

Abstract
INTRODUCTION
Breast cancer being a multifaceted disease constitutes a wide spectrum of histological and molecular variability in tumors. However, the task for the identification of these variances is complicated by the interplay between inherited genetic and epigenetic aberrations. Therefore, this study provides an extrapolate outlook to the sinister partnership between DNA methylation and single-nucleotide polymorphisms (SNPs) in relevance to the identification of prognostic markers in breast cancer. The effect of these SNPs on methylation is defined as methylation quantitative trait loci (meQTL).
MATERIALS AND METHODS
We developed a novel method to identify prognostic gene signatures for breast cancer by integrating genomic and epigenomic data. This is based on the hypothesis that multiple sources of evidence pointing to the same gene or pathway are likely to lead to reduced false positives. We also apply random resampling to reduce overfitting noise by dividing samples into training and testing data sets. Specifically, the common samples between Illumina 450 DNA methylation, Affymetrix SNP array, and clinical data sets obtained from the Cancer Genome Atlas (TCGA) for breast invasive carcinoma (BRCA) were randomly divided into training and test models. An intensive statistical analysis based on log-rank test and Cox proportional hazard model has established a significant association between differential methylation and the stratification of breast cancer patients into high- and low-risk groups, respectively.
RESULTS
The comprehensive assessment based on the conjoint effect of CpG–SNP pair has guided in delaminating the breast cancer patients into the high- and low-risk groups. In particular, the most significant association was found with respect to cg05370838–rs2230576, cg00956490–rs940453, and cg11340537–rs2640785 CpG–SNP pairs. These CpG–SNP pairs were strongly associated with differential expression of ADAM8, CREB5, and EXPH5 genes, respectively. Besides, the exclusive effect of SNPs such as rs10101376, rs140679, and rs1538146 also hold significant prognostic determinant.
CONCLUSIONS
Thus, the analysis based on DNA methylation and SNPs have resulted in the identification of novel susceptible loci that hold prognostic relevance in breast cancer.
Keywords: DNA methylation, single-nucleotide polymorphism, meQTLs, overall survival
Introduction
Variation in gene expression transforms cellular programming from normal to diseased state. The multiple genetic circuits within a cell create a characteristic signature profile of gene expression endorsing each cell a unique identity. These gene expression-based signatures have been successfully implemented in classifying breast cancer into different subtypes.1,2 Similarly, approaches based on genome-wide DNA methylation profiling identified breast cancer-specific methylation signatures that correlate with specific clinical outcomes.3 In addition to diagnostic potential, aberrations in DNA methylation profile regulate gene expression, dictating tumor recurrence and overall survival (OS) in breast cancer and their subtypes.4–9 The prognostic potential of genes, mainly FLRT2 and SFRP1, has been identified to be regulated by DNA methylation, and these genes are enriched in ER1/luminal B of breast cancer. However, the expression of specific genes related to immune function, such as CD3D, CD79B, CD6, HCLS1, HLA-A, and lAX1, have been identified to be consistently associated with recurrence-free survival and OS in subtypes of breast cancer.10,11 Furthermore, the combination of methylated genes such as GSTP1, FOXC1, and ABCB1 have been correlated with respect to the survival of patients.12 The deregulation of DNA methylation has been significantly correlated with the expression of BCAP31 and OGG1 genes and has shown significant association to survival in a large cohort of breast cancer patients.13 Besides, differential methylation of CpG islands proximal to the genes regulating cell cycle and proliferation (HDAC4, KIF2C, Ki-67, and UBE2C), angiogenesis (BTG1, KLF5, and VEGF), and cell fate determination (LHX2, LXH2, OLIG2, and SPRY1) possesses significant prognostic values independent of the subtypes and clinical features.14
Genome-wide association studies (GWASs) have identified a large number of genomic variants associated with complex diseases, including breast cancer.15–17 However, most of the disease-associated genomic variants that have been reported in the literature so far are predominantly located in the intergenic or intronic regions of the genome.18 Furthermore, numerous studies have noted that GWAS haplotypes are enriched in regulatory elements that are concordant with the disease phenotypes.19 Therefore, it is highly likely that most of the disease-causing genomic variations act by altering gene regulation, such as transcription factor binding and DNA methylation, rather than directly affecting protein function.
Despite the advances in sequencing and availability of multi-omics data sets,20,21 finding causative and prognostic genetic variants for complex diseases, such as breast cancer, remains challenging. Thus, a robust method of associating genomic variants, such as SNPs, in regulatory regions, such as CpG islands, with corresponding DNA methylation alterations is required.22 The influence of these genetic variants on DNA methylation level was referred to as cis-methylation quantitative trait loci (cis-meQTLs).23,24 Here, we report the joint effect of meQTLs on clinicopathological variables for the identification of prognostic biomarkers, their clinical validity, and the extent to which they capture the pathological difference between breast cancer prognostic groups using these external independent studies.
Materials and Methods
Data set retrieval from TCGA repository
Genotype and epitype data for breast invasive carcinoma (BRCA) were obtained from the Cancer Genome Atlas (TCGA) consortium. In total, 746 DNA methylation, 1076 SNP array, and 1035 clinical sample details for tumors were obtained from TCGA. Besides, RNAseq data set for 1056 tumors and 112 matched normal samples were also retrieved to study the effect of methylation and genotype on fold change (FC) in gene expression.
Illumina 450 k DNA methylation data
Level 3 data set pertaining to 746 tumor samples, such that 740 were obtained from the primary tumor, while the remaining six samples pertaining to metastatic class were filtered out. Each of these normalized data sheets incorporated the details for genomic coordinates and beta values for each CpG site, while the associated gene information was optional. Sixty-five non-random SNPs were excluded, and 485,512 CpG sites were processed for further studies. These methylation files were processed to interrogate the SNPs associated with each CpG locus. The entire set of SNPs information were based on the Affymetrix Genome-Wide Human SNP Array 6.0 genotypic platform.
Affymetrix SNP array data set preparation
Level 1 SNP array data were normalized, and the genotype call for each sample was based on the “Corrected Robust Linear Model with Maximum Likelihood Distance” algorithm.25 The algorithm estimates the genotype using linear mixture model, and for each SNP–genotype combination, the uncertainty parameter was corrected using HapMap samples. In order to process the large data set, the crlmm package was substantiated with ff package to reduce memory footprint (http://cran.at.r-project.org/web/packages/ff/index.html). The algorithm was implemented to decode the genotype calls for SNPs as 1 (AA/Reference allele), 2 (AB/Heterozygous allele), and 3 (BB/Alternate allele). The genotype calls at the threshold of 0.05 were filtered, while those having more than 25% low-confidence calls were excluded. Data normalization and filtration resulted in 905,422 variants.
RNAseq data set preparation
Level 3 RNAseq data set for gene expression was processed, and quality control was brought about by Broad Institute TCGA workgroup (http://gdac.broadinstitute.org/). The reference gene transcript set was based on HG19 UCSC track (http://hgdownload.cse.ucsc.edu/downloads.html). Furthermore, MapSplice was used to carry out the alignment, and the quantification was done using RNA-Seq by Expectation-Maximization (RSEM).26,27 Finally, the upper quantile normalized RSEM count estimates were downloaded.
Procedure for the identification of CpG–SNP pair associated with the prognosis in breast cancer
Figure 1 outlines the procedure for the identification of regulatory CpG–SNP pair involved in the risk associated with the survival of breast cancer patients. The details are described in the following steps.
Figure 1.

Detailed outline for identifying a significant effect of CpG–SNP pairs on the overall survival. It also includes in finding the candidate risk SNPs in the breast cancer prognosis. The individual CpG sites and SNPs have also been correlated with the gene expression. This process utilizes DNA methylation, SNP array, RNAseq, and clinical data.
Step 1
About 660 tumor samples overlapping with DNA methylation, SNPs, and clinical data set were randomly split into training and test models in order to study the synergistic effect of methylation and associated SNPs on survival of breast cancer patients (Fig. 2A). The caret package of R (http://caret.r-forge.r-project.org/) was implemented to group 3/4 of samples (486) into training and 1/4 (164) as testing based on the vital status (dead or alive) of the patients outlined in the clinical data set (Fig. 2B).
Figure 2.

