


Abstract
Aptamers directed against human thrombin can selectively bind to two different exosites on the protein surface. The simultaneous use of two DNA aptamers, HD1 and HD22, directed to exosite I and exosite II respectively, is a very powerful approach to exploit their combined affinity. Indeed, strategies to link HD1 and HD22 together have been proposed in order to create a single bivalent molecule with an enhanced ability to control thrombin activity. In this work, the crystal structures of two ternary complexes, in which thrombin is sandwiched between two DNA aptamers, are presented and discussed. The structures shed light on the cross talk between the two exosites. The through-bond effects are particularly evident at exosite II, with net consequences on the HD22 structure. Moreover, thermodynamic data on the binding of the two aptamers are also reported and analyzed.
INTRODUCTION
Simultaneous interaction of multiple ligands with biomolecules often finely modulates biological processes. New strategies for the design of pharmaceutical agents could possibly take into account the synergistic effect of polyvalent binding (1). Among others, the case of human α-thrombin (thrombin) is particularly interesting. Thrombin plays a pivotal role in the coagulation cascade maintaining blood hemostasis by balancing pro- and anti-coagulant actions (2–4). Its activity is regulated and controlled by the binding of several cofactors and substrates on two electropositive surfaces, called exosite I and II (Figure 1), which, together with the catalytic site, make this protein unique in the pancreatic trypsin family of serine proteases.
Figure 1.

Surface representation of thrombin with its two exosites colored in orange. The active site is covalently inhibited by PPACK (in blue).
In particular, exosite I is involved in binding to fibrinogen, platelet receptor PAR-1, thrombomodulin and to endogenous and exogenous inhibitors, while exosite II interacts with heparin, F2 prothrombin fragment and physiological inhibitors such as antithrombin III and nexin-I (5). Ligand binding to either exosite I or exosite II may influence the organization of the active site and/or the reactivity of thrombin. For these reasons, considerable efforts are currently being made to identify effectors of the enzyme that are able to regulate the onset and progression of cardiovascular diseases (6).
A special class of thrombin synthetic ligands is represented by DNA aptamers, which are DNA oligonucleotides that bind specific target molecules (7,8). They are identified by in vitro selection from large random sequence libraries, through a process also known as SELEX. Several properties of aptamers make them very attractive as therapeutic compounds. They have little immunogenicity and a well-established synthesis protocol and chemical modification technology, which allow a fine-tuning of their bioavailability and pharmacokinetics. They usually bind their target with dissociation constants in the low-nanomolar range. Moreover, complementary oligonucleotide antidotes can reverse aptamer actions facilitating the control over their activity in vivo (9).
The ability of oligonucleotides to adopt different three-dimensional structures allows them to form complementary shapes that very well fit or embrace the recognition site of their target. However, only a few structures are available to show how aptamers can assume complex conformations that enable specific binding to proteins that do not normally interact with nucleic acids (10–19).
Two DNA aptamers, HD1 (7) and HD22 (20), directed to exosite I and exosite II, respectively, are by far the most studied thrombin binding aptamers both for therapeutic and for biosensing purposes (21,22). We have recently unraveled the structures of thrombin in complex with these aptamers showing that HD1 adopts an antiparallel G-quadruplex architecture (23–26), whereas HD22 presents a mixed duplex–quadruplex folding (27). Since the interaction of each aptamer is mediated by different protein sub-domains, it is possible to enhance their activity just by linking them together thus generating a bivalent aptamer with improved affinity and specificity (28–38). In particular, a relevant enhancement of functional affinity has been obtained by using linkers based on PEG-chains (32), randomized DNA sequences (36) or DNA weave tiles (33,37,38).
The strategy adopted to conjugate the two aptamers does not require a detailed knowledge of the protein–aptamer interaction at the two exosites. However, the elucidation at the atomic level of the way in which both aptamers bind to the protein in a ternary complex may suggest new approaches for the design of thrombin inhibitors with enhanced specificity. It should be noted that while the positioning of HD1-like aptamers at exosite I is well established (24–26,39), HD22 was found to adhere to exosite II in an unexpected bent conformation (27), whose details could be finely regulated by the presence of the aptamer bound at exosite I.
Here we report the crystal structure of two ternary complexes, which differ for the HD1-like aptamer bound at exosite I. The resulting structures, which are embedded in a different packing organization, fully confirm the conformation of HD22 already found in the binary complex (27), but also display small differences that can be interpreted in term of a long-range communication between the two exosites. Thus, the present data can represent a good starting model for a computational analysis aimed at a better understanding of the cooperative mechanisms generated by the simultaneous binding of ligands at the two exosites (40–43). Isothermal titration calorimetry (ITC) data on the formation of the binary and ternary complexes are also reported and discussed.
MATERIALS AND METHODS
Sample preparation
Thrombin (Haemtech Technologies, Essex Junction, VT, USA) inhibited by PPACK (D-Phe-Pro-Arg-chloromethylketone) was equilibrated against a 0.75 M KCl solution using a Centricon mini-concentrator (Vivaspin 500, Sartorius, Goettingen, Germany) and a refrigerated centrifuge (Z216MK, Hermle Labortechnik, Wehingen, Germany). The two HD1-like aptamers, lacking the nucleobase of T3 (ΔT3) and T12 (ΔT12), were synthesized as previously described (44). More information about the sequence of both aptamers is reported in Supplementary Data. Each aptamer was dissolved in 10 mM sodium phosphate buffer pH 7.1 up to a concentration of 2 mM. In order to induce the correct fold of the aptamers, a process of annealing is mandatory. Thus, both solutions were heated for 10 min at 85°C, slowly cooled down and stored at 4°C overnight.
HD22 was purchased from Sigma-Genosys and handled in the same way. Its sequence is reported in Supplementary Data. A solution of the aptamer at a concentration of 4 mM was prepared using 10 mM sodium phosphate pH 7.1 as buffer. An annealing process was necessary to induce its correct fold.
