

ABSTRACT
Biguanides, including metformin (widely used in diabetes treatment) and phenformin, are AMP-activated protein kinase (AMPK) activators and potential drugs for cancer treatment. A more in-depth understanding of how cancer cells adapt to biguanide treatment may provide important therapeutic implications to achieve more effective and rational cancer therapies. NBR2 is a glucose starvation-induced long non-coding RNA (lncRNA) that interacts with AMPK and regulates AMPK activity upon glucose starvation. Here we show that phenformin treatment induces NBR2 expression, and NBR2 deficiency sensitizes cancer cells to phenformin-induced cell death. Surprisingly, unlike glucose starvation, phenformin does not induce NBR2 interaction with AMPK, and correspondingly, NBR2 deficiency does not affect phenformin-induced AMPK activation. We further reveal that NBR2 depletion attenuates phenformin-induced glucose transporter GLUT1 expression and glucose uptake. GLUT1 deficiency sensitizes cancer cells to phenformin-induced cell death, whereas GLUT1 restoration in NBR2 deficient cells rescues the increased cell death upon phenformin treatment. Together, the results of our study reveal that NBR2-GLUT1 axis may serve as an adaptive response in cancer cells to survive in response to phenformin treatment, and identify a novel mechanism coupling lncRNA to biguanide-mediated biology.
KEYWORDS: AMPK, biguanide, GLUT1, long non-coding RNA, NBR2, phenformin
Introduction
Biguanides, including metformin and phenformin, are inhibitors of mitochondrial respiratory chain complex I, and have been shown to exhibit antitumor effects.1-7 Metformin is one of the most widely prescribed drugs for the treatment of type II diabetes. Both retrospective analyses and subsequent clinical trials support the beneficial effect of metformin in cancer prevention and treatment.8-11 Currently, metformin is being tested in multiple clinical trials as an anticancer agent (www.clinicaltrials.gov). Phenformin is a much more potent inhibitor of mitochondrial respiratory chain complex I than metformin. Although the use of phenforminin in patients has been suspended by the FDA due to lactic acidosis associated with its use, such side effects have occurred in only a very small subset of patients, mainly in elderly patients with renal failure.12 Moreover, it was recently reported that supplementation of 2-deoxyglucose with phenformin may avoid the risk of lactic acidosis.13 Therefore, there has been increasing interest to re-examine phenformin as a potential agent in cancer prevention and treatment.1-3,14
At least 3 mechanisms have been proposed to explain the antitumor effects of biguanides2,15: the first mechanism relates to the effect of biguanides to decrease blood glucose and insulin levels. Specifically, biguanides inhibits hepatic gluconeogenesis and increases glucose uptake in skeletal muscle, leading to decreases of blood glucose and insulin levels, which in turn suppresses tumor growth through systemic effect. Second, biguanides inhibit respiratory chain complex I in the mitochondria, resulting in ATP depletion and energy stress induction in tumor cells, which leads to the activation of AMP activated protein kinase (AMPK). AMPK, a critical sensor of cellular energy status, is activated under energy stress conditions with decreases of ATP concentrations and corresponding increases of AMP concentrations.16 AMP binds to AMPK and leads to allosteric activation of AMPK. In addition, the upstream kinase LKB1 phosphorylates AMPK in response to energy stress, which further substantially activates AMPK. Once activated, AMPK phosphorylates a diverse array of downstream substrates to inhibit ATP-consuming anabolic processes and activate ATP-generating catabolic processes, and eventually to restore energy balance in response to energy stress.16,17 Since anabolic processes, such protein or lipid synthesis, are required to support tumor growth, AMPK activation inhibits tumor development in many cancers, and several components in AMPK pathway, such as LKB1, are mutated in a variety of human cancers.18 Correspondingly, it has been proposed that biguanides inhibit tumor development at least in part through biguanides-mediated AMPK activation.2,15 It should be noted that other studies have documented that biguanides can also exert their antitumor or other biological effects through AMPK-independent mechanisms.19-22
Third, some tumor cells with certain genetic alterations lack the ability to appropriately respond and adapt to energy stress induced by biguanides, rendering such tumor cells particularly sensitive to biguanides-induced cell death. For example, it has been shown that LKB1 deficiency sensitizes non-small cell lung cancer cells or tumors to biguanide treatment, leading to the suggestion that LKB1 status may be used to select lung cancer patients for biguanide treatment.23 It has also been shown that hyperglycemia-induced metabolic compensation affects metformin sensitivity in cancer cells.24 Finally, a recent study showed that cancer cells with impaired oxidative phosphorylation or glucose uptake/utilization are sensitive to biguanides, and proposed that mutations in genes involved in oxidative phosphorylation or glucose uptake/utilization may serve as biomarkers to identify cancer patients for biguanide treatment.25 However, whether these proposed biomarkers can indeed be used as predictive markers for biguanide treatment in cancer patients awaits further clinical investigations. In addition, a better understanding of how cancer cells respond and adapt to biguanide treatment may yield other important therapeutic implications.
Long non-coding RNAs (lncRNAs) are the type of non-coding RNAs that are more than 200 nucleotides.26-28 Many studies have shown that lncRNAs have critical biological functions, ranging from regulating cell proliferation/growth/migration to controlling stem cell homeostasis and metabolism. LncRNAs exert biological functions through their interactions with other cellular macromolecules, such as chromatin DNA, RNA, or protein.29 Dysregulation of lncRNAs has been associated with many human diseases, including cancer and metabolic diseases.30,31 However, the potential role of lncRNAs in biguanide-mediated biological processes remains largely unexplored.
NBR2 (neighbor of BRCA1 gene 2) is a non–protein coding gene that resides adjacent to tumor suppressor gene BRCA1,32 and its potential biological function had remained unknown for many years since its initial discovery.33-35 Our recent study identified NBR2 as a lncRNA that is induced by glucose deprivation.36,37 We further showed that, upon glucose starvation, NBR2 interacts with AMPK and promotes AMPK kinase activity. Correspondingly, NBR2 deficiency dampens glucose starvation-induced AMPK activation and AMPK-mediated downstream biological processes.36 NBR2 deficient cells share some similarities to cells with defective AMPK pathway, such as LKB1 deficient cells. For example, both NBR2 and LKB1 deficient cells are more sensitive to glucose starvation-induced cell death.36,38 As discussed above, since defective AMPK activation, at least in some cellular contexts, renders cells more sensitive to biguanide-induced cell death, in this study we examined the potential role of NBR2 in mediating cellular response to biguanide treatment. We revealed that NBR2 deficiency renders cancer cells more sensitive to phenformin-induced cell death. Surprisingly, NBR2 knockdown does not affect phenformin-induced AMPK activation. Instead, we showed that NBR2 deficiency inhibits phenformin-induced GLUT1 expression and glucose uptake, and NBR2 regulates cell survival under phenformin treatment through its regulation of GLUT1 expression.