(A) Venn diagram details about the DNA methylation, SNP array, and clinical samples across the tumor patients. (B) The tumor sample overlapping across the three data sets is grouped into the 75% training set and 25% test set based on the vital status.
Step 2
The training model was built across 486 samples for each of the 7970 CpG–SNP pairs located at 50 nt upstream of downstream of CpG sites. The significant association between the beta value with respect to each CpG site and the variable genotype associated with each SNP was computed based on the non-parametric one-way analysis of variance (ANOVA). Here, the β-values were modeled as a linear function with respect to alleles (AA, AB, and BB). The complete analysis was carried out at R-interface at a threshold P-value of 0.05. Each of the SNPs having a significant association between DNA methylation was labeled as meQTL. The differential methylation of CpG site was a consequence of these meQTLs. The finding of these meQTLs in the training model was validated in the test model to reduce false-positive rates.
Step 3
Each of the differentially methylated CpG sites from step 2 was further analyzed to speculate their effect on gene expression. Spearman’s correlation test could establish the effect of methylation on log2-tranformed FC in gene expression, such that FC = log2 (T/N), where T is the estimated expression value of a tumor sample and N is the median expression of normal samples.
Step 4
For each of the CpG–SNP pairs from step 2, SNPs were evaluated to visualize their significant effect on gene expression. We extracted the variable genotype (AA, AB, BB) associated with each SNP and log2-tranformed FC in gene expression. One-way ANOVA was applied to assess the statistical significance between each SNP genotype and its neighboring gene expression.28 Moreover, the mean FC in gene expression was calculated with respect to the genotype associated with each SNP. This association between the differential gene expressions with respect to allele was labeled as expression quantitative trait loci (eQTLs).
Step 5
Each of the significant CpGs from CpG–SNP pairs obtained from step 2 was further evaluated for their association with the risk to the OS of the breast cancer patients. The complete analysis was based on the univariate and multivariate Cox proportional hazard (PH) models.29–31 The survival analysis was based on the clinical details in the form of vital status (patient alive or dead) and the date for the last follow-up and days to death. Besides, the SNPs from the overlapSelect were exclusively evaluated for their effect on OS. The training model was built for each of the SNPs across 486 patients. The findings in the training model were validated in the test model. Complete analysis was carried out based on the log-rank test. All the significant SNPs identified in the test model were subjected to multivariate Cox regression analysis to visualize their cumulative effect on OS.
Results
Identification of methylated probes or loci differing in genotypes
In this analysis, we mainly elaborate the pattern of polymorphic allele distribution (AA, AB, and BB) and their influence on DNA methylation exclusively in breast cancer patients. Considering the close proximity between the genetic variability and DNA methylation, the comprehensive analysis of the overlapping layers expands our knowledge in understanding the association of genetic variability with disease etiology. Realizing the fact that a large portion of cancer-related SNPs are positioned in the non-coding region holds substantial functional impact, the coaxial analysis of genotype–epitype interactions will facilitate identification of novel prognostic markers. The training data set comprising 484 samples were interrogated to locate CpG–SNP pairs at an interval of 50 bp upstream and downstream of a given CpG site. For each of the benchmark data set, its training and test data sets were used as exclusive subsets. The predictive model was built in training data set and validated over the test data set bearing 164 samples. Based on the analysis carried out by overlapSelect tool, a total of 7970 CpG–SNP pairs were identified at a base interval of 50 bp. Of the total 7970 CpG–SNP pairs, there were 1820 CpG loci being influenced by the individual genotype resulting in differential methylation patterns in the predictive training model. These loci across the same chromosome are called cismeQTLs and can influence the methylation pattern across the extended genomic regions. In the cis-meQTL analysis of 1820 CpG–SNP pairs, 489 polymorphic alleles were identified to be significantly associated with differential methylation in the test data set (P < 0.05). However, only 392 and 243 SNPs were detected to have remarkable effect on methylation at a stringency of 0.01 and 0.001, respectively. The majority of these cis-meQTLs were mapped to the intronic regions (50%–60%) though a limited number were associated with synonymous (1.2%–1.7%) or non-synonymous coding SNPs (3%–4%). Some of these SNPs being associated with one or more CpG loci suggest that they not only influence the methylation status to the associated CpG loci but also affect the surroundings at very close proximity. Genome-wide localization of cis-meQTLs identified in the test model (P < 0.05) and their loci on the respective chromosome have been depicted in Manhattan plot (Fig. 3). From Figure 3, it is evident that the meQTL density is high on chromosome 2.
Figure 3.

Dots within the Manhattan plot display the identification of significant SNPs in the vicinity of CpG site leading to the meQTLs in the test model; the x-axis represents genomic position of SNPs, while the y-axis represents the −log P-value of the association between the SNPs and CpG sites. The red and blue lines indicate the threshold −log10(1 × 10−4) and −log10(0.05), respectively, for genome-wide statistical significance.
In particular, the association of breast risk alleles, rs1570056 and rs11154883, with DNA methylation levels (cg18287222) and MAP3K5 gene (P < 0.001) is an interesting case, because the gene encodes for mitogen-activated protein kinase (MAPK) protein that activates signaling cascade. The downstream protein kinases that are activated include MAPK or extracellular signal-regulated kinase (ERK), MAPK kinase (MKK or MEK), and MAPK kinase kinase (MAPKKK). These kinases are highly conserved, and the homologs exist in yeast, Drosophila, and mammalian cells.32 While the differential distribution of major allele (T) and minor allele (C) (SNP: rs1570056) regulates the DNA methylation of the CpG site cg18287222 (Fig. 4A), the mutation in the allele G → A associated with SNP rs1154883 simultaneously regulates the same CpG loci. These alleles influenced DNA methylation at significant P-values of 5.8 × 10−5 (<0.001) and 0.0002, respectively (Fig. 4A and B). Thus, it presents an interesting fact that the alleles of the respective SNP act in a differential manner in regulating DNA methylation. Finally, we examined the overlap in regulatory variation affecting both methylation and gene expression based on RNAseq data. The differentially methylated CpG site was negatively associated (r = −0.53) with MAP3K5 gene expression at a P-value of <0.01 (Fig. 5). We also tested the association of these SNPs with the expression level of the gene. The variable allele associated with each SNP regulated the quantitative expression of MAP3K5 gene at P-values of 0.028 and 0.012 with respect to rs1570056 and rs11154883 SNPs, respectively (Fig. 6A and B). The polymorphism associated with differential messenger RNA (mRNA) expression level is referred to as eQTL. In summary, our result clearly demonstrates the meQTLs and eQTLs.
Figure 4.

Breast cancer SNPs rs1570056 and rs11154883 are associated with differential CpG methylation. Cis-association between the SNPs (A) rs1570056 and (B) rs11154883 regulates the methylation of CpG probe cg18287222. These SNPs are at loci within an intron variant of MAP3K5 gene. The box plot shows the distribution of the methylation levels in each genotype category with error bars representing the 25% and 75% quantiles.
Figure 5.
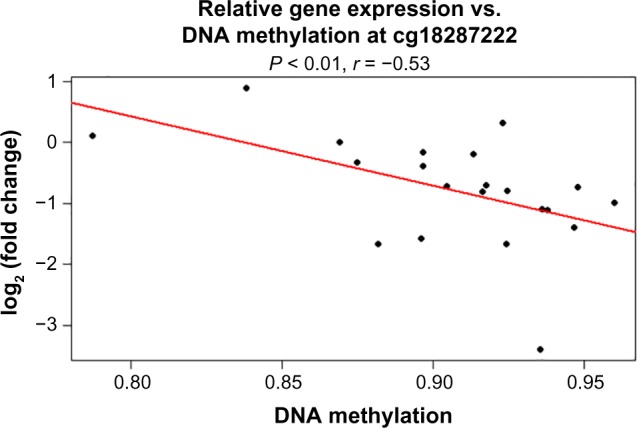
Spearman’s correlation with respect to FC in gene expression and DNA methylation in breast cancer. (a) DNA methylation residuals at probe cg18287222 is negatively associated (r = −0.53) with MAP3K5 expression in breast cancer patients at a P-value of <0.01. The regression line (red line) depicts the linear association between DNA methylation residuals and gene expression residuals.
Figure 6.