The ternary complex was then prepared depositing a twofold molar excess of HD22 onto a frozen sample of thrombin. The system was left for about 3 h at 4°C. Then the solution was frozen again and a 2-fold molar excess of the ΔT3 (or ΔT12) solution was deposited onto it. The system was let equilibrating at 4°C overnight. The final solution was then diluted and extensively washed to remove the excess of the aptamers and was finally concentrated to about 0.2 mM. The final buffer was changed to 25 mM sodium phosphate buffer pH 7.1, 0.1 M NaCl.
Crystallization and data collection
Crystallization conditions for the two complexes were identified after extensive screening of sitting-drop crystallization experiments at 20°C in 96-well plates (Greiner Bio-One, Monroe, NC, USA) using an Automated Protein Crystallization Workstation (Hamilton Robotics) and precipitant solutions of commercially available crystallization screens (Hampton Research Crystal Screen 1, Crystal Screen 2 and Index).
Crystals suitable for X-ray diffraction data collection were obtained after very fine optimization of the crystallization conditions in the hanging drop vapor diffusion method mixing 0.5 μl complex solution with 0.5 μl reservoir solution at 20°C. Crystals of the ternary complex of thrombin with HD22 and ΔT3 grew in 28% MPEG 2000, 0.1 M Bis/Tris pH 6.5 and belong to space group I23. Crystals of the ternary complex of thrombin with HD22 and ΔT12 grew in 18% PEG 3350, 0.2 M sodium formate, acetonitrile 2% v/v and belong to space group C2.
Crystals were then cryoprotected with 20% (v/v) glycerol and flash-cooled in liquid nitrogen. Data were collected at the CNR Institute of Biostructures and Bioimages, Naples, Italy, using a Saturn944 CCD detector equipped with CuKα X-ray radiation from a Rigaku Micromax 007 HF generator. Datasets were processed using HKL2000 software (45). Matthews coefficient calculations suggested in both cases the presence of a 1:1:1 ternary complex in the asymmetric unit. Detailed statistics of data collection are reported in Supplementary Table S1.
Structural solution and refinement
The phase problem was solved by molecular replacement using PHASER (46). Thrombin heavy and light chains and HD22 from PDB structure 4I7Y, and HD1 variants from 4LZ1 and 4LZ4 were used as independent search models. Crystallographic refinement was performed using PHENIX (47) and REFMAC5 (48), while manual model building was handled with COOT (49). After the inclusion of low-resolution data and bulk solvent correction, the final R/Rfree values were 15.5/20.8 for TerΔT3 and 20.8/26.8 for TerΔT12. The structures were analyzed using MolProbity (50) and NucPlot (51). A summary of refinement statistics is show in Supplementary Table S1. Molecular graphics figures were prepared with PyMOL (DeLano Scientific, Palo Alto, CA, USA).
ITC measurements and binding assays
Binding energies for the interactions between aptamers and thrombin were determined in a buffer of 25 mM sodium phosphate (pH 7.1), 100 mM NaCl using ITC on a MicroCal iTC200 (GE Healthcare, Tokyo, Japan). Degassed thrombin solutions (4-5 μM) were titrated with each aptamer solution (40–50 μM). Control experiments were carried out to calculate the heat of dilution for aptamers. The thermogram for the interaction was determined by subtracting the heats of sample experiments from those of the control experiment. The thermograms were fitted to obtain the KA value analyzed with Origin 7 software (MicroCal Inc., Northampton, MA, USA) for models that assume a single set of identical binding sites. Thermodynamic parameters governing protein–aptamer interactions were estimated by the following standard relationships [Equations (1) and (2)]:
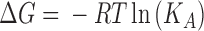 |
(1) |
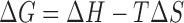 |
(2) |
where ΔG is the binding free energy change, ΔH is the binding enthalpy change, ΔS is the binding entropy change, R is the gas constant, and T is the temperature in Kelvin.
RESULTS
Crystallization of the two ternary complexes
To our knowledge, the crystallization of a protein in complex with two different DNA aptamers has never been accomplished. In order to succeed in this task, an extremely careful set-up of experimental conditions is always crucial (52) but sometimes it is just not enough. In particular, our attempts to crystallize a ternary complex between thrombin, HD1 and HD22 have not been successful despite the hundreds of crystallization trials. On one side, the flexibility of the nucleic acid chains and, on the other, the two modes of binding of HD1 (26) make the solution a heterogeneous mixture of species, thus hampering the crystallization process. In order to overcome this problem, crystallization trials were performed using either one of two HD1 variants, named ΔT3 and ΔT12, which lack the T3 or T12 nucleobase, respectively (see Supplementary Data and Figure S1). Indeed, in a previous paper (26) we have shown that the removal of one base from a TT loop also removes the degeneracy of the two HD1 binding modes. Indeed the use of the HD1 variants was successful; the two ternary complexes, hereafter referred to as TerΔT3 and TerΔT12, formed ordered and well-diffracting crystals suitable for X-ray diffraction data collection (Supplementary Figure S2).
Overall crystal structure of the ternary complexes
The structure of the two complexes was solved at 2.95 and 3.58 Å resolution with Rfactor/Rfree values of 0.155/0.208 and 0.208/0.268 for TerΔT3 and TerΔT12, respectively. Statistics of data collection and refinement for both complexes are reported in Supplementary Table S1 (Supplementary Data).
In both cases, HD22 binds thrombin exosite II, while the HD1 variant binds exosite I on the opposite side of thrombin molecule. A surface representation of the two complexes is shown in Figure 2. The two ternary complexes present a similar architecture, with the main structural differences arising from the orientation of the HD1 variant: with respect to the protein ΔT3 and ΔT12 are related by a 180° rotation around the quadruplex axis of the aptamer. Due to the higher resolution of crystallographic data of TerΔT3 than TerΔT12, hereafter we will mainly refer to TerΔT3.
Figure 2.

