

Myasthenia gravis (MG) is an antibody-mediated autoimmune disorder affecting neuromuscular junctions, which is strongly associated with thymoma. Usually, the target of the autoimmune attack is the acetylcholine receptor (AChR) of the postsynaptic skeletal muscle endplate. However, non-AChR components, such as muscle-specific receptor tyrosine kinase (MuSK) and low-density lipoprotein receptor related protein 4 (LRP4), both of which are involved in the endplate maturation, may also serve as targets for autoantibodies.1 Myasthenia gravis may develop secondary to follicular dendritic cell sarcoma (FDCS), a rare malignant neoplasm of follicular dendritic cells (FDCs).2 However, therapeutic options for these MG patients are unclear. We herein illustrated the therapeutic effect of Rituximab on an anti-MuSK- and anti-AChR-positive MG patient concomitant with FDCS.
A 60-year-old woman was admitted to the in-patient department with 20 days symptoms of exertion dysphagia and dyspnea. She has a 6-month history of fatigable limb weakness, fluctuating ptosis, and diplopia, which worsened with activity and ameliorated with rest. Her fluctuating symptoms were responsive to intramuscular neostigmine. She had been diagnosed with recurrent FDCS during the past 15 years, and lymphadenectomy had been performed for 4 times. Upon physical examination, she was fully alert and oriented. Bilateral ptosis, weakness of eyelid closure without lower facial involvement, weakened lifting of bilateral soft palates, and head drop were noted while the other cranial nerves were intact. The muscle strength grade was Medical Research Council (MRC) Grade-4 in the limbs and tendon reflexes were normal. The Babinski signs were negative bilaterally. Lymph nodes were palpable in her left cervical and axillary regions. The laboratory investigations including routine blood test, liver functions, and renal, serum cancer marker (carcino-embryonic antigen, CA199, CA153, CA724, CA242, neuron-specific enolase, free beta-human chorionic gonadotrnpin, squamous cell carcinoma antigen, alpha fetoprotein) and paraneoplastic neurological syndrome (PNS) screening were unremarkable. The antibodies to AChR (optical density [OD] value=0.442, reference value: OD value <0.367) and MuSK (OD=0.846, reference value: OD <0.512) were positive. Thoracic and abdominal CT revealed no thymic abnormality, or any mediastinal or distant sites of mass suggestive of possible extranodal lesion. Instead, lymphadenectasis of her left cervical and axillary region was reported. Needle biopsy of the lymph node and pathology confirmed a diagnosis of FDCS (Figure 1). The Hematoxylin and Eosin staining revealed diffuse proliferation of spindled cells with nuclear atypia (Figure 1A) and eosinophilic cytoplasm (Figure 1B). Scattered small lymphoid cells and perivascular (arrow) clusters of small lymphocytes (Figure 1C) are also marked. Immunohistochemistry demonstrates positivity of tumor cells for CD21 (Figure 1D), CD35 (Figure 1E), vimentin (Figure 1F), S-100 (Figure 1G), CD3 (Figure 1H) and CD1a (not shown) while negativity for EMA, CK, CD20, lysozyme, MPO, CD34, CD30, and HMB45 (not shown). The Ki-67 labeling index of these cells reached approximately 20%. Based on the work-up, recurrent FDCS associated MG were diagnosed. On day 7 after admission, respiratory failure requiring intubation and ventilation indicative of myasthenic crisis led to her transfer to the Neurological Intensive Care Unit (N-ICU). Intravenous immunoglobulin (ivIg) was administered (400 mg/kg/day for 5 days) followed by tapered methylprednisolone (1000 mg, 500 mg, 250 mg, and 120 mg methylprednisolone, and each dose of steroids were used for 3 days before the one mg/kg/day oral methylprednisolone) was prescribed, which resulted in gradual recovery and extubation 30 days later. However, on day 38 of hospitalization, bilateral ptosis, weakness of eyelid closure and head drop were noted again, and the myasthenic crisis recurred subsequently without any identified causes like infections, or abrupt reduction of oral methylprednisolone. Considering the exacerbation and myasthenic crisis, together with the positive antibodies associated with MG, rituximab (375 mg/m2 iv injection per week for 4 consecutive weeks) was immediately prescribed in addition to oral methylprednisolone (one mg/kg/day). Gradually, she regained spontaneous respiration with a maximum of 10 hours after the first administration, and succeeded in extubation on day 53 of hospitalization.