FCs in gene expression with respect to variable genotype was identified to be significant at P-values of 0.028 and 0.012 with respect to SNPs rs1570056 and rs11154883, respectively. (A) FC in gene expression is evaluated in the presence of SNP rs1570056. Homozygous dominant allele “TT” causes comparatively more downregulation in gene expression in comparison to heterozygous (TC) and homozygous recessive (CC) allele. (B) FC in gene expression is evaluated in the presence of SNP rs11154883. Homozygous recessive allele AA causes results in more downregulation in comparison to heterozygous (GA) and homozygous dominant (GG) alleles.
Prognostic potential of differentially methylated CpGs on survival of breast cancer patients
Breast cancer has displayed an increasing incidence, and more importantly, the steady mortality rate in a past decade. While the clinical screening has attributed to the enhanced survival of breast cancer patients, still improvised markers are required to assess accurately patient prognosis at the time of diagnosis. The disease heterogeneity and limited specificity and the clinicopathological variables are used in prognostication and staging of breast cancer. Thus, the development of complementary biomarkers with more specific prognostic potential will allow to assess the risk of developing recurrent and/or metastatic disease. We report for the first time the association between the differentially methylated CpG site and OS of breast cancer patients. Univariate and multivariate Cox PH regression analyses have been implemented to establish the prognostic potential of differentially methylated CpGs. Of the total 7970 overlap CpG–SNP pairs, 489 CpGs were identified to be significantly associated with the survival of breast cancer patients in the training model. The prognostic potential of these CpGs was validated in the test model of 164 patients.
To test the association of risk in 164 breast cancer patients for OS, we first began our analysis using univariate Cox PH model. On evaluating 489 CpGs based on the clinicopathological variables of vital status and last follow-up days, 18 covariates were found to be significantly associated with OS of breast cancer patients in the test model. The most significant association with OS was observed for cg04003327: rs1054641 on chromosome 2q37.3 (hazard ratio [HR] = 0.01, additive P = 0.003), cg14033170: rs177595 on chromosome 7p15.1 near CREB5 gene (HR = 158.94, additive P = 0.004), and cg00902464: rs17403618 on chromosome 1p21.2 (HR = 0.02, additive P = 0.016) (Table 1). The risk allele associated with CpG sites cg11340537, cg00956490, cg04586622, and cg14033170 has already been identified in GWAS phenotypes. The genotypic variation associated with SNP rs2640785 regulates differential methylation of the CpG site cg11340537 located in the exonic region of the EXPH5 gene. The missense variation (GAG → GTG) associated with this risk allele is of greater significance as it is conjointly associated with differential methylation, gene expression, and survival of breast cancer patients. A similar explanation can be associated with synonymous risk variant rs940453 (ATA → ATC) that regulates methylation of the CpG site cg00956490 and simultaneously influences ZNF775 gene expression and OS. However, the risk allele rs2384061 is an intron variant that is associated with the CpG site cg0458662 and regulates the expression of ADCY3 gene. The SNP rs2230576 mapped to the 3′-untranslated region (3′-UTR) variant is correlated with differential methylation of the CpG site cg05370838 and gene expression of ADMA8 gene. The differentially methylated CpG site holds significance in regulating the OS of breast cancer patients (HR = 0.008, additive P = 0.049).
Table 1.
Univariate analysis to depict the associations between differentially methylated CpGs and the overall survival of the breast cancer patients.
| CpG ID | SNP ID | GENE | HR | 95% OF CI | P-VALUE |
|---|---|---|---|---|---|
| cg04003327 | rs1054641 | ESPNL; SCLY | 0.011948 | 0.00076–0.18 | 0.0032 |
| cg14033170 | rs177595 | CREB5 | 158.9545 | 3.10816–8129.1 | 0.0038 |
| cg00902464 | rs17403618 | LOC100128787 | 0.023795 | 0.00178–0.32 | 0.0167 |
| cg03383184 | rs6988652 | Intergenic | 52.99806 | 1.4918–1882.7 | 0.0170 |
| cg00101629 | rs6660333 | KIAA1026 | 0.050101 | 0.00378–0.667 | 0.0173 |
| cg03521812 | rs4620521 | Intergenic | 0.023186 | 0.00107–0.498 | 0.0177 |
| cg17378966 | rs2431663 | DUSP1 | 13.45278 | 1.2033–150.39 | 0.0262 |
| cg08937612 | rs12409375 | VSIG8 | 0.003215 | 2.63E-05–0.39 | 0.0270 |
| cg26901096 | rs17444979 | LOC254312 | 13.55016 | 1.32054–139.1 | 0.0292 |
| cg13558682 | rs9424283 | LRRC47 | 0.024577 | 0.001227–0.49 | 0.0366 |
| cg16774160 | rs3088007 | HSPA12B | 0.000191 | 2.23E-07–0.16 | 0.0384 |
| cg06099459 | rs10505956 | C12orf77 | 0.002645 | 1.64E-05–0.426 | 0.0416 |
| cg05370838 | rs2230576 | ADAM8 | 0.008903 | 0.000201–0.395 | 0.0498 |
| cg11340537 | rs2640785 | EXPH5 | 0.031486 | 0.00135–0.733 | 0.0528 |
| cg00956490 | rs940453 | ZNF775 | 0.001156 | 3.58E-06–0.373 | 0.0645 |
| cg04586622 | rs2384061 | ADCY3 | 0.008966 | 0.000116–0.693 | 0.0648 |
| cg00889709 | rs16923085 | FAM110B | 0.061646 | 0.00400–0.948 | 0.0652 |
| cg14798310 | rs738806 | SLC2A11, MIF DQ574115 | 0.000387 | 2.06E-07–0.725 | 0.0793 |
Abbreviations: CI, confidence interval for the hazard ratio; HR, hazard ratio.
The univariate analysis was followed by the multivariate regression model to assess the risk associated with 18 covariables validated in the test model obtained from the univariate study. This multivariate Cox PH analysis leads to the identification of eight CpGs having a significant association with OS of the breast cancer patients (Table 2). Among these, the most substantial findings were observed for cg04003327 (HR = 0.016; 95% confidence interval [95% CI] = 0.0003–0.86; P = 0.04), cg11340537 (HR = 0.28; 95% CI = 0.005–14.49; P = 0.05), and cg00956490 (HR = 0.0005; 95% CI = 1.36 × 10−7−2.44; P = 0.08). These eight covariates showed the clear demarcation of the patients into high- (84 patients) and low-risk (84 patients) groups, respectively, at a significant P-value of 0.04 (Fig. 7).
Table 2.
Summary for univariate and multivariate analyses of the associations between the CpGs and the overall risk based on the Cox PH model.
| SNP ID | CpG ID | GENE | LOCUS | UNIVARIATE HR 95% OF CI p | MULTIVARIATE HR 95% OF CI p |
|---|---|---|---|---|---|
| rs1054641 | cg04003327 | ESPNL; SCLY | 2q37.3 | 0.012 (0.001–0.18) 0.003 | 0.016 (0.0003–0.86) 0.04 |
| rs2640785 | cg11340537 | EXPH5 | 11q22.3 | 0.031 (0.001–0.73) 0.05 | 0.28 (0.005–14.49) 0.05 |
| rs940453 | cg00956490 | ZNF775 | 7q36.1 | 0.001 (3.58E-06–0.37) 0.06 | 0.0005 (1.36E-07–2.44) 0.08 |
| rs2230576 | cg05370838 | ADAM8 | 10q26.3 | 0.0008 (0.002–0.39) 0.049 | 0.028 (0.0001–4.5) 0.17 |
| rs6660333 | cg00101629 | KIAA1026 | 1p36.21 | 0.05 (0.003–0.66) 0.17 | 0.88 (0.02–37.57) 0.95 |
| rs177595 | cg14033170 | CREB5 | 7p15.1 | 158.94 (3.1–8129.07) 0.003 | 213 (1.7–25740) 0.028 |
| rs4620521 | cg03521812 | Intergenic | 1q31.2 | 0.02 (0.001–0.49) 0.018 | 0.04 (0.001–1.8) 0.098 |
| rs9424283 | cg13558682 | LRRC47 | 1p36.32 | 0.024 (0.001–0.49) 0.036 | 0.336 (0.001–101.1) 0.71 |
Abbreviations: CI, confidence interval for the hazard ratio; HR, hazard ratio.
Figure 7.