Surface representation of TerΔT3 and TerΔT12. The main difference is the orientation of the HD1 variant (in green) at exosite I. It can easily be seen that the TGT loop of HD1 points down in TerΔT3 and up in TerΔT12.
Thrombin
Protein heavy chain (residues 16–246) and light chain (residues 1B–14J) are well defined in the electron-density maps as well as PPACK into the active site and the sugar at the glycosylation site, with the only exception of the disordered autolysis loop (residues 146–150). In comparison to the aptamer-free protein (PDB code: 1PPB) the rmsd, after the superposition of all the Cα protein atoms, is 1.0 Å, very close to the values found for thrombin/aptamer binary complexes (26,27). The two aptamers bury a very large protein surface area (Table 1).
Table 1. The interaction area (in Å2) is reported for aptamers in complex with thrombin in the two ternary structures and in the three reference structures of thrombin in complex with each single aptamer.
| Interaction area (Å2) of thrombin with: | HD1-like at exosite I | HD22 at exosite II |
|---|---|---|
| TerΔT3 (5EW1) | 501.3 | 1116.2 |
| TerΔT12 (5EW2) | 533.6 | 1044.2 |
| thrombin-ΔT3 (4LZ4) | 512.3 | |
| thrombin-ΔT12 (4LZ1) | 509.8 | |
| thrombin-HD22 (4I7Y) | 1079.5 |
HD1 variants and exosite I
The HD1 variants have the canonical antiparallel G-quadruplex architecture that coordinates a sodium ion at the center of the G-core. It presents only minor differences with that found for the binary complexes, in which the quadruplex motif coordinates a potassium ion. The highly flexible TGT loop, which is placed far away from the protein interface, lacks tight packing interactions and is partially disordered.
In both complexes the TT and TN loops (N stands for the DNA spacer, replacing T3 and T12 in ΔT3 and ΔT12 variants, respectively) of the HD1 variants grab a protruding region of exosite I: the TT loop interacts with the A-region of exosite I (Arg75, Glu77, Arg77A, Asn78 and Ile79) whereas the defective TN loop interacts with the B-region (Arg75 and Tyr76) (Supplementary Figure S3).
Thus in the two variants the aptamer is rotated by 180° around the pseudo molecular twofold axis normal to the quadruplex structure: this operation interchange the TT loop with the TN loop and allows the TT loop to interact in both cases with the protein A-region; the TGT loop also acquires a different orientation. These findings strengthen our previous suggestion that the interaction of the TT loop with the A-region of the protein is the most effective for the stabilization of the complex (26). The interface of thrombin with HD1 variants involves four bases, for a total of about nine H-bonds. One important interaction for the recognition process (26) involves residue Glu77 and the first thymine of the TT loop (T3 in TerΔT12, T12 in TerΔT3).
The guanidinium groups of Arg75 and Arg77A and the nucleobases of the loop residues T4 and T13 are linked together by a cyclic network of hydrogen bonds in a roughly planar arrangement (Figure 3A). This hybrid tetrad formed by both protein and oligonucleotide residues stacks onto the guanines of the first tetrad (G2-G5-G11-G14) at an average distance of about 3.4 Å. The interactions of the two Arg residues are typical cation-π/H-bond stair motifs (53); all together, these interactions build up a peculiar G-quadruplex (Figure 3B).
Figure 3.

(A) Schematic representation of cyclic H-bond pattern between residues Arg75, T4, Arg77A and T13. The arrangement stack onto a G-tetrad and is characterized by two cation-π/H-bond stair motifs (53). (B) View of the hybrid quadruplex with the protein–DNA tetrad highlighted in orange.
HD22 and exosite II
All residues of HD22 could be fitted in the electron density maps except for the 5′ terminal guanine residue of TerΔT3 and for both terminal residues of TerΔT12. The overall structure of HD22 (Figure 4A) is very similar for the two complexes (the rmsd is 0.98 Å), and presents interesting new features with respect to the characteristic mixed duplex/quadruplex architecture highlighted by Russo Krauss et al. for HD22 (27) in the binary complex (the rmsd is 1.24 Å for TerΔT3 and 1.39 Å for TerΔT12). Similarly to the binary complex the two tetrads G8-G11-G17-G20 (tetrad I) and G5-G7-G12-G16 (tetrad II) of the quadruplex domain stack on top of each other to form the eight-guanine core. The two tetrads are spaced by four loops, which contribute to the stability of this local conformation by forming two inter-loop Watson–Crick (WC) base pairs that stack on the guanine residues of the G-tetrads.
Figure 4.

(A) Cartoon representation of HD22. The duplex is colored in gray, the junction residues in black, the G-core in red, and the loops in yellow. The sodium ion is represented as a red sphere. (B) Cartoon representation of HD22 in interaction with thrombin at exosite II. Residues of the aptamer interacting with the protein are colored in yellow. (C) Electron density map of the two tetrads in HD22. The sodium ion is clearly visible between them.
The duplex domain presents the same three canonical WC base pairs (T2-A26, C3-G25 and C4-G23) found in the binary complex. Moreover, the transition from the duplex to the quadruplex region occurs in one strand without intermediate residues, whereas in the second strand it is mediated by two residues, G21 and G22. The former (G21) forms two strong hydrogen bonds with the phosphate group of G5 (G-fork). The bulging out of the two residues forces the duplex axis to adopt an almost orthogonal orientation with the quadruplex axis (Figure 4A), thus creating an interaction surface contributed by the quadruplex and duplex motifs that extensively covers exosite II of the protein (Figure 4B).
The protein–aptamer interactions provide a structural basis for the interpretation of the mutagenesis experiments performed by Mayer and collaborators (54), which could not be explained on the basis of the previous model (20). Indeed, the complete loss of binding activity resulting from the mutations T6C, G8A, G23A can be explained by the fact that T6, G8, G23 are all involved in key intramolecular interactions that would be disrupted upon the substitution. On the other hand, the lack of any effect on the binding for the mutation G22A is expected. Although G22 is involved in stacking interactions with the adjacent residues G21 and G23 and is hydrogen bonded to the nearby Arg233, both these interactions would remain basically unchanged upon mutation with an adenine (see also Supplementary Data for details).