Figure 1.
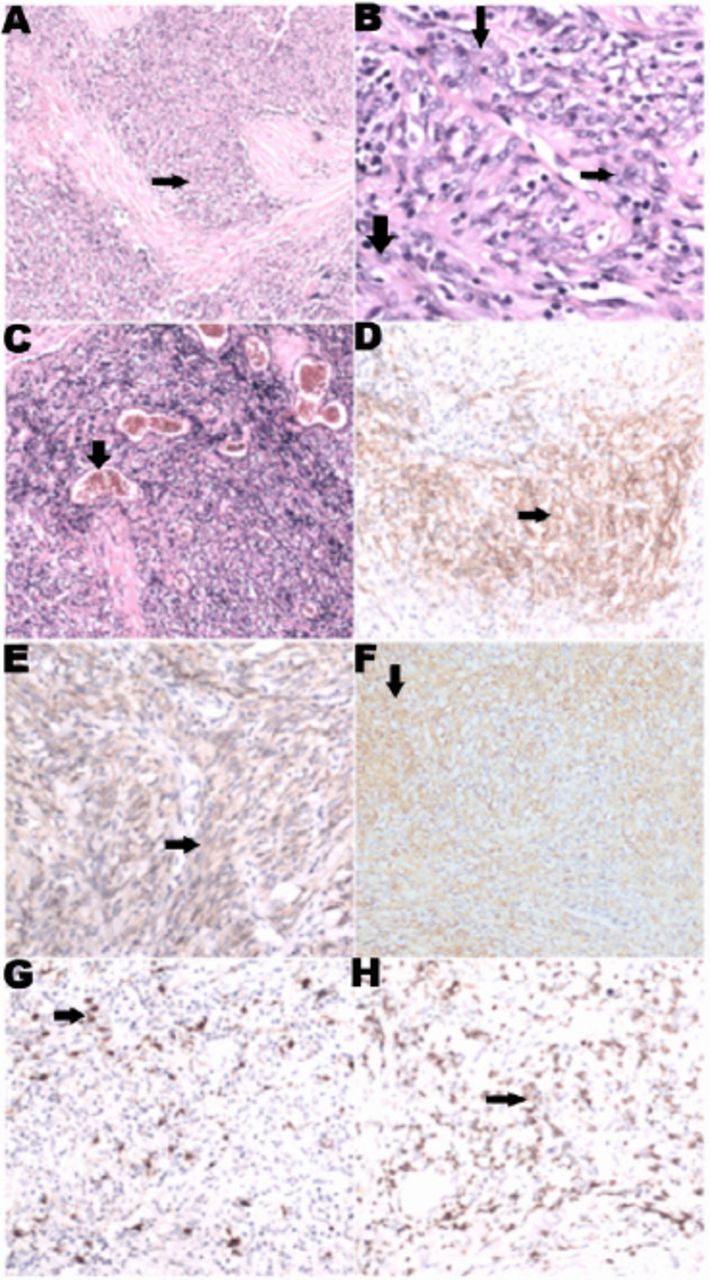
Pathological and immunohistochemical features of the follicular dendritic cell sarcoma (FDCS). The Hematoxylin and Eosin staining reveal diffuse proliferation of spindled cells with nuclear atypia (arrows, A to C, ×40). Eosinophilic cytoplasm with indistinct cytoplasmic borders and vesicular nuclei of the spindled cells are shown (B×200, arrows). Scattered small lymphoid cells and perivascular (arrow) clusters of small lymphocytes (C×40) are also marked. Immunohistochemistry demonstrates positivity of tumor cells for CD21 (D×100), CD35 (E×100), vimentin (F×100), S-100 (G×100), CD3 (H×100) and CD1a (arrows).