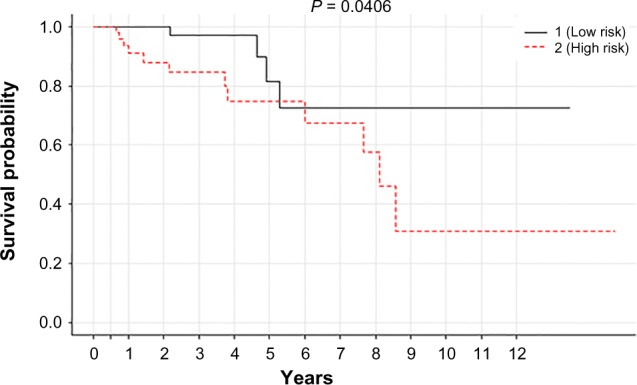
Kaplan–Meier plot associated with CpGs to classify 164 tumor patients (test set) into high- (84 patients) and low-risk (84 patients) groups, respectively, at a threshold P-value of 0.041.
Besides, the exclusive effect of SNPs was also evaluated on OS of breast cancer patients. In the following section, we explain about the variable allele distribution and their association with OS.
Probing the association of SNPs with OS of breast cancer patients
Genetic variation characterized by single-nucleotide polymorphism (SNP) offers promising surrogate biomarkers to predict therapeutic responses and prognosis in breast cancer patients. In this study, we investigated the risk associated with the individual SNP and in cumulative fashion on the OS. A probabilistic framework was developed for predicting and prioritizing the candidate SNPs in the training data set and validated across test set constituting 164 samples. The complete survival analysis was based on the homozygous dominant and recessive allele and heterozygous allele distribution available for each SNP.
The univariate survival analysis associated with the individual SNP was based on the log-rank test at a threshold P-value of 0.05. Of the total 7970 CpG–SNP pairs, 492 SNPs were significantly associated with the OS in the training set of breast cancer patients. Each individual SNP was validated in the test model to reduce the false-positive rate. Of the total significant SNPs in the training set, 23 were substantially associated with survival in the test model and their respective P-value ranged from ≤0.0001 to ≤0.05 (Table 3). These SNPs hold a variable distribution across the genome. In the midst of significant findings, seven SNPs (rs2880556, rs17006586, rs876701, rs41470747, rs2967798, rs11804125, and rs1548373) were present as an intro variant and five SNP loci (rs12085531, rs12653167, rs12591432, rs940482, and rs1532272) were in the intergenic region. Similarly, three SNPs (rs16943263, rs9325443, and rs1538146) were localized in the upstream region, while each of the two SNPs (rs7117026 and rs10101376) was associated with non-coding transcript variant and synonymous variant (rs17142291 and rs140679) and remaining one SNP (rs1862372) was associated with 5′UTR variant. Moreover, the SNPs highlighted in the table are already mentioned in GWAS in relevance to cancer and other diseases.
Table 3.
Summary of SNPs associated with OS of breast cancer patients using log-rank test.
| CpG_ID | SNP_ID | P-VALUE | GENE | A | B | AA | AB | BB |
|---|---|---|---|---|---|---|---|---|
| cg11929693 | rs2880556 | 2.29E-24 | LOC340073 | G | T | 153 | 9 | 2 |
| cg09939673 | rs7117026 | 2.55E-12 | DQ592890 | A | T | 1 | 10 | 153 |
| cg00067528 | rs17006586 | 1.47E-05 | ATP6V1B1 | C | T | 140 | 21 | 3 |
| cg01711124 | rs12085531 | 9.05E-05 | Intergenic | C | T | 4 | 24 | 136 |
| cg09573435 | rs1862372 | 0.000594 | SEMA6A | C | T | 111 | 43 | 10 |
| cg22675791 | rs876701 | 0.000627 | DGKZ | A | G | 6 | 36 | 122 |
| cg20705812 | rs2286218 | 0.001795 | DLGAP2 | A | G | 143 | 16 | 5 |
| cg08980697 | rs41470747 | 0.006462 | RASGEF1B | C | A | 1 | 12 | 151 |
| cg14584565 | rs16943263 | 0.006649 | LOC283761 | G | C | 152 | 8 | 4 |
| cg04513214 | rs12653167 | 0.008100 | Intergenic | T | G | 162 | 1 | 1 |
| cg22422090 | rs2967798 | 0.008121 | KLHL3 | T | A | 102 | 44 | 18 |
| cg24310780 | rs11804125 | 0.008351 | LMX1A | G | T | 122 | 30 | 12 |
| cg03339247 | rs1548373 | 0.013806 | ZFHX3 | C | T | 106 | 38 | 20 |
| cg25203310 | rs10101376 | 0.014656 | LOC286083 | G | A | 59 | 47 | 58 |
| cg20214734 | rs17142291 | 0.016161 | ASB13 | G | A | 4 | 9 | 151 |
| cg15179472 | rs12591432 | 0.018465 | Intergenic | C | T | 123 | 33 | 8 |
| cg15461663 | rs940482 | 0.029081 | Intergenic | C | T | 99 | 53 | 12 |
| cg22514112 | rs1532272 | 0.031189 | Intergenic | A | G | 94 | 52 | 18 |
| cg04966682 | rs140679 | 0.033337 | GABRG3 | C | T | 57 | 67 | 40 |
| cg02576753 | rs140679 | 0.033336 | GABRG3 | C | T | 57 | 67 | 40 |
| cg20896197 | rs9325443 | 0.037904 | KIF20B | A | C | 91 | 59 | 14 |
| cg24540569 | rs574095 | 0.041499 | Intergenic | A | G | 3 | 26 | 135 |
| cg15398976 | rs1538146 | 0.049488 | TRPC4 | G | T | 65 | 54 | 45 |
Abbreviations: AA, reference allele; AB, heterozygous allele; BB, alternate allele.
The Kaplan–Meier plot for the significant SNPs having nearly equal genotypic frequency is displayed in Figure 8. While the presence of heterozygous allele “GA” associated with SNP rs10101376 is detrimental, the homozygous dominant alleles “CC” and “TT” concomitant with SNPs rs140679 and rs1538146 affect the survival of the breast cancer patient at a threshold P-value cutoff of 0.05. The homozygous dominant allele “TT” (rs1538146) has loci along upstream of the TRPC4 gene. The transient receptor potential cation channel (TRCP4) gene encodes a member of a canonical subfamily of transient receptor potential cation channels. This encoded protein forms a non-selective calcium-permeable cation channel that is activated by a Gq-coupled receptor and tyrosine kinase. The polymorphism associated with TRCP4 gene is deleterious, as it is conjointly linked with gene expression and regulates the OS. Similarly, the allele “CC” associated with SNP rs1538146 regulates the expression of gamma-aminobutyric acid (GABA)-A receptor gene and is deleterious and will affect the survival of breast cancer patients. Besides the log-rank test, these 23 significant SNPs were also subjected to univariate Cox PH regression analysis. The most significant association in the univariate model for survival was observed for rs7117026 located on chromosome 11p11.2 (HR = 0.109, additive P < 0.001) as a non-coding transcript variant of DQ582890 gene, rs1548373 on chromosome 16q22.3 (HR = 2.35 and additive P = 0.0096) as an intron variant of ZFHX3 gene, rs140679 on chromosome 15q12 (HR = 0.359, additive P = 0.016) as a non-synonymous variant of GABRG3 gene, rs876701 on chromosome 11p11.2 (HR = 0.371, additive P = 0.038) as an intron variant of DGKZ gene, and rs41470747 on chromosome 4q21.21 (HR = 0.357, additive P = 0.039) as an intron variant of RAS-GEF1B gene. Additionally, borderline-associated risk variants included rs574095, rs12653167, rs2286218, and rs1538145 at an additive threshold of P = 0.1. Besides, SNP rs16943263 associated with CpG locus cg14584565 (HR = 2.44 and P = 0.17) is also identified in classifying the patients in high- and low-risk groups (Table 4).
Figure 8.

Kaplan–Meier survival plot for SNPs: (A) rs10101376, (B) rs140679, and (C) rs1538146. The survival analysis has been done with respect to reference (solid black line), heterozygous (red dotted line), and alternate alleles (solid green line) at a threshold P-value of <0.05.
Table 4.
