


Abstract
CRISPR–Cas system epitomizes prokaryote-specific quintessential adaptive defense machinery that limits the genome invasion of mobile genetic elements. It confers adaptive immunity to bacteria by capturing a protospacer fragment from invading foreign DNA, which is later inserted into the leader proximal end of CRIPSR array and serves as immunological memory to recognize recurrent invasions. The universally conserved Cas1 and Cas2 form an integration complex that is known to mediate the protospacer invasion into the CRISPR array. However, the mechanism by which this protospacer fragment gets integrated in a directional fashion into the leader proximal end is elusive. Here, we employ CRISPR/dCas9 mediated immunoprecipitation and genetic analysis to identify Integration Host Factor (IHF) as an indispensable accessory factor for spacer acquisition in Escherichia coli. Further, we show that the leader region abutting the first CRISPR repeat localizes IHF and Cas1–2 complex. IHF binding to the leader region induces bending by about 120° that in turn engenders the regeneration of the cognate binding site for protospacer bound Cas1–2 complex and brings it in proximity with the first CRISPR repeat. This appears to guide Cas1–2 complex to orient the protospacer invasion towards the leader-repeat junction thus driving the integration in a polarized fashion.
INTRODUCTION
Archaea and Bacteria defend themselves from the assault of phages and plasmids by employing CRISPR–Cas adaptive immune system (1–5). CRISPR constitutes an array of direct repeats (each of ∼30–40 bp) that are intervened by similarly sized variable spacer sequences. The spacer sequences are captured from the invading foreign DNA and they serve as immunological memory—akin to antibodies in higher organisms—to mount retaliation during recurrent infection (6,7). Several studies in the recent past revealed that CRISPR interference proceeds via three stages: (i) adaptation, (ii) maturation and (iii) interference. Immunological memory is generated during adaptation wherein short stretches of DNA from invaders (protospacer) is acquired and incorporated into the CRISPR locus. This is followed by the transcription and processing of the pre-CRISPR RNA transcript that generates the mature CRISPR RNA (crRNA) onto which several Cas proteins assemble to form a ribonucleoprotein (RNP) surveillance complex. The crRNA within the RNP guides the target recognition by base complementarity whereas the protein components facilitate the cleavage of the target DNA (2–5,8).
While the adaptation constitutes the cornerstone of CRISPR–Cas system by expanding the immunological memory, it is also the less well-understood process than the other two (8,9). The adaptation process can be envisaged to encompass two subsets of events: the uptake of protospacer fragments from the foreign DNA and their subsequent insertion into the CRISPR array. The generation of protospacer fragments from the foreign DNA in Escherichia coli (Type I-E) involves the RecBCD nuclease activity (10); however, it appears that only those fragments of about 33 bp DNA that border the protospacer adjacent motif (PAM) are captured and integrated into the CRISPR array (11–15). In Type-I, Type-II and Type-V CRISPR–Cas systems (4), the PAM comprises of short stretch (2–5 nucleotides) of conserved sequence present on either upstream or downstream of acquired protospacer element (9,13,16–19). This sequence is varied among different species and assist in discriminating self- versus non-self during interference step. Point mutations in PAM and protospacer of invading nucleic acid elements lead to imperfect pairing and abrogate target cleavage by interference complex. This mismatched priming leads to acquisition of new spacers more rapidly and efficiently from the mutated invader by a process termed as ‘primed acquisition’. This feedback loop mechanism in addition to naïve adaptation (or non-primed adaptation) effectively aids the bacteria to counter mutated phages (9,11,19,20).
Two of the highly conserved Cas proteins, Cas1 and Cas2 form a complex (Cas1–2 complex) that captures the protospacer element and promotes its insertion into the CRISPR array. Here, Cas1 is shown to function like an integrase and Cas2 provides a structural scaffold that stimulates the catalytic activity of Cas1 (21–23). This complex structure acts as a molecular ruler that appears to determine the length of the acquired protospacer element (21,23). Nucleophilic attack mediated by free 3′-OH ends of protospacers integrates them into the repeat-spacer array (21,22,24). In Type I-E, in order to mediate naïve adaptation, Cas1–2 complex alone is sufficient whereas the active interference complex is indispensable for primed acquisition (11,13). On the contrary, Type-IB, Type-IF and Type-II systems require all the Cas proteins (including maturation and interference proteins) for the incorporation of new spacer in vivo (25–28). In addition to the involvement of Cas proteins, recent studies have highlighted the importance of host-encoded proteins in CRISPR immunity. A nucleoid protein H-NS was shown to control CRISPR immunity by regulating Cas operon expression (29). Recent study also demonstrated the requirement of genome stability proteins like RecG helicase and PriA in E. coli primed acquisition (30). Physical and genetic interaction studies performed on E. coli Cas1 revealed its interaction with various DNA repair pathway proteins, viz., RuvB, RecB, RecC and others (31).
While Cas1–2 complex seems to be essential, it is not sufficient for the spacer uptake in vivo. Sequences upstream of the first CRISPR repeat (referred as leader) are shown to harbor DNA elements critical for adaptation process (13,32,33). Despite the presence of several repeat-spacer units in the CRISPR array, the site of integration of new protospacer has always been at the leader-repeat junction resulting in the integration of the protospacer and concomitant duplication of the first repeat (11–15). This polarization preserves the chronology of the integration events such that the newest protospacer is closer to the leader proximal end and the oldest protospacer at the distal end. Intriguingly, while Cas1–2 complex alone is sufficient for the integration of protospacer elements in E. coli and shown to have intrinsic sequence specificity in vitro (24), it lacks the homing site specificity towards the leader-repeat junction leading to integration at all CRISPR repeats (22). This hints at the involvement of accessory factors that bring in specificity towards the integration site for the invading protospacers. Recently, Integration Host Factor (IHF) was shown to act as an essential accessory factor that determines the specificity of protospacer acquisition in E. coli (34). Here, based on CRISPR/dCas9 mediated immunoprecipitation (35) and biochemical analysis, we were independently led to identify IHF as an essential factor in protospacer acquisition. We further show that the leader region harbours binding site for IHF and that it participates in the protospacer acquisition by bending the leader region by about 120° to produce a reversal in the direction of DNA. This brings the Cas1–2 complex, which is also localized adjacent to IHF, into proximity with the first CRIPSR repeat favouring the nucleophilic attack of the invading protospacer on the leader-repeat junction.
