

ABSTRACT
The DNA damage checkpoint, when activated in response to genotoxic damage during S phase, arrests cells in G2 phase of the cell cycle. ATM, ATR, Chk1 and Chk2 kinases are the main effectors of this checkpoint pathway. The checkpoint kinases prevent the onset of mitosis by eliciting well characterized inhibitory phosphorylation of Cdk1. Since Cdk1 is required for the recruitment of condensin, it is thought that upon DNA damage the checkpoint also indirectly blocks chromosome condensation via Cdk1 inhibition. Here we report that the G2 damage checkpoint prevents stable recruitment of the chromosome-packaging-machinery components condensin complex I and II onto the chromatin even in the presence of an active Cdk1. DNA damage-induced inhibition of condensin subunit recruitment is mediated specifically by the Chk2 kinase, implying that the condensin complexes are targeted by the checkpoint in response to DNA damage, independently of Cdk1 inactivation. Thus, the G2 checkpoint directly prevents stable recruitment of condensin complexes to actively prevent chromosome compaction during G2 arrest, presumably to ensure efficient repair of the genomic damage.
KEYWORDS: Cell Cycle, Checkpoint, Chromosome condensation, DNA damage, Mitosis
Introduction
Genotoxic insults induce a set of cellular responses that concertedly permit dividing cells to efficiently repair the injuries before partitioning of the duplicated chromosomes during mitosis. Sensing of the damage, halting cell cycle progression and activating the repair pathways are central to this multipronged response. DNA damage checkpoints are specialized mechanisms that impose cell cycle arrest once the damage is detected and processed. The G1 (or G1/S) checkpoint of which tumor suppressor p53 is an important effector, responds to damage incurred during G1 and prevents entry into S phase. The G2 checkpoint, on the other hand, is activated in cells that have suffered chromosomal injuries during S phase and causes cells to arrest in G2 phase thereby inhibiting the onset of mitosis.1 The phosphoinositol 3-kinase (PI3K) family kinases ATM and ATR and the distal serine/threonine kinases Chk1 and Chk2 are the critical effectors of both checkpoint controls.2 The key target of the G2 checkpoint is the major mitosis-promoting kinase Cdk1. During the normal cycle, activation of Cdk1 requires its dephosphorylation by the Cdc25 family of phosphatases and destabilisation of Cdk1 inhibitory kinase Wee1.3 Upon DNA damage, Chk1/Chk2-mediated cytoplasmic sequestration and inhibition of Cdc25, together with stabilization of Wee1, precludes activation of the Cdk1/cyclin B complex and thus prevents entry into mitosis.4-6 p53 also contributes to the long term silencing of Cdk1/cyclin B during G2 checkpoint-imposed arrest via expression of p21, GADD45 and 14-3-3σ proteins.7 However, some cell types that lack p53 activity still accumulate in G2 upon DNA damage during S phase, suggesting the presence of additional mechanisms such as BRCA1-stimulated expression of p21 and GADD45 for the long-term inactivation of Cdk1 in the absence of p53.7
While checkpoint-imposed arrest allows cells sufficient time to execute DNA repair before cell cycle progression is resumed, chromosomes must also remain in a decondensed state during the arrest in order to be accessible to the repair machinery. Chromosome condensation (organization of chromosomal DNA into higher ordered structure) is associated with the onset of mitosis and is critical for proper segregation of the chromosomes.8-10 Condensins play an important part in the chromosome-compaction process and exist in 2 forms: condensin complexes I and II.11,12 Both complexes are composed of SMC and non-SMC subunits. The core SMC subunits, SMC2 and SMC4, are common to both complexes but the non-SMC subunits differ. The condensin complex I contains 3 non-SMC subunits, namely, CAP-G, CAP-H and CAP-D2, whereas condensin complex II contains non-SMC subunits CAP-G2, CAP-H2 and CAP-D3.11,13 The condensin complex II is nuclear throughout the cell cycle but associates with chromatin in mitosis. Condensin I, on the other hand, is predominantly but not exclusively cytoplasmic during interphase14 and gathers on chromatin upon nuclear envelope breakdown.15,16 Condensin II associates stably with the chromosomes whereas binding of condensin I is more dynamic,16 suggesting that the 2 complexes may have different roles in chromosome compaction.13 There is growing evidence that for mitotic chromosomes condensin I is required more for lateral condensation and condensin II for axial condensation.10,17,18
Cdk1-mediated activation of condensins is essential for DNA condensation.19,20 Indeed, prolonged Cdk1 activity in mitosis causes hyper condensation of mitotic chromosomes and expression of non-degradable cyclin B1 severely delays chromosome decondensation.21 In HeLa cells, phosphorylation of condensin II CAP-D3 subunit on Thr1415 by Cdk1 promotes early stages of chromosome condensation.22,23 Mitotic chromatids can be reconstituted in vitro by a minimum set of purified factors including the core histones, nucleoplasmin, Nap1, FACT, topoisomerase II and condensin I. Strikingly, Cdk1 phosphorylation of condensin I is the sole mitosis-specific modification required for chromatid reconstitution using these factors.24 A condensin independent role of Cdk1 in chromosome condensation has also been proposed; however, the mechanism of its involvement is not known.25 Aurora B and polo-like kinases are also reported to phosphorylate and regulate condensins.26,27 Details of how different mitotic kinases cooperate to regulate the mitotic chromosome architecture still remain largely unclear.
An increasing body of evidence is highlighting that condensins have additional key roles outside mitotic chromosome condensation. It has been reported that CAP-G2 and CAP-H2 progressively accumulate on the replicated region of the chromosomes during S phase, leading to the suggestion that condensin II may promote chromatid resolution of the duplicated chromosomes during S phase in preparation for the chromosome condensation and segregation during mitosis.28 Condensins have been implicated in an additional interphase function related to DNA damage and repair. In fission yeast, a mutation in cut14/SMC2 leads to a defect in both DNA damage repair and chromosome condensation.29 These defects are alleviated by a mutation in the ssDNA binding protein replication protein A (RPA).29 A mutation in the Cnd2/CAP-H subunit also leads to similar phenotypic consequences.30 In vertebrate cells, condensin I is preferentially recruited to the DNA damage sites enriched for base damage and interacts with poly (ADP-ribose) polymerase I.31 Recently, it has also been shown that depletion of both condensins in neural stem cells can lead to DNA damage and trigger p53-induced-apoptosis.32 These studies imply potential links between condensins and DDR; however, the nature of these links is not clear at present.
Since Cdk1 is one of the major regulators of chromosome condensation, abrogation of its activity by the G2 DNA damage checkpoint would not only prevent onset of mitosis but also indirectly precludes other mitosis-associated events. However, the possibility that the DNA damage checkpoint directly inhibits not only Cdk1 but also other major mitotic events such as chromosome compaction machinery has not been tested. Direct regulation of chromosome condensation by the checkpoint would be obscured by inhibition of the onset of mitosis via Cdk1 inactivation.
In this study, we have investigated this possibility in cells stably expressing constitutively active Cdk1 (Cdk1AF) from a conditional promoter. We find that Cdk1AF cells activate the G2 checkpoint in response to DNA damage but proceed to mitosis without arresting in G2. However, unlike mitotic cells, they fail to stably recruit condensin complex I and II to the chromatin despite the presence of an active Cdk1. We show that the failure to recruit condensins in these cells is due to an active G2 checkpoint since abrogation of Chk2 activity restores the recruitment of condensin complex subunits to the chromatin. Interestingly, down-regulation of Chk1 fails to reverse Cdk1AF cells' inability to recruit the condensin complexes, suggesting that inhibition of condensin complex recruitment is specifically mediated by Chk2 kinase. Our results reveal a novel Chk2-mediated inhibitory regulation imposed by the G2 DNA damage checkpoint on chromosome compaction, in addition to the one enforced indirectly by the inactivation of Cdk1.
Results
Cell cycle progression and DNA damage response in Cdk1AF cells
Since our aim was to determine the mitotic processes inhibited directly by the DNA damage checkpoint independently of Cdk1 inhibition, we generated 2 independent HeLa cell clones stably expressing Flag-tagged, constitutively active Cdk1AF (Thr14 →Ala; Tyr15 →Phe) under the control of doxycycline (DOX) inducible Tet-promoter (Tet-ON Cdk1AF, clone B and clone D). Addition of doxycycline induces expression of constitutively active Cdk1AF. To test if normal cell cycle progression is adversely affected by Cdk1AF expression, HeLa cells carrying stably integrated Tet-ON construct (referred to ‘HeLa cells’ hereafter) or Tet-ON Cdk1AF construct (‘HeLa Cdk1AF cells or Cdk1AF cells’ hereafter) were synchronised in early S phase using a serum starvation/thymidine protocol and then released in the presence or absence of doxycycline (+/− DOX). Flow cytometric analysis and the appearance of phospho-Histone 3 (pH3) signals in western blots show that the cell cycle progression profiles of cells without or with the expression of Cdk1AF are comparable in 2 different clones (Figs. 1A and S1A). This is consistent with a previous report that Cdk1AF over-expressing HeLa cells carry out a relatively normal first cycle.33-36 As these 2 Cdk1AF clones exhibit comparable cell cycle profile, we arbitrarily chose clone D for the subsequent experiments.
Figure 1.

Mitotic timing and the checkpoint response in HeLa cells remains largely unaffected by inducible expression of Cdk1AF (A) HeLa Tet-ON and HeLa Tet-ON Cdk1AF cells were synchronised by a combined serum starvation and thymidine protocol (Materials and Methods), then washed and cultured in the presence (+DOX) or absence (-DOX) of doxycycline (DOX, 1 μg/ml). Cells at 0, 4, 8, 12 and 16 hours (h) post-release were collected for FACS and Western blotting. FACS profiles are shown in the left panel. Cdk1AF-Flag (FLAG), phosphorylated H3 (pH3) and Actin (Actin) at different time points detected by western immunoblotting are shown in the right panel. (B) HeLa Tet-ON and HeLa Tet-ON Cdk1AF cells were synchronised as before. Immediately upon release (T=0), HeLa Tet-ON Cdk1AF cells were induced with DOX which remained in the media for the rest of the experiment. DNA damage was induced with Adriamycin (Adr: 300 nM) at 4 h (T = 4) post release. Aliquots of HeLa Tet-ON cells were concurrently treated with 5 mM caffeine (Caff) (T=4). To inhibit apoptosis, pan-caspase inhibitor Z-VAD was added to all samples at T=4. Cells were harvested for biochemical analysis or fixed for immunofluorescence at T=16. FACS profiles are shown in the left and middle panels. Levels of phospho-Chk1 (pChk1), Chk1, phospho-Chk2 (pChk2), Chk2 and Actin are shown in the right panel, with vertical boxes indicating protein levels at 8 h and 24 h. (C) Synchronised HeLa Tet-ON and HeLa Tet-ON Cdk1AF cells were treated as described in Fig. 1B. After 4 h exposure to Adriamycin (T=8), cells were stained with γH2AX (red) and DAPI (blue) (left panel). Inset shows mitotic cells, confirmed by the presence of biopolar spindles with β- tubulin (green) staining. For both γH2AX and Rad51 quantification: n> 500 for interphase; n > 200 for mitotic cells. Scale bar represents 5 μm. (D) Cells were collected at indicated time points and lysed. Histone H1 kinase activity was measured in immunoprecipitates obtained with anti-Cdk1 antibodies (to pull down Cdk1, left panel) or anti-FLAG (to pull down Cdk1-FLAG, right panel). An autoradiogram of Histone H1 with P-32 phosphate in the in vitro kinase assay is shown (Materials and Methods). Untreated cells (labeled as release) or cells treated with thymidine (Thy) or Nocodazol (Noc) were used as controls.