Despite the close similarity in the overall organization of HD22 with respect to the binary complex, the present data show new interesting details that throw further light on the flexibility and adaptability of the aptamer structure. Indeed, in the ternary complexes a sodium ion is present between the two tetrads (Figure 4C). The ion is 2.6 Å from the least square plane through the bases of tetrad II and only 0.9 Å from that of tetrad I. The bases G8-G11-G17-G20 present the classical anti-syn-anti-syn conformation sequence, and a regular arrangement of Hoogsteen-like H-bonds (Supplementary Figure S4). The four O6 atoms point toward the quadruplex axis and the cation coordinates the O6 atoms at a distance of 2.1 Å in a square pyramidal geometry. Tetrad I and tetrad II are more planar with respect to the binary complex, where their puckering decreases the space available for the coordination of the cation. Moreover, the four bases of tetrad II, which in the binary complex show the non-canonical anti-syn-anti-anti conformational sequence and a unique H-bond pattern, also have a non-canonical conformation with all residues in the anti conformation. In this novel organization the four nucleobases direct alternatively N2 and O6 toward the quadruplex axis forming a cyclic pattern of hydrogen bonds (Supplementary Figure S4).
The tertiary structure of the molecule opens a through-space communication pathway between the duplex and quadruplex regions via the G-fork. This pathway is clearly elucidated by the present structures. Indeed, the modified base orientations and hydrogen bond scheme of tetrad II causes through the G-fork a small reorganization at the C3-G25, and C4-G23 region of the duplex (Figure 5). This results in a slight contraction of the quadruplex–duplex mouth, which allows a better bite of the aptamer on exosite II.
Figure 5.

Cartoon representation of the recognition between HD22 and exosite II in the binary (blue) and TerΔT3 (red) complexes.
In the correspondence to the shift of the duplex moiety, correlated movements are also observed on the protein side. In particular, a small rotation of the external helical region 125–129 and a shift of the loop 162–182, which form a cavity on the protein surface, allows a better fit of the T24 backbone. This residue, which bulges out from the duplex strand and is an anchor point of HD22 on thrombin surface, moves about 1.5 Å more deeply buried inside the cavity (Figure 5). With respect to the binary complex additional contacts between the double helix of HD22 and the thrombin surface are also observed (Supplementary Table S2). At the interface between the quadruplex region and the protein, the modifications are less evident and most of the interactions present in the binary complex are generally well preserved. Despite their completely different packing organization, the above differences are common to the ternary complexes TerΔT3 and TerΔT12, suggesting that they are not merely an artifact of the crystal packing. Interestingly, they are observed in the presence of PPACK bound at the active site that is known to reduce the protein flexibility (55). It should be noted, however, that the protein maintains some degree of flexibility even in the active-site liganded form (56).
ITC data on DNA aptamer binding
In addition to the structural data, thermodynamic parameters for the binding of the aptamers to human thrombin have been obtained by ITC analysis (Supplementary Figure S4), according to the scheme reported in Figure 6. Experiments have been performed at 25°C using high purity thrombin samples with the active site covalently inhibited by PPACK, in order to minimize possible artifacts caused by the autocatalytic degradation of the enzyme. In agreement with previous suggestions (57,58), the results strongly indicate that all three aptamers (HD1, ΔT3 and HD22) form with thrombin a complex with a 1:1 stoichiometry (Table 2 and Figure 6A–C). The ΔH and −TΔS values for thrombin and ΔT3 were similar with those for thrombin and HD1 indicating that the loss of the π-π stacking interactions between Tyr76 and the missing nucleobase did not significantly affect the change of ΔH and −TΔS. As far as HD22 is concerned, the finding that the enthalpy contribution to the binding is smaller than that observed in the case of the interaction of HD1 is unexpected. Indeed, the value of the aptamer/thrombin contact area is doubled in the case of HD22 with respect to that found for HD1 (Table 1) (27). The smaller value of ΔH, however, could be the result of the conformational flexibility of HD22 in solution, which should be much higher than that of HD1. Some conformers have to change structure to be able to bind exosite II with probably an enthalpy penalty. On the other hand, the low value of −TΔS is likely associated to a more substantial dehydration occurring with the binding of HD22 compared to the binding of HD1. Indeed, the binding site at exosite II of HD22 is twice as large as the binding site at exosite I of HD1, it is richer in polar charged residues and more densely hydrated. It is worth noting that ΔG is practically unchanged with respect to HD1 or ΔT3 complexes.
Figure 6.

Scheme of all reactions studied by ITC. For each reaction, thermodynamic parameters (ΔH, −TΔS and ΔG) have been calculated and are reported in Table 2.
Table 2. Thermodynamic parameters for the reaction presented in Figure 6.
| A | B | C | D | E | F | G | |
|---|---|---|---|---|---|---|---|
| N | 0.98 ± 0.06 | 0.83 ± 0.10 | 0.96 ± 0.03 | 0.98 ± 0.06 | 1.02 ± 0.14 | 0.95 ± 0.11 | 1.04 ± 0.02 |
| KA | 7.8 ± 0.1 | 5.2 ± 0.8 | 11 ± 2 | 14 ± 7 | 18 ± 15 | 6.2 ± 2.4 | 9.9 ± 0.7 |
| ΔH | −21.3 ± 0.9 | −20.4 ± 4.6 | −14.8 ± 0.2 | −14.1 ± 1.2 | −14.8 ± 2.2 | −21.4 ± 3.2 | −22.6 ± 0.2 |
| −TΔS | 10.6 ± 0.9 | 9.9 ± 4.7 | 3.8 ± 0.2 | 3.1 ± 1.4 | 3.8 ± 2.7 | 10.8 ± 3.4 | 11.7 ± 0.1 |
| ΔG | −10.7 ± 0.1 | −10.5 ± 0.1 | −11.0 ± 0.1 | −11.0 ± 0.3 | −11.0 ± 0.5 | −10.6 ± 0.2 | −10.9 ± 0.0 |
Abbreviations: N, binding stoichiometry of oligonucleotides to thrombin; KA (107 M−1), binding constant; ΔH, −TΔS and ΔG (kcal mol−1) changes in binding enthalpy, binding entropy and binding free energy at 25°C, respectively.