MATERIALS AND METHODS
Construction of bacterial strains and plasmids
Descriptions of the strains, plasmids and oligonucleotides are listed in supplementary Tables S2–S4, respectively. Escherichia coli IYB5101 (referred as Wt) (13) was used as parental strain for all the genomic manipulations, unless specified otherwise. Knock-out strains of ihfα (ΔIHFα) and ihfβ (ΔIHFβ) were created using λ Red recombineering (36). Keio collection strains (37) carrying deletions of ihfα and ihfβ were used as templates for amplification of kanamycin resistant cassettes along with 100–130 bp flanking sequence. Amplified cassettes were used to transform λ Red recombinase expressing E. coli IYB5101 to create ΔIHFα and ΔIHFβ strains.
pdCas9-bacteria (38) was modified with the construct encoding 3XFLAG-dCas9-StrepII. Overlap extension PCR was used to generate a 166 bp DNA fragment encoding a gRNA complementary to a region that is 86 bp upstream of first CRISPR repeat in E. coli BL21-AI (NCBI accession: NC_012947.1, nucleotide positions: 1002800–1003800). This region was inserted in between SpeI and HindIII sites of the pgRNA-bacteria (38) to create the pgRNA-leader.
pCSIR-T (14) was used as template to amplify Wt array, IHF binding site mutants (IBS and ΔIBS) as well as Cas binding site variants of the leader regions denoted as CBS1 (−34 to −45 nt), CBS2 (−46 to −57 nt), CBS3 (−58 to −69 nt), CBS2(L) (−54 to −57 nt), CBS2(C) (−50 to −53 nt) and CBS2(R) (−46 to −49 nt)—the nucleotide positions are from the leader-repeat junction. All the amplified Wt and mutant arrays were individually inserted in between KpnI/PstI sites in pOSIP-CT (39) and subsequently integrated into Phi 21 (P21) locus of E. coli IYB5101 strain by a one step process of cloning and integration into attB locus termed as ‘clonetegration’.
To generate pBend-Wt and pBend-CBS2, 81 bp complementary oligos (encompassing 69 bp of leader sequence) corresponding to Wt and CBS2 leader sequences were annealed and end filled by PCR, phosphorylated using T4 polynucleotide kinase and inserted into pBend5 using HpaI site (40).
Escherichia coli K-12 MG1655 genomic DNA was used as template to amplify genes encoding IHFα, IHFβ, Cas1 and Cas2. To generate p8R-IHFαβ and p1R-IHFαβ, a bicistronic cassette encoding IHFα and IHFβ was amplified and inserted into p8R and p1R using SspI site. Whereas p13SR-Cas1 and p1S-Cas2 were generated by inserting the region encoding Cas1 and Cas2 into p13SR and p1S, respectively, using SspI site. All constructs were verified by sequencing.
Expression and purification of proteins
Escherichia coli BL21(DE3) harbouring p1R-IHFαβ was grown in terrific broth supplemented with 100 μg/ml kanamycin at 37°C till the OD600 reaches 0.6. At this point, IHF expression was induced with addition of 0.5 mM IPTG and the cells were allowed to grow for 4 h at 37°C. Thereafter, the cells were harvested and resuspended in IHF binding buffer (20 mM Tris–Cl pH 8, 150 mM NaCl, 10% glycerol, 1 mM PMSF and 6 mM β-mercaptoethanol). The cells were then subjected to lysis by sonication and clarified soluble extract was loaded on to 5 ml StrepTrap HP column (GE Healthcare). After loading, column was washed with IHF binding buffer and proteins were eluted with IHF binding buffer containing 2.5 mM d-desthiobiotin (Sigma). Eluted protein fractions were pooled up and loaded on to 5 ml HiTrap Heparin HP column (GE Healthcare). The column was washed with IHF binding buffer and bound proteins were eluted with a linear gradient of 0.15 – 2 M NaCl in IHF binding buffer. Purified fractions were pooled and dialyzed against IHF binding buffer. Dialyzed protein was concentrated, flash frozen and stored at −80°C until required.
In order to express Cas1, E. coli BL21(DE3) harbouring p13SR-Cas1 was grown until OD600 = 0.6 at 37°C in auto-induction media supplemented with 100 μg/ml spectinomycin. Thereafter, growth and induction were continued for 16 h more at 16°C. Subsequently, cells were harvested and resuspended in Buffer 1A (20 mM HEPES–NaOH pH 7.4, 500 mM KCl, 10% glycerol, 1 mM PMSF and 1 mM DTT) and lysed by sonication. The clarified soluble cell extract was loaded on to 5ml StrepTrap HP column, which was then washed with Buffer 1A. Proteins were eluted with Buffer 1A containing 2.5 mM d-desthiobiotin. Eluted protein fractions were dialyzed against Buffer 1B (20 mM HEPES–NaOH pH 7.4, 50 mM KCl, 10% glycerol and 1 mM DTT) and loaded onto 5 ml HiTrap Heparin HP column. Protein loaded columns were washed with Buffer 1B and bound proteins were eluted with a linear gradient of 0.05–2 M KCl in Buffer 1B. Purified fractions were pooled up and dialyzed against buffer containing 20 mM HEPES–NaOH pH 7.4, 150 mM KCl, 10% glycerol and 1 mM DTT. Dialyzed protein was concentrated, snap frozen and stored at −80°C until required.
