [PubMed] [Google Scholar]
- Formey D. et al. Comparative analysis of mitochondrial genomes of Rhizophagus irregularis - syn. Glomus irregulare - reveals a polymorphism induced by variability generating elements. New Phytologist 196, 1217–1227 (2012). [DOI] [PubMed] [Google Scholar]
- Sung G.-H. Complete mitochondrial DNA genome of the medicinal mushroom Cordyceps militaris (Ascomycota, Cordycipitaceae). Mitochondrial DNA 26, 789–790 (2015). [DOI] [PubMed] [Google Scholar]
- Zhang Y. et al. Comparison of mitochondrial genomes provides insights into intron dynamics and evolution in the caterpillar fungus Cordyceps militaris. Fungal Genetics and Biology 77, 95–107 (2015). [DOI] [PubMed] [Google Scholar]
- Zhang Y. J., Zhang S., Liu X. Z., Wen H. A. & Wang M. A simple method of genomic DNA extraction suitable for analysis of bulk fungal strains. Letters in Applied Microbiology 51, 114–118 (2010). [DOI] [PubMed] [Google Scholar]
- Tamura K., Stecher G., Peterson D., Filipski A. & Kumar S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol 30, 2725–2729 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agapow P. M. & Burt A. Indices of multilocus linkage disequilibrium. Molecular Ecology Notes 1, 101–102 (2001). [Google Scholar]
- Librado P. & Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452 (2009). [DOI] [PubMed] [Google Scholar]
- Charleston M. A. TreeMap 3b, Available at: https://sites.google.com/site/cophylogeny (2011).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.