

When cells face genome instability, DNA damage surveillance mechanisms deploy a wide range of responses to stop the cell cycle and facilitate the repair of DNA lesions. Once the genome has been repaired, cells recover and return to a normal proliferative state.1 In contrast, if the lesions are not repaired, sustained activation of checkpoint mechanisms will often result in apoptosis. There is, however, a different and often overlooked cellular response to unrepairable DNA damage: checkpoint adaptation (reviewed in1). This process allows cells to re-enter the cell cycle and continue proliferating despite the presence of a limited number of unrepaired lesions. This process was initially described in Saccharomyces cerevisiae by Toczyski and Hartwell in 1997.2 More recently, checkpoint adaptation has been observed in higher organisms –including humans– and is thought to be an important driver of tumorigenesis.1,3
In their recent paper published in Cell Cycle, Rawal and colleagues found that phosphorylation of Cdc5 T-loop stimulates its kinase activity, and that this event represents an important regulatory step for adaptation to DNA damage.4 Cdc5 is the sole Polo-like kinase (PLK) in S. cerevisiae and is a key regulator of cell cycle transitions. PLKs are also critical regulators of the DNA damage response, but their role in this process is complex and not fully understood. In addition to promoting checkpoint adaptation, PLKs are also involved in the recovery phase of the DNA damage response, and in the direct modulation of DNA repair pathways.1
Although the involvement of Cdc5 in the DNA damage response is firmly established, the nature of the signal that acts upstream of Cdc5 to activate and/or redirect its kinase activity toward specific DNA damage functions has remained a mystery. The recent study by Rawal and colleagues addresses this issue by investigating the regulation of Cdc5 by phosphorylation. They demonstrate that phosphorylation of Thr238 in Cdc5 T-loop is crucial to maintain full kinase activity in mitosis.4 The fact that this residue is not essential for cell survival allowed the authors to study its contribution to checkpoint adaptation. They demonstrated that introduction of a phosphoablative T238A mutation in Cdc5 significantly reduces its kinase activity, completely prevents checkpoint adaptation, and alters the regulation of the Mus81-Mms4 resolvase. These defects have important physiological consequences since cdc5-T238A cells experience elevated levels of chromosome loss and rearrangements.4
Rawal and colleagues thus provide important mechanistic insights into the nature of the signal that promotes Cdc5 activity during the DNA damage response. Moreover, their analysis dovetails nicely with a recent investigation that addresses the importance of Cdc5 phosphorylation by Cdk1. Indeed, Rodriguez-Rodriguez et al have found that the Cdk1-dependent phosphorylation of Cdc5 on its T-loop is essential to promote full kinase activity.5 They also show that mutations preventing this Cdk1-dependent phosphorylation disrupt Cdc14 activation in early anaphase. This suggests that Cdc5 might promote adaptation to DNA damage at least in part by stimulating late mitotic events, consistent with a recent study by Valerio-Santiago and colleagues.6
The study by Rawal and colleagues also raises the important issue of the target of Cdc5 during the cellular response to DNA damage. They provide convincing evidence that the phosphorylation of Mms4 is reduced in cdc5-T238A cells4, but the fact that another adaptation defective mutant, cdc5-ad, shows normal Mms4 phosphorylation suggests that this protein is not a critical target of Cdc5 during the adaptation response. Interestingly, recent work by Ratsima and colleagues7 has shed some light on the nature of the proteins acting downstream of Cdc5 during adaptation. In particular, they have shown that Cdc5 acts in a polo box domain (PBD)-dependent manner with the RSC complex to promote checkpoint adaptation and genome stability. Surprisingly, they also showed that Cdc5 must localize to spindle pole bodies (SPBs) to promote adaptation, thereby suggesting that SPB components might act as a platform or target for Cdc5 in this process. Taken together with the study by Rawal and colleagues, these observations raise the interesting possibility that Cdk1 might promote Cdc5 activity during the adaptation response through 2 independent mechanisms: first by promoting full Cdc5 kinase activity via Thr238 phosphorylation4,5, and second by stimulating a phospho-dependent interaction between Cdc5 PBD and its downstream effectors7 (Fig. 1). Further work will be required to determine the relative contribution of these 2 mechanisms to Cdc5-dependent checkpoint adaptation.