For purification of C-terminal Strep-II tagged Cas2, E. coli BL21(DE3) harbouring p1S-Cas2 was grown until OD600 = 0.6 in auto-induction media supplemented with 100 μg/ml kanamycin at 37°C. Thereafter, growth and induction were continued for 16 h more at 16°C. Subsequently, the cells were harvested and resuspended in Buffer 2A (20 mM HEPES–NaOH pH 7.4, 500 mM KCl, 10% glycerol, 10 mM Imidazole, 1 mM PMSF and 1 mM DTT) and lysed by sonication. The clarified soluble extract was loaded onto 5 ml HiTrap IMAC HP column (GE Healthcare). After loading, column was washed with Buffer 2A and proteins were eluted using a linear gradient of imidazole (0.01–0.5 M) in Buffer 2A. Purified fractions were pooled up and mixed with TEV protease (in 10:1 ratio of His-SUMO-Cas2-strep: TEV) and incubation was continued during dialysis with Buffer 2A at 4°C overnight. Dialyzed protein mixture was loaded onto 5 ml HiTrap IMAC HP column 5× times to allow binding of His-tagged SUMO-Cas2-Strep, SUMO and TEV protease. Subsequently, a 5 ml StrepTrap HP column was connected tandemly and protein mixture was allowed to pass 5× more times. The C-terminal strep-tagged Cas2 was later eluted with Buffer 2B (20 mM HEPES–NaOH pH 7.4, 500 mM KCl, 10% glycerol, 2.5 mM d-desthiobiotin and 1 mM DTT). Eluted fractions were pooled up and dialyzed against buffer containing 20 mM HEPES–NaOH pH 7.4, 150 mM KCl, 10% glycerol and 1 mM DTT. Dialyzed protein was concentrated, snap frozen and stored at −80°C until required.
CRISPR/dCas9 mediated immunoprecipitation
Escherichia coli BL21-AI was transformed with p3XF-dCas9, pgRNA-leader and pCas1–2[K] (14) and was allowed to grow in a shaker operated at 180 rpm till OD600 = 0.6 at 37°C in LB media supplemented with 0.2% L-arabinose, 0.1 mM IPTG, 25 μg/ml chloramphenicol, 100 μg/ml ampicillin and 50 μg/ml spectinomycin. 100 ng/ml anhydrotetracycline was added to induce the expression of 3× FLAG-tagged dCas9 and growth was continued for four more hours to allow dCas9–gRNA complex to anchor on its target site, i.e. upstream region of CRISPR leader. Chemical crosslinking and cell lysis were performed as described previously (41) with few modifications. Formaldehyde was added to a final concentration of 1% to crosslink proximally interacting nucleic acids and proteins. Crosslinking was continued for 20 min at 25°C with gentle rocking. Glycine was added to a final concentration of 0.5 M and incubation was continued for 5 min at 25°C to quench the crosslinking reaction. 10 ml cells were centrifuged at 2500 g at 4°C for 5 min and the pellet was washed twice with equal volume of buffer W (20 mM Tris–Cl pH 7.5 and 150 mM NaCl). Pelleted cells were resuspended in 1ml buffer L (10 mM Tris–Cl pH 8.0, 20% sucrose, 50 mM NaCl, 10 mM EDTA, 10 mg/ml lysozyme) and incubated at 37°C for 30 min. Lysate was resuspended in 4 ml of buffer R (50 mM HEPES–KOH pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% Triton-X 100, 0.1% sodium deoxycholate, 1 mM PMSF and 0.1% SDS). The cells were subjected to sonication for four rounds of 15 × 1 s pulses with 2 min pause between each round in Vibra-cell probe sonicator that was set at 33% amplitude. Clarified supernatant containing sheared DNA-protein complex was separated by centrifugation. 800 μl of supernatant was mixed with 200 μl of protein G dyna beads (Life technologies) conjugated with 20 μg of anti-FLAG M2 antibody (Sigma) and rocked gently at 4°C overnight. Incubated beads were separated by centrifugation and washed twice each with 1 ml of Low Salt Wash Buffer (20 mM Tris–Cl pH 8.0, 150 mM NaCl, 2 mM EDTA, 1% TritonX-100, 0.1% SDS), High Salt Wash Buffer (20 mM Tris–Cl pH 8.0, 500 mM NaCl, 2 mM EDTA, 1% TritonX-100, 0.1% SDS), LiCl Wash Buffer (10 mM Tris–Cl pH 8.0, 250 mM LiCl, 1 mM EDTA, 0.5% Nonidet P-40 (NP-40), 0.5% sodium deoxycholate), and TBS Buffer (50 mM Tris, pH 7.5, 150 mM NaCl) with 0.1% NP-40 as described previously (35). In the final step, beads were separated by centrifugation and resuspended in 100 μl of buffer containing 20 mM Tris–Cl pH 8 and 150 mM NaCl. 30 μl of resuspended beads were mixed with 10 μl of 4× SDS sample buffer and heated at 95°C for 30 min to reverse crosslink and denature the proteins. Heated mixture was loaded on to SDS-PAGE and electrophoresed to enter stacking gel. The part of stacking gel containing the proteins was sliced and analyzed by mass spectrometry for the identification of protein factors in the sample.
Spacer acquisition assays
In vivo acquisition assays were performed as described earlier (13). Briefly, three cycles of growth and induction was performed with E. coli IYB5101 (Wt) or its variants carrying pCas1–2[K] (14) at 37°C for 16 h in LB media supplemented with 50 μg/ml spectinomycin, 0.2% l-arabinose and 0.1 mM IPTG. In between each cycle, cultures were diluted to 1:300 times with fresh LB media containing aforementioned supplements and growth was continued for 16 h. For IHF complementation experiments, ΔIHFα and ΔIHFβ strains were transformed with p8R-IHFαβ and pCas1–2[K] and 3 cycles of inductions were performed as discussed above. To monitor CRISPR array expansion, 200 μl of induced cells were collected after cycle 3 and washed thrice and resuspended in distilled water. These cells were used as template for PCR to monitor CRISPR array expansion either in CRISPR 2.1 array (in case of Wt, ΔIHFα and ΔIHFβ strains) or P21 locus integrated CRISPR DNA (in case of Wt, IBS, ΔIBS, CBS1-3, CBS2(L), CBS2(C) and CBS2(R) strains). All the PCR amplified samples were separated on 1.5% agarose gels to identify parental and expanded arrays (parental array + 61 bp).
Electrophoretic mobility shift assays
Wt or mutant leader DNA (IBS, ΔIBS and CBS1-3) was PCR amplified from the strain carrying the respective construct that is integrated into P21 locus. 14 nM of amplified DNA was incubated with increasing concentration of purified IHF (0, 0.2, 0.3, 0.4, 0.5, 0.6, 0.8, 1.0, 1.2, 1.4 and 1.6 μM) in buffer containing 0.5X TBE (50 mM Tris–Cl pH 8.3, 50 mM boric acid and 1mM EDTA), 100 mM KCl, 10% glycerol and 5 μg/ml BSA for 30 min at 25°C. Post-incubation samples were directly loaded on 8% native acrylamide gel and electrophoresed in 1X TBE at 4°C. Gels were post-stained with ethidium bromide (EtBr) and DNA bands were visualized in gel documentation system (Bio-Rad).