Finally, it has to be underlined that the present study represents the first complete comparative thermodynamic analysis of the binding of each aptamer alone as well as of the binding of each aptamer when the other exosite is occupied (Table 2 and Figure 6D–G). It should be noted that these thermodynamic parameters do not manifest the presence of exosite-exosite interactions, which have been clearly evidenced in literature by other methods (40–43).
DISCUSSION
The existence of two binding sites on the surface of thrombin allows protein activity to be finely regulated; indeed, recent data on the allosteric interplay among thrombin functional sites (the two exosites and the catalytic site) have shown that the simultaneous presence of HD1 and HD22 significantly enhances thrombin-catalyzed hydrolysis of a peptide substrate (59). However in the ternary complexes, whose structures we report here, the HD1-like aptamer located at exosite I and HD22, the mixed duplex/quadruplex aptamer that binds at exosite II, are more than 30 Å apart, too far away for a through-space interaction between them to occur. Moreover, the thermodynamic data do not indicate the presence of significant cooperative effects in the binding of the two aptamers. Nonetheless, the structural details of the ternary complexes in comparison to the corresponding binary ones (26,27) indicate that, at least for HD22, small but intriguing differences are observed. In particular, the reorganization of tetrad II with a different orientation of G12 and the more massive presence of the sodium ion, which is present only at a very low level, if any, in the binary complex, slightly displaces the duplex region and causes the whole aptamer to improve the adhesion to exosite II. Alternatively, it can be said that small modifications at the protein/HD22 interaction surface may ultimately influence the reorganization of the quadruplex region. We surmise that this may be due to the binding of the ligand at exosite I that is likely to modify the dynamics of the whole protein skeleton.
Incidentally, also in the thrombin-HD22 binary complex crystallized in the presence of K+, the ion is not present at an appreciable level in the quadruplex moiety (data not published).
A through-bond propagation effect has already been suggested on the basis of various studies (60–62). The results indicate that the binding of a ligand at one site reduces the dynamics of the whole protein so that a smaller number of conformations are accessible for the binding of a second ligand (62–64). The crystallographic data reveal that the binding of HD22 at exosite II does not produce visible effects on the binding of the HD1-like aptamer at exosite I. Vice versa, the presence of a ligand at exosite I causes small but significant effects for the binding of HD22. With respect to the former, this aptamer is endowed with a much higher conformational flexibility due to its composite nature. In particular, the bent active conformation of HD22 displays intriguing intramolecular duplex–quadruplex interactions. The protein surface covered by HD22 is much broader than that covered by HD1 and includes the C-terminus of the heavy chain and residues 96–97A that were found to be very dynamic by nuclear magnetic resonance and molecular dynamics methods (56). This peculiarity, in addition to the bimodular nature of HD22 and to the role played by the sodium cation may render this aptamer more prone to reveal the subtle effects of the protein dynamics and the perturbation exerted by the presence of a ligand at exosite I.
CONCLUSIONS
The two structures reported in this paper represent the first example of a protein simultaneously bound to two aptamers in a ternary complex. In particular, the case of thrombin represents an intriguing example of how ligand binding to more than one exosite can control enzymatic activity.
In the structures of the two ternary complexes, the presence in the protein active site of PPACK, which was used to avoid the heterogeneity of protein solution induced by autoproteolysis, stabilizes the functional core of the protein and reduces the backbone dynamics (55). This prevents a marked structural identification of an interplay between the active site and the two exosites. However, few clues regarding site–site interaction clearly emerge from our data and can be a useful starting point for the computational analysis of dynamics and long-range effects connected to the simultaneous binding at the two exosites, a question that remains still open and controversial. Furthermore, although various bivalent aptamers have been already produced, we believe that the present structures may have direct implications on novel strategies to design direct thrombin inhibitors with enhanced specificity.
ACCESSION NUMBERS
Coordinates and structure factors for TerΔT3 and TerΔT12 have been deposited in the Protein Data Bank under ID codes 5EW1 and 5EW2, respectively.
Acknowledgments
Giosuè Sorrentino and Maurizio Amendola (Institute of Biostructures and Bioimaging, CNR, Naples, Italy) are gratefully acknowledged for technical assistance during data collection.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Grants-in-Aid for Scientific Research from a Ministry of Education, Culture, Sports, Science and Technology (MEXT) (in part); MEXT-Supported Program for the Strategic Research Foundation at Private Universities (2014-2019), Japan; Hirao Taro Foundation of KONAN GAKUEN for Academic Research; Okazaki Kazuo Foundation of KONAN GAKUEN for Advanced Scientific Research; Chubei Itoh Foundation. Funding for open access charge: Grants-in-Aid for Scientific Research from a Ministry of Education,Culture, Sports, Science and Technology (MEXT).
Conflict of interest statement. None declared.