Figure 1.
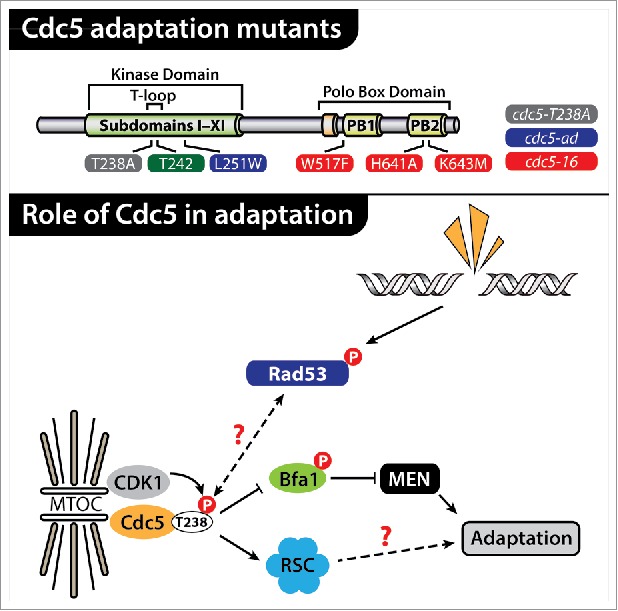
Cdc5 promotes adaptation to persistent DNA damage. Cdc5 mutations leading to adaptation defects are shown at the top of the figure.2,4,5,7 Note that Thr242 is another Cdk1-dependent phosphosite in Cdc5 T-loop.5 The lower part of the figure depicts factors acting upstream (Cdk15) and downstream (Bfa16 and RSC complex7) of Cdc5 during the adaptation response to persistent DNA damage.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- [1].Serrano D, D'Amours D. When genome integrity and cell cycle decisions collide: roles of polo kinases in cellular adaptation to DNA damage. Syst Synth Biol 2014; 8:195-203; PMID:25136381; http://dx.doi.org/ 10.1007/s11693-014-9151-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Toczyski DP, Galgoczy DJ, Hartwell LH. CDC5 and CKII control adaptation to the yeast DNA damage checkpoint. Cell 1997; 90:1097-106; PMID:9323137; http://dx.doi.org/ 10.1016/S0092-8674(00)80375-X [DOI] [PubMed] [Google Scholar]
- [3].Swift LH, Golsteyn RM. Cytotoxic amounts of cisplatin induce either checkpoint adaptation or apoptosis in a concentration-dependent manner in cancer cells. Biol Cell 2016; 108:127-48; PMID:26871414; http://dx.doi.org/ 10.1111/boc.201500056 [DOI] [PubMed] [Google Scholar]
- [4].Rawal CC, et al.. Reduced kinase activity of polo kinase Cdc5 affects chromosome stability and DNA damage response in S. cerevisiae. Cell Cycle 2016; 15(21):2906-2919; PMID:27565373; http://dx.doi.org/ 10.1080/15384101.2016.1222338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Rodriguez-Rodriguez JA, Moyano Y, Játiva S, Queralt E. Mitotic Exit Function of Polo-like Kinase Cdc5 Is Dependent on Sequential Activation by Cdk1. Cell Rep 2016; 15:2050-62; PMID:27210759; http://dx.doi.org/ 10.1016/j.celrep.2016.04.079 [DOI] [PubMed] [Google Scholar]
- [6].Valerio-Santiago M, de Los Santos-Velázquez AI, Monje-Casas F. Inhibition of the mitotic exit network in response to damaged telomeres. PLoS Genet 2013; 9:e1003859; PMID:24130507; http://dx.doi.org/ 10.1371/journal.pgen.1003859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Ratsima H, Serrano D, Pascariu M, D'Amours D. Centrosome-Dependent Bypass of the DNA Damage Checkpoint by the Polo Kinase Cdc5. Cell Rep 2016; 14:1422-34; PMID:26832404; http://dx.doi.org/ 10.1016/j.celrep.2016.01.014 [DOI] [PubMed] [Google Scholar]