FRET-based monitoring of DNA bending
A 35 bp DNA encompassing leader sequence (−4 to −38 from the leader-repeat junction) of Wt (or Wt without quencher or ΔIBS) was assembled from three oligos by annealing and end labeled with 3′,6-FAM and 5′ Iowa Black (IDT). 222 nM of DNA was incubated with increasing concentrations of purified IHF (0, 0.3, 0.6, 0.9, 1.2, 1.5 and 1.8 μM) in buffer containing 0.5× TBE, 100 mM KCl, 10% glycerol and 5 μg/ml BSA for 20 min at 25°C. Post-incubation samples were excited at 495 nm and emission was monitored from 500 to 600 nm, with averaging over three scans in FluoroMax-4 spectrofluorometer (Horiba Scientific, Edison, NJ, USA). The slit width used for excitation and emission was 2 and 7 nm, respectively. In order to further ascertain that the enhanced quenching is due to IHF mediated DNA bending; a fluorescence recovery assay was designed. In this assay, buffer (0.5× TBE, 100 mM KCl, 10% glycerol and 5 μg/ml BSA) containing 222 nM DNA was excited at 495 nm and emission was captured for 200 s at 520 nm. To this sample, IHF was added to a final concentration of 1.8 μM and fluorescence emission was recorded till 600 s. Thereafter, IHF degradation and DNA release were initiated by addition of proteinase K to a final concentration of 1 mg/ml and emission was monitored for another 400 s. After background correction, the fluorescence intensity of DNA in the presence of IHF is normalized relative to that of DNA alone.
Estimation of bending angles by circular permutation gel retardation assay
pBend-Wt and pBend-CBS2 were digested with HindIII and EcoRI to produce a 329 bp DNA fragment. This fragment was gel purified as per the manufacturer's instruction (Qiagen) and digested with BamHI, KpnI, SspI, EcoRV, SpeI, BglII and MluI in separate reactions. All the digested DNA samples were further purified (Qiagen) and 21 nM of each DNA was incubated individually with 0.7 μM IHF in buffer containing 0.5× TBE, 100 mM KCl, 10% glycerol and 5 μg/ml BSA for 30 min at 25°C. Post-reaction samples were directly loaded on 8% native acrylamide gel and electrophoresed in 1× TBE at 4°C. Gels were post-stained with EtBr and DNA bands were visualized in gel documentation system. IHF bending angles were calculated as described previously (42). Mobilities of IHF bound DNA complex (Rb) and the respective free DNA (Rf) were calculated for all the restriction-digested fragments. Rb values were normalized to the respective Rf values and were plotted against flexure displacement (length from the middle of the binding site to the 5′ end of the restriction fragment/total restriction fragment length). The resulting plot was fitted to a quadratic equation: 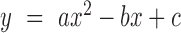 , where
, where 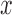 and
and 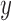 denotes flexure displacement and Rb/Rf, respectively. The bending angle (α) was calculated using the relationship
denotes flexure displacement and Rb/Rf, respectively. The bending angle (α) was calculated using the relationship 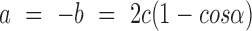 . Here, we have represented the bending angle (α) as the average value that was calculated from the parameters
. Here, we have represented the bending angle (α) as the average value that was calculated from the parameters 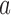 and
and 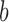 .
.
Circular permutation gel retardation assay in the presence of Cas1–2 was performed as described above with few modifications. 210 nM of Cas1–2 was incubated with 21 nM of digested pBend-Wt fragments in the presence or absence of 0.7 μM IHF in buffer containing 20 mM HEPES–NaOH pH 7.5, 25 mM KCl, 10 mM MgCl2 and 1 mM DTT for 30 min at 25°C. Post-reaction samples were directly loaded on 8% native acrylamide gel and electrophoresed in 1× TBE at 4°C. Gels were post-stained with EtBr and DNA bands were visualized in gel documentation system.
In vitro integration assay
Wt or mutant leader DNA (IBS, ΔIBS and CBS1-3) was PCR amplified from the strain carrying the respective construct that is integrated into P21 locus. The constructs for repeat variants (Wt and Rep1-2) were synthesized (Genscript) and extracted from the plasmid by restriction digestion using the respective restriction sites (EcoRI/BamHI/XhoI). Various types of protospacers (Supplementary Figure S7A) were prepared by annealing the corresponding oligos. In vitro integration assays employing Cas1 and Cas2 were performed as previously described (22) with few modifications. 210 nM of Cas1 and Cas2 were mixed and incubated at 4°C for 15 min. 550 nM of desired protospacer DNA was added to the mixture and incubation at 4°C was continued for another 15 min. To this complex, 21 nM of CRISPR DNA substrate (Wt or Mutant leader or Mutant repeat) was added along with 0.7 μM IHF in duplicates and incubated at 37°C for 60 min in buffer containing 20 mM HEPES–NaOH pH 7.5, 25 mM KCl, 10 mM MgCl2 and 1 mM DTT. First set of reaction mixtures were directly loaded and electrophoresed on 8% native acrylamide gel in 1× TBE at 4°C. Whereas second set of reaction mixtures was treated with 1 mg/ml proteinase K for 30 min at 37°C prior to electrophoresis. Electrophoresed gels were post-stained with EtBr and imaged in gel documentation system.
The sizes of the integrated products were analyzed by denaturing capillary electrophoresis. DNA with 6-FAM labeled on 5′ end of the leader (L*) or on 5′ end of spacer2 (R*) was used as substrate (Supplementary Figure S5). Integration reactions were performed as describe above. Post-reaction samples were treated with 1 mg/ml proteinase K and separated by capillary electrophoresis. The intensities of the fragments were visualized using GeneMapper (Thermo Fisher Scientific) after loading the corresponding .fsa files.