Summary of univariate and multivariate analyses of SNP associations with overall risk based on the Cox PH model.
| SNP ID | CpG ID | GENE | LOCUS | UNIVARIATE HR 95% OF CI p | MULTIVARIATE HR 95% OF CI p |
|---|---|---|---|---|---|
| rs1862372 | cg09573435 | SEMA6A | 5q23.1 | 1.66 (0.66–4.15) 0.28 | 1.15 (0.2–6.3) 0.87 |
| rs2880556 | cg11929693 | LOC340073 | 5q31.3 | 2.23 (0.5–9.8) 0.29 | 59.7 (2.52–14.12) 0.011 |
| rs1548373 | cg03339247 | ZFHX3 | 16q22.3 | 2.35 (1.23–4.17) 0.0096 | 5.99 (1.84–19.49) 0.0029 |
| rs12591432 | cg15179472 | Intergenic | 15q23 | 1.32 (0.48–3.6) 0.59 | 4.50 (0.67–30.21) 0.12 |
| rs12653167 | cg04513214 | Intergenic | 5p15.1 | 2.96 (0.95–9.24) 0.062 | 4.85 (0.45–52.28) 0.19 |
| rs16943263 | cg14584565 | LOC283761 | 15q26.1 | 2.44 (0.68–8.6) 0.17 | 0.06 (0.0007–6.6) 0.25 |
| rs12085531 | cg01711124 | Intergenic | 1p36.12 | 0.52 (0.20–1.3) 0.17 | 1.4 (0.44–4.87) 0.53 |
| rs1538145 | cg15398976 | TRPC4 | 13q13.3 | 0.558 (0.29–1.07) 0.081 | 0.58 (0.21–1.54) 0.28 |
| rs41470747 | cg08980697 | RASGEF1B | 4q21.21 | 0.35 (0.13–0.94) 0.039 | 0.38 (0.047–3.07) 0.37 |
| rs140679 | cg04966682 | GABRG3 | 15q12 | 0.359 (0.15–0.82) 0.016 | 0.11 (0.018–0.7) 0.019 |
| rs17142291 | cg20214734 | ASB13 | 10p15.1 | 0.56 (0.15–2.0) 0.38 | 11.80 (0.47–291.84) 0.13 |
| rs11804125 | cg24310780 | LMX1A | 1q23.3 | 1.12 (0.56–2.2) 0.75 | 1.38 (0.34–5.47) 0.64 |
| rs7117026 | cg09939673 | DQ5982890 | 11p11.2 | 0.109 (0.03–0.3) 3.00 × 10−4 | 0.00198 (0.00003–0.12) 0.0034 |
| rs876701 | cg22675791 | DGKZ | 11p11.2 | 0.371 (0.14–0.94) 0.038 | 0.29 (0.07–1.27) 0.1 |
| rs574095 | cg24540569 | Intergenic | 1p31.3 | 0.445 (0.19–1.0) 0.058 | 0.25 (0.036–1.70) 0.16 |
| rs2286218 | cg20705812 | DLGAP2 | 8p23.3 | 2.76 (0.88–8.5) 0.08 | 0.97 (0.05–19.20) 0.99 |
Abbreviations: CI, confidence interval for the hazard ratio; HR, hazard ratio.
Finally, conjoint analysis of 23 SNPs was carried out to assess the cumulative effect of the genetic variant on OS. We performed multivariate Cox regression analysis between the SNPs and clinical variance. Of the total 23 variables from log-rank test, 16 SNPs in the test model could classify the patients into high- and low-risk groups, respectively, at a threshold P-value of 0.05 (Fig. 9A). However, top nine SNPs presented a clear demarcation for 164 patients at a P-value of 0.005 (Fig. 9B). The delineation was such that 84 patients (test sample) survived for a longer duration, while the remaining were prone to poor prognosis, and their survival probability was identified to be 81/2 years. Most of these genetic variants are germline and have shown significant association with OS. Thus, the Cox proportional model conjointly with clinical features suggests the association between the genetic variants and the risk to the survival of breast cancer patients, which may also modulate the cancer prognosis.
Figure 9.
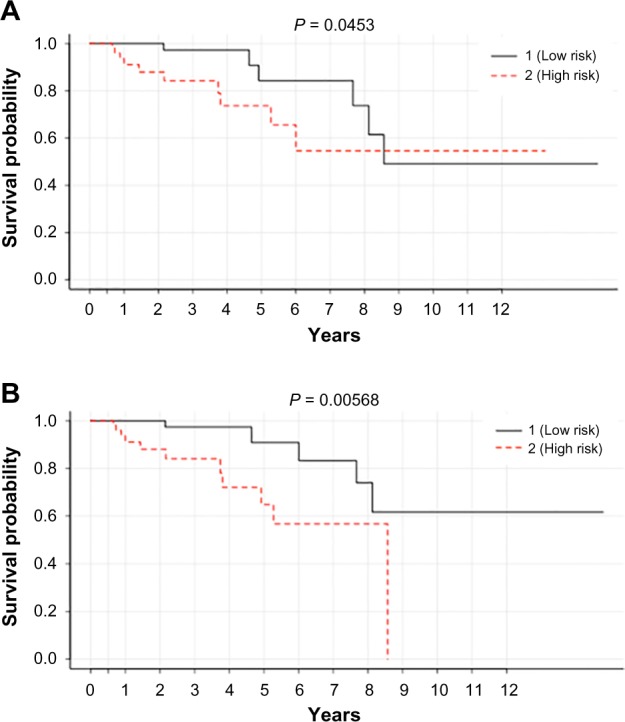
Kaplan–Meier curve associated with (A) top 16 SNPs and (B) top 9 SNPs (listed Table 3) in classifying 164 tumor patients (test set) into high- (84 patients) and low-risk (84 patients) groups at a threshold of P < 0.05.
Discussion and Conclusions
Molecular understanding of intertumor heterogeneity is key to effective cancer treatment and personalized medicine. Analysis of high-throughput molecular profiling data has revealed the extent of intertumor heterogeneity in breast cancer. The identification of different levels (subtypes) of tumor heterogeneity and the most appropriate treatment strategies for each subtype is expected to radically improve the treatment practices for the optimal clinical management of breast cancers.33
GWASs have led to the identification of a large number of genetic variants that confer susceptibility to different types of cancers. However, the risk conferred by an individual variant is not sufficient to uphold the individual risk prediction. Assessing the genetic variability by incorporating multiple SNPs into a predictive model could achieve improved risk discrimination that may be useful for prognostic stratification of breast cancer patients.34,35 It is often a challenge to assess the functional impact of non-coding genetic variants, for example, the effect of SNPs transcriptional activity and the associated disease risk.