Our experience from this case favors use of rituximab in FDCS-associated anti-MuSK- and anti-AChR-positive MG. Interestingly, CD3 and CD1a, which are negative in most FDCS were positive in this case. This variant subtype has been scarcely reported to be related to the onset of MG.3 In the anti-AChR-positive MG, antibodies of Ig G1 and IgG3 isotype may potentially activate complement and attract lymphocytes, which results in the loss of functional AChRs.1 While in anti-MuSK-positive MG, the non-complement-binding IgG4 subclass interferes with the cellular agrin-MuSK signaling cascade and contributes to the formation of AChR-rapsyn clusters, which result in a reduction of the functional AChRs without the loss of junctional folds or AChR density.1 Moreover, patients with anti-MuSK-positive MG usually manifest with atypical severe clinical symptoms with a predilection of middle-aged woman.1 The occurrence of positive antibodies to MuSK and AChR, alleged double-positive MG, has been documented in literature.3 In this case, anti-MuSK MG is associated mostly with bulbar symptoms and limb weakness is uncommon. Also, the association between thymoma and anti-AchR antibodies is uncommon.
The pathogenesis of MG complicated with FDCS remains largely unknown. FDCS might mediate aberrant immune activation through different mechanisms, such as disturbing cytokine production and leading to the imbalance of the immunological microenvironment.
Rituximab is a monoclonal antibody targeting CD20 antigen and may deplete B cells, mainly pre-B and mature B cells, via cell-mediated or complement-dependent cytotoxicity.4 In addition to its confirmatory effects on B cell lymphoma, rituximab has been occasionally tested in various antibody-mediated autoimmune disorders, including neuromyelitis optica and MG.1 Rituximab proved to be effective in treating MG, particularly in dealing with refractory MG.3 The positive anti-MuSK- and anti-AChR autoantibodies in our patient indicated a pathogenic role of antibody-producing B cells. Given her recurrent myasthenic crisis and poor response to the methylprednisolone therapy, we prescribed rituximab which led to a marked improvement. Although the pathogenesis of MG concurrent with FDCS is unclear, rituximab appears to be a promising alternative in the treatment of these patients with refractory MG.
In summary, refractory MG might be associated with FDCS. Rituximab appears effective in dealing with patients with refractory MG.
References
- 1.Meriggioli MN, Sanders DB. Autoimmune myasthenia gravis:emerging clinical and biological heterogeneity. Lancet Neurol. 2009;8:475–490. doi: 10.1016/S1474-4422(09)70063-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kairouz S, Hashash J, Kabbara W, McHayleh W, Tabbara IA. Dendritic cell neoplasms:an overview. Am J Hematol. 2007;82:924–928. doi: 10.1002/ajh.20857. [DOI] [PubMed] [Google Scholar]
- 3.Zouvelou V, Zisimopoulou P, Psimenou E, Matsigkou E, Stamboulis E, Tzartos SJ. AChR-myasthenia gravis switching to double-seropositive several years after the onset. J Neuroimmunol. 2014;267:111–112. doi: 10.1016/j.jneuroim.2013.12.012. [DOI] [PubMed] [Google Scholar]
- 4.Yang CS, Yang L, Li T, Zhang DQ, Jin WN, Li MS, et al. Responsiveness to reduced dosage of rituximab in Chinese patients with neuromyelitis optica. Neurology. 2013;81:710–713. doi: 10.1212/WNL.0b013e3182a1aac7. [DOI] [PMC free article] [PubMed] [Google Scholar]