To determine the effect of Cdk1AF expression on DNA damage response and G2 checkpoint activation, control HeLa cells (HeLa Tet-ON) and HeLa Cdk1AF cells were synchronised as described above and released in the presence or absence of DOX. 4 h after the release, Adriamycin (Adr), a topoisomerase II (topo II) inhibitor, was added to induce DNA damage. In a parallel culture, caffeine was also added to Adr-treated cells. This allowed us to observe the effects of eliminating the DNA damage checkpoint, since it is abrogated by caffeine treatment. Both HeLa and HeLa Cdk1AF cells exhibited sustained 4N DNA content in response to Adr treatment (Fig. 1B, left and middle panel). Appearance of phospho-Chk1 and phospho-Chk2 shows that the DNA damage checkpoint is activated in both HeLa and HeLa Cdk1AF cells (Fig. 1B, right panel lane 5 and 11). However, while HeLa cells remained arrested in G2, HeLa Cdk1AF cells entered mitosis (Fig. 2A, left panel) implying that Adriamycin (Adr)-treated HeLa Cdk1AF cells enter mitosis with an activated checkpoint. The presence of uniformly stained γH2AX positive nuclei in HeLa Cdk1AF cells suggests that DNA damage remains unrepaired in these cells as they enter mitosis due to the expression of Cdk1AF, just as in the caffeine-treated HeLa cells (Fig. 1C, left panel). In contrast, both HeLa and HeLa Cdk1AF cells with or without DOX treatment (not treated with Adr) showed no significant difference (NS) in the accumulation of γH2AX foci, with ∼93% mitotic and anaphase cells having less than 5 γH2AX foci, indicating that the observed accumulation of γH2AX foci (Fig. 1C, left panel) is specific due to Adriamycin rather than DOX-inducible Cdk1AF expression (Fig. S1B). Rad51 is also a useful marker in this context. As expected, moderate levels of Rad51 foci can be detected in both HeLa and HeLa Cdk1AF interphase cells without DOX treatment (Fig. S1C). Upon Adriamycin treatment, Rad51 (red) foci are induced in interphase nuclei of Cdk1AF cells but not as intensely in mitotic cells (Fig. 1C, right most panel). As activation of the G2 DNA damage checkpoint is known to cause a reduction in Cdk1 activity, level of Cdk1 activity is also a good indicator of checkpoint activation. Activation of the checkpoint by Adr treatment led to a dramatic decline in native Cdk1 activity in HeLa control cells at 10 hrs after release from thymidine (Fig. 1D, left panel lane 4). This decline in the Cdk1 activity was abrogated in the presence of caffeine (Fig. 1D, left panel lane 6). Upon induction by DOX, Cdk1AF cells shows robust Cdk1AF (flag tagged) activity (Fig. 1D, right panel lanes 4 and 5). As expected, DOX-induced Cdk1AF activity in HeLa Cdk1AF cells was not affected significantly by Adr treatment within the same time period (Fig. 1D left panel lane 9 vs lane 8; Fig. 1D, right panel lanes 6, 7 and 8 vs lanes 4 and 5). Collectively, these results suggest that despite the expression of Cdk1AF, the checkpoint response in DNA damaged HeLa Cdk1AF cells is intact and the checkpoint is largely unable to inactivate the constitutively active Cdk1AF.
Figure 2.

Mitotic entry and aberrant mitosis in damaged Cdk1AF cells. (A) HeLa Tet-ON and HeLa Tet-ON Cdk1AF cells were synchronised as described in Figure 1B. After the release from thymidine arrest, HeLa Tet-ON Cdk1AF cells were treated with Adriamycin in the presence doxycycline (Adr+Dox), while HeLa Tet-ON cells were treated with Adriamycin (Adr) or Adriamycin and Caffeine (Adr+Caffeine) as controls. The cells were trapped in mitosis with nocodazole treatment as described in Figure 1B. Mitotic cells were harvested by shake-off at indicated time points from the same plates and were analyzed by FACS (Materials and Methods). The shake-off cell numbers were counted and cumulative mitotic cell numbers were plotted (left panel) as a ratio of mitotic cells to all viable cells collected (both floating and attached) by the end of experiment at T=24 h or T=45 h. Three independent experiments were performed. Samples from the same experiment were also subjected to MPM2-FACS analysis. Percentages of accumulative MPM2 positive cells are shown in the right panel. (B) Representative Immunofluorescence micrographs indicating mitotic hallmarks exhibited by HeLa Tet-ON Cdk1AF cells with DNA damage (synchronised and treated as described in Figure 1B). After 4 h exposure to Adriamycin, damaged Cdk1AF cells began entering mitosis asynchronously. Immunofluorescence staining for mitotic spindles (β-tubulin, green), condensed chromosomes (pH3, red); cyclin B (red); lamin A/C (green) and Giantin (red) are shown. Scale bar represents 5 μm. (C) Synchronised HeLa Tet-ON and HeLa Tet-ON Cdk1AF cells were treated as described in Figure 1B. HeLa Tet-ON cells treated with Adriamycin and caffeine (Adr+caffeine) or HeLa Tet-ON Cdk1AF cells treated with Adriamycin and doxycycline (Adr+DOX) were collected at T=16 and stained for DNA (DAPI, blue), kinetochores (ACA, green) and mitotic spindles (β-tubulin, red). Representative images are shown in the left panel. White arrows indicate prematurely decondensed chromosomes in metaphase, white arrowheads indicate chromosomes trapped in the cleavage furrow in late anaphase and telophase, and yellow arrows indicate fragmented chromatin in metaphase. The numbers (aberrant cells/total cells) indicate the frequency of occurrence of aberrant mitotic phenotypes in one experiment. Box plot (right panel) shows the quantification of aberrant mitotic figures. Cells in metaphase, anaphase and telophase/cytokinesis were scored according to chromosomal and kinetochore status/alignment/segregation, and collated. * indicates p<0 .005. Error bars represent 95% confidence intervals (CI). A total of 200 cells were counted for each condition in 3 independent experiments. (D) To inhibit ATR or Chk2, asynchronous HeLa Tet-ON Cdk1AF cells were transfected with siATR or siChk2 4 h prior to the synchronisation as described in Figure 1B respectively, then released and treated with Adr+DOX. To inhibit ATM, synchronised HeLa Tet-ON Cdk1AF cells were treated with KU-55933 4 h after release from thymidine. To inhibit Chk1, synchronised HeLa Tet-ON Cdk1AF cells were treated with PF477736 4 h after release from thymidine. Adr and DOX were added as described in Figure 1B. Cells were collected for vizualization and quantification of aberrant mitosis. Box plot shows rescuing of aberrant mitosis in damaged Cdk1AF cells by siRNA targeting ATR (siATR) and KU-55933 (ATM inhibitor). A total of 200 cells were counted for each condition in 3 independent experiments. * indicates p < 0 .005. Error bars represent 95% confidence intervals (CI).
Aberrant mitosis in HeLa Cdk1AF cells following Adriamycin treatment
To determine the proportion of Adriamycin-treated cells that initiate mitosis upon expression of Cdk1AF, in comparison with Adr+caffeine treatment, cells were synchronised as before and released from early S phase arrest. HeLa cells were released in the presence of Adr or Adr+caffeine, whereas HeLa Cdk1AF cells were released in the presence of Adr+DOX. Mitotic cells were shaken-off, harvested and analyzed cytologically for condensed chromosomes and analyzed by FACS to determine the fraction of MPM2- positive cells. MPM2 is a monoclonal antibody that recognizes mitosis specific epitopes. The accumulative mitotic index (as described in Materials and Methods) showed that just as caffeine treatment allowed the majority of Adr-treated HeLa cells to proceed to mitosis, Cdk1AF expression (Adr + DOX) also induced a significant proportion of HeLa Cdk1AF cells to enter mitosis, though not highly synchronously (Fig. 2A). It is noted that the accumulated percentage of MPM2-positive cells in HeLa Cdk1AF cells with Adr+DOX treatment was relatively lower than that of HeLa cells with Adr+Caffeine treatment (Fig. 2A), which could be due to difficulty in detaching mitotic cells in the shake-off step.37 Consistent with this, both HeLa and HeLa Cdk1AF cells with or without DOX treatment also showed a relatively lower percentage of MPM2 cells (Fig. S2A). To determine if these cells progress through various stages of mitosis, early S phase-synchronised HeLa Cdk1AF cells were released in the presence of Adr+DOX. After 4 h of release, damaged HeLa Cdk1AF cells began to enter mitosis asynchronously and exhibited many hallmarks of mitotic entry such as phosphorylation of histone H3, nuclear localization of cyclin B, breakdown of nuclear envelope (ascertained by disassembly of nuclear scaffold by lamin staining and Golgi fragmentation by giantin staining), assembly of the mitotic spindle (β-tubulin staining) (Fig. 2B). Some of these cells, deemed to be in early mitosis, showed clear presence of cyclin B, securin (regulator of chromosome segregation), Shugoshin (protector of centromeric cohesins) and Plk1 (a prominent mitotic regulator), while others had progressed further in mitosis as indicated by diminished presence of these proteins, presumably due to proteasome dependent proteolysis (Fig. S2B). These cells also showed 2 well-separated centrosomes as visualised by immunostaining with anti-pericentrin antibodies, a typical hallmark of mitosis (Fig. S2C, right panel). Thus, Cdk1AF expression allows DNA damaged HeLa Cdk1AF cells to enter into and progress through mitosis. However, we noticed that some features of these mitoses were abnormal. These included prematurely decondensed chromosomes, chromosomes trapped in the cleavage furrow and fragmented chromatin (Fig. S2B). Both HeLa Cdk1AF clones showed similar mitotic defects, suggesting there was no clonal bias (Fig. S2D). We suspected that these abnormalities may be caused by the cells' entry into mitosis in the presence of an activated checkpoint. To test this, we compared the mitotic features of early S-phase synchronised Cdk1AF cells released in the presence of Adr or Adr+caffeine. Since caffeine treatment abrogates the DNA damage checkpoint, caffeine-treated cells will enter mitosis without the activated checkpoint. The caffeine-treated, DNA-damaged Cdk1AF cells condensed their chromosomes, showed normal congression at the metaphase plate, progressed to telophase and were almost indistinguishable from the control HeLa Cdk1AF cells that were not treated with Adr. Overall, caffeine treatment significantly reduced the proportion of Cdk1AF cells with abnormal mitotic features (Fig. 2C). These results suggest that an abnormal mitotic feature caused by Adr treatment of Cdk1AF cells is due to progression through mitosis in the presence of an active checkpoint.