Results
NBR2 deficiency renders cancer cells more sensitive to phenformin-induced cell death
Our previous study identified NBR2 as an energy stress-induced lncRNA.36 Specifically, both glucose deprivation and 2-deoxy-glucose (2DG) treatment upregulated the expression of NBR2 in a panel of cancer cell lines. Glucose provides the major energy source for the majority of cells. After uptaken into cells or metalized from other nutrients, glucose first enters into glycolysis pathway in order to generate ATP eventually. 2DG, an analog of glucose, inhibits hexokinase and blocks the first rate-limiting step in glycolysis. Thus both glucose starvation and 2DG treatment decrease ATP level and increase AMP level, and induce energy stress.39 The biguanide compound phenformin is an inhibitor of mitochondrial respiratory chain complex I, and phenformin treatment in cells also decreases ATP concentration and thus induces energy stress.2,15 Therefore, we tested whether phenformin treatment, similar to glucose starvation or 2DG treatment, also induced NBR2 expression. Indeed, our analysis revealed that phenformin treatment induced NBR2 expression in a variety of cell lines (Fig. 1A). Since our previous study showed that NBR2 is down-regulated in kidney and breast cancers,36 we have used 786-O cells (a kidney cancer cell line) and MDA-MB-231 cells (a breast cancer cell line) in our following studies.
Figure 1.
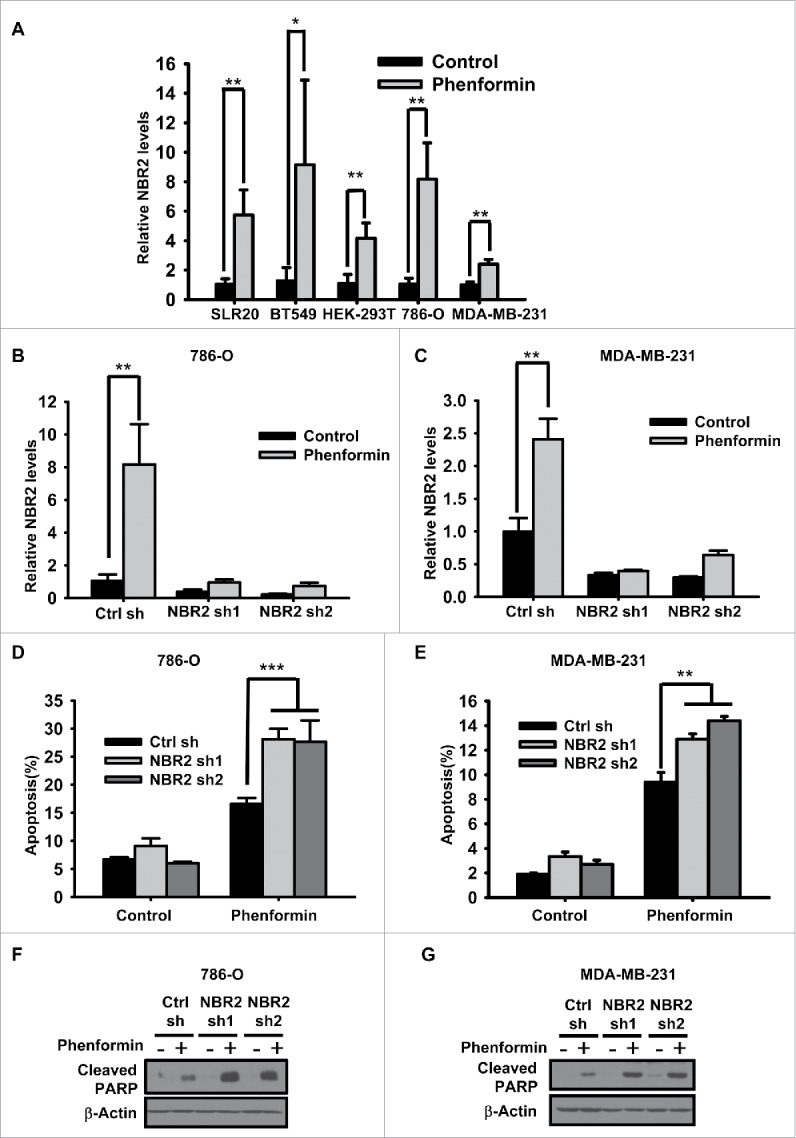
Phenformin induces NBR2 expression and NBR2 deficiency renders cancer cells more sensitive to phenformin-induced cell death. (A) Various cell lines were treated with 0 or 2mM phenformin for 18–24 hours, and then subjected to real-time PCR analysis to measure NBR2 expression. Three independent experiments were performed and the values were expressed as the mean ± SD, *: P< 0.05, **: P < 0.01. (B and C) Bar graph showing NBR2 shRNA-mediated knockdown efficiency by real-time PCR analysis under basal and phenformin treatment in 786-O (B) and MDA-MB-231 cells (C). Three independent experiments were performed and the values were expressed as the mean ± SD, **: P < 0.01. (D and E) Control shRNA or NBR2 shRNA-infected 786O cells (D) or MDA-MB-231 cells (E) were treated with 0 or 2 mM phenformin for 18 hours, then subjected to Annexin V/PI staining followed by FACS analysis to measure the percentages of Annexin V positive/PI negative cells. Three independent experiments were performed and the values were expressed as the mean ± SD, **: P < 0.01, ***: P < 0.001. (F and G) Control shRNA or NBR2 shRNA-infected 786O cells (F) or MDA-MB-231 cells (G) were treated with 0 or 2 mM phenformin for 18 hours. Cell lysates were then analyzed by Western blotting to measure PARP cleavage.
Our previous study showed that, whereas NBR2 deficiency did not affect apoptosis under normal culture condition, NBR2 knockdown sensitized cells to glucose starvation-induced apoptosis.36 We thus examined whether NBR2 deficiency also affected phenformin treatment-induced cell death. We established 786-O and MDA-MB-231 cells with stable knockdown of NBR2 by 2 independent NBR2 shRNAs. Real-time PCR confirmed efficient knockdown of NBR2 under both basal and phenformin treatment conditions (Fig. 1B–C). Annexin V staining analysis revealed that, in both 786-O and MDA-MB-231 cells, NBR2 knockdown rendered cells more sensitive to phenformin treatment-induced apoptosis (Fig. 1D–E). Western blotting also showed that phenformin treatment increased PARP cleavage, a biochemical surrogate for apoptosis measurement, and NBR2 knockdown further increased PARP cleavage under phenformin treatment (Fig. 1F–G). Together, our data revealed that phenformin treatment induces NBR2 expression and that NBR2 deficiency promotes phenformin treatment-induced apoptosis, suggesting that phenformin-induced NBR2 expression may serve as an adaptive response to maintain cell survival in response to phenformin treatment.