Spacer disintegration assay
The reaction mixture from integration assay was purified using PCR purification kit (Qiagen) as per the manufacturer's instruction. 210 nM of Cas1 and Cas2 were mixed and incubated at 4°C for 15 min. To this complex, 21 nM purified integration product was mixed with or without 0.7 μM IHF and incubated at 37°C for 60 min in buffer containing 20 mM HEPES–NaOH pH 7.5, 25 mM KCl, 10 mM MgCl2 and 1 mM DTT. Subsequently, proteinase K was supplemented to a final concentration of 1 mg/ml concentration and incubated for 30 min at 37°C. Sample was mixed with 6× DNA loading dye and electrophoresed on 8% native acrylamide gel in 1× TBE at 4°C. Electrophoresed gels were post-stained with EtBr and imaged in gel documentation system.
Genome analysis
The lists comprising of type I-E and other type I (excluding type I-E) organisms were compiled from previous study (4). Using IHF-α as a query, we initiated blastp (43) search against the genomes harbouring type I-E and other type I (non-type I-E) CRISPR systems. Hits were considered bonafide if the E-value is less than 0.005 and the alignment coverage with respect to the query is at least 60%. Based on these criteria, we estimated the distribution of IHF across the species. Since HU and IHF are structurally similar and also shares similarity at the sequence level (44), we relied on the annotation to distinguish between the two. The multiple sequence alignment corresponding to the leader region for type I-E CRISPR system was obtained from the CRISPRleader database (45). The conservation profile was generated using WebLogo 3 (46).
RESULTS
CRISPR/dCas9 based immunoprecipitation detects the participation of IHF as accessory factor for adaptation in vivo
In order to identify the potential host factors that are likely to promote the directional insertion of protospacer fragment, we exploited the CRIPSR/dCas9 based immunoprecipitation (35). Here, we expressed the Cas1–2 complex together with the inactive form of FLAG-tagged Cas9 (dCas9) and gRNA that is targeted towards the leader region of the CRISPR array in E. coli BL21-AI (NCBI accession: NC_012947.1, nucleotide positions: 1002800–1003800). Subsequent to the chemical crosslinking, the DNA bound protein factors that are localized into the leader region were selectively pulled down using the anti-FLAG coated beads against the FLAG-tagged dCas9 (Figure 1A and Supplementary Figure S1). The immunoprecipitated protein factors were analyzed using mass spectrometry to identify the associated factors with the DNA. We hypothesized that since the Cas1–2 complex shows integrase-like activity, the presence of host factors that are previously characterized to facilitate the integration of DNA elements would be prospective candidates. Most of the identified factors belong to ribosomal proteins and translational factors – an aspect characteristic of their omnipresence due to their housekeeping functions. Remarkably, a few of the identified factors were mapped to Cas proteins including Cas1 and Cas2 that bolstered the utility of this approach. Among others, we also noted the presence of the DNA architectural proteins such as H-NS and IHF and DNA repair proteins such as RecA (vide. Supplementary Table S1). We filtered out the factors if they were previously shown not to be involved or essential in determining the protospacer integration (10,12,29) or functionally unrelated such as chaperones, proteases, metabolic enzymes, etc. For example, though the DNA architectural protein H-NS was identified with high score than that of Cas1 and Cas2, it was previously shown that it is not essential for CRISPR adaptation and that it acts as a repressor of cas operon in E. coli (12,29). Therefore, we didn't pursue further with H-NS and similar such rationale was exercised to exclude other factors. On the other hand, though another architectural protein IHF scored lower than H-NS, its role in site-specific recombination championed by λ integrase as well as in DNA transposition was well characterized (47,48). Given that Cas1–2 complex functions like an integrase – akin to λ integrase – and since the role of IHF in CRISPR adaptation is not previously characterized, we were tempted to probe the involvement of IHF in protospacer acquisition.
Figure 1.

Identification and characterization of CRISPR associated host factors. (A) CRISPR DNA encompassing leader (green), repeats (R1–R4 as red diamonds), spacers (S1–S3 in blue, magenta and cyan, respectively) and upstream and downstream regions (in pink) is shown. Heterologously expressed Cas1 (in orange circle) and Cas2 (in grey circle) complex bound to the protospacer DNA (in black) is displayed. 3× FLAG tagged dCas9-gRNA (green ovals) complex is shown bound to the upstream region of the leader. During the active spacer acquisition process, Cas proteins and the unknown host factors (in yellow circle) are cross-linked (black dotted lines) to the proximal DNA region by the addition of formaldehyde. Subsequent to the isolation and fragmentation of this cross-linked protein bound DNA, protein bound CRISPR regions were selectively pulled down using anti-FLAG antibody coated beads (in grey circles) against FLAG tagged dCas9. Purified CRISPR DNA-protein complexes were reverse cross-linked and analyzed by mass spectrometry for identification of the unknown host factors. (B) PCR products from spacer acquisition assay performed in E. coli IYB5101 (referred as Wt, lanes 1–2), ihfα knockout (ΔIHFα, lanes 3–4) and ihfβ knockout (ΔIHFβ, lanes 5–6) strains are shown. IHFαβ complementation in ΔIHFα (lanes 7–8) and ΔIHFβ (lanes 9–10) strains was achieved using plasmid p8R-IHFαβ. Absence (–) or presence (+) of plasmids and inducers is indicated on top of each lane. Positions corresponding to parental and expanded arrays are indicated on the left whereas DNA marker (M) positions are displayed on the right. (C) CRISPR locus encompassing leader (black), repeats (R1–R2 as orange diamonds) and spacers (S1–S2 in blue and cyan, respectively) is shown. Numbering on top of the schema represents the position of nucleotides with respect to leader-repeat junction. Putative IHF binding site corresponding to the wild type (Wt), mutated (IBS) and deleted (ΔIBS) region is shown. Conserved motifs were underlined and highlighted in different colours. (D) PCR products from spacer acquisition assay performed in E. coli harbouring the CRISPR locus integrated into the P21 attB site are shown. Wt (lanes 1–2), IBS (lanes 3–4) and ΔIBS (lanes 5–6) are indicated. Absence (–) or presence (+) of plasmids and inducers is shown on top of each lane. Positions corresponding to parental and expanded arrays are indicated on left whereas DNA marker (M) positions are represented on the right.
IHF is essential for protospacer acquisition into the leader proximal end in vivo
IHF is a heterodimer comprising of α and β subunits. To test the involvement of IHF in protospacer acquisition, we created a null mutant of IHF devoid of either α or β subunits in E. coli IYB5101. It was found that deletion of either α or β subunit abrogates the acquisition of protospacer elements (Figure 1B). In order to reinforce this, we complemented the null mutant with plasmid borne IHFα and IHFβ that restored the expansion of CRISPR 2.1 array in E. coli IYB5101 (Figure 1B). This strengthened our conjecture that the acquisition of protospacer requires the participation of IHF in vivo.