Questions still remain for the prognostic biomarkers identified for cancer using data-mining approaches. The first question is that there is little overlap among numerous prognostic signatures generated from different studies. Another question is that most signatures generated do not have clear biological meanings as why these prognostic genes may affect patient outcome, which leads to the clinical application of such signatures still under debate. In this study, we developed a novel method to identify prognostic gene signatures for breast cancer by integrating genomic and epigenomic data. This is based on the hypothesis that multiple sources of evidence pointing to the same gene or pathway are likely to lead to reduced false positives. We also apply random resampling to reduce overfitting noise by dividing samples into training and testing data sets. In this analysis, TCGA BRCA overlapping data set between DNA methylation, Affymetrix SNP array, and clinical samples were randomly divided into two subsets based on the vital status obtained from clinical data. The predictive model was trained based on certain features, mainly the beta values and genotypes associated with methylation and SNP, respectively. The robustness of the features were evaluated statistically in the training subset and validated in an exclusive and independent test subset. The significant association between methylation and genotype was calculated based on one-way ANOVA at a threshold P-value of 0.05. Each SNP encoded for variable homozygous and heterozygous genotypic (allele) frequency across the breast cancer samples. Localization of each SNP was interrogated at 50 bp upstream and downstream of each CpG site. Thus, for a window size of 50 bp, we investigated CpG–SNP pairs to enlist their statistical significance such that a minimum of one SNP is associated with one CpG locus. This evidence of correlation between genetic variants at specific loci and DNA methylation led to the identification of meQTLs. Of the total distribution of 7970 CpG–SNP pairs in the window size of 50 bp, 1874 SNPs were significantly associated with differential methylation in the predictive training model. Out of these 1874 CpG–SNP pairs, 489 SNPs were significantly correlated with differential methylation in the test model. These CpG–SNP pairs enlighten the plausible mechanism through which SNPs have influence on the phenotype. In one of the scenarios, presence of an SNP in the vicinity of CpG loci prevents the binding of CpG methyl-binding proteins, which as a consequence affects DNA methylation.36 In another scenario, these SNPs may affect the transcriptional silencing via differential DNA methylation. Indeed, it has also been reported that DNA methylation plays a significant role in the regulation of splicing and aids in distinguishing exons from introns.37,38 Thus, genetic variants characterized by the presence of SNPs in the intronic region causes differential methylation leading to a different set of spliceosomes.39 Interestingly, in our recent analysis, we have identified MAP3K5 variants that constitute two variable SNPs (rs1570056 and rs11154883) located in the intronic region and affect gene function through gene silencing. These variants act in contrary such that the differential distribution of major allele (T) and minor allele (C) with respect to SNP rs1570056 and A and G alleles associated with SNP rs11154883 causes differential methylation pattern. This differential distribution landmarks the presence of a specific meQTL. Besides overlap with meQTL, these SNPs lead to eQTLs in the cis-regulatory region. Thus, the meQTLs have been identified to be enriched in eQTLs. Moreover, MAP3K5 (MAPK) is an essential component of MAPK signal transduction pathway and plays a crucial role in the apoptosis.40,41 Characterizing the genetic control of methylation and its association with the regulation of MAP3K5 gene expression presents signature marks that can resolve in understanding the underlying biology behind the complex phenotype in breast cancer. We have also reported for the first time the association between CpGs and the risk in the survival of breast cancer patients. The high mortality rate associated with metastasis in breast cancer urge for the development of more personalized prognostic algorithms that will complement the general, clinical predictors. We have systematically investigated the risk associated with host-related BRCA traits that may serve as a biomarker for disease prognosis. In this study, we have implemented model selection framework composed of linear statistical techniques of univariate analysis based on log-rank test and multivariate Cox regression model. We examined a comprehensive panel of 489 differentially methylated CpGs obtained from the previous analysis in association with clinicopathological characteristics to assess the OS. Based on the univariate regression analysis, 18 CpGs were identified as the landmark risk loci for OS in the test model. However, the conjoint multivariate regression analysis of these differentially methylated CpGs led to the identification of eight CpG sites as promising candidates having significant prognostic potential. These noteworthy biomarkers clearly demarcated 164 breast cancer patients of the test sample into high- and low-risk groups. The most interesting fact is that the SNPs (rs2640785, rs940753, and rs2230576) associated with respective differentially methylated CpG sites (cg11340537, cg00956490, and cg04586622) have been already reported in GWAS phenotypes. We explored the potential mechanism by which differentially methylated CpG site cg11340537 directs OS in breast cancer patients. The missense variant (GAG → GTG) associated with SNP rs2640785 dictates differential methylation of the CpG site cg11340537 and mRNA expression of EXPH5 (Exophilin 5) gene. EXPH5 gene shares homology with Rab-GTPase and play a significant role in vesicle trafficking.42,43 The active participation of this gene has been reported in colorectal cancer.44 The differential methylation associated with the CpG site cg14033170 also holds greater significance. SNP rs177595, an intron variant located in the vicinity of the CpG site cg14033170, dictates the differential methylation and subsequently deregulates CREB5 gene expression. CREB5 gene encodes for cAMP responsive element-binding protein 5. Previous studies have suggested that CREB5 gene plays a fundamental role in a metastatic signal network in colorectal cancer.45 Moreover, it has been reported that eQTL associated with CREB5 gene causes colorectal, prostate, and nasopharyngeal cancers.46–48 On a similar account, meQTL associated with the CpG site cg00956490 holds prognostic significance. The risk variant rs940453 linked to CpG loci regulates the mRNA expression of ZNF775 gene. The gene encodes for zinc finger protein 775.49 It has been identified to be involved in transcriptional regulation. SNP rs2230576 is a 3′-UTR variant that has been mapped to the vicinity of differentially methylated CpG site cg05370838 and ADAM metallopeptidase domain 8 (ADAM8) gene. The differentially methylated CpG site is associated with high risk in breast cancer patients. ADMA8 gene localized in the vicinity of the CpG site encodes for membrane-anchored protein that has been implicated in several biological processes including cell–cell interactions, cell–matrix interactions, and neurogenesis.50 It has been reported that ADMA8 is aberrantly expressed in breast tumors, specific in triple-negative breast cancers (TNBCs). The aberrant expression of ADAM8 gene has been correlated with poor prognosis in breast cancer patients and concomitantly with increased number of circulating tumor cells and metastasis.51 The anomalous expression of the ADAM8 gene is also associated with poor survival in colorectal, lung, gastric, and pancreatic cancers, hepatocellular and gastrointestinal carcinomas, and gliomas.52–55
Studies done so far correlate with the conjoint effect of significant CpG–SNP pair in regulating the differential methylation and OS of breast cancer patients. Recent studies have illustrated the upshot of genetic variants in regulating the overall risk associated with breast cancer patients. However, the cumulative effect is still to be disclosed. In the next section, we detailed about the prognostic potential of individual SNP and their cumulative action. In our study, we have comprehensively analyzed the TCGA SNP array data mapped to methylated loci and concomitantly evaluated its association with the breast cancer survival. Of the total 7970 CpG–SNP pairs, 492 SNPs in the training model were predicted to be significantly associated with OS. However, the univariate analysis based on the log-rank test mapped 23 SNPs to be significant across the test data set. Most of these SNPs have been highlighted in GWASs. In this study, we have mainly displayed Kaplan–Meier plot for the SNP having higher and nearly equal allelic distribution in breast cancer population. The heterozygous allele “GA” associated with SNP rs10101376 is detrimental and is related to poor prognosis. Similarly, the homozygous dominant allele “TT” linked to rs140679 SNP disrupts the mRNA expression of GABA A receptor (GABRG3)56 and subsequently deteriorates survival probability in breast cancer patients. Presence of homozygous genetic variant “TT” with respect to SNP rs1538146 at 1349 bp upstream of TRPC4 gene (transient receptor potential cation channel, subfamily C) reduces the OS and has a significant prognostic determinant. The canonical transient receptor potential (TRPC) channels are permeable to Ca2+ cationic channels and regulate Ca2+ influx in response to G protein-coupled receptor.57 Overexpression of TRPC4 gene results in anomalous cell proliferation and has been reported in the prostate, ovarian, and lung cancers and renal cell carcinoma.58–61 Our findings have demonstrated the potential importance of assessing prognosis in breast cancer based on the univariate model of SNP distribution. Finally, we assembled these SNPs to construct logistic regression model and evaluated their cumulative effect on OS of breast cancer. Of the total 23 SNPs, 18 SNPs had significant prognostic potential and could classify 164 breast cancer patients into poor prognostic (high-risk) and good prognostic groups (low-risk). However, the conjoint effect of nine SNPs holds more clear vision on demarcation.
In summary, the comprehensive assessment of CpG–SNP pairs has led to the identification of loci that hold the risk to the OS of breast cancer patients. The novel findings are highly promising and strongly support the identification of these loci in the clinical visualization of breast cancer progression. Such prognostic scans at the genome-wide level will likely be not only beneficial for the identification of novel prognostic biomarkers but will also open a new horizon to the novel pathways involved in breast cancer progression, directing to the potential targets for more efficient treatment strategies.
Abbreviations
- SNP
single-nucleotide polymorphism
- meQTLs
methylation quantitative trait loci
- TCGA
the Cancer Genome Atlas
- BRCA
breast invasive carcinoma
- ADAM8
A disintegrin and metalloproteinase domain 8
- CREB5
cAMP responsive element-binding protein 5
- ZNF775
zinc finger protein
- MAP3K5
mitogen-activated protein kinase
- GWASs
genome-wide association studies
Footnotes
ACADEMIC EDITOR: J. T. Efird, Editor in Chief
PEER REVIEW: Four peer reviewers contributed to the peer review report. Reviewers’ reports totaled 2205 words, excluding any confidential comments to the academic editor.
FUNDING: This work was partially funded by the National Library of Medicine of the National Institutes of Health (NIH) under the Award Number R01LM011297 to RVD. The authors confirm that the funder had no influence over the study design, content of the article, or selection of this journal.