To test further that these mitotic aberrations were indeed precipitated by the active DNA damage checkpoint, the activities of various effectors (ATM, ATR, Chk2 and Chk1) of the ATM/Chk2 or ATR/Chk1 axis were inhibited using either small molecule inhibitors or in the case of siRNA mediated knockdown (for summary of known effector activity see Fig. S3A). In case of siRNA-mediated knockdown, HeLa Cdk1AF cells were transfected with siRNA targeting ATR or Chk2 24 h before synchronisation. Cells were synchronised in early S phase as before, released in the presence of Adr with or without inhibitors and various mitotic features were scored (Fig. 2D). As before, caffeine treatment significantly restored the mitotic features back to normal in >90 % cells treated with Adr. Among the various effectors of the checkpoint pathway, inactivation of ATM alone (by KU-55933) or in combination with Chk1 (by PF477736) or with ATR (by siATR) (∼18%, ∼23% and ∼16% cells, respectively) showed the most noticeable and significant restoration of mitotic features to normal compared to the control damaged Cdk1AF cells (∼9%), though significantly less than the caffeine treatment (∼85%) (Fig. 2D). Since caffeine abrogates checkpoint control completely, a high level of phenotypic rescue is indeed expected in caffeine-treated cells compared to cells in which one or 2 components are inactivated. These results argue that mitotic defects in DNA-damaged HeLa Cdk1AF cells are, to a significant extent, due to uncoordinated mitotic progression caused by cells' attempt to execute mitotic events in the presence of an active checkpoint.
DNA damaged Cdk1AF cells are unable to maintain chromosome condensation during mitosis
A possible source of the uncoordinated mitotic progression in DNA-damaged Cdk1AF cells could be the antagonistic action of Cdk1AF and the checkpoint such that while constitutively active Cdk1AF drives ‘forward’ many aspects of mitosis, the active checkpoint may still continue to inhibit some key mitotic processes. As mentioned in the previous section, one mitotic feature that is most conspicuously aberrant in DNA-damaged Cdk1AF cells is their failure to maintain chromosome condensation, giving rise to prematurely decondensed chromosomes. Since condensin complex I and II are intimately involved in the process of chromosome condensation, we examined the recruitment of SMC2 to chromosomes, a subunit common to both condensin complexes, in synchronised HeLa and HeLa Cdk1AF cells under various conditions. HeLa Cdk1AF cells clone B and clone D were synchronised in early S phase as described above and released in the presence of DOX or Adr+DOX or Adr+DOX+caffeine, with untreated HeLa Tet-ON cells as a control. Nocodazole was added to trap mitotic cells that had escaped G2 arrest. As expected, SMC2 shows clear axial localization in the untreated HeLa cells and DOX-treated Cdk1AF cells (Fig. 3A, panel 1 and 2). In Adr-treated HeLa Cdk1AF cells, SMC2 is displaced from the decondensed chromatin or associates loosely with chromatin (Fig. 3A, panel 3 and 6). However, caffeine treatment of these cells restores the localization of condensin subunits to the chromatin for both HeLa Cdk1AF clone B and clone D cells (Fig. 3A, panel 4 and 7). Since caffeine inactivates the DNA damage checkpoint and allows damaged cells to enter mitosis, restoration of SMC2 localization may be considered due simply to the onset of mitosis. However, Adr-treated HeLa Cdk1AF cells enter mitosis due to the expression of constitutively active Cdk1AF (Fig. 2), yet fail to localize SMC2 properly (Fig. 3A, panel 3 and 6).
Figure 3.

Loss of axial condensin localization in damaged Cdk1AF cells. (A) Synchronised HeLa Tet-ON, HeLa Tet-ON Cdk1AF cells clone B and clone D were treated as described in Figure 1B. As controls, damaged HeLa Tet-ON Cdk1AF cells clone B and clone D were also treated with Caffeine. These cells, trapped in mitosis using nocodazole, were harvested, hypotonically-swollen, fixed with ice cold methanol-acetone (1:1) and stained for the condensin subunit SMC2 (green), with chromosomes counterstained with DAPI. Representative images are shown. Scale bar represents 5 μm. Insert shows zoom-in of individual chromosomes. SMC2 intensity in the nuclei was quantitated using the threshold signal in DAPI staining to define the nuclear periphery (Materials and Methods). Box plots show the ratio of SMC2 to DAPI intensity (relative intensity) in the right panel. Error bars represent 95% confidence intervals (CI), n = 20, *indicates p < 0 .001; NS indicates no significant difference. (B) Synchronised HeLa Tet-ON, HeLa Tet-ON Cdk1AF cells clone B and clone D were treated as described in Figure 3A. Cells were collected and fixed with methanol: acetic acid (3:1) (Materials and Methods). Chromosome spreads were stained with SMC2 (red) with DNA mounted with DAPI (blue). Representative images are shown. Scale bar represents 5 μm. Insert shows zoom-in of individual chromosomes. (C) Synchronised HeLa Tet-ON, HeLa Tet-ON Cdk1AF clone D cells were treated as described in Figure 3A. Cells were stained for CAP-D2 (green), CAP-G2 (red) and DNA (DAPI). Representative images are shown. Scale bar represents 5 μm. Insert shows zoom-in of individual chromosomes. (D) Synchronised HeLa Tet-ON and HeLa Tet-ON Cdk1AF cells were treated as described in Figure 1B and trapped in mitosis using nocodazole. Mitotic chromosomes from nocodazole-trapped cells were isolated and subjected to SDS-PAGE. The levels of SMC2, CAP-D2, CAP-G2, CAP-G, Histone H3 and ORC2 are shown in the left panel. Levels of SMC2, CAP-D2 and CAP-G2 in whole nuclear extracts (containing both nucleoplasmic and chromosomal materials) with the mitotic chromosomes from damaged HeLa Tet-ON cells treated with caffeine and damaged HeLa Tet-ON Cdk1AF cells treated with DOX are shown in the middle panel. Synchronised HeLa Tet-ON and HeLa Tet-ON Cdk1AF cells were treated as described in Figure 1B, trapped with nocodazole and harvested by mitotic shake-off 12 h after adriamycin treatment. Whole cell lysates were prepared and immunoprecipitated (IP) with anti-CAP-D2, or anti-SMC2 or anti-CAP-G2 antibodies respectively. The immuno-complexes were resolved by SDS-PAGE and Western blotting were performed to detect SMC2, CAP-D2, and CAP-G2 respectively (right panel).
The loss of axial localization of SMC2 in Adr-treated HeLa Cdk1AF cells was further confirmed by immune-staining of spread metaphase-chromosomes with anti-SMC2 antibodies (Fig. 3B and S3B). Adr-treated HeLa Cdk1AF cells also show the loss of SMC4 localization (Fig. S3C). Adr treatment causes loss of not only common condensin subunits SMC2 and SMC4 but also non-SMC subunits CAP-D2 (condensin I subunit) and CAP-G2 (condensin II subunit) localization to the chromosomes in both HeLa and HeLa Cdk1AF cells (Fig. 3C). As with SMC2, caffeine restores CAP-D2 and CAP-G2 localization in HeLa cells. In addition, Adr + DOX treatment results in loss of localization of CAP-H2 (a condensin II subunit) in both Cdk1AF clones, but caffeine restores it (Fig. S4). Collectively, these results suggest that the chromosome condensation defect in Adr-treated Cdk1AF cells is due to their failure to recruit condensin complexes to the chromatin.
To further investigate the failure of condensin recruitment in Adr-treated Cdk1AF cells, the presence of condensin complex I and II components on mitotic chromosomes isolated by cell fractionation was examined biochemically (Fig. 3D). HeLa Cdk1AF cells were synchronised as described before, released in the presence or absence of Adr and trapped in mitosis using nocodazole (Noc). The control HeLa cells were released in the presence or absence of Adr, with or without caffeine and cells escaping G2 arrest were trapped in mitosis using Noc. Cells were harvested by mitotic shake-off 12 h after Adr treatment and mitotic chromosomes were isolated using a fractionation protocol. The presence of SMC2, CAP-D2, CAP-G2, CAP-G, Histone H3 and ORC2 in the nuclear extract (containing both nucleoplasm and chromatin) and in the chromatin fraction was detected by Western blotting. In control HeLa and HeLa Cdk1AF cells, SMC2, CAP-D2, CAP-G2 and CAP-G are clearly present in the mitotic chromosome ‘extract’. These condensin subunits are also detected in the HeLa cells treated with Adr+caffeine (Fig. 3D, left panel; lane 1, 2, 3, and 4). However, SMC2, CAP-D2 and CAP-G are seen at significantly lower levels in the mitotic chromosome ‘extracts’ prepared from Adr-treated HeLa Cdk1AF cells (Fig. 3D, left panel; lane 5). This was further confirmed by a comparison between the nuclear extracts and mitotic chromosome extracts. In HeLa cells treated with Adr+caffeine, SMC2, CAP-D2 and CAP-G2 subunits are present in both nuclear extracts and mitotic chromosome ‘extracts’ (Fig. 3D, middle panel; lane 1 and 2). However, in Adr-treated HeLa Cdk1AF cells, while the SMC2, CAP-D2 and CAP-G2 are present in the nuclear extracts, SMC2 and CAP-D2 are barely detectable in the mitotic chromosome ‘extracts’ (Fig. 3D, middle panel; lanes 3 and 4). Next, we asked if Adr treatment prevents only the recruitment of condensin complexes or also affects assembly of the condensin complexes. To this end, we determined if SMC2 interacts with CAP-D2 and CAP-G2 in Adr or Adr+caffeine treated cells using a co-immunoprecipitation assay. The interaction between SMC2 and CAP-D2 or CAP-G2 could be detected in both HeLa and HeLa Cdk1AF cells under all experimental conditions (Fig. 3D, right panel) implying that the DNA damage checkpoint may inhibit the recruitment, rather than the assembly of condensin complexes. Together, these observations suggest that DNA-damaged Cdk1AF cells are unable to efficiently recruit condensin complexes to the chromosomes despite their progression into mitosis catalyzed by constitutively active Cdk1AF.
To ensure that these observations are not cell-type specific, U2OS cells without or with Cdk1AF expression were subjected to the same experimental regimes described above. The state of Chk1 and Chk2 phosphorylation was monitored by Western blotting and mitotic defects were visualised by immuno-fluorescence microscopy. U2OS Cdk1AF cells responded to Adr-induced damage in a manner similar to HeLa Cdk1AF cells (Fig. 4A). Like HeLa Cdk1AF cells, Adr-treated U2OS Cdk1AF cells entered mitosis with activated checkpoint and exhibited mitotic defects similar to those observed in HeLa Cdk1AF cells (Fig. 4B).
Figure 4.