NBR2 does not regulate phenformin-induced AMPK activation and mTORC1 inactivation
It is well known that phenformin treatment induces the activation of AMPK activation and the inactivation of mammalian target of rapamycin complex 1 (mTORC1, also called mechanistic TORC1), and at least in some cellular contexts, cells with defective AMPK signaling pathway are more sensitive to phenformin treatment-induced cell death. For example, phenformin selectively induced apoptosis in lung cancer cells with deficiency of LKB1, which encodes the major upstream kinase of AMPK in response to energy stress.23 Since our previous study showed that NBR2 deficiency attenuates glucose starvation-induced AMPK activation and mTORC1 inactivation,36 we also tested whether NBR2 deficiency similarly affected AMPK-mTORC1 pathway under phenformin treatment. As expected, phenformin treatment activated AMPK pathway as shown by the increased phosphorylation of AMPK or AMPK substrate acetyl-CoA carboxylase (ACC), and inhibited mTORC1 pathway as revealed by the decrease of S6 phosphorylation (Fig. 2A and 2B). To our surprise, NBR2 knockdown did not significantly affect phenformin treatment-induced AMPK activation or mTORC1 inactivation in either 786-O or MDA-MB-231 cells (Fig. 2A and 2B).
Figure 2.

NBR2 does not regulate phenformin-induced AMPK activation and mTORC1 inactivation. (A and B) Control shRNA or NBR2 shRNA-infected 786O cells (A) or MDA-MB-231 cells (B) were treated with 0 or 2mM phenformin for 12 hours. Cell lysates were then analyzed by Western blotting. (C) In vitro-synthesized biotinylated sense (S) NBR2 were incubated with protein lysates from 786-O cells which had been cultured in 25 or 0 mM glucose-containing medium for 24 hours, or treated with 2 mM phenformin for 18 hours. Precipitation reactions were conducted using streptavidin beads and then subjected to Western blotting. (D) 786-O cells were cultured in 0 or 25 mM glucose-containing medium for 24 hours, or treated with 2 mM phenformin for 18 hours. Protein lysates were prepared and immunoprecipitated with AMPK α antibody or IgG. The RNA levels of NBR2 in immunoprecipitates or cell lysates (input) were measured by real-time PCR. Three independent experiments were performed and the values were expressed as the mean ± SD. *: P < 0.05.
Our previous study showed that NBR2 promotes AMPK activation through a glucose starvation-induced NBR2 interaction with AMPK.36 Thus, we examined whether phenformin similarly affected NBR2-AMPK interaction by RNA-pulldown assay. To this end, we incubated in vitro-synthesized biotinylated sense (S) NBR2 or antisense (AS) NBR2 (as a negative control) with protein lysates from cells which had been cultured in 25 or 0 mM glucose-containing medium, or treated with phenformin, followed by precipitation with streptavidin beads. Such analysis confirmed that glucose starvation induced the interaction of NBR2 with endogenous AMPKα; notably, phenformin treatment did not induce NBR2 interaction with AMPKα (Fig. 2C). We also performed RNA immunoprecipitation assay to examine NBR2-AMPK interaction at the endogenous levels. Consistent with our previous report,36 the analysis confirmed that glucose starvation substantially increased the enrichment of NBR2 in AMPKα precipitates; however, phenformin treatment did not affect the enrichment of NBR2 in AMPKα precipitates (Fig. 2D). Together, both RNA-pulldown and RNA immunoprecipitation assays revealed that, in contrast to glucose starvation, phenformin treatment did not regulate NBR2 interaction with AMPK, which provides a likely explanation on the differential effects of NBR2 on AMPK activation under glucose starvation and phenformin treatment conditions (see Discussion).
NBR2 regulates GLUT1 expression and glucose uptake in response to phenformin treatment
Recent studies showed that glucose uptake and utilization affect cancer cell sensitivity to phenformin treatment.25 Specifically, low expression of glucose transporters, including GLUT1 and GLUT3, in certain cancer cells correlated with the defective glucose uptake/utilization and increased sensitivity to phenformin treatment in those cancer cells. Importantly, restoration of GLUT expression attenuated the phenformin sensitivity in the corresponding cancer cells.25 We thus examined whether NBR2 deficiency affected glucose uptake under phenformin treatment. As shown in Figure 3A–B, phenformin treatment significantly increased glucose uptake in both 786-O and MDA-MB-231 cells, which is in line with the observations made in other cellular contexts that biguanides promote glucose uptake 19,40,41 While NBR2 knockdown did not affect the basal glucose uptake, NBR2 deficiency attenuated phenformin-induced glucose uptake. We also observed that NBR2 knockdown led to moderate decrease of lactate production under phenformin treatment (Fig. 3C–D).
Figure 3.

NBR2 regulates GLUT1 expression and glucose uptake in response to phenformin treatment. (A and B) 786O cells (A) or MDA-MB-231 cells (B) were treated with 0 or 2 mM phenformin for 12 hours, and then subjected to analyses to measure glucose uptake. Three independent experiments were performed and the values were expressed as the mean ± SD. ***: P< 0.001. (C and D) 786O cells (C) or MDA-MB-231 cells (D) were treated with 0 or 2 mM phenformin for 12 hours, and then subjected to analyses to measure lactate production. Three independent experiments were performed and the values were expressed as the mean ± SD. *: P < 0.05. (E and F) 786O cells (E) or MDA-MB-231 cells (F) were treated with 0 or 2 mM phenformin for 12 hours, and then subjected to real-time PCR analysis to measure the expression of indicated glucose transporters. Three independent experiments were performed and the values were expressed as the mean ± SD. ***: P < 0.001. (G and H) 786O cells (G) or MDA-MB-231 cells (H) were treated with 0 or 2 mM phenformin for 18 hours. Cell lysates were then analyzed by Western blotting.
The aforementioned data raised the possibility that NBR2 may regulate glucose transporter in response to phenformin treatment. To this end, we first examined whether NBR2 deficiency affected the expression levels of GLUT1-4, the 4 major glucose transporters,42 under phenformin treatment. Real-time PCR analysis revealed that phenformin treatment increased the expression of GLUT1, 3 and 4 (we could not detect GLUT2 expression in cell lines used in this study); notably, phenformin-induced expression of GLUT1, but not GLUT3 or GLUT4, was significantly attenuated upon NBR2 knockdown (Fig. 3E–F). We further confirmed that NBR2 deficiency decreased phenformin-induced GLUT1 expression by western blotting (Fig. 3G–H). Collectively, our data showed that NBR2 deficiency inhibits phenformin treatment-induced GLUT1 expression and glucose uptake. Importantly, GLUT1 is the glucose transporter with the most relevance to cancer biology with overexpression in many human cancers.43
NBR2 regulates phenformin-induced cell death at least partly through GLUT1
Next we studied whether NBR2-mediated GLUT1 expression plays any causal role in NBR2 regulation of cell survival under phenformin treatment. To address this question, we first examined whether GLUT1 deficiency, similar to NBR2 deficiency, also sensitized cells to phenoformin-induced apoptosis. We identified 2 GLUT1 siRNAs that exhibited significant knockdown efficiency in 786O and MDA-MB-231 cells (Fig. 4A). Further analysis revealed that GLUT1 knockdown indeed potentiated phenformin-induced apoptosis in these cells, as revealed by both Annexin V staining and cleaved PARP western blotting (Fig. 4B–4C).
Figure 4.