REFERENCES
- 1.Mammen M., Choi S.-K., Whitesides G.M. Polyvalent interactions in biological systems: implications for design and use of multivalent ligands and inhibitors. Angew. Chem. Int. Ed. Engl. 1998;37:2754–2794. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 2.Di Cera E., Dang Q., Ayala Y. Molecular mechanisms of thrombin function. Cell. Mol. Life Sci. 1997;53:701–730. doi: 10.1007/s000180050091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Crawley J., Zanardelli S., Chion C., Lane D. The central role of thrombin in hemostasis. J. Thromb. Haemost. 2007;5:95–101. doi: 10.1111/j.1538-7836.2007.02500.x. [DOI] [PubMed] [Google Scholar]
- 4.Di Cera E. Thrombin as procoagulant and anticoagulant. J. Thromb. Haemost. 2007;5:196–202. doi: 10.1111/j.1538-7836.2007.02485.x. [DOI] [PubMed] [Google Scholar]
- 5.Di Cera E., Gruber A. Thrombin: Structure, Functions, and Regulation. In: Maragoudakis EM, Tsopanoglou EN, editors. Thrombin: Physiology and Disease. NY: Springer; 2009. pp. 1–18. [Google Scholar]
- 6.Adams T.E., Huntington J.A. Thrombin-cofactor interactions: structural insights into regulatory mechanisms. Arterioscler. Thromb. Vasc. Biol. 2006;26:1738–1745. doi: 10.1161/01.ATV.0000228844.65168.d1. [DOI] [PubMed] [Google Scholar]
- 7.Macaya R.F., Schultze P., Smith F.W., Roe J.A., Feigon J. Thrombin-binding DNA aptamer forms a unimolecular quadruplex structure in Solution. Proc. Natl. Acad. Sci. U.S.A. 1993;90:3745–3749. doi: 10.1073/pnas.90.8.3745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Paborsky L., McCurdy S.N., Griffin L.C., Toole J.J., Leung L. The single-stranded DNA aptamer-binding site of human thrombin. J. Biol. Chem. 1993;268:20808–20811. [PubMed] [Google Scholar]
- 9.Nimjee S.M., Rusconi C.P., Sullenger B.A. Aptamers: an emerging class of therapeutics. Annu. Rev. Med. 2005;56:555–583. doi: 10.1146/annurev.med.56.062904.144915. [DOI] [PubMed] [Google Scholar]
- 10.Huang R.-H., Fremont D.H., Diener J.L., Schaub R.G., Sadler J.E. A structural explanation for the antithrombotic activity of ARC1172, a DNA aptamer that binds von Willebrand factor domain A1. Structure. 2009;17:1476–1484. doi: 10.1016/j.str.2009.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davies D.R., Gelinas A.D., Zhang C., Rohloff J.C., Carter J.D., O'Connell D., Waugh S.M., Wolk S.K., Mayfield W.S., Burgin A.B., et al. Unique motifs and hydrophobic interactions shape the binding of modified DNA ligands to protein targets. Proc. Natl. Acad. Sci. U.S.A. 2012;109:19971–19976. doi: 10.1073/pnas.1213933109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheung Y.-W., Kwok J., Law A.W.L., Watt R.M., Kotaka M., Tanner J.A. Structural basis for discriminatory recognition of Plasmodium lactate dehydrogenase by a DNA aptamer. Proc. Natl. Acad. Sci. U.S.A. 2013;110:15967–15972. doi: 10.1073/pnas.1309538110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gelinas A.D., Davies D.R., Edwards T.E., Rohloff J.C., Carter J.D., Zhang C., Gupta S., Ishikawa Y., Hirota M., Nakaishi Y., et al. Crystal structure of interleukin-6 in complex with a modified nucleic acid ligand. J. Biol. Chem. 2014;289:8720–8734. doi: 10.1074/jbc.M113.532697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jarvis Thale C., Davies Douglas R., Hisaminato A., Resnicow Daniel I., Gupta S., Waugh Sheela M., Nagabukuro A., Wadatsu T., Hishigaki H., Gawande B., et al. Non-helical DNA triplex forms a unique aptamer scaffold for high affinity recognition of nerve growth factor. Structure. 2015;23:1293–1304. doi: 10.1016/j.str.2015.03.027. [DOI] [PubMed] [Google Scholar]
- 15.Yatime L., Maasch C., Hoehlig K., Klussmann S., Andersen G.R., Vater A. Structural basis for the targeting of complement anaphylatoxin C5a using a mixed L-RNA/L-DNA aptamer. Nat. Commun. 2015;6:6481. doi: 10.1038/ncomms7481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Das K., Balzarini J., Miller M.T., Maguire A.R., DeStefano J.J., Arnold E. Conformational states of HIV-1 reverse transcriptase for nucleotide incorporation vs pyrophosphorolysis—binding of foscarnet. ACS Chem. Biol. 2016;11:2158–2164. doi: 10.1021/acschembio.6b00187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kato K., Ikeda H., Miyakawa S., Futakawa S., Nonaka Y., Fujiwara M., Okudaira S., Kano K., Aoki J., Morita J., et al. Structural basis for specific inhibition of Autotaxin by a DNA aptamer. Nat. Struct. Mol. Biol. 2016;23:395–401. doi: 10.1038/nsmb.3200. [DOI] [PubMed] [Google Scholar]
- 18.Miller M.T., Tuske S., Das K., DeStefano J.J., Arnold E. Structure of HIV-1 reverse transcriptase bound to a novel 38-mer hairpin template-primer DNA aptamer. Protein Sci. 2016;25:46–55. doi: 10.1002/pro.2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mullins N.D., Maguire N.M., Ford A., Das K., Arnold E., Balzarini J., Maguire A.R. Exploring the role of the [small alpha]-carboxyphosphonate moiety in the HIV-RT activity of [small alpha]-carboxy nucleoside phosphonates. Org. Biomol. Chem. 2016;14:2454–2465. doi: 10.1039/c5ob02507a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tasset D.M., Kubik M.F., Steiner W. Oligonucleotide inhibitors of human thrombin that bind distinct epitopes. J. Mol. Biol. 1997;272:688–698. doi: 10.1006/jmbi.1997.1275. [DOI] [PubMed] [Google Scholar]