COMPETING INTERESTS: Authors disclose no potential conflicts of interest.
Paper subject to independent expert blind peer review. All editorial decisions made by independent academic editor. Upon submission manuscript was subject to anti-plagiarism scanning. Prior to publication all authors have given signed confirmation of agreement to article publication and compliance with all applicable ethical and legal requirements, including the accuracy of author and contributor information, disclosure of competing interests and funding sources, compliance with ethical requirements relating to human and animal study participants, and compliance with any copyright requirements of third parties. This journal is a member of the Committee on Publication Ethics (COPE).
Author Contributions
Conceived and designed the experiments: SKP, RVD. Analyzed the data: AS, YB, SJ. Wrote the first draft of the manuscript: AS. Contributed to the writing of the manuscript: YB, SJ, RVD. Agree with manuscript results and conclusions: SA, YB, SJ, SKP, RVD. Jointly developed the structure and arguments for the paper: AS, YB, SJ, RVD. Made critical revisions and approved final version: YB, RVD. All authors reviewed and approved of the final manuscript.
REFERENCES
- 1.Ali HR, Rueda OM, Chin SF, et al. Genome-driven integrated classification of breast cancer validated in over 7,500 samples. Genome Biol. 2014;15:431. doi: 10.1186/s13059-014-0431-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Haibe-Kains B, Desmedt C, Loi S, et al. A three-gene model to robustly identify breast cancer molecular subtypes. J Natl Cancer Inst. 2012;104:311–25. doi: 10.1093/jnci/djr545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Widschwendter M, Jones PA. DNA methylation and breast carcinogenesis. Oncogene. 2002;21:5462–82. doi: 10.1038/sj.onc.1205606. [DOI] [PubMed] [Google Scholar]
- 4.Cancer Genome Atlas Network Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jovanovic J, Ronneberg JA, Tost J, Kristensen V. The epigenetics of breast cancer. Mol Oncol. 2010;4:242–54. doi: 10.1016/j.molonc.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ronneberg JA, Fleischer T, Solvang HK, et al. Methylation profiling with a panel of cancer related genes: association with estrogen receptor, TP53 mutation status and expression subtypes in sporadic breast cancer. Mol Oncol. 2011;5:61–76. doi: 10.1016/j.molonc.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fleischer T, Frigessi A, Johnson KC, et al. Genome-wide DNA methylation profiles in progression to in situ and invasive carcinoma of the breast with impact on gene transcription and prognosis. Genome Biol. 2014;15:435. doi: 10.1186/s13059-014-0435-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fackler MJ, Umbricht CB, Williams D, et al. Genome-wide methylation analysis identifies genes specific to breast cancer hormone receptor status and risk of recurrence. Cancer Res. 2011;71:6195–207. doi: 10.1158/0008-5472.CAN-11-1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stirzaker C, Zotenko E, Song JZ, et al. Methylome sequencing in triple-negative breast cancer reveals distinct methylation clusters with prognostic value. Nat Commun. 2015;6:5899. doi: 10.1038/ncomms6899. [DOI] [PubMed] [Google Scholar]
- 10.Schnitt SJ. Classification and prognosis of invasive breast cancer: from morphology to molecular taxonomy. Mod Pathol. 2010;23(suppl 2):S60–4. doi: 10.1038/modpathol.2010.33. [DOI] [PubMed] [Google Scholar]
- 11.Liu Z, Zhang XS, Zhang S. Breast tumor subgroups reveal diverse clinical prognostic power. Sci Rep. 2014;4:4002. doi: 10.1038/srep04002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dejeux E, Ronneberg JA, Solvang H, et al. DNA methylation profiling in doxorubicin treated primary locally advanced breast tumours identifies novel genes associated with survival and treatment response. Mol Cancer. 2010;9:68. doi: 10.1186/1476-4598-9-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fleischer T, Edvardsen H, Solvang HK, et al. Integrated analysis of high-resolution DNA methylation profiles, gene expression, germline genotypes and clinical end points in breast cancer patients. Int J Cancer. 2014;134:2615–25. doi: 10.1002/ijc.28606. [DOI] [PubMed] [Google Scholar]
- 14.Kamalakaran S, Varadan V, Giercksky Russnes HE, et al. DNA methylation patterns in luminal breast cancers differ from non-luminal subtypes and can identify relapse risk independent of other clinical variables. Mol Oncol. 2011;5:77–92. doi: 10.1016/j.molonc.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Darabi H, McCue K, Beesley J, et al. Polymorphisms in a putative enhancer at the 10q21.2 breast cancer risk locus regulate NRBF2 expression. Am J Hum Genet. 2015;97:22–34. doi: 10.1016/j.ajhg.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Palmer JR, Ruiz-Narvaez EA, Rotimi CN, et al. Genetic susceptibility loci for subtypes of breast cancer in an African American population. Cancer Epidemiol Biomarkers Prev. 2013;22:127–34. doi: 10.1158/1055-9965.EPI-12-0769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pirie A, Guo Q, Kraft P, et al. Common germline polymorphisms associated with breast cancer-specific survival. Breast Cancer Res. 2015;17:58. doi: 10.1186/s13058-015-0570-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Andersson R, Gebhard C, Miguel-Escalada I, et al. An atlas of active enhancers across human cell types and tissues. Nature. 2014;507:455–61. doi: 10.1038/nature12787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Whyte WA, Orlando DA, Hnisz D, et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153:307–19. doi: 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tomczak K, Czerwinska P, Wiznerowicz M. The cancer genome atlas (TCGA): an immeasurable source of knowledge. Contemp Oncol (Pozn) 2015;19:A68–77. doi: 10.5114/wo.2014.47136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ma CX, Ellis MJ. The Cancer Genome Atlas: clinical applications for breast cancer. Oncology (Williston Park) 2013;27:1263–9. 1274–69. [PubMed] [Google Scholar]
- 22.Costa V, Aprile M, Esposito R, Ciccodicola A. RNA-Seq and human complex diseases: recent accomplishments and future perspectives. Eur J Hum Genet. 2013;21:134–42. doi: 10.1038/ejhg.2012.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heyn H, Sayols S, Moutinho C, et al. Linkage of DNA methylation quantitative trait loci to human cancer risk. Cell Rep. 2014;7:331–8. doi: 10.1016/j.celrep.2014.03.016. [DOI] [PubMed] [Google Scholar]
- 24.Zhi D, Aslibekyan S, Irvin MR, et al. SNPs located at CpG sites modulate genome-epigenome interaction. Epigenetics. 2013;8:802–6. doi: 10.4161/epi.25501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carvalho B, Bengtsson H, Speed TP, Irizarry RA. Exploration, normalization, and genotype calls of high-density oligonucleotide SNP array data. Biostatistics. 2007;8:485–99. doi: 10.1093/biostatistics/kxl042. [DOI] [PubMed] [Google Scholar]
- 26.Wang K, Singh D, Zeng Z, et al. MapSplice: accurate mapping of RNA-seq reads for splice junction discovery. Nucleic Acids Res. 2010;38:e178. doi: 10.1093/nar/gkq622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gentile JR, Roden AH, Klein RD. An analysis-of-variance model for the intrasubject replication design. J Appl Behav Anal. 1972;5:193–8. doi: 10.1901/jaba.1972.5-193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Prentice RL, Kalbfleisch JD. Hazard rate models with covariates. Biometrics. 1979;35:25–39. [PubMed] [Google Scholar]
- 30.Harrell FE, Jr, Lee KL, Califf RM, Pryor DB, Rosati RA. Regression modelling strategies for improved prognostic prediction. Stat Med. 1984;3:143–52. doi: 10.1002/sim.4780030207. [DOI] [PubMed] [Google Scholar]