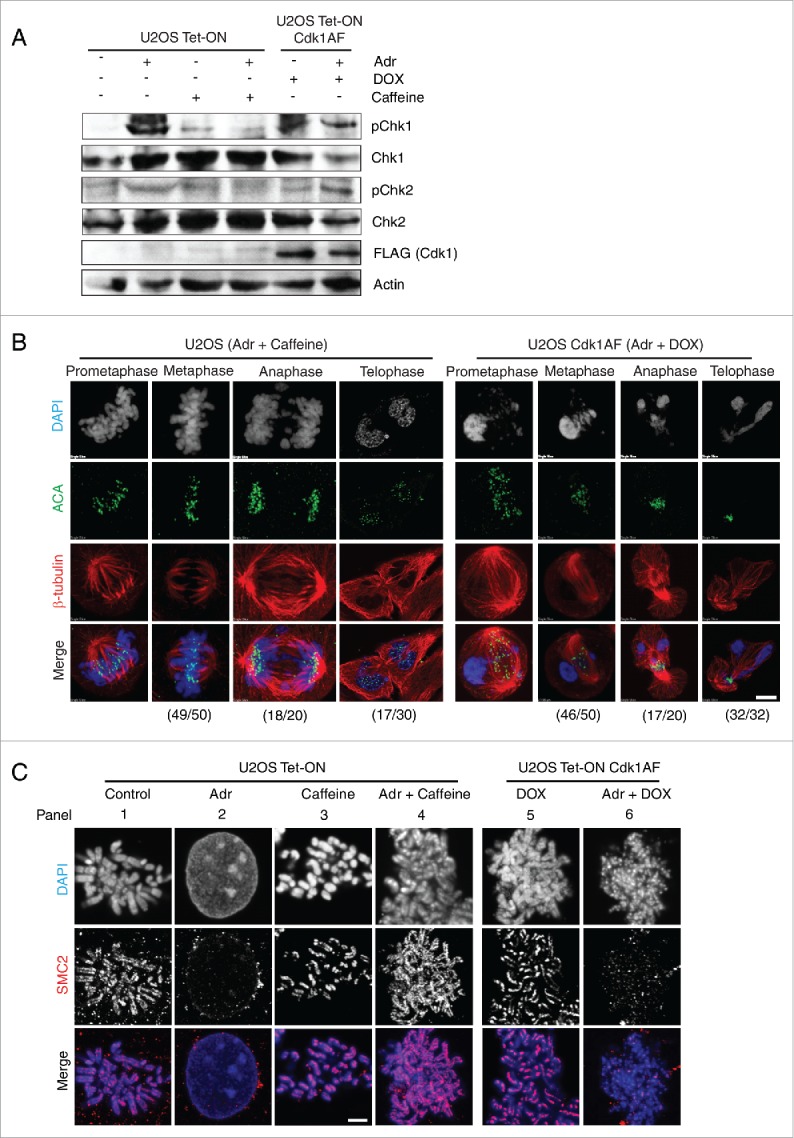
DNA damage response in U2OS Tet-ON Cdk1AF cells. (A) U2OS Tet-ON and U2OS Tet-ON Cdk1AF cells were treated in an identical manner to HeLa Tet-ON and HeLa Tet-ON Cdk1AF cells as described in Figure 1. Cells were collected at T=24 h. The state of pChk1, Chk1, pChk2 and Chk2 was monitored by Western blotting. (B) At T=16 h, U2OS Tet-ON Cdk1AF cells experienced an aberrant mitosis similar to that observed in HeLa Tet-ON Cdk1AF cells, whereas damaged U2OS Tet-ON cells treated with caffeine underwent normal mitosis (left panel). Cell were stained for mitotic spindle (β-tubulin, red), kinetochores (ACA, green) and DNA with (DAPI, blue). The numbers (aberrant cells/total cells) indicate the frequency of occurrence of aberrant mitotic phenotypes in one experiment. Representative images are shown. Scale bar represents 5 μm. (C) Synchronised U2OS Tet-ON, U2OS Tet-ON Cdk1AF cells were treated as described in Figure 1B. As controls, damaged U2OS Tet-ON Cdk1AF cells were also treated with Caffeine. These cells, trapped in mitosis using nocodazole, were harvested, hypotonically-swollen, fixed and stained for the condensin subunit SMC2 (red), with chromosomes counterstained with DAPI. As in the case of HeLa Tet-ON Cdk1AF cells, damaged U2OS Tet-ON Cdk1AF cells lose axial SMC2 localization which is retained in DNA-damaged U2OS Tet-ON cells treated with caffeine. Representative immunofluorescence staining images of anti- SMC2 (red) and counterstaining with DAPI are shown. Scale bar represents 5 μm.
We also examined the recruitment of SMC2, a subunit common to both condensin complexes, in synchronised U2OS Tet-ON and U2OS Tet-ON Cdk1AF cells after Adr treatment. U2OS Tet-ON Cdk1AF cells were synchronised in early S phase as described above and released in the presence of DOX or Adr+DOX. U2OS Tet-ON cells either untreated or treated with Adr or Adr+caffeine served as controls. As expected, SMC2 shows clear axial localization in the untreated U2OS Tet-ON cells and DOX-treated Cdk1AF cells (Fig. 4C, panel 1 and 5). However, in Adr-treated U2OS Tet-ON Cdk1AF cells, SMC2 was displaced from the decondensed chromatin or associate loosely with chromatin (Fig. 4C, pane 6), despite Cdk1AF-induced entry into mitosis just as in Adr-treated HeLa Cdk1AF cells. These results from U2OS cells correspond to parallel experiments in Hela cells and suggest that the mitotic defects we observed in Adr-treated HeLa Cdk1AF cells are not cell-type specific.
Inhibition of Chk2 kinase restores SMC2 recruitment to mitotic chromosomes
The failure of DNA damaged Cdk1AF cells to recruit condensin complexes to the chromosomes despite their progression into mitosis is intriguing. Since the DNA damage checkpoint is active in Adr-treated Cdk1AF cells, it is possible that the activated checkpoint effectors continue to prevent condensin localization to the chromosomes even as these cells proceed to mitosis. As both ATR-Chk1 and ATM-Chk2 axes of the DNA damage checkpoint contribute to the overall inhibition of the onset of mitosis, we wished to determine the control point predominantly responsible for inhibiting condensin recruitment. HeLa Cdk1AF cells were transfected with either control siRNA or siRNA targeting Chk1, Chk2 or ATR 24 h prior to synchronisation as described above (Fig. 5A, left panel). Cells were then released in the presence of Adr and nocodazole was added to the culture medium to trap cells in mitosis. The mock-treated cells were released in Adr+caffeine as a ‘positive control’ (complete inactivation of the checkpoint). The presence of SMC2 was visualised by immuno-staining using anti-SMC2 antibodies. While the majority of Adr-treated cells failed to localize SMC2 to the chromosomes, almost all Adr+caffeine treated cells were able to localize SMC2 to the chromatin (Fig. 5A, bar graph and micrographs). Interestingly, knockdown of ATR (siATR) did not restore SMC2 localization, while Chk1 downregulation restored SMC2 localization to the chromosomes in ∼20% of the cells. The down-regulation of Chk2 exhibited the most significant effect with almost 60% of the Adr-treated HeLa Cdk1AF cells being able to recruit SMC2 to the chromosomes (Fig. 5A). We do note, however, that siChk2 (unlike siATR and siChk1) only partially knocked down Chk2, which may explain lack of further penetrance. These observations indicate that Chk2 is most likely the major DNA damage checkpoint effector that prevents recruitment of condensin complexes to the chromosomes in response to DNA damage.
Figure 5.

Inhibition of the ATM-Chk2 pathway restores SMC2 association with mitotic chromosomes. (A) Asynchronous HeLa Tet-ON Cdk1AF cells were transfected, respectively, with siRNA targeting Chk1, Chk2, ATR, and control siRNA (targeting a non-relevant gene), or mock transfected 24 h prior to synchronisation as described in Figure 2D. The efficacy of various siRNAs are shown (left panel). Cells were trapped in mitosis using nocodazole, fixed and stained with anti-SMC2 and DAPI. The histogram shows SMC2 axial localization in damaged HeLa Tet-ON Cdk1AF cells due to the knockdown of the targeted protein (central panel). Representative immunofluorescence micrographs illustrating different degrees of recruitment are shown in right panel. Scale bar represents 5 μm. Insert shows zoom-in of individual chromosomes. Three independent experiments were performed with similar results and data is shown for one experiment. 200∼400 cells were counted for each condition. Downregulation of respective proteins was confirmed by Western blotting analysis (left panel). Actin for siChk2 experiment was used as loading control. (B) Asynchronous HeLa Tet-ON Cdk1AF cells were transfected with siRNA targeting Chk2 24 h prior to synchronisation as described in Figure 2D, treated with or without specific inhibitor to Aurora B (ZM447439) 4 h after release from thymidine in the presence of Adr and DOX as described in Figure 1B. The histogram shows SMC2 axial localization in damaged HeLa Tet-ON Cdk1AF cells (right panel). Representative immunofluorescence micrographs illustrate that the Aurora B inhibitor (ZM447439) abrogates siRNA Chk2 mediated-recruitment of SMC2 localization (left panel). Scale bar represents 5 μm. Insert shows zoom-in of individual chromosomes.
The notion that Chk2 is the main inhibitor of chromosome condensation in HeLa Cdk1AF cells also implies that it either prevents the loading of condensin complexes directly or via other cell cycle regulators (other than Cdk1) involved in chromosome condensation. Cell cycle kinases such as Aurora B have been implicated in the regulation of chromosome condensation.26 In HeLa cells, Aurora B inhibition has been reported to reduce the loading of condensin I but not of condensin II in prometaphase.38 Therefore, we tested the involvement of Aurora kinases in Chk2-mediated inhibition of condensin recruitment. HeLa Cdk1AF cells transiently transfected with siChk2 or control siRNA were synchronised as described above and released in the presence of Adr. Nocodazole was added to trap cells that entered mitosis. In a duplicate experiment, Aurora kinase inhibitor ZM447439 (inhibits both Aurora A and Aurora B) was added 4 h after the release from thymidine-mediated arrest. SMC2 localization was visualised by immune-staining with anti-SMC2 antibodies. As expected, while Adr treatment drastically reduced SMC2 recruitment to the chromosomes, Chk2 downregulation restored the SMC2 localization in ∼60% of the cells (Fig. 5B, bar graph). Interestingly, Aurora kinase inhibitor ZM447439 dramatically abrogated SMC2 recruitment in siChk2 cells. In a control experiment, ZM447439 treatment alone reduced SMC2 intensity on mitotic chromosomes of HeLa cells, but did not affect SMC2 localization (Fig. 5B, left panel 3rd column). While these experiments do not resolve the question whether Chk2-imposed inhibition of SMC2 localization is mediated through Aurora kinase, they do indicate an involvement of Aurora kinases in some capacity in the Chk2-induced inhibition of SMC2 localization.