- 21.Trapaidze A., Bancaud A., Brut M. Binding modes of thrombin binding aptamers investigated by simulations and experiments. Appl. Phys. Lett. 2015;106:043702. [Google Scholar]
- 22.Trapaidze A., Hérault J.-P., Herbert J.-M., Bancaud A., Gué A.-M. Investigation of the selectivity of thrombin-binding aptamers for thrombin titration in murine plasma. Biosens. Bioelectron. 2016;78:58–66. doi: 10.1016/j.bios.2015.11.017. [DOI] [PubMed] [Google Scholar]
- 23.Russo Krauss I., Merlino A., Randazzo A., Mazzarella L., Sica F. Crystallization and preliminary X-ray analysis of the complex of human α-thrombin with a modified thrombin-binding aptamer. Acta Cryst. F. 2010;66:961–963. doi: 10.1107/S1744309110024632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Russo Krauss I., Merlino A., Giancola C., Randazzo A., Mazzarella L., Sica F. Thrombin-aptamer recognition: a revealed ambiguity. Nucleic Acids Res. 2011;39:7858–7867. doi: 10.1093/nar/gkr522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Russo Krauss I., Merlino A., Randazzo A., Novellino E., Mazzarella L., Sica F. High-resolution structures of two complexes between thrombin and thrombin-binding aptamer shed light on the role of cations in the aptamer inhibitory activity. Nucleic Acids Res. 2012;40:8119–8128. doi: 10.1093/nar/gks512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pica A., Russo Krauss I., Merlino A., Nagatoishi S., Sugimoto N., Sica F. Dissecting the contribution of thrombin exosite I in the recognition of thrombin binding aptamer. FEBS J. 2013;280:6581–6588. doi: 10.1111/febs.12561. [DOI] [PubMed] [Google Scholar]
- 27.Russo Krauss I., Pica A., Merlino A., Mazzarella L., Sica F. Duplex–quadruplex motifs in a peculiar structural organization cooperatively contribute to thrombin binding of a DNA aptamer. Acta Cryst. D. 2013;69:2403–2411. doi: 10.1107/S0907444913022269. [DOI] [PubMed] [Google Scholar]
- 28.Müller J., Wulffen B., Pötzsch B., Mayer G. Multidomain targeting generates a high-affinity thrombin-inhibiting bivalent aptamer. Chembiochem. 2007;8:2223–2226. doi: 10.1002/cbic.200700535. [DOI] [PubMed] [Google Scholar]
- 29.Hasegawa H., Taira K.-I., Sode K., Ikebukuro K. Improvement of aptamer affinity by dimerization. Sensors. 2008;8:1090–1098. doi: 10.3390/s8021090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Müller J., Freitag D., Mayer G., Pötzsch B. Anticoagulant characteristics of HD1-22, a bivalent aptamer that specifically inhibits thrombin and prothrombinase. J. Thromb. Haemost. 2008;6:2105–2112. doi: 10.1111/j.1538-7836.2008.03162.x. [DOI] [PubMed] [Google Scholar]
- 31.Rinker S., Ke Y., Liu Y., Chhabra R., Yan H. Self-assembled DNA nanostructures for distance-dependent multivalent ligand–protein binding. Nat. Nanotechnol. 2008;3:418–422. doi: 10.1038/nnano.2008.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tian L., Heyduk T. Bivalent ligands with long nanometer-scale flexible linkers†. Biochemistry. 2008;48:264–275. doi: 10.1021/bi801630b. [DOI] [PubMed] [Google Scholar]
- 33.Hansen M.N., Zhang A.M., Rangnekar A., Bompiani K.M., Carter J.D., Gothelf K.V., LaBean T.H. Weave tile architecture construction strategy for DNA nanotechnology. J. Am. Chem. Soc. 2010;132:14481–14486. doi: 10.1021/ja104456p. [DOI] [PubMed] [Google Scholar]
- 34.Rakhmetova S.Y., Radko S., Gnedenko O., Bodoev N., Ivanov A., Archakov A. Photoaptameric heterodimeric constructs as a new approach to enhance the efficiency of formation of photocrosslinks with a target protein. Biochemistry (Moscow) 2010;4:68–74. doi: 10.18097/pbmc20105601072. [DOI] [PubMed] [Google Scholar]
- 35.Müller J., Becher T., Braunstein J., Berdel P., Gravius S., Rohrbach F., Oldenburg J., Mayer G., Pötzsch B. Profiling of active thrombin in human blood by supramolecular complexes. Angew. Chem. Int. Ed. Engl. 2011;50:6075–6078. doi: 10.1002/anie.201007032. [DOI] [PubMed] [Google Scholar]
- 36.Ahmad K.M., Xiao Y., Soh H.T. Selection is more intelligent than design: improving the affinity of a bivalent ligand through directed evolution. Nucleic Acids Res. 2012;40:11777–11783. doi: 10.1093/nar/gks899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rangnekar A., Zhang A.M., Li S.S., Bompiani K.M., Hansen M.N., Gothelf K.V., Sullenger B.A., LaBean T.H. Increased anticoagulant activity of thrombin-binding DNA aptamers by nanoscale organization on DNA nanostructures. Nanomed. Nanotechnol. 2012;8:673–681. doi: 10.1016/j.nano.2011.08.011. [DOI] [PubMed] [Google Scholar]
- 38.Rangnekar A., Nash J.A., Goodfred B., Yingling Y.G., LaBean T.H. Design of potent and controllable anticoagulants using DNA aptamers and nanostructures. Molecules. 2016;21:202. doi: 10.3390/molecules21020202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Russo Krauss I., Spiridonova V., Pica A., Napolitano V., Sica F. Different duplex/quadruplex junctions determine the properties of anti-thrombin aptamers with mixed folding. Nucleic Acids Res. 2015;44:983–991. doi: 10.1093/nar/gkv1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fredenburgh J.C., Stafford A.R., Weitz J.I. Evidence for allosteric linkage between exosites 1 and 2 of thrombin. J. Biol. Chem. 1997;272:25493–25499. doi: 10.1074/jbc.272.41.25493. [DOI] [PubMed] [Google Scholar]