Unlike some of the related DNA architectural proteins such as HU, IHF exhibits sequence specific DNA binding. It recognizes the consensus sequence 5′-WATCAANNNNTTR-3′ (where W – A/T, N – A/T/G/C, R – A/G). Therefore, we searched for potential IHF binding site abutting the CRISPR 2.1 locus in E. coli IYB5101 as well as in related strains (Supplementary Figure S2). This search led to the identification of a putative binding site adjacent to the first CRISPR repeat (Figure 1C). We wondered whether this region could act as a potential binding site for IHF. To test this, we deleted the binding site partially (ΔIBS in Figure 1C) and assayed for the acquisition. Interestingly, we found no expansion of the array (Figure 1D). Similarly, mutation of the key binding nucleotides (IBS in Figure 1C) also abolished the acquisition (Figure 1D). This suggests that the identified site for IHF binding indeed impacts the adaptation process and these findings are also in concurrence with the recent report (34).
IHF binding induces bending of the leader region
The structure of IHF–DNA complex shows that the IHF α and β subunits form an intertwined compact body from which two β structures protrude out clamping the DNA (49). This induces bending of DNA by about 160° leading to the reversal of the direction of DNA (Figure 2A). This prompted us to investigate whether IHF binds and bends the putative site (IBS) in the leader region. Towards this, we purified the IHF from E. coli and tested the DNA binding using EMSA (Figure 2B). This showed retardation of DNA mobility in the presence of IHF indicating that indeed IHF binds the leader region (Figure 2B). In line with the recent study (34), substitution or deletion of key binding nucleotides drastically reduced the IHF binding (Supplementary Figure S3).
Figure 2.

IHF bends CRISPR leader. (A) Structure of IHF in complex with λ DNA H’ site (PDB ID: 1IHF) is shown. IHFα and IHFβ are represented in cyan and blue ribbons, respectively, whereas λ DNA H’ strands are shown in red. (B) EMSA was performed by incubating 14 nM DNA with increasing concentration of IHF (0, 0.2, 0.3, 0.4, 0.5, 0.6, 0.8, 1.0, 1.2, 1.4 and 1.6 μM). The corresponding positions of unbound and IHF bound DNA are represented on the left, whereas DNA marker (M) positions are shown on the right. (C) IHF induced DNA bending was monitored using FRET. One end of the DNA (in red) was labeled with 6-FAM (F) and the other end with Iowa Black (Q) or left unlabeled. IHF (in blue and cyan ovals) and proteinase K (in magenta pie) that were added at various time points are indicated. Addition of IHF to DNA leads to quenching of fluorescence, which was later restored by the addition of proteinase K. Solid line indicates intensities corresponding to fluorescent DNA with quencher whereas the dotted line indicates intensities of fluorescent DNA without quencher. (D) Schematic representation of substrates generated for circular permutation gel retardation assay is presented. Seven restriction enzymes (in black) were used to generate equal sized DNA fragments carrying CRISPR leader (in grey) and IHF binding site (in magenta) in different positions. EcoRI and HindIII (in green) were used to excise the cassette from pBend-Wt. (E) The migration of IHF bound DNA is shown. Enzymes labeled on each lane were used to generate the respective restriction DNA fragments. Positions of bent nucleoprotein complexes are shown on the left, whereas DNA marker (M) positions are labeled on the right. (F) Plot of the relative mobilities (Rb/Rf) of restricted fragments against the flexure displacement for the assay performed in (E) is shown. Positions corresponding to Rb/Rf values of the respective restriction fragments are indicated.
Motivated by the IHF binding to the leader region, we wondered whether the binding leads to bending of the DNA. To assess this, we designed a FRET based assay wherein one end of the IHF binding region is tagged with a fluorophore (6-FAM) and the other end with the quencher (Iowa Black). In the linear DNA, the fluorophore and the quencher will be sequestered and hence this won't quench the fluorescence. However, if IHF bends the DNA, this brings both the fluorophore and quencher into proximity leading to quenching of the fluorescence. Indeed, we observed that addition of IHF led to drastic reduction in the fluorescence intensity (Solid line in Figure 2C and Supplementary Figure S4). However, in the same reaction, when a protease was added to remove IHF, it restored the fluorescence (solid line in Figure 2C). On the contrary, similar experiment performed with the 6-FAM labeled DNA, albeit without the quencher, showed that despite addition of IHF the intensity of the fluorescence remained constant (dotted line in Figure 2C). This allows us to exclude the possibility that the quenching of fluorescence is not caused by IHF binding alone and it is indeed the DNA bending effected by the IHF that brings the two ends into proximity leading to quenching of fluorescence.
Having established the fact that IHF indeed bends the leader region, we were interested in investigating the extent to which IHF bends the leader DNA. To address this, we utilized the bending vector pBend5, which contains circularly permuted duplicated restriction sites (40). Cloning of the IHF binding site (IBS) into pBend5 and subsequent digestion using the restriction enzymes ensue fragments with same length but with the binding site distributed to different positions, either in the middle or towards the end (Figure 2D). When the DNA undergoes bending due to protein binding, the fragment that harbors the binding site in the middle migrates slower than the one with the binding site at the end. From this mobility differences, it is possible to estimate the bending angle, which is defined as the angle by which the DNA deviates from the linearity (vide. methods). We estimated that IHF bends DNA by ∼120° suggesting that the sharp deformation could result in the reversal of the DNA direction (Figure 2E and F).