DNA damage induces post-translational modification of condensin subunits
Since active Chk2 kinase inhibits chromatin recruitment of condensin complex subunits, we determined if any of these subunits undergo posttranslational modifications in response to DNA damage. HeLa (Tet-ON) cells were synchronised in early S phase as described before and then released in the presence or absence of Adr or Adr+caffeine. HeLa Cdk1AF cells were treated in an identical manner except that Cdk1AF was induced by addition of DOX. Nocodazole (Noc) was added to both cultures to trap cells in mitosis. Cell extracts were analyzed by Western blotting on standard SDS-PAGE gels or high-crosslinking SDS-PAGE (Materials and Methods) using anti-condensin subunit antibodies. Of the various condensin subunits analyzed, CAP-D2 was most clearly detected as 2 protein bands on high-crosslinking gels. In HeLa Tet-ON (control cells), the low mobility (upper) band is seen in cells treated with Noc, caffeine or Adr+caffeine (Fig. 6A, left panel lanes 2, 4 and 5). Since under these conditions cells enter mitosis, the upper band appears to be M phase-specific. However, in Adr-treated cells (Lane 2), only a high mobility (lower) band is present suggesting that this mobility shift is due to checkpoint activation. In HeLa Cdk1AF cells expressing constitutively active and ‘checkpoint resistant’ Cdk1AF, only the upper band is seen (Lane 6). However, unlike HeLa cells (Lane 3), Adr treatment results in partial mobility shift in HeLa Cdk1AF cells (Lane 7). The CAP-D2 mobility shift in these experiments is most likely due to differential phosphorylation since treatment of the extracts with calf intestinal phosphatase (CIP) completely eliminated the low mobility (Upper) bands in all samples (Fig. 6A, right panel). A low mobility band (upper band) is seen in control HeLa cells without Adr treatment (no checkpoint) or when treated with Adr+caffeine (checkpoint inactivation) and also in HeLa Cdk1AF cells with or without Adr treatment, suggesting that phosphorylation of this CAP-D2-specific band is checkpoint independent and could in part be due to Cdk1 activity. The higher mobility (lower) band, on the other hand, is checkpoint dependent since it appears in both Adr-treated HeLa (control) and Adr-treated HeLa Cdk1AF cells. The CAP-G2 subunit also showed DNA damage-correlated high mobility band but not as clearly as CAP-D2. One possible interpretation of these results is that checkpoint activation prevents mitosis-specific phosphorylation of CAP-D2 and continues to do so even when HeLa Cdk1AF cells enter mitosis due to the expression of constitutively active Cdk1AF.
Figure 6.
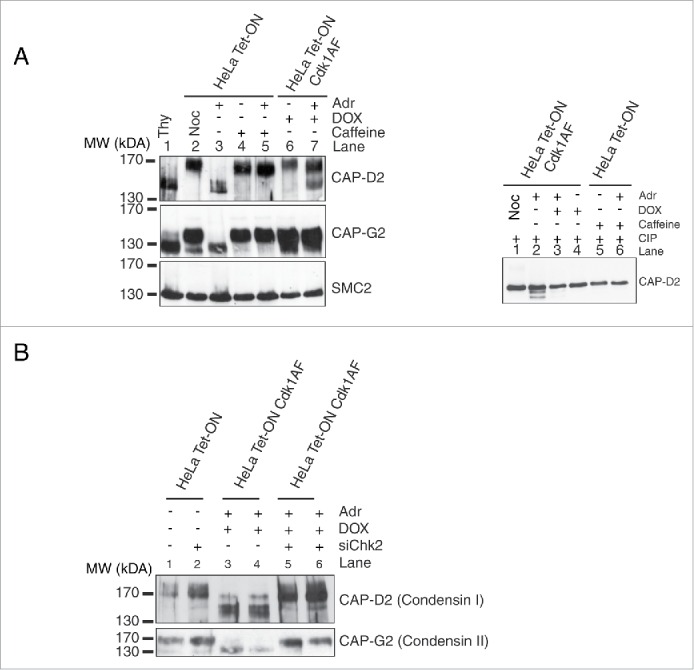
Post-translational modifications of condensin subunits induced by DNA damage. (A) Synchronised HeLa Tet-ON and HeLa Tet-ON Cdk1AF cells were treated as described in Figure 1B, trapped in mitosis using nocodazole and harvested by mitotic shake-off 16 h after Adriamycin treatment. Whole cell lysates were resolved on an 8% high cross-linking SDS-PAGE. Cells treated only with Adriamycin or nocodazole or thymidine were used as controls. The electrophoretic mobility shifts of CAP-D2, CAP-G2 and SMC2 are shown. The same lysates were treated with calf intestinal phosphatases (CIP) at 37°C for 20 minutes, and then subjected to high cross-linking SDS-PAGE, The mobility change of CAP-D2 is shown in the right panel. (B) Hela Tet-ON Cdk1AF cells transiently transfected with siChk2 were synchronised as described in Figure 2D, arrested with nocodazole and harvested by mitotic shake off. Western blotting was performed for the vizualization of the CAP-D2 and CAP-G2 bands.
The appearance of a CAP-D2 high mobility (lower) band upon Adr treatment of HeLa Cdk1AF cells (Fig. 6A) correlated well with their inability to recruit condensin complex subunits to the chromatin (Fig. 3). Since knockdown of Chk2 by siRNA significantly restores condensin subunit recruitment (Fig. 5A), we determined if the appearance of CAP-D2 high mobility bands is linked to Chk2 activity. Adr-treated HeLa and Hela Cdk1AF cells were transfected with siChk2 as described before and cell extracts were analyzed by Western blotting. Interestingly, the higher mobility (lower) CAP-D2 band seen in HeLa and HeLa Cdk1AF cells upon Adr treatment are not detected in cells transfected with siChk2 (Fig. 6B, compare lanes 3 and 4 with lanes 5 and 6). CAP-G2 exhibits a pattern similar to CAP-D2. These observations argue that the CAP-D2/CAP-G2 mobility shift in response to checkpoint activation is Chk2 dependent. How Chk2 kinase alters the status of CAP-D2/CAP-G2 and how this modification influences chromosome compaction warrants further investigation.
Discussion
DNA-damage induced inactivation of Cdk1 has been established as the major control node through which G2 DNA damage checkpoint prevents onset of mitosis and thus, all M-phase related events that dependent on Cdk1. However, the question whether the checkpoint and its effectors directly inhibit mitotic events has not been investigated extensively. In this study, we show that activation of the G2 checkpoint inhibits chromosome condensation, a major mitotic event, independently of Cdk1 inhibition. This inhibition of chromosome condensation by the checkpoint is not apparent in cells normally arrested in G2 upon DNA damage since mitotic entry and all mitosis-specific events are inhibited via inactivation of Cdk1. However, it is revealed in cells expressing constitutively active Cdk1AF. As Cdk1AF is not inactivated by the G2 checkpoint, DNA-damaged Cdk1AF cells enter mitosis and still proceed with major mitotic events (bipolar spindle assembly, breakdown of nuclear envelope (NEBD), etc). However, these cells fail to condense their chromosomes properly (Figs. 1 and 2) despite the presence of constitutively active Cdk1 (one of the main regulators of chromosome condensation) and chromatin being accessible to condensin complex I and II upon NEBD. This phenomenon is not cell-type specific as U2OS Cdk1AF cells also behave in a similar manner (Fig. 4). It is also not a clone specific effect as 2 different HeLa Cdk1AF clones exhibit similar behavior (Figs. 3, S1, S2, and S4). Our results suggest that the failure of DNA-damaged Cdk1AF cells to condense their chromosomes is due to their inability to recruit condensin complex components such as SMC2, CAP-D2 and CAP-G2 to the chromatin (Fig. 3). Since DNA-damaged Cdk1AF cells enter mitosis in the presence of an active checkpoint (Fig. 1), it is possible that checkpoint effectors continue to prevent condensin subunit recruitment. This notion is consistent with our observation that siRNA-mediated knockdown of Chk2 restores the recruitment of condensin subunits (Fig. 5).
The regulatory link between the checkpoint and condensin complexes is further emphasized by checkpoint-dependent differential phosphorylation of CAP-D2 and CAP-G2 (Fig. 6). While the low mobility (upper) bands are present in control cells, the higher mobility (lower) bands appear in DNA damaged cells, implying that the checkpoint activation results in diminished phosphorylation of CAP-D2 and CAP-G2. As DOX-induced expression of Cdk1AF in Adr-treated cells leads to only partial inhibition of CAP-D2 phosphorylation (Fig. 6A, left panel; lane 7) as opposed to the ‘complete’ inhibition in Adr-treated control cells (Fig. 6A, left panel; lane 3) and since Cdk1 has been reported to phosphorylate condensin subunits,22 it is likely that CAP-D2 and CAP-G2 phosphorylation observed in our experiments may predominantly be due to Cdk1. However, contributions by other mitotic kinases cannot be ruled out as condensin subunits are also known to be modified by Aurora B and Polo-like kinase.26,27 In C. elegans, for instance, condensin I has been postulated to be recruited by Aurora BAIR-2 to resolve the chromatin obstructions and maintain a delay in cytokinesis to prevent aneuploidy or DNA damage in the abscission checkpoint pathway.39 Recent studies have indicated that Histone H3 phosphorylation of threonine 118 mediated by Aurora-A promotes timely condensin I disassociation.40 Our experiments using Aurora inhibitor further implicate Aurora kinases in DNA damage checkpoint-mediated inhibition of condensin subunit recruitment (Fig. 5B). Aurora kinases may be necessary for phosphorylation of condensin I/CAP-D2 and condensin II/CAP-G2 when Chk2 is active, an area that warrants future investigation.
The DNA damage checkpoint-imposed inhibition of condensin subunits recruitment to the chromatin appears to be mediated by Chk2. Taken together with lower phosphorylation of CAP-D2 and CAP-G2 in DNA damaged cells, this suggests that Chk2 (or ATM-Chk2 axis) prevents Cdk1-mediated phosphorylation of the condensin subunits in response to DNA damage. One possibility is that this inhibition is direct such that Chk2 may phosphorylate the condensin subunits rendering them resistant to Cdk1-mediated phosphorylation (or other kinases), thus precluding condensin recruitment and therein chromosome condensation even in the presence of constitutively active Cdk1. A detailed analysis of the phosphorylation sites and the corresponding kinase would be required to test this notion.
Thus, there appear to be 2 major negative controls that the DNA damage checkpoint imposes on chromosome condensation: (i) the well-established inhibition of Cdk1 and (ii) Chk2-mediated inhibition of condensin subunit recruitment to the chromatin. The latter mechanism, though not apparent during normal checkpoint-imposed arrest in G2, is revealed in this study through cells expressing constitutively active, checkpoint-resistant Cdk1AF. What could be the physiological utility of the second inhibitory mechanism when chromosome condensation can be effectively precluded by Cdk1 inactivation? While the condensin complex I is sequestered largely in the cytoplasm during interphase with a small fraction found associated with the interphase nucleus,14 condensin II is reported to be localized in the nucleus throughout the cell cycle. 41 In chicken DT40 cells at least, association of condensin II with DNA seems to increase dramatically as cells enter G1 phase.14 It has been suggested that condensin II initiates the first step in condensation in preparation for the second step which is triggered by condensin complex I.10,41 Interestingly, the condensin II complex is also implicated in DSB repair and response.10,42 It is plausible that condensin II's functions in DSB repair/response and chromosome condensation are mutually exclusive such that during checkpoint imposed arrest in G2, checkpoint effectors (such as Chk2) suppress its function in the first step of condensation, making it available for DSB repair/response. Thus, when cells expressing Cdk1AF enter mitosis with an active checkpoint (this study), the condensin complex II is unable to localize to fulfil its function in chromosome condensation.