- 41.Baglin T.P., Carrell R.W., Church F.C., Esmon C.T., Huntington J.A. Crystal structures of native and thrombin-complexed heparin cofactor II reveal a multistep allosteric mechanism. Proc. Natl. Acad. Sci. U.S.A. 2002;99:11079–11084. doi: 10.1073/pnas.162232399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Verhamme I.M., Olson S.T., Tollefsen D.M., Bock P.E. Binding of exosite ligands to human thrombin—re-evaluation of allosteric linkage between thrombin exosite I and II. J. Biol. Chem. 2002;277:6788–6798. doi: 10.1074/jbc.M110257200. [DOI] [PubMed] [Google Scholar]
- 43.Nimjee S.M., Oney S., Volovyk Z., Bompiani K.M., Long S.B., Hoffman M., Sullenger B.A. Synergistic effect of aptamers that inhibit exosites 1 and 2 on thrombin. RNA. 2009;15:2105–2111. doi: 10.1261/rna.1240109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nagatoishi S., Sugimoto N. Interaction of water with the G-quadruplex loop contributes to the binding energy of G-quadruplex to protein. Mol. Biosyst. 2012;8:2766–2770. doi: 10.1039/c2mb25234a. [DOI] [PubMed] [Google Scholar]
- 45.Otwinowski Z., Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 46.McCoy A.J., Grosse-Kunstleve R.W., Adams P.D., Winn M.D., Storoni L.C., Read R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Adams P.D., Afonine P.V., Bunkoczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W., et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Cryst. D. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vagin A.A., Steiner R.A., Lebedev A.A., Potterton L., McNicholas S., Long F., Murshudov G.N. REFMAC5 dictionary: organization of prior chemical knowledge and guidelines for its use. Acta Cryst. D. 2004;60:2184–2195. doi: 10.1107/S0907444904023510. [DOI] [PubMed] [Google Scholar]
- 49.Emsley P., Lohkamp B., Scott W.G., Cowtan K. Features and development of Coot. Acta Cryst. D. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Davis I.W., Leaver-Fay A., Chen V.B., Block J.N., Kapral G.J., Wang X., Murray L.W., Arendall W.B., Snoeyink J., Richardson J.S. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–W383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Luscombe N.M., Laskowski R.A., Thornton J.M. NUCPLOT: a program to generate schematic diagrams of protein-nucleic acid interactions. Nucleic Acids Res. 1997;25:4940–4945. doi: 10.1093/nar/25.24.4940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Russo Krauss I., Merlino A., Vergara A., Sica F. An overview of biological macromolecule crystallization. Int. J. Mol. Sci. 2013;14:11643–11691. doi: 10.3390/ijms140611643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rooman M., Liévin J., Buisine E., Wintjens R. Cation–π/H-bond stair motifs at protein–DNA interfaces. J. Mol. Biol. 2002;319:67–76. doi: 10.1016/s0022-2836(02)00263-2. [DOI] [PubMed] [Google Scholar]
- 54.Mayer G., Müller J., Mack T., Freitag D.F., Höver T., Pötzsch B., Heckel A. Differential regulation of protein subdomain activity with caged bivalent ligands. Chembiochem. 2009;10:654–657. doi: 10.1002/cbic.200800814. [DOI] [PubMed] [Google Scholar]
- 55.Koeppe J.R., Komives E.A. Amide H/2H exchange reveals a mechanism of thrombin activation. Biochemistry. 2006;45:7724–7732. doi: 10.1021/bi060405h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fuglestad B., Gasper P.M., Tonelli M., McCammon J.A., Markwick P.R., Komives E.A. The dynamic structure of thrombin in solution. Biophys. J. 2012;103:79–88. doi: 10.1016/j.bpj.2012.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nallagatla S.R., Heuberger B., Haque A., Switzer C. Combinatorial synthesis of thrombin-binding aptamers containing iso-guanine. J. Comb. Chem. 2009;11:364–369. doi: 10.1021/cc800178m. [DOI] [PubMed] [Google Scholar]
- 58.Treuheit N.A., Beach M.A., Komives E.A. Thermodynamic compensation upon binding to exosite 1 and the active site of thrombin. Biochemistry. 2011;50:4590–4596. doi: 10.1021/bi2004069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tan X., Dey S.K., Telmer C., Zhang X., Armitage B.A., Bruchez M.P. Aptamers act as activators for the thrombin mediated-hydrolysis of peptide substrates. Chembiochem. 2014;15:205–208. doi: 10.1002/cbic.201300693. [DOI] [PubMed] [Google Scholar]
- 60.Petrera N.S., Stafford A.R., Leslie B.A., Kretz C.A., Fredenburgh J.C., Weitz J.I. Long range communication between exosites 1 and 2 modulates thrombin function. J. Biol. Chem. 2009;284:25620–25629. doi: 10.1074/jbc.M109.000042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lechtenberg B.C., Johnson D.J.D., Freund S.M.V., Huntington J.A. NMR resonance assignments of thrombin reveal the conformational and dynamic effects of ligation. Proc. Natl. Acad. Sci. U.S.A. 2010;107:14087–14092. doi: 10.1073/pnas.1005255107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Malovichko M.V., Sabo T.M., Maurer M.C. Ligand binding to anion-binding exosites regulates conformational properties of thrombin. J. Biol. Chem. 2013;288:8667–8678. doi: 10.1074/jbc.M112.410829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Koeppe J.R., Seitova A., Mather T., Komives E.A. Thrombomodulin tightens the thrombin active site loops to promote protein C activation. Biochemistry. 2005;44:14784–14791. doi: 10.1021/bi0510577. [DOI] [PubMed] [Google Scholar]
- 64.Fuglestad B., Gasper P.M., McCammon J.A., Markwick P.R.L., Komives E.A. Correlated motions and residual frustration in thrombin. J. Phys. Chem. B. 2013;117:12857–12863. doi: 10.1021/jp402107u. [DOI] [PMC free article] [PubMed] [Google Scholar]