IHF induced bending of the linear DNA facilitates protospacer integration
Since IHF deforms the linear DNA, we were interested in deciphering the mechanism by which it influences the integration of the protospacer into the CRISPR locus. Further, our analysis of CRISPR leader sequences from organisms harbouring type I-E system showed that the identified IHF binding site (–9 to –35 nt; boxed in solid line in Figure 3A) along with another region (–44 to –59 nt; boxed in dotted line in Figure 3A) is highly conserved across other species as well. Therefore, we designed a linear DNA construct encompassing the above mentioned leader region and two units of repeat-spacer segments (Figures 1C and 3A). When IHF was added to the CRISPR DNA, we noted a single slow migrating band suggesting that the IHF induced DNA bending retards the mobility of the CRISPR DNA (lane 4 in Figure 3B). Subsequent addition of Cas1–2 complex and protospacer fragment resulted in the appearance of a super-shifted band (lane 12 in Figure 3B). Strikingly, this band was not observed in the absence of IHF (lane 11 in Figure 3B). This hints that Cas1–2 complex associates with the CRISPR DNA only when IHF is present. When the DNA bound proteins were removed using proteinase K treatment, we spotted a slow migrating band, whose size seemed to be larger than the CRISPR DNA (lane 12 in Figure 3C). Remarkably, this band appeared only from the proteinase K treated reaction mixture consisting of CRISPR DNA, protospacer fragment, IHF and Cas1–2 complex (lane 12 in Figure 3C). To further probe the requirement of IHF for the formation of super-shifted band, we performed the experiment with IHF binding site variants. This showed that either deletion (ΔIBS) or mutation of the IHF binding site (IBS) completely abolished the appearance of super-shifted band (lanes 6 and 9 in Figure 3D and E). This suggests the possibility that the slow migrating band represents the protospacer integrated into the CRISPR DNA.
Figure 3.

IHF binding stimulates Cas1–2 mediated protospacer integration into the linear CRISPR DNA. (A) Multiple sequence alignment of CRISPR leader sequences derived from 17 representative type I-E organisms that belong to F1B1 cluster (45). The annotations mentioned on the left side of the alignment were derived from CRISPRmap (64). Positions with respect to leader-repeat junction are labeled on top of the sequences. Residues in the alignment are highlighted in blue based on the extent of conservation using Jalview (65). Conservation profile depicted in the lower section is generated using WebLogo 3 (46). Two conserved regions in the CRISPR leader are highlighted using dotted and solid boxes (IHF binding site). (B) Native gel depicting spacer integration assay performed with Wt is displayed. Absence (–) or presence (+) of each reaction component is indicated on top of each lane. Positions of super-shifted nucleoprotein complex, IHF-DNA complex, CRISPR DNA substrate and protospacer are indicated on the left, whereas positions of the DNA marker (M) and integration product are shown on the right. (C) Native gel showing proteinase K treated samples from assay in (B) is displayed. (D) Spacer integration assay performed with CRISPR DNA substrates harbouring IHF binding site disrupting mutations (IBS, lanes 4–6) and deletion (ΔIBS, lanes 7–9) is shown. (E) Native gel representing proteinase K treated samples from assay in (D) is presented.
In order to ascertain the protospacer integration, we designed 5′ 6-FAM labeled linear CRISPR DNA constructs and repeated the aforementioned experiment. The proteinase K treated reaction mixture corresponding to the slow migrating band was resolved by denaturing capillary electrophoresis. The fragments were analyzed in comparison with the fluorescently labeled standards to estimate their sizes. This showed that when the label was at the 5′ leader proximal end, the size of the fragment was estimated to be ∼161 and 63 nt, whereas when the label was at the 5′ distal end, the size of the fragment was ∼168 nt (Supplementary Figure S5). Since the 63 nt fragment maps the cleavage around the leader-repeat junction, this suggests that the protospacer integration has taken place proximal to the leader region in the top strand leading to half-site integration intermediate (vide. Supplementary Figure S5). In agreement with recent work (34), this further suggests that IHF indeed stimulates the protospacer incorporation into the linear CRISPR DNA. Further, since it was reported that half-site integration intermediate is selectively excised by the Cas1–2 complex (22,24), we reasoned that this could serve as an additional diagnosis for the existence of half-site integration intermediate. Therefore, we purified the reaction mixture containing the half-site integration intermediate and monitored disintegration in the presence of Cas1–2 complex. Indeed, we observed that the presence of Cas1–2 complex led to the disappearance of the integrated product (Supplementary Figure S6). Interestingly, the disintegration activity of Cas1–2 complex is significantly inhibited in the presence of IHF (Supplementary Figure S6). Taken together, it is possible to reiterate that the protospacer integration occurs into the leader-repeat junction in the top strand. Further, it appears that once the site of invasion is marked, in line with earlier reports (21,22,24), we recognized that it is the 3′ –OH of the protospacer with 3′ overhang that mounts the nucleophilic attack on the CRISPR DNA (Supplementary Figure S7).
Cas1–2 complex is localized upstream of IHF binding site
It was shown that up to 60 bp leader segment adjoining the first CRISPR repeat is essential for spacer acquisition (13). However, the IHF binding region falls within the 35 bp from the first CRISPR repeat (boxed in solid line in Figure 3A). Given the importance of this region, we wondered what the function of the remaining 25 bp could be in the leader region. Intriguingly, we also noted high conservation of sequence upstream to that of IHF binding site (boxed in dotted line in Figure 3A). Therefore, we randomly mutated the 36 bp leader region upstream of the IHF binding site, 12 bp at a time (CBS1–3), and tested whether this modified region could support acquisition (Figure 4A). We observed that though CBS1 [–34 to –45] and CBS3 [–58 to –69] had no effect on the spacer acquisition, surprisingly, no expansion was seen for the CBS2 [–46 to –57] (Figure 4A and B). This led us to wonder whether any of these mutated sequences affects the binding of IHF and thereby leading to the abrogation of acquisition. Hence we tested the binding of IHF to leader region containing CBS1-3. We noted that while CBS1 reduces the IHF binding, perhaps owing to its marginal overlap with the cognate binding site (Figure 4A), the other two showed only minor effect on the IHF binding (Supplementary Figure S9). Interestingly, we also found that the IHF mediated DNA bending is not affected for CBS2 (Supplementary Figure S10). This suggests that the impairment of spacer acquisition due to CBS2 is not effected by IHF.
Figure 4.