Chromosome shattering is a phenomenon that can occur when cells prematurely enter mitosis. 43,44 At first glance, the splayed and decondensed appearance of the chromatin in Adr-treated Cdk1AF cells may appear overtly similar to chromosome shattering. However, we believe this not to be the case. It has been found that chromosome shattering is only evident cytologically in air dried methanol/acetic acid spreads and not in PFA fixed cells.45 While the chromosomes figures we see in DNA damaged cells forced into mitosis (i.e. Fig. 3A, panels 3 and 6) may be reminiscent of shattered chromosomes morphologically, they are more individualised and dishevelled rather than shattered. Furthermore, chromosome shattering is known to occur more frequently in S phase as a result of the condensation machinery colliding with the replication apparatus 14 and not in mitosis.
The overall phenotype of DNA-damaged Cdk1AF cells we have documented here is similar to what has been termed as ‘premature entry into mitosis’. 46 The ‘premature entry’ phenotype is generally thought to be caused by uncoordinated progression through mitosis. We suggest that this loss of coordination is due to the antagonistic effects of Cdk1AF and the active checkpoint on mitotic processes: while Cdk1AF initiates many major mitotic processes and promotes cells’ progression through mitosis, the active checkpoint continues to inhibit some of these events such as chromosome condensation, eventually resulting in grossly discordant mitotic progression. This notion is corroborated by the observation that upon inactivation of the checkpoint by caffeine, Adr-treated Cdk1AF cells, though they enter mitosis with unrepaired chromosomes, perform various mitotic functions (spindle assembly, chromosome condensation, NEBD, formation of metaphase plate etc) in a coordinated fashion (Figs. 2 and S2). Hence, in addition to the well-established checkpoint-mediated negative regulation of chromosome condensation via inactivation of Cdk1, this study uncovers condensin subunits as a direct target of the DNA damage checkpoint. The finer details of how the checkpoint kinase Chk2 prevents recruitment of condensin subunits to the chromatin to preclude chromosome condensation remain to be elucidated.
Conclusion
The DNA damage checkpoint arrests cells in G2 phase of the cell cycle in response to DNA damage incurred during S phase. The checkpoint imposes G2 arrest by inhibiting the activity of the key mitotic regulator Cdk1. This keeps chromosomes in a decondensed state which is necessary for efficient DNA repair during the arrest. Since Cdk1 activity is required for chromosome condensation during the normal cell cycle, it is generally believed that the checkpoint prevents chromosome condensation indirectly by inhibiting Cdk1. However, this study shows that checkpoint kinase Chk2 prevents recruitment of condensin complexes to the chromosome in response to DNA damage. Thus the DNA damage checkpoint precludes chromosome condensation during G2 arrest not only by inhibiting Cdk1 activity but also by preventing condensin complexes recruitment to the damaged chromosomes via Chk2 kinase.
Materials and methods
Cell culture, synchronisation and drug treatment
HeLa Tet-ON cells were maintained in high glucose DMEM (pH 7.4) medium supplemented with 10% Tet-negative (EU approved) fetal bovine serum (Cat No A15-169; PAA), penicillin-streptomycin-glutamine (Invitrogen) and G418 (200 μg/ml; Invitrogen). U2OS Tet-ON cells were maintained in similarly supplemented McCoy's 5A medium. Media used for maintaining inducible cells lines were further supplemented with puromycin (0.5–2.0 μg/ml; Invitrogen). All cells were incubated under humidified atmosphere containing 5% CO2 and 95% air at 37˚C.
Cells were synchronised at G1/S phase by combining serum starvation and a thymidine block. Cells were maintained in serum-free medium for 24 hours, followed by a further 24 hours treatment with thymidine (3 mM) in medium supplemented with 10% fetal bovine serum (FBS). Cells were then washed in PBS and released from G1/S arrest into either normal media or media with doxycycline (DOX; 1 μg/ml) to induce expression of Cdk1AF. Doxycycline alters the conformation of the transactivator protein (rtTA) to promote constitutive expression of the transgene in a binary manner. Unless otherwise stated, cells were treated with the following reagents 4 hours after release (when cells had progressed to G2 phase) at the indicated final concentrations: Adriamycin (300 nM), caffeine (5 mM), ATM inhibitor KU-005933 (10 mM; KuDOS Pharmaceuticals), Chk1 inhibitor PF477736 (1 mM), Checkpoint (Chk) kinase inhibitor UCN-01 (300 nM) and Aurora kinase inhibitor ZM447439 (2 mM; Tocris). The pan-caspase inhibitor Z-VAD-fmk (100 nM; Bachem) was used to inhibit apoptosis in all experiments. In some experiments, cells were trapped in early metaphase with nocodazole (100 ng/ml).
Transfection and establishment of stable cell lines
Stable transfectants were obtained using FuGENE 6 (Roche Applied Science), according to the manufacturer's instructions. HeLa Tet-ON Advanced and U2OS Tet-ON cells (Cat No 632110 and 630919 respectively; Clontech) were used to establish puromycin-resistant stable cell-lines, inducibly expressing constitutively active Cdk1 (Cdk1AF). The FLAG tagged Cdk1AF (Thr14 →Ala; Tyr15 →Phe) construct was a generous gift from Dr. Randy Poon (Hong Kong University of Science and Technology).
Expression of targeted genes was knocked down by transient expression of siRNA (Dharmacon) directed against human ATR (5′-AAGCCAAGACAAATTCTGTGT-3′, A, and 5′-AACCTCCGTGATGTTGCTTGA-3′, B;47 human Chk1 (5′- AAGCGTGCCGTAGACTGTCCA -3; 48 and human Chk2 (5′- GAACCTGAGGACCAAGAAC -3′). Cells treated with Oligofectamine or transfected with a control nonspecific siRNA duplex VIII (Dharmacon; ACUCUAUCUGCACGCUGACUU) were used as controls for a direct comparison. Transfections were performed with 150 nM siRNA oligonucleotide duplexes with Oligofectamine (Invitrogen) according to the manufacturer's instructions.
For siRNA transfections, cells were plated at least 16 hours prior to transfection in 6-well plates to yield ∼70% confluency at the time of transfection. At 24 hours post-transfection, cells were subjected to the synchronisation protocol described above.
Determination of DNA content and mitotic index
Mitotic cells were harvested by shake-off at indicated time points from the same plates without trypsinisation. To determine the mitotic index,49 the shake-off cells at each time point from the same plates were fixed with 80% ethanol and incubated for 2 hours on ice with anti-MPM2 antibodies (1:200; Upstate Technology) diluted in a blocking solution (PBS, 0.2% Triton-X, 2% BSA and 0.1% sodium azide), washed twice, then incubated for 2 hours on ice with Alexa 488-conjugated goat anti-mouse IgG (1:200; Invitrogen). Cells were washed in PBS containing 0.1% Triton X-100, then treated with RNase (1 mg/ml) and stained with propidium iodide (50 μg/ml) for subsequent analysis by a FACS Calibur (Becton Dickinson). The cell numbers with 4N DNA content at each time point were analyzed by FlowJo software. The accumulative percentage of mitotic cells collected at each time point was expressed as a ratio of accumulative mitotic cells to all viable cells collected (both detached and attached) by the end of experiment at T = 24 h or T = 45 h. Similarly, MPM2-positive cell numbers were determined and expressed as accumulative MPM2-positive cells to all collected viable cells at T = 24 h or T = 45 h.
Immunofluorescence microscopy
For immunofluorescence staining with antibodies, cells grown on cover-slips were fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) for 20 minutes at room-temperature. For vizualization of SMC2, CAP-D2 and SMC4, cultured cells on slides were trapped in mitosis with nocodazole (100 ng/ml for 12 hours), hypotonically swollen in 75 mM KCl at 37°C for 5 minutes, and fixed with ice cold methanol-acetone (1:1) at −20°C for 5 minutes.15,50 Alternatively, the hypotonically swollen cells were fixed with methanol: acetic acid (3:1) before dropping onto slides for immunofluorescence staining.
For chromosome spreads, mitotic cells were collected by shake-off, treated with 75 mM KCl at 37°C for 30 minutes, spun onto slides by cytospin. The chromosome spreads were then fixed with 2% paraformaldehyde in PBS (pH 7.4) for 15 minutes, permeabilised in 0.5% Triton X-100 in PBS for 5 minutes, and post-fixed with 100% methanol at −20°C for 10 minutes.13
For CAP-G, CAP-G2 and CAP-D2 staining, mitotic cells were swollen in hypotonic solution, then fixed with 4% (w/v) paraformaldehyde for 15 minutes at room temperature.51
Fixed cells were permeabilised with 0.25% Triton X-100 in PBS for 10 minutes and blocked with 5% FBS/2%BSA/0.1% sodium azide in PBS/0.1% Tween-20 for 30 minutes at room temperature. Incubation with primary antibodies was performed overnight at 4°C (primary antibodies used are listed in Fig. S5), after which cells were washed 3 times and incubated with the appropriate Alexa Fluor secondary antibodies (1:1000; Invitrogen) for 1 hour at room temperature. Coverslips were mounted with Vectashield mounting medium containing DAPI (Vector Laboratories). Images were acquired with a confocal microscope (Olympus FV1000 upright confocal microscope and Zeiss LSM 780 confocal microscope) with a 40× or 100× objective. For SMC2 intensity measurements, all images were processed in the Fiji distribution of ImageJ.52 Details of the analysis are as follows: Channels for SMC2 and DAPI were separated. A Gaussian blur (sigma=1) was applied to the DAPI channel and a specific cell of interest was selected using the wand tool. The selected cell was binarised and used as a mask to analyze the SMC2 intensity. Both mean and integrated intensity for each channel were recorded and their ratios were calculated.
Box plots were generated using Beeswarm R package (https://cran.r-project.org/web/packages/beeswarm/index.html). Line graphs were generated using gplots (https://cran.r-project.org/web/packages/gplots/index.html). Statistical analyses were conducted using Student's t test (unpaired). p values less than 0.001 were considered to indicate statistically significant differences.