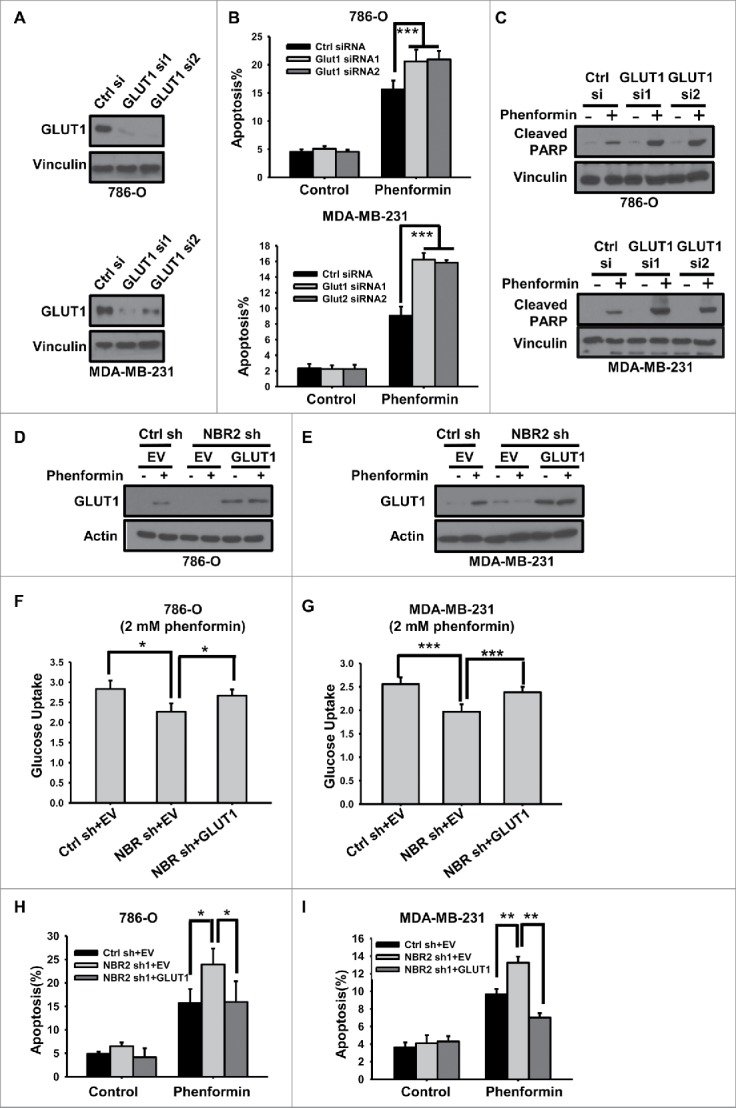
NBR2 regulates phenformin-induced cell death at least partly through GLUT1. (A) 786O cells (upper panel) or MDA-MB-231 cells (lower panel) were transfected with control or 2 independent GLUT1 siRNAs. Protein lysates were prepared and analyzed by Western blotting as indicated. (B) 786O cells (upper panel) or MDA-MB-231 cells (lower panel) transfected with control or GLUT1 siRNAs were treated with 0 or 2 mM phenformin for 18 hours, and then subjected to Annexin V/PI staining followed by FACS analysis to measure the percentages of Annexin V positive/PtdIns negative cells. Three independent experiments were performed and the values were expressed as the mean ± SD. ***: P< 0.001. (C) 786O cells (upper panel) or MDA-MB-231 cells (lower panel) transfected with control or GLUT1 siRNAs were treated with 0 or 2 mM phenformin for 18 hours, protein lysates were prepared and analyzed by Western blotting to detect PARP cleavage. (D and E) 786O cells (D) or MDA-MB-231 cells (E) with stable expression of control shRNA or NBR2 shRNA were infected with empty vector (EV) or GLUT1. Protein lysates were prepared and analyzed by Western blotting. (F and G) 786O cells (F) or MDA-MB-231 cells (G) with indicated genotypes were treated with 2 mM phenformin for 12 hours, and then subjected to analyses to measure glucose uptake. Three independent experiments were performed and the values were expressed as the mean ± SD. *: P < 0.05, ***: P < 0.001. (H and I) 786O cells (H) or MDA-MB-231 cells (I) with indicated genotypes were treated with 0 or 2 mM phenformin for 18 hours, and then subjected to Annexin V/PI staining followed by FACS analysis to measure the percentages of Annexin V positive/PI negative cells. Three independent experiments were performed and the values were expressed as the mean ± SD. *: P < 0.05, **: P < 0.01.
As a complementary approach, we also tested whether restoration of GLUT1 in NBR2 deficient cells would normalize the increase of phenformin-induced cell death afforded by NBR2 knockdown. GLUT1 was stably expressed in 786-O or MDA-MB-231 cell lines with NBR2 knockdown. Western blotting showed that the expression level of ectopically expressed GLUT1 in NBR2 knockdown cells was comparable to or slightly higher than that of phenformin-induced endogenous GLUT1 in control shRNA-infected cells (Fig. 4D–E). As expected, GLUT1 over-expression restored defective glucose uptake in NBR2 deficient cells under phenformin treatment condition (Fig. 4F–G). Importantly, GLUT1 re-expression suppressed phenformin-induced apoptosis in NBR2 deficient cells to the level similar to that in control shRNA-infected cells (Fig. 4H–I). Taken together, our data strongly suggest that the enhanced apoptosis in response to phenformin treatment observed in NBR2 deficient cells is largely mediated by the defective phenformin-induced GLUT1 expression in these cells, suggesting that NBR2 promotes cell survival under phenformin treatment at least partly through upregulating GLUT1 expression.
Discussion
Our current and previous studies36 together reveal that NBR2 exhibits differential effects on AMPK activation induced by different AMPK agonists. Specifically, while NBR2 knockdown attenuated glucose starvation, 2DG, or AMPK activator A769662-induced AMPK activation,36 NBR2 deficiency did not affect phenformin-induced AMPK activation (Fig. 2A–B), which is likely due to the differential effects of glucose starvation and phenformin treatment on NBR2 interaction with AMPK (Fig. 2C–D). Although both glucose starvation and phenformin treatment decrease cellular ATP concentration and induce energy stress (as revealed by AMPK activation), glucose starvation also induces many other biological effects which may not be affected by phenformin treatment (such as the effect on NADPH production through the pentose phosphate pathway). Future studies will be directed to study the underlying mechanisms by which glucose starvation, but not phenformin treatment, induces NBR2-AMPK interaction. It should be noted that other studies have shown that, in many contexts, biguanide treatment is not equivalent to glucose starvation or AMPK activator treatment, and biguanides can exert biological effects through AMPK-independent mechanisms.19-21 The observations from our study are in line with this notion.