Leader region upstream of IHF binding site influences protospacer integration. (A) Linear CRISPR DNA substrate encompassing leader (in black), two repeats (R1–R2 as orange diamonds), 33 bp spacer1 (S1 in blue), 19 bp spacer2 (S2 in cyan) and IHF binding site (in grey) is shown. Region upstream and downstream to the CRISPR locus is depicted in purple. Numbering on top of the schema represents the position of the nucleotides with respect to leader-repeat junction. Mutated residues of CRISPR leader (CBS1–3) are highlighted in red. (B) PCR products from spacer acquisition assay performed in E. coli harbouring the CRISPR locus integrated into the P21 attB site are shown. Wt (lanes 1–2), CBS1 (lanes 3–4), CBS2 (lanes 5–6) and CBS3 (lanes 7–8) are indicated. Absence (–) or presence (+) of plasmids and inducers is indicated on top of each lane. Positions corresponding to parental and expanded arrays are indicated on the left, whereas DNA marker (M) positions are shown on the right. (C) Spacer integration assay performed with Wt (lanes 1–3), CBS1 (lanes 4–6), CBS2 (lanes 7–9) and CBS3 (lanes 10–12) CRISPR DNA substrates is shown. Positions of super-shifted nucleoprotein complex, IHF-DNA complex, CRISPR DNA substrate and protospacer are indicated on the left, whereas positions of the DNA marker (M) and integration product are shown on the right. (D) Native gel showing proteinase K treated samples from assay in (C) is displayed.
Since CBS2 does not impact the IHF binding to the leader region significantly, we hypothesized that CBS2 could be a binding site for Cas1–2 complex. Therefore, we conducted the integration experiments involving CBS1–3. This showed that the super-shifted band that was seen in wild type CRISPR DNA (lane 3 in Figure 4C) appeared in CBS1 and CBS3 too (lanes 6 and 12 in Figure 4C). Intriguingly, this band was absent when CBS2 was utilized (lane 9 in Figure 4C). Moreover, IHF dependent mobility shift was prominently seen for Wt and CBS2, albeit it was weak for CBS1. For CBS3, the IHF dependent mobility shift was not prominent despite the presence of super-shifted band. This suggests that except for CBS1—that overlaps partly with the IHF binding site—the IHF binding is not impaired in others. In line with this, all except CBS2 showed the presence of integration product (Figure 4D).
Highly conserved sub-motif region within the CBS2 is crucial for protospacer integration
In order to probe the CBS2 further, we made three constructs, viz., CBS2(L), CBS2(C) and CBS2(R). In each of these constructs, 4 bp were mutated with respect to CBS2 (Figure 5A). We tested each of these constructs for their ability to support protospacer acquisition in vivo. Remarkably, we found that except CBS2(C), other two constructs showed expansion of CRISPR array suggesting that the 4 bp (GTGG) in the middle of CBS2 (−50 to −53 nt) are crucial for protospacer acquisition (Figure 5B). To assess how these residues are impacting the protospacer acquisition, we conducted integration assays involving these constructs. In line with the acquisition assay in vivo, we observed that both CBS(L) and CBS(R) showed shift in the mobility of the CRISPR DNA in the presence of IHF and Cas1–2 complex (lanes 9 and 15 in Figure 5C). Surprisingly, in case of CSB2(C), there was no super-shifted complex even in the presence of IHF and Cas1–2 complex (lane 12 in Figure 5C). In line with this, the integration product was absent from the proteinase K treated CBS2(C) sample (lane 12 in Figure 5D). On the contrary, the integration products were observed for CBS2 (L) and CBS2 (R) (lanes 9 and 15 in Figure 5D). This suggests that CBS2(C) residues are essential for the integration of protospacer fragment into the leader-repeat junction. In tune with this, we also noted high conservation of residues corresponding to CBS2(C) in organisms harbouring type I-E system (boxed in dotted line in Figure 3A). Taken together, the disappearance of super-shifted band despite the presence of IHF related band in CBS2 and CBS2(C) led us to reason that the CBS2(C) is likely to harbour the binding site for Cas1–2 complex (see Discussion). We refer to residues (-50 to -53 nt) corresponding to CBS2(C) as integrase anchoring site (IAS).
Figure 5.

Short DNA motif within CBS2 guide protospacer incorporation. (A) Linear CRISPR DNA substrate encompassing leader (in black), two repeats (R1–R2 as orange diamonds), 33 bp spacer1 (S1 in blue), 19 bp spacer2 (S2 in cyan) and IHF binding site (in grey) is shown. Region upstream and downstream to the CRISPR locus is depicted in purple. Numbering on top of the schema represents the position of the nucleotides with respect to leader-repeat junction. Mutated residues of CRISPR leader (CBS2, CBS2(L), CBS2(C) and CBS2(R)) are highlighted in red and demarcated by a solid rectangle. (B) PCR products from spacer acquisition assay performed in E. coli harbouring the CRISPR locus integrated into the P21 attB site are shown. Wt (lanes 1–2), CBS2 (lanes 3–4), CBS2(L) (lanes 5–6), CBS2(C) (lanes 7–8) and CBS2(R) (lanes 9–10) are indicated. Absence (−) or presence (+) of plasmids and inducers is indicated on top of each lane. Positions corresponding to parental and expanded arrays are indicated on the left, whereas DNA marker (M) positions are shown on the right. (C) Spacer integration assay performed with Wt (lanes 1–3), CBS2 (lanes 4–6), CBS2(L) (lanes 7–9), CBS2(C) (lanes 10–12) and CBS2(R) (lanes 13–15) CRISPR DNA substrates is shown. Positions of super-shifted nucleoprotein complex, IHF–DNA complex, CRISPR DNA substrate and protospacer are indicated on the left, whereas positions of the DNA marker (M) and integration products are shown on the right. (D) Native gel showing proteinase K treated samples from assay in (C) is displayed.
DISCUSSION
An outstanding question regarding CRISPR adaptation pertains to the mechanism regulating specific integration of new protospacers into the leader-repeat junction amidst the presence of several repeat regions. Unlike in vivo, the integration of protospacer in vitro occurs in other repeats too (22). This observation led us to hypothesize the involvement of specific host proteins in defining the site of protospacer invasion in vivo. Our genome wide search by employing the CRISPR/dCas9-based immunoprecipitation (35) led us to recognize the participation of DNA architectural protein IHF in specifying the directional insertion of protospacer elements into the leader proximal end of CRIPSR array. IHF is known to specifically recognize its binding region and induce sharp DNA bends thereby facilitating site-specific recombination and DNA transposition (48,50,51). IHF mediated positioning of distantly oriented low affinity core site and high affinity attachment site of λ integrase into proximity facilitates bacteriophage λ integration into the genome of E. coli (51). Here, DNA deformation is utilized by IHF