- 31.Bradburn MJ, Clark TG, Love SB, Altman DG. Survival analysis part II: multivariate data analysis – an introduction to concepts and methods. Br J Cancer. 2003;89:431–6. doi: 10.1038/sj.bjc.6601119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang XS, Diener K, Jannuzzi D, et al. Molecular cloning and characterization of a novel protein kinase with a catalytic domain homologous to mitogen-activated protein kinase kinase kinase. J Biol Chem. 1996;271:31607–11. doi: 10.1074/jbc.271.49.31607. [DOI] [PubMed] [Google Scholar]
- 33.Zardavas D, Fouad TM, Piccart M. Optimal adjuvant treatment for patients with HER2-positive breast cancer in 2015. Breast. 2015;24(suppl 2):S143–8. doi: 10.1016/j.breast.2015.07.034. [DOI] [PubMed] [Google Scholar]
- 34.Murray JL, Thompson P, Yoo SY, et al. Prognostic value of single nucleotide polymorphisms of candidate genes associated with inflammation in early stage breast cancer. Breast Cancer Res Treat. 2013;138:917–24. doi: 10.1007/s10549-013-2445-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cheng Q, Chang JT, Geradts J, et al. Amplification and high-level expression of heat shock protein 90 marks aggressive phenotypes of human epidermal growth factor receptor 2 negative breast cancer. Breast Cancer Res. 2012;14:R62. doi: 10.1186/bcr3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Taqi MM, Bazov I, Watanabe H, et al. Prodynorphin CpG-SNPs associated with alcohol dependence: elevated methylation in the brain of human alcoholics. Addict Biol. 2011;16:499–509. doi: 10.1111/j.1369-1600.2011.00323.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Oberdoerffer S. A conserved role for intragenic DNA methylation in alternative pre-mRNA splicing. Transcription. 2012;3:106–9. doi: 10.4161/trns.19816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Osmark P, Hansson O, Jonsson A, Ronn T, Groop L, Renstrom E. Unique splicing pattern of the TCF7L2 gene in human pancreatic islets. Diabetologia. 2009;52:850–4. doi: 10.1007/s00125-009-1293-z. [DOI] [PubMed] [Google Scholar]
- 39.Shukla S, Kavak E, Gregory M, et al. CTCF-promoted RNA polymerase II pausing links DNA methylation to splicing. Nature. 2011;479:74–9. doi: 10.1038/nature10442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Prickett TD, Zerlanko B, Gartner JJ, et al. Somatic mutations in MAP3K5 attenuate its proapoptotic function in melanoma through increased binding to thioredoxin. J Invest Dermatol. 2014;134:452–60. doi: 10.1038/jid.2013.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu JS, Kim AK. Platycodin D induces reactive oxygen species-mediated apoptosis signal-regulating kinase 1 activation and endoplasmic reticulum stress response in human breast cancer cells. J Med Food. 2012;15:691–9. doi: 10.1089/jmf.2011.2024. [DOI] [PubMed] [Google Scholar]
- 42.McGrath JA, Stone KL, Begum R, et al. Germline mutation in EXPH5 implicates the Rab27B effector protein Slac2-b in inherited skin fragility. Am J Hum Genet. 2012;91:1115–21. doi: 10.1016/j.ajhg.2012.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pigors M, Schwieger-Briel A, Leppert J, et al. Molecular heterogeneity of epidermolysis bullosa simplex: contribution of EXPH5 mutations. J Invest Dermatol. 2014;134:842–5. doi: 10.1038/jid.2013.373. [DOI] [PubMed] [Google Scholar]
- 44.Liu F, Ji F, Ji Y, et al. Dissecting the mechanism of colorectal tumorigenesis based on RNA-sequencing data. Exp Mol Pathol. 2015;98:246–53. doi: 10.1016/j.yexmp.2015.01.004. [DOI] [PubMed] [Google Scholar]
- 45.Nomura N, Zu YL, Maekawa T, Tabata S, Akiyama T, Ishii S. Isolation and characterization of a novel member of the gene family encoding the cAMP response element-binding protein CRE-BP1. J Biol Chem. 1993;268:4259–66. [PubMed] [Google Scholar]
- 46.Dong B, Kim S, Hong S, et al. An infectious retrovirus susceptible to an IFN antiviral pathway from human prostate tumors. Proc Natl Acad Sci U S A. 2007;104:1655–60. doi: 10.1073/pnas.0610291104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Su WH, Yao Shugart Y, Chang KP, Tsang NM, Tse KP, Chang YS. How genome-wide SNP-SNP interactions relate to nasopharyngeal carcinoma susceptibility. PLoS One. 2013;8:e83034. doi: 10.1371/journal.pone.0083034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Qi L, Ding Y. Involvement of the CREB5 regulatory network in colorectal cancer metastasis. Yi Chuan. 2014;36:679–84. doi: 10.3724/SP.J.1005.2014.0679. [DOI] [PubMed] [Google Scholar]
- 49.Wang KS, Liu X, Zhang Q, Aragam N, Pan Y. Parent-of-origin effects of FAS and PDLIM1 in attention-deficit/hyperactivity disorder. J Psychiatry Neurosci. 2012;37:46–52. doi: 10.1503/jpn.100173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yoshiyama K, Higuchi Y, Kataoka M, Matsuura K, Yamamoto S. CD156 (human ADAM8): expression, primary amino acid sequence, and gene location. Genomics. 1997;41:56–62. doi: 10.1006/geno.1997.4607. [DOI] [PubMed] [Google Scholar]
- 51.Romagnoli M, Mineva ND, Polmear M, et al. ADAM8 expression in invasive breast cancer promotes tumor dissemination and metastasis. EMBO Mol Med. 2014;6:278–94. doi: 10.1002/emmm.201303373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shen Z, Kauttu T, Seppanen H, et al. Both macrophages and hypoxia play critical role in regulating invasion of gastric cancer in vitro. Acta Oncol. 2013;52:852–60. doi: 10.3109/0284186X.2012.718444. [DOI] [PubMed] [Google Scholar]
- 53.Li SQ, Zhu S, Wan XD, Xu ZS, Ma Z. Neutralization of ADAM8 ameliorates liver injury and accelerates liver repair in carbon tetrachloride-induced acute liver injury. J Toxicol Sci. 2014;39:339–51. doi: 10.2131/jts.39.339. [DOI] [PubMed] [Google Scholar]
- 54.Yang Z, Bai Y, Huo L, et al. Expression of A disintegrin and metalloprotease 8 is associated with cell growth and poor survival in colorectal cancer. BMC Cancer. 2014;14:568. doi: 10.1186/1471-2407-14-568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Errico A. Gastrointestinal cancer: ADAM8 provides new hope in pancreatic cancer. Nat Rev Clin Oncol. 2015;12:126. doi: 10.1038/nrclinonc.2015.22. [DOI] [PubMed] [Google Scholar]
- 56.Gole L, Crolla JA, Thomas SN, Jacobs PA, Dennis NR. Characterization of breakpoints in the GABRG3 and TSPY genes in a family with a t(Y;15) (p11.2;q12) Am J Med Genet A. 2004;125A:177–80. doi: 10.1002/ajmg.a.20482. [DOI] [PubMed] [Google Scholar]
- 57.Freichel M, Tsvilovskyy V, Camacho-Londono JE. TRPC4- and TRPC4-containing channels. Handb Exp Pharmacol. 2014;222:85–128. doi: 10.1007/978-3-642-54215-2_5. [DOI] [PubMed] [Google Scholar]
- 58.Vanden Abeele F, Lemonnier L, Thebault S, et al. Two types of store-operated Ca2+ channels with different activation modes and molecular origin in LNCaP human prostate cancer epithelial cells. J Biol Chem. 2004;279:30326–37. doi: 10.1074/jbc.M400106200. [DOI] [PubMed] [Google Scholar]
- 59.Veliceasa D, Ivanovic M, Hoepfner FT, Thumbikat P, Volpert OV, Smith ND. Transient potential receptor channel 4 controls thrombospondin-1 secretion and angiogenesis in renal cell carcinoma. FEBS J. 2007;274:6365–77. doi: 10.1111/j.1742-4658.2007.06159.x. [DOI] [PubMed] [Google Scholar]
- 60.Zeng B, Yuan C, Yang X, Atkin SL, Xu SZ. TRPC channels and their splice variants are essential for promoting human ovarian cancer cell proliferation and tumorigenesis. Curr Cancer Drug Targets. 2013;13:103–16. [PubMed] [Google Scholar]
- 61.Jiang HN, Zeng B, Zhang Y, et al. Involvement of TRPC channels in lung cancer cell differentiation and the correlation analysis in human non-small cell lung cancer. PLoS One. 2013;8:e67637. doi: 10.1371/journal.pone.0067637. [DOI] [PMC free article] [PubMed] [Google Scholar]