Immunoprecipitation
Cell pellets were washed once with cold PBS, resuspended in 200 μl lysis buffer [ProteoJet mammalian cell lysis reagent (K0301; Fermentas) supplemented with 0.1 mM Na-orthovanadate; 10 μg/ml pepstatin A; 40 mM β-glycerophosphate; 1 mM NaF and complete protease inhibitors (Roche)], then subjected to homogenization (Micro Smash MS-100) for 20 minutes at 2000 rpm. After centrifugation at 12000 rpm for 10 minutes at 4°C, the supernatant was recovered for protein concentration measurement. 500 μg protein extract was used for immunoprecipitation. Antibodies were added according to the manufacturers’ protocol. After 3 hours incubation at 4°C, 60 μl Protein A Sepharose CL-4B beads (GE 17-0780-01) pretreated with PBS-BSA was added. The lysate-bead mixture was incubated under rotary agitation for 4 hours at 4°C. The beads were then washed 3 times with non-denaturing lysis buffer (20 mM Tris-HCl pH 8; 137 mM NaCl; 10% glycerol; 1% NP-40; 2 mM EDTA and complete protease inhibitors). After the last supernatant was removed, 15 μl of 2X loading buffer was added for Western blotting.
Kinase assay
After immunoprecipitation, beads were washed 3 times with RIPA buffer (1% TritonX-100; 1% sodium deoxycholate; 0.1% SDS; 150 mM NaCl; 50 mM Tris-HCl pH7.2) and twice with 25 mM MOPS. After the last wash, 4 μl of HBII buffer (60 mM β-glycerophosphate; 25 mM MOPS, pH7.2; 15 mM p-nitrophenylphosphate; 15 mM MgCl2; 5 mM EGTA; 1 mM DTT; 1 mM PMSF; 20 μg/ml leupeptin; 40 μg/ml aprotinin; and 0.1 mM Na-orthovanadate) was added and the cell suspension was placed on ice. Ten μl of equal volumes mixture of Solution 1 (20 μl histone H1 [Roche; Cat # 11004875001; used at 1:1000] + 10 μl 250 mM MOPS + 20 μl H20) and Solution 2 (1.1 μl 0.1 M ATP + 275 μl H20 + 3 μl gamma 32P ATP) was added to the immunoprecipated beads. After incubating for 10 min at room temperature, 3 μl of 5X loading buffer was added to stop the reaction. Samples were loaded onto a 12.5% SDS-gel. After the gels were fixed, they were dried at 80°C for 2 hours and were exposed to films at room temperature for 1 hour.
Isolation of mitotic chromosomes
Mitotic shake-off cells were resuspended in 50 ml pre-warmed 1X swelling buffer [10X swelling buffer: 100 mM Hepes, pH 7.4; 400 mM KCl; 50 mM EGTA; 40 mM MgSO4; complete protease inhibitors (Roche)], then centrifuged for 7 minutes at 200 g. Cell pellets were put on ice for 5 minutes, then lysed in 10 ml chromosome buffer (60 mM Pipes; 25 mM Hepes, pH 7.0; 10 mM EGTA, 4 mM MgSO4; 1 mM DDT and protease inhibitors) by repetitive, aggressive pipetting where fluid expelled from the pipette is forced between the tip of the pipette tip and the centrifuge tube ≥ 10 times. After centrifugation at 200 g for 5 minutes, the supernatant (containing chromosomes) was removed and subjected to centrifugation at 400 g for 7 minutes to pellet nuclei and larger pieces of cellular debris. The supernatant was then recovered and centrifuged at 1200 g for 10 minutes to pellet chromosomes. Pellets were resuspended in 500 μl chromosome buffer for protein concentration measurement and Western blotting.
Nuclear extracts
Cells harvested by either trypsin treatment or mitotic shake-off were pelleted by centrifugation. The pellet was resuspended in 400 μl lysis buffer [10 mM Hepes pH 7.5, 2 mM MgCl2; 1 mM EGTA; 1 mM EDTA; 10 mM KCl; 1 mM DTT; 10 mM NaF; 0.1 mM Na- orthovanadate; complete protease inhibitors (Roche)], then incubated for 15 minutes on ice with frequent gentle mixing. 25 μl of 10% NP40 was added prior to homogenization for 6 cycles of 10 second. Nuclei were pelleted by 30 seconds centrifugation at 13000 rpm. 50 μl NEB (25 mM Hepes pH 7.5, 500 mM NaCl, 1 mM DTT, 10 mM NaF, 10% Glycerol, 0.2% NP-40 and 5 mM MgCl2) was added to resuspend the pellet, followed by 3 pulses of sonication (power 15). After centrifugation at 13000 rpm for 5 minutes, the supernatant (nuclear extract) was recovered for protein concentration measurement and Western blotting.
Western blotting
As described in the Immunoprecipitation section, after protein concentration measurement, 40 μg of proteins from each samples were subjected to SDS-PAGE. Primary antibodies used are listed in Fig. S5.
Supplementary Material
Abbreviations
- Adr
Adriamycin
- CIP
calf intestinal phosphatase
- DDR
DNA Damage Response
- DOX
doxycycline
- DSB
Double-Strand Breaks
- h
hour
- FACS
Fluorescence Activated Cell Sorting, also known as Flow cytometry
- HeLa
HeLa Tet-ON
- HeLa Cdk1AF
HeLa Tet-ON Cdk1AF
- IF
immunofluorescence staining
- IP
immunoprecipitation
- MPM2
Anti-phospho-Ser/Thr-Pro MPM-2 antibody, also known as Mitotic Protein #2
- NEBD
Nuclear Envelope Breakdown
- Noc
nocodazole
- NS
No significant difference
- pH3
phospho-Histone 3
- Thy
thymidine
- WB
Western blotting
- Z-VAD
carbobenzoxy-valyl-alanyl-aspartyl-[O-methyl]- fluoromethylketone
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We are grateful to Dr Randy Poon (Hong Kong University of Science and Technology) for the Cdk1AF construct.
Funding
This study was supported by the Biomedical Research Council, Singapore. A part of this work was supported by NHMRC (Australia) project grant GNT1030358 and GNT1047009 and by the Victorian Government's Operational Infrastructure Support Program.
Author Contributions
Conceived and designed the experiments: TZ US. Performed the experiments: TZ SLS. Analyzed the data: TZ, DFH, US. Wrote the paper: TZ DFH US.
References
- [1].Zhou BB, Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature 2000; 408:433-9; PMID:11100718; http://dx.doi.org/ 10.1038/35044005 [DOI] [PubMed] [Google Scholar]
- [2].Bartek J, Lukas J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell 2003; 3:421-9; PMID:12781359; http://dx.doi.org/ 10.1016/S1535-6108(03)00110-7 [DOI] [PubMed] [Google Scholar]
- [3].Domingo-Sananes MR, Kapuy O, Hunt T, Novak B. Switches and latches: a biochemical tug-of-war between the kinases and phosphatases that control mitosis. Philos Trans R Soc Lond B Biol Sci 2011; 366:3584-94; PMID:22084385; http://dx.doi.org/ 10.1098/rstb.2011.0087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Falck J, Petrini JH, Williams BR, Lukas J, Bartek J. The DNA damage-dependent intra-S phase checkpoint is regulated by parallel pathways. Nat Genet 2002; 30:290-4; PMID:11850621; http://dx.doi.org/ 10.1038/ng845 [DOI] [PubMed] [Google Scholar]
- [5].Lee J, Kumagai A, Dunphy WG. Positive regulation of Wee1 by Chk1 and 14-3-3 proteins. Mol Biol Cell 2001; 12:551-63; PMID:11251070; http://dx.doi.org/ 10.1091/mbc.12.3.551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Blasina A, de Weyer IV, Laus MC, Luyten WH, Parker AE, McGowan CH. A human homologue of the checkpoint kinase Cds1 directly inhibits Cdc25 phosphatase. Curr Biol 1999; 9:1-10; PMID:9889122; http://dx.doi.org/ 10.1016/S0960-9822(99)80041-4 [DOI] [PubMed] [Google Scholar]
- [7].Taylor WR, Stark GR. Regulation of the G2/M transition by p53. Oncogene 2001; 20:1803-15; PMID:11313928; http://dx.doi.org/ 10.1038/sj.onc.1204252 [DOI] [PubMed] [Google Scholar]
- [8].Belmont AS. Mitotic chromosome structure and condensation. Curr Opin Cell Biol 2006; 18:632-8; PMID:17046228; http://dx.doi.org/ 10.1016/j.ceb.2006.09.007 [DOI] [PubMed] [Google Scholar]
- [9].Kschonsak M, Haering CH. Shaping mitotic chromosomes: From classical concepts to molecular mechanisms. BioEssays 2015; 37:755-66; PMID:25988527; http://dx.doi.org/ 10.1002/bies.201500020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Hirano T. Condensins: universal organizers of chromosomes with diverse functions. Genes Dev 2012; 26:1659-78; PMID:22855829; http://dx.doi.org/ 10.1101/gad.194746.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Hirano T, Kobayashi R, Hirano M. Condensins, chromosome condensation protein complexes containing XCAP-C, XCAP-E and a Xenopus homolog of the Drosophila Barren protein. Cell 1997; 89:511-21; PMID:9160743; http://dx.doi.org/ 10.1016/S0092-8674(00)80233-0 [DOI] [PubMed] [Google Scholar]
- [12].Hirano T, Mitchison TJ. A heterodimeric coiled-coil protein required for mitotic chromosome condensation in vitro. Cell 1994; 79:449-58; PMID:7954811; http://dx.doi.org/ 10.1016/0092-8674(94)90254-2 [DOI] [PubMed] [Google Scholar]
- [13].Ono T, Losada A, Hirano M, Myers MP, Neuwald AF, Hirano T. Differential contributions of condensin I and condensin II to mitotic chromosome architecture in vertebrate cells. Cell 2003; 115:109-21; PMID:14532007; http://dx.doi.org/ 10.1016/S0092-8674(03)00724-4 [DOI] [PubMed] [Google Scholar]
- [14].Zhang T, Paulson JR, Bakhrebah M, Kim JH, Nowell C, Kalitsis P, Hudson DF. Condensin I and II behaviour in interphase nuclei and cells undergoing premature chromosome condensation. Chromosome Res 2016; 24:243-69; PMID:27008552; http://dx.doi.org/ 10.1007/s10577-016-9519-7 [DOI] [PubMed] [Google Scholar]
- [15].Hirota T, Gerlich D, Koch B, Ellenberg J, Peters JM. Distinct functions of condensin I and II in mitotic chromosome assembly. J Cell Sci 2004; 117:6435-45; PMID:15572404; http://dx.doi.org/ 10.1242/jcs.01604 [DOI] [PubMed] [Google Scholar]
- [16].Gerlich D, Hirota T, Koch B, Peters JM, Ellenberg J. Condensin I stabilizes chromosomes mechanically through a dynamic interaction in live cells. Curr Biol 2006; 16:333-44; PMID:16488867; http://dx.doi.org/ 10.1016/j.cub.2005.12.040 [DOI] [PubMed] [Google Scholar]