Our data further showed that NBR2 regulates cancer cell sensitivity to phenformin treatment at least partly through NBR2-mediated upregulation of glucose transporter GLUT1 in response to phenformin treatment. There are at least 14 members of glucose transporters, among which GLUT1-4 are the best characterized glucose transporters.44 These glucose transporters function to transport glucose across the plasma membrane with different regulatory and kinetic properties as well as differential expression patterns.42 For example, GLUT2 represents the major hepatocyte glucose transporter and mainly functions to uptake excess glucose from blood into hepatocytes, while GLUT4 mainly mediates insulin-regulated glucose uptake in adipocytes and muscle cells. GLUT3 primarily mediates glucose uptake in neurons; GLUT1, on the other hand, exhibits relatively ubiquitous expression pattern and mediates basal glucose uptake in most cell lines. GLUT1 is also upregulated in many types of human cancers, and its high expression often correlates with poor prognosis in those cancers.43 It is well documented that many cancers exhibit increased glucose uptake in order to meet their increased bioenergetics and biosynthetic needs, and the enhanced glucose uptake is at least partly caused by the increased GLUT1 expression in cancer cells. Indeed, such observation has been applied in the clinic to use 18F-deoxyglucose positron emission tomography (FDG-PET) to monitor glucose uptake in tumors and to detect tumors in cancer patients.45 It has been shown that, in colorectal cancer cells, BRAF or KRAS mutations lead to upregulation of GLUT1 expression, which serves to promote glucose uptake and survival under low-glucose conditions.46 Another study showed that low expression of GLUT1 sensitizes cancer cells to phenformin treatment.25 Phenformin-induced expression of GLUT1, 3, and 4, as revealed from our study, likely represents an adaptive response of cancer cells to promote glucose uptake and compensate for the energy stress imposed by phenformin treatment. Notably, NBR2 knockdown only affected phenformin-induced GLUT1 expression, which is in line with the well-established role of GLUT1 in cancer biology. How NBR2 regulates GLUT1 expression will be the subject of future studies. Our data showed that, under phenformin treatment, NBR2 localized in both cytoplasm and nucleus (Fig. 5). Thus, it is possible that NBR2 may control the transcription of GLUT1 through NBR2 interaction with other proteins involved in transcriptional regulation.
Figure 5.

The subcellular localization of NBR2 under phenformin treatment. (A) Western blotting analysis for Vinculin (cytoplasmic marker) and Lamin A/C (nuclear marker) in total, cytoplasmic (Cyt) and nuclear (Nul) fractions of 786-O cells. (B) Real-time PCR showing the relative expression levels for GAPDH, U1 and NBR2 in cytoplasmic and nuclear fractions of 786-O cells. Three independent experiments were performed and the values were expressed as the mean ± SD. **: P < 0.01.
Our discovery that NBR2 deficiency renders cancer cells more susceptible to phenformin treatment may also have clinical application. It is possible to use NBR2 status as a biomarker to predict the response of cancer patients to biguanide treatment. Based on this scenario, cancer patients with low expression levels of NBR2 may respond better to biguanide treatment than those with high expression levels of NBR2. Since our current study has focused on phenformin, it is important to emphasize that several lines of evidence strongly support the utility of phenformin in cancer treatment in the near future: first, phenformin is a much more potent inhibitor of mitochondrial complex I, with more than 50 fold greater potency, than metformin.47,48 Thus, presumably phenformin will be a more potent drug for cancer treatment than metformin. Second, since metformin uptake into cells largely depends on transporter OCT1, the variable expression levels of OCT1 in tumors will likely affect the clinical outcome in cancer patients with metformin treatment. Phenformin, on the other hand, is more lipophilic and does not depend on transporter to cross the plasma membrane. Third, although phenformin is associated with more risk of lactic acidosis than metformin, such side effects only occurred in about 0.06% patients (64 cases per 100,000 patients per year for phenformin treatment vs 3 cases per 100,000 patients per year for metformin treatment).12 Indeed, it has been argued that the side effects of phenformin actually are even milder than those of many other cancer drugs currently used in clinic, and the toxicity of phenformin can be minimized by adjusting the treatment dosage and duration in cancer patients.14 Finally, whether NBR2 plays any role in metformin treatment-induced cell death remains to be investigated in the future studies.
Together, our results identify NBR2 as a phenformin-induced lncRNA, and show that NBR2 promotes cancer cell survival in response to phenformin through upregulating GLUT1. Our study thus reveals an unanticipated role of lncRNAs in biguanide-mediated biological processes and suggests that NBR2 may serve as a novel biomarker to predict biguanide treatment response in cancer patients.
Materials and methods
Cell culture studies
Human kidney cancer cell lines, human breast cancer cell lines, and human embryonic kidney 293 (HEK293T) cells used in this study were mostly obtained from American Type Culture Collection (ATCC). All of the cell lines were free of mycoplasma contamination (tested by the vendors using the MycoAlert kit from Lonza). No cell lines used in this study are found in the database of commonly misidentified cell lines (ICLAC and NCBI Biosample) based on short tandem repeats (STR) profiling performed by vendors. siRNA and plasmid transfections were performed using Lipofectamine 3000 (Life Technologies). Lentiviruses were produced in HEK293T cells with packing mix (ViraPower Lentiviral Expression System, Invitrogen) and used to infect target cells as per manufacturer's instruction. For glucose starvation experiments, cells were cultured in DMEM with different concentrations of glucose (0, or 25 mM) + 10% dialyzed FBS. To measure apoptosis, the cells were stained by Annexin V: FITC Apoptosis Detection Kit I (BD pharmingen, 556547) as previously described.49-51 Briefly, treated cells were washed with PBS twice and then 1×106 cells were resuspended in 100μL of 1x binding buffer. FITC-labeled Annexin V and propidium iodide were added to samples and incubated in dark for 15 minutes at room temperature. Subsequently cells were subjected to FACS analysis. Cell proliferation was assessed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assays as previously described.52
Constructs and reagents
shRNAs targeting human NBR2 (NM_005821.2-615s1c1, NM_005821.2-514s1c1) were purchased from Sigma. siRNA targeting GLUT1 were purchased from Sigma (EHU028011, NM_006516). GLUT1 cDNA-containing lentiviral plasmid was obtained from MD Anderson Cancer Center shRNA and ORFeome Core Facility. Phenformin was purchased from Sigma (P7045).
Glucose uptake, and lactate production assays
To measure glucose uptake, cells were seeded in 6-well plate in triplicate and then treated with or without phenformin for 24 hours. Culture medium was then removed from each well and replaced with 1ml of fresh culture medium containing 50 μM fluorescent 2-NBDG (Molecular Probes-Invitrogen). The cells were incubated at 37°C with 5% CO2 for 30 minutes. The cells were then washed twice with cold phosphate-buffered saline (PBS) and collected for flow cytometry analysis. To measure lactate production, cells were seeded in 6-well plate in triplicate and then treated with or without phenformin for 24 hours. Culture medium was harvested and lactate concentration was detected by lactate test strips and Lactate Plus Meter (Nova Biomedical). Lactate production was normalized by cellular protein mass.
Quantitative real-time PCR and RIP assay
Quantitative real-time PCR was performed as previously described.53,54 Briefly, total RNA was extracted from cells using RNeasy (Qiagen) and 1st strand cDNA was prepared with High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, ABI). Real-time PCR was performed using QuantiTect SYBR Green PCR kit (Qiagen) or TaqMan Universal PCR Master Mix (ABI), and was run on Stratagene MX3000P. For quantification of gene expression, the 2−ΔΔCt method was used. GAPDH expression was used for normalization. RIP assay was performed with Magna RIP RNA-Binding Protein Immuno-precipitation Kit (Millipore). Briefly, cells were lysed in RIP lysis buffer. Then the lysates were immuno-precipitated with antibody or IgG along with protein magnetic beads. After proteinase K digestion, the RNAs pulled down with proteins were purified by phenol chloroform extraction and precipitated in ethanol. The RNAs were then re-suspended in RNAse-free water and cDNA was synthesized and subjected to real-time-PCR to detect NBR2 or GAPDH (internal control) transcripts. The RNA level was normalized with input (10%).
RNA pull-down assays
Biotin labeled RNAs were synthesized by Scientific TranscriptAid T7 High Yield Transcription Kit (Thermo). PCR primers with T7 promoters were used to amplify DNA templates for RNA synthesis, RNA transcribed in vitro with biotin RNA labeling mix and T7 RNA polymerase, treated with RNase-free DNase I (Roche), and purified with the RNeasy Mini Kit (Qiagen). Cells lysates or purified proteins were incubated with biotin-labeled RNAs overnight. The proteins associated with biotin-labeled RNAs were then pulled down with Streptavidin Magnetic Beads (Thermo) after 1-hour incubation. The proteins was then washed and used for Western blot analysis.
Western blot analysis
Cultured cells were lysed with NP40 buffer (150 mM sodium chloride, 1.0% NP-40, 50 mM Tris, pH 8.0) containing complete mini protease inhibitors (Roche) and phosphatase inhibitor cocktail (Calbiochem). Western blots were obtained utilizing 20 to 40 µg of lysate protein. The following antibodies were used in this study: Monoclonal Anti-Vinculin antibody (Sigma-Aldrich, V4505, 1:5000 dilution), Phospho-Acetyl-CoA Carboxylase (Ser79) Antibody (Cell Signaling Technology, 3661S, 1:1,000 dilution), Acetyl-CoA Carboxylase Antibody (Cell Signaling Technology, 3662S, 1:1,000 dilution), Phospho-AMPKα (Thr172) (40H9) Rabbit mAb (Cell Signaling Technology, 2535S, 1:1,000 dilution), AMPKα (D63G4) Rabbit mAb (Cell Signaling Technology, 5832S, 1:1,000 dilution), Phospho-S6 Ribosomal Protein (Ser240/244) (D68F8) XP® Rabbit mAb (Cell Signaling Technology, 5364S, 1:5,000 dilution), S6 Ribosomal Protein (5G10) Rabbit mAb (Cell Signaling Technology, 2217S, 1:5,000 dilution), Cleaved PARP (Asp214) Antibody (Human Specific) (Cell Signaling Technology, 9541S, 1:2,000 dilution), Cleaved Caspase-3 (Asp175) (5A1E) Rabbit mAb (Cell Signaling Technology, 9664S, 1:500 dilution), β-Actin(Cell Signaling Technology,4967S, 1:2500 dilution), Lamin A/C(Cell Signaling Technology,4777S, 1:1,000 dilution ), GLUT1 (Santa Cruz, sc-7903, 1:500 dilution).
Subcellular fractionation
Cells were harvested by trypsin and washed twice with PBS. Cell pellets were lysed in buffer I containing 20 mM HEPES, 10 mM KCL, 2 mM MgCl2 and 0.5% NP40. After centrifugation, supernatants were collected as cytoplasmic lysis. Pellets were further lysed in buffer II containing 0.5 M NaCl, 20 mM HEPES, 10 mM KCL, 2 mM MgCl2 and 0.5% NP40. Supernatants were collected as nuclear lysis by centrifugation. Cytoplasmic and nuclear fractions were split for RNA extraction and real-time PCR or protein extraction and Western blotting. Vinculin and Lamin A/C were used as markers of cytoplasm and nucleus in Western blotting. GAPDH and U1 were used as markers of cytoplasm and nucleus in real-time PCR.
Oligonucleotide sequences, probes and primers (forward and reverse)
qPCR primers for gene expression and RIP:
NBR2-Forward: 5′-GGAGGTCTCCAGTTTCGGTA-3′
NBR2-Reverse: 5′-TTGATGTGTGCTTCCTGGG-3′
GAPDH-Forward: 5′-CCATGGGGAAGGTGAAGGTC-3′
GAPDH -Reverse: 5′-GAAGGGGTCATTGATGGCAAC-3′
U1-Forward: 5′-TCCCAGGGCGAGGCTTATCCATT-3′
U1-Reverse: 5′-GAACGCAGTCCCCCACTACCACAAAT-3′
Glut1-Forward: 5′-ATTGGCTCCGGTATCGTCAAC-3′
Glut1-Reverse: 5′-GCTCAGATAGGACATCCAGGGTA-3′
Glut2-Forward: 5′-GACAGTGAAAACCAGGGTCC-3′
Glut2-Reverse: 5′-TGTGCCACACTCACACAAGA-3′
Glut3-Forward: 5′-TTGAACACCTGCATCCTTGA-3′
Glut3-Reverse: 5′-GACAGCCCATCATCATTTCC-3′
Glut4-Forward: 5′-AGCACCGCAGAGAACACAG-3′
Glut4-Reverse: 5′-GTCGGGCTTCCAACAGATAG-3′
Primers for RNA pull down assay
NBR2-sense-Forward: 5′-TAATACGACTCACTATAGGG AGGGGTCCAGTTGCGGCTTAT-3′
NBR2-sense-Reverse: 5′-AGTTT ACTTA CTATT GCTGA-3′
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank all members of the Gan laboratory for their advice and technical assistance.
Funding
This research has been supported by grants from MD Anderson Cancer Center, Cancer Prevention & Research Institute of Texas (RP130020), National Institutes of Health (CA181196 and CA190370), Ellison Medical Foundation (AG-NS-0973-13), and Gabrielle's Angel Foundation for Cancer Research (to B. G.). B. G. is a Kimmel Scholar and an Ellison Medical Foundation New Scholar. B. G. is a member of the M.D. Anderson Cancer Center, and is supported by the National Institutes of Health Core Grant CA016672.