- [17].Green LC, Kalitsis P, Chang TM, Cipetic M, Kim JH, Marshall O, Turnbull L, Whitchurch CB, Vagnarelli P, Samejima K, et al.. Contrasting roles of condensin I and condensin II in mitotic chromosome formation. J Cell Sci 2012; 125:1591-604; PMID:22344259; http://dx.doi.org/ 10.1242/jcs.097790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Shintomi K, Hirano T. The relative ratio of condensin I to II determines chromosome shapes. Genes Dev 2011; 25:1464-9; PMID:21715560; http://dx.doi.org/ 10.1101/gad.2060311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Kimura K, Hirano M, Kobayashi R, Hirano T. Phosphorylation and activation of 13S condensin by Cdc2 in vitro. Science 1998; 282:487-90; PMID:9774278; http://dx.doi.org/ 10.1126/science.282.5388.487 [DOI] [PubMed] [Google Scholar]
- [20].Kimura K, Cuvier O, Hirano T. Chromosome condensation by a human condensin complex in Xenopus egg extracts. J Biol Chem 2001; 276:5417-20; PMID:11136719; http://dx.doi.org/ 10.1074/jbc.C000873200 [DOI] [PubMed] [Google Scholar]
- [21].Parry DH, O'Farrell PH. The schedule of destruction of three mitotic cyclins can dictate the timing of events during exit from mitosis. Curr Biol 2001; 11:671-83; PMID:11369230; http://dx.doi.org/ 10.1016/S0960-9822(01)00204-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Abe S, Nagasaka K, Hirayama Y, Kozuka-Hata H, Oyama M, Aoyagi Y, Obuse C, Hirota T. The initial phase of chromosome condensation requires Cdk1-mediated phosphorylation of the CAP-D3 subunit of condensin II. Genes Dev 2011; 25:863-74; PMID:21498573; http://dx.doi.org/ 10.1101/gad.2016411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Bakhrebah M, Zhang T, Mann JR, Kalitsis P, Hudson DF. Disruption of a conserved CAP-D3 threonine alters condensin loading on mitotic chromosomes leading to chromosome hypercondensation. J Biol Chem 2015; 290:6156-67; PMID:25605712; http://dx.doi.org/ 10.1074/jbc.M114.627109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Shintomi K, Takahashi TS, Hirano T. Reconstitution of mitotic chromatids with a minimum set of purified factors. Nat Cell Biol 2015; 17:1014-23; PMID:26075356; http://dx.doi.org/ 10.1038/ncb3187 [DOI] [PubMed] [Google Scholar]
- [25].Bazile F, St-Pierre J, D'Amours D. Three-step model for condensin activation during mitotic chromosome condensation. Cell Cycle 2010; 9:3243-55; PMID:20703077; http://dx.doi.org/ 10.4161/cc.9.16.12620 [DOI] [PubMed] [Google Scholar]
- [26].Nakazawa N, Mehrotra R, Ebe M, Yanagida M. Condensin phosphorylated by the Aurora-B-like kinase Ark1 is continuously required until telophase in a mode distinct from Top2. J Cell Sci 2011; 124:1795-807; PMID:21540296; http://dx.doi.org/ 10.1242/jcs.078733 [DOI] [PubMed] [Google Scholar]
- [27].St-Pierre J, Douziech M, Bazile F, Pascariu M, Bonneil E, Sauve V, Ratsima H, D'Amours D. Polo kinase regulates mitotic chromosome condensation by hyperactivation of condensin DNA supercoiling activity. Mol Cell 2009; 34:416-26; PMID:19481522; http://dx.doi.org/ 10.1016/j.molcel.2009.04.013 [DOI] [PubMed] [Google Scholar]
- [28].Ono T, Yamashita D, Hirano T. Condensin II initiates sister chromatid resolution during S phase. J Cell Biol 2013; 200:429-41; PMID:23401001; http://dx.doi.org/ 10.1083/jcb.201208008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Akai Y, Kurokawa Y, Nakazawa N, Tonami-Murakami Y, Suzuki Y, Yoshimura SH, Iwasaki H, Shiroiwa Y, Nakamura T, Shibata E, et al.. Opposing role of condensin hinge against replication protein A in mitosis and interphase through promoting DNA annealing. Open biology 2011; 1:110023; PMID:22645654; http://dx.doi.org/ 10.1098/rsob.110023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Aono N, Sutani T, Tomonaga T, Mochida S, Yanagida M. Cnd2 has dual roles in mitotic condensation and interphase. Nature 2002; 417:197-202; PMID:12000964; http://dx.doi.org/ 10.1038/417197a [DOI] [PubMed] [Google Scholar]
- [31].Heale JT, Ball AR Jr., Schmiesing JA, Kim JS, Kong X, Zhou S, Hudson DF, Earnshaw WC, Yokomori K. Condensin I interacts with the PARP-1-XRCC1 complex and functions in DNA single-strand break repair. Mol Cell 2006; 21:837-48; PMID:16543152; http://dx.doi.org/ 10.1016/j.molcel.2006.01.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Nishide K, Hirano T. Overlapping and non-overlapping functions of condensins I and II in neural stem cell divisions. PLoS Genet 2014; 10:e1004847; PMID:25474630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Jin P, Gu Y, Morgan DO. Role of inhibitory CDC2 phosphorylation in radiation-induced G2 arrest in human cells. J Cell Biol 1996; 134:963-70; PMID:8769420; http://dx.doi.org/ 10.1083/jcb.134.4.963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Blasina A, Paegle ES, McGowan CH. The role of inhibitory phosphorylation of CDC2 following DNA replication block and radiation-induced damage in human cells. Mol Biol Cell 1997; 8:1013-23; PMID:9201712; http://dx.doi.org/ 10.1091/mbc.8.6.1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Jin P, Hardy S, Morgan DO. Nuclear localization of cyclin B1 controls mitotic entry after DNA damage. J Cell Biol 1998; 141:875-85; PMID:9585407; http://dx.doi.org/ 10.1083/jcb.141.4.875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Ma Y, Yuan X, Wyatt WR, Pomerening JR. Expression of constitutively active CDK1 stabilizes APC-Cdh1 substrates and potentiates premature spindle assembly and checkpoint function in G1 cells. PLoS One 2012; 7:e33835; PMID:22479455; http://dx.doi.org/ 10.1371/journal.pone.0033835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Campbell AE, Hsiung CC, Blobel GA. Comparative analysis of mitosis-specific antibodies for bulk purification of mitotic populations by fluorescence-activated cell sorting. BioTechniques 2014; 56:90-1, 3-4; PMID:24502799; http://dx.doi.org/ 10.2144/000114137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Lipp JJ, Hirota T, Poser I, Peters JM. Aurora B controls the association of condensin I but not condensin II with mitotic chromosomes. J Cell Sci 2007; 120:1245-55; PMID:17356064; http://dx.doi.org/ 10.1242/jcs.03425 [DOI] [PubMed] [Google Scholar]
- [39].Bembenek JN, Verbrugghe KJ, Khanikar J, Csankovszki G, Chan RC. Condensin and the spindle midzone prevent cytokinesis failure induced by chromatin bridges in C. elegans embryos. Curr Biol 2013; 23:937-46; PMID:23684975; http://dx.doi.org/ 10.1016/j.cub.2013.04.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Wike CL, Graves HK, Hawkins R, Gibson MD, Ferdinand MB, Zhang T, Chen Z, Hudson DF, Ottesen JJ, Poirier MG, et al.. Aurora-A mediated histone H3 phosphorylation of threonine 118 controls condensin I and cohesin occupancy in mitosis. eLife 2016; 5:e11402; PMID:26878753; http://dx.doi.org/ 10.7554/eLife.11402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Ono T, Fang Y, Spector DL, Hirano T. Spatial and temporal regulation of Condensins I and II in mitotic chromosome assembly in human cells. Mol Biol Cell 2004; 15:3296-308; PMID:15146063; http://dx.doi.org/ 10.1091/mbc.E04-03-0242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Sakamoto T, Inui YT, Uraguchi S, Yoshizumi T, Matsunaga S, Mastui M, Umeda M, Fukui K, Fujiwara T. Condensin II alleviates DNA damage and is essential for tolerance of boron overload stress in Arabidopsis. Plant Cell 2011; 23:3533-46; PMID:21917552; http://dx.doi.org/ 10.1105/tpc.111.086314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Johnson RT, Rao PN. Mammalian cell fusion: induction of premature chromosome condensation in interphase nuclei. Nature 1970; 226:717-22; PMID:5443247; http://dx.doi.org/ 10.1038/226717a0 [DOI] [PubMed] [Google Scholar]
- [44].Ravi M, Nivedita K, Pai GM. Chromatin condensation dynamics and implications of induced premature chromosome condensation. Biochimie 2012; 95:124-33; PMID:23079335 [DOI] [PubMed] [Google Scholar]
- [45].Hubner B, Strickfaden H, Muller S, Cremer M, Cremer T. Chromosome shattering: a mitotic catastrophe due to chromosome condensation failure. Euro Biophys J 2009; 38:729-47; PMID:19536536; http://dx.doi.org/ 10.1007/s00249-009-0496-z [DOI] [PubMed] [Google Scholar]
- [46].Lu KP, Hunter T. Evidence for a NIMA-like mitotic pathway in vertebrate cells. Cell 1995; 81:413-24; PMID:7736593; http://dx.doi.org/ 10.1016/0092-8674(95)90394-1 [DOI] [PubMed] [Google Scholar]
- [47].Casper AM, Nghiem P, Arlt MF, Glover TW. ATR regulates fragile site stability. Cell 2002; 111:779-89; PMID:12526805; http://dx.doi.org/ 10.1016/S0092-8674(02)01113-3 [DOI] [PubMed] [Google Scholar]
- [48].Zhao H, Watkins JL, Piwnica-Worms H. Disruption of the checkpoint kinase 1/cell division cycle 25A pathway abrogates ionizing radiation-induced S and G2 checkpoints. Proc Natl Acad Sci U S A 2002; 99:14795-800; PMID:12399544; http://dx.doi.org/ 10.1073/pnas.182557299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Vogel C, Kienitz A, Hofmann I, Muller R, Bastians H. Crosstalk of the mitotic spindle assembly checkpoint with p53 to prevent polyploidy. Oncogene 2004; 23:6845-53; PMID:15286707; http://dx.doi.org/ 10.1038/sj.onc.1207860 [DOI] [PubMed] [Google Scholar]
- [50].Hudson DF, Ohta S, Freisinger T, Macisaac F, Sennels L, Alves F, Lai F, Kerr A, Rappsilber J, Earnshaw WC. Molecular and genetic analysis of condensin function in vertebrate cells. Mol Biol Cell 2008; 19:3070-9; PMID:18480406; http://dx.doi.org/ 10.1091/mbc.E08-01-0057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Takemoto A, Maeshima K, Ikehara T, Yamaguchi K, Murayama A, Imamura S, Imamoto N, Yokoyama S, Hirano T, Watanabe Y, et al.. The chromosomal association of condensin II is regulated by a noncatalytic function of PP2A. Nat Struct Mol Biol 2009; 16:1302-8; PMID:19915589; http://dx.doi.org/ 10.1038/nsmb.1708 [DOI] [PubMed] [Google Scholar]
- [52].Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al.. Fiji: an open-source platform for biological-image analysis. Nat Methods 2012; 9:676-82; PMID:22743772; http://dx.doi.org/ 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.