References
- [1].Pernicova I, Korbonits M. Metformin–mode of action and clinical implications for diabetes and cancer. Nat Rev Endocrinol 2014; 10:143-56; PMID:24393785; http://dx.doi.org/ 10.1038/nrendo.2013.256 [DOI] [PubMed] [Google Scholar]
- [2].Foretz M, Guigas B, Bertrand L, Pollak M, Viollet B. Metformin: from mechanisms of action to therapies. Cell Metab 2014; 20:953-66; PMID:25456737; http://dx.doi.org/ 10.1016/j.cmet.2014.09.018 [DOI] [PubMed] [Google Scholar]
- [3].Pollak M. Potential applications for biguanides in oncology. J Clin Invest 2013; 123:3693-700; PMID:23999444; http://dx.doi.org/ 10.1172/JCI67232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Menendez JA, Oliveras-Ferraros C, Cufi S, Corominas-Faja B, Joven J, Martin-Castillo B, Vazquez-Martin A. Metformin is synthetically lethal with glucose withdrawal in cancer cells. Cell Cycle 2012; 11:2782-92; PMID:22809961; http://dx.doi.org/ 10.4161/cc.20948 [DOI] [PubMed] [Google Scholar]
- [5].Picone P, Vilasi S, Librizzi F, Contardi M, Nuzzo D, Caruana L, Baldassano S, Amato A, Mule F, San Biagio PL, et al.. Biological and biophysics aspects of metformin-induced effects: cortex mitochondrial dysfunction and promotion of toxic amyloid pre-fibrillar aggregates. Aging 2016; 8:1718-34; PMID:27509335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Tseng CH. Metformin reduces gastric cancer risk in patients with type 2 diabetes mellitus. Aging 2016; 8:1636-49; PMID:27587088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Wurth R, Pattarozzi A, Gatti M, Bajetto A, Corsaro A, Parodi A, Sirito R, Massollo M, Marini C, Zona G, et al.. Metformin selectively affects human glioblastoma tumor-initiating cell viability: A role for metformin-induced inhibition of Akt. Cell Cycle 2013; 12:145-56; PMID:23255107; http://dx.doi.org/ 10.4161/cc.23050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. Bmj 2005; 330:1304-5; PMID:15849206; http://dx.doi.org/ 10.1136/bmj.38415.708634.F7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Higurashi T, Hosono K, Takahashi H, Komiya Y, Umezawa S, Sakai E, Uchiyama T, Taniguchi L, Hata Y, Uchiyama S, et al.. Metformin for chemoprevention of metachronous colorectal adenoma or polyps in post-polypectomy patients without diabetes: a multicentre double-blind, placebo-controlled, randomised phase 3 trial. Lancet Oncol 2016; 17(4):475-83; PMID:26947328 [DOI] [PubMed] [Google Scholar]
- [10].Bodmer M, Meier C, Krahenbuhl S, Jick SS, Meier CR. Long-term metformin use is associated with decreased risk of breast cancer. Diabetes Care 2010; 33:1304-8; PMID:20299480; http://dx.doi.org/ 10.2337/dc09-1791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Moiseeva O, Deschenes-Simard X, Pollak M, Ferbeyre G. Metformin, aging and cancer. Aging 2013; 5:330-1; PMID:23660016; http://dx.doi.org/ 10.18632/aging.100556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Crofford OB. Metformin. N Engl J Med 1995; 333:588-9; PMID:7623910; http://dx.doi.org/ 10.1056/NEJM199508313330910 [DOI] [PubMed] [Google Scholar]
- [13].Lea MA, Chacko J, Bolikal S, Hong JY, Chung R, Ortega A, desbordes C. Addition of 2-deoxyglucose enhances growth inhibition but reverses acidification in colon cancer cells treated with phenformin. Anti Cancer Res 2011; 31:421-6 [PubMed] [Google Scholar]
- [14].Pollak M. Metformin and other biguanides in oncology: advancing the research agenda. Cancer Prevention Res 2010; 3:1060-5; PMID:20810670; http://dx.doi.org/ 10.1158/1940-6207.CAPR-10-0175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hardie DG. The LKB1-AMPK pathway-friend or foe in cancer? Cancer Cell 2013; 23:131-2; PMID:23410967; http://dx.doi.org/ 10.1016/j.ccr.2013.01.009 [DOI] [PubMed] [Google Scholar]
- [16].Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol 2012; 13:251-62; PMID:22436748; http://dx.doi.org/ 10.1038/nrm3311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Hardie DG, Schaffer BE, Brunet A. AMPK: An energy-sensing pathway with multiple inputs and outputs. Trends Cell Biol 2016; 26:190-201; PMID:26616193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer 2009; 9:563-75; PMID:19629071; http://dx.doi.org/ 10.1038/nrc2676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Kalender A, Selvaraj A, Kim SY, Gulati P, Brule S, Viollet B, Kemp BE, Bardeesy N, Dennis P, Schlager JJ, et al.. Metformin, independent of AMPK, inhibits mTORC1 in a rag GTPase-dependent manner. Cell Metab 2010; 11:390-401; PMID:20444419; http://dx.doi.org/ 10.1016/j.cmet.2010.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Vincent EE, Coelho PP, Blagih J, Griss T, Viollet B, Jones RG. Differential effects of AMPK agonists on cell growth and metabolism. Oncogene 2015; 34:3627-39; PMID:25241895; http://dx.doi.org/ 10.1038/onc.2014.301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Guigas B, Bertrand L, Taleux N, Foretz M, Wiernsperger N, Vertommen D, Andreelli F, Viollet B, Hue L. Five-Aminoimidazole-4-carboxamide-1-β-D-ribofuranoside and metformin inhibit hepatic glucose phosphorylation by an AMP-activated protein kinase-independent effect on glucokinase translocation. Diabetes 2006; 55:865-74; PMID:16567505; http://dx.doi.org/ 10.2337/diabetes.55.04.06.db05-1178 [DOI] [PubMed] [Google Scholar]
- [22].Foretz M, Hebrard S, Leclerc J, Zarrinpashneh E, Soty M, Mithieux G, Sakamoto K, Andreelli F, Viollet B. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J Clin Invest 2010; 120:2355-69; PMID:20577053; http://dx.doi.org/ 10.1172/JCI40671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Shackelford DB, Abt E, Gerken L, Vasquez DS, Seki A, Leblanc M, Wei L, Fishbein MC, Czernin J, Mischel PS, et al.. LKB1 inactivation dictates therapeutic response of non-small cell lung cancer to the metabolism drug phenformin. Cancer Cell 2013; 23:143-58; PMID:23352126; http://dx.doi.org/ 10.1016/j.ccr.2012.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Litchfield LM, Mukherjee A, Eckert MA, Johnson A, Mills KA, Pan S, Shridhar V, Lengyel E, Romero IL. Hyperglycemia-induced metabolic compensation inhibits metformin sensitivity in ovarian cancer. Oncotarget 2015; 6:23548-60; PMID:26172303; http://dx.doi.org/ 10.18632/oncotarget.4556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Birsoy K, Possemato R, Lorbeer FK, Bayraktar EC, Thiru P, Yucel B, Wang T, Chen WW, Clish CB, Sabatini DM. Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides. Nature 2014; 508:108-12; PMID:24670634; http://dx.doi.org/ 10.1038/nature13110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Ulitsky I, Bartel DP. lincRNAs: genomics, evolution, and mechanisms. Cell 2013; 154:26-46; PMID:23827673; http://dx.doi.org/ 10.1016/j.cell.2013.06.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Consortium EP. An integrated encyclopedia of DNA elements in the human genome. Nature 2012; 489:57-74; PMID:22955616; http://dx.doi.org/ 10.1038/nature11247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Xiao ZD, Zhuang L, Gan B. Long non-coding RNAs in cancer metabolism. BioEssays 2016; 38(10):991-6; PMID:27550823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Guttman M, Rinn JL. Modular regulatory principles of large non-coding RNAs. Nature 2012; 482:339-46; PMID:22337053; http://dx.doi.org/ 10.1038/nature10887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Batista PJ, Chang HY. Long noncoding RNAs: cellular address codes in development and disease. Cell 2013; 152:1298-307; PMID:23498938; http://dx.doi.org/ 10.1016/j.cell.2013.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Schmitt AM, Chang HY. Long noncoding RNAs in cancer pathways. Cancer Cell 2016; 29:452-63; PMID:27070700; http://dx.doi.org/ 10.1016/j.ccell.2016.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Xiao ZD, Liu X, Zhuang L, Gan B. NBR2: A former junk gene emerges as a key player in tumor suppression. Mol Cell Oncol 2016; 3:e1187322; PMID:27652330; http://dx.doi.org/ 10.1080/23723556.2016.1187322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Xu CF, Chambers JA, Solomon E. Complex regulation of the BRCA1 gene. J Biol Chem 1997; 272:20994-7; PMID:9261099; http://dx.doi.org/ 10.1074/jbc.272.34.20994 [DOI] [PubMed] [Google Scholar]
- [34].Xu CF, Brown MA, Nicolai H, Chambers JA, Griffiths BL, Solomon E. Isolation and characterisation of the NBR2 gene which lies head to head with the human BRCA1 gene. Hum Mol Genet 1997; 6:1057-62; PMID:9215675; http://dx.doi.org/ 10.1093/hmg/6.7.1057 [DOI] [PubMed] [Google Scholar]
- [35].Jin H, Selfe J, Whitehouse C, Morris JR, Solomon E, Roberts RG. Structural evolution of the BRCA1 genomic region in primates. Genomics 2004; 84:1071-82; PMID:15533724; http://dx.doi.org/ 10.1016/j.ygeno.2004.08.019 [DOI] [PubMed] [Google Scholar]
- [36].Liu X, Xiao ZD, Han L, Zhang J, Lee SW, Wang W, Lee H, Zhuang L, Chen J, Lin HK, et al.. LncRNA NBR2 engages a metabolic checkpoint by regulating AMPK under energy stress. Nat Cell Biol 2016; 18:431-42; PMID:26999735; http://dx.doi.org/ 10.1038/ncb3328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Liu X, Xiao ZD, Gan B. An lncRNA switch for AMPK activation. Cell Cycle 2016; 15(15):1948-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Shaw RJ, Kosmatka M, Bardeesy N, Hurley RL, Witters LA, DePinho RA, Cantley LC. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci U S A 2004; 101:3329-35; PMID:14985505; http://dx.doi.org/ 10.1073/pnas.0308061100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Lin A, Yao J, Zhuang L, Wang D, Han J, Lam EW, Network TR, Gan B. The FoxO-BNIP3 axis exerts a unique regulation of mTORC1 and cell survival under energy stress. Oncogene 2014; 33:3183-94; PMID:23851496; http://dx.doi.org/ 10.1038/onc.2013.273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, et al.. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest 2001; 108:1167-74; PMID:11602624; http://dx.doi.org/ 10.1172/JCI13505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Sakamoto K, McCarthy A, Smith D, Green KA, Grahame Hardie D, Ashworth A, Alessi DR. Deficiency of LKB1 in skeletal muscle prevents AMPK activation and glucose uptake during contraction. EMBO J 2005; 24:1810-20; PMID:15889149; http://dx.doi.org/ 10.1038/sj.emboj.7600667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Bell GI, Kayano T, Buse JB, Burant CF, Takeda J, Lin D, Fukumoto H, Seino S. Molecular biology of mammalian glucose transporters. Diabetes Care 1990; 13:198-208; PMID:2407475; http://dx.doi.org/ 10.2337/diacare.13.3.198 [DOI] [PubMed] [Google Scholar]
- [43].Macheda ML, Rogers S, Best JD. Molecular and cellular regulation of glucose transporter (GLUT) proteins in cancer. J Cell Physiol 2005; 202:654-62; PMID:15389572; http://dx.doi.org/ 10.1002/jcp.20166 [DOI] [PubMed] [Google Scholar]
- [44].Thorens B, Mueckler M. Glucose transporters in the 21st century. Am J Physiol Endocrinol Metab 2010; 298:E141-5; PMID:20009031; http://dx.doi.org/ 10.1152/ajpendo.00712.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 2009; 324:1029-33; PMID:19460998; http://dx.doi.org/ 10.1126/science.1160809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Yun J, Rago C, Cheong I, Pagliarini R, Angenendt P, Rajagopalan H, Schmidt K, Willson JK, Markowitz S, Zhou S, et al.. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science 2009; 325:1555-9; PMID:19661383; http://dx.doi.org/ 10.1126/science.1174229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Dykens JA, Jamieson J, Marroquin L, Nadanaciva S, Billis PA, Will Y. Biguanide-induced mitochondrial dysfunction yields increased lactate production and cytotoxicity of aerobically-poised HepG2 cells and human hepatocytes in vitro. Toxicol Applied Pharmacol 2008; 233:203-10; PMID:18817800; http://dx.doi.org/ 10.1016/j.taap.2008.08.013 [DOI] [PubMed] [Google Scholar]
- [48].Owen MR, Doran E, Halestrap AP. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J 2000; 348 Pt 3:607-14; PMID:10839993; http://dx.doi.org/ 10.1042/bj3480607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Lin A, Piao HL, Zhuang L, Sarbassov DD, Ma L, Gan B. FoxO transcription factors promote AKT Ser473 phosphorylation and renal tumor growth in response to pharmacological inhibition of the PI3K-AKT pathway. Cancer Res 2014; 74(6):1682-93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Gan B, Hu J, Jiang S, Liu Y, Sahin E, Zhuang L, Fletcher-Sananikone E, Colla S, Wang YA, Chin L, et al.. Lkb1 regulates quiescence and metabolic homeostasis of haematopoietic stem cells. Nature 2010; 468:701-4; PMID:21124456; http://dx.doi.org/ 10.1038/nature09595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Gan B, Sahin E, Jiang S, Sanchez-Aguilera A, Scott KL, Chin L, Williams DA, Kwiatkowski DJ, DePinho RA. mTORC1-dependent and -independent regulation of stem cell renewal, differentiation, and mobilization. Proc Natl Acad Sci U S A 2008; 105:19384-9; PMID:19052232; http://dx.doi.org/ 10.1073/pnas.0810584105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Liu XW, Cai TY, Zhu H, Cao J, Su Y, Hu YZ, He QJ, Yang B. Q6, a novel hypoxia-targeted drug, regulates hypoxia-inducible factor signaling via an autophagy-dependent mechanism in hepatocellular carcinoma. Autophagy 2014; 10:111-22; PMID:24220190; http://dx.doi.org/ 10.4161/auto.26838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Lee H, Dai F, Zhuang L, Xiao ZD, Kim J, Zhang Y, Ma L, You MJ, Wang Z, Gan B. BAF180 regulates cellular senescence and hematopoietic stem cell homeostasis through p21. Oncotarget 2016; 7:19134-46; PMID:26992241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Gan B, Lim C, Chu G, Hua S, Ding Z, Collins M, Hu J, Jiang S, Fletcher-Sananikone E, Zhuang L, et al.. FoxOs enforce a progression checkpoint to constrain mTORC1-activated renal tumorigenesis. Cancer Cell 2010; 18:472-84; PMID:21075312; http://dx.doi.org/ 10.1016/j.ccr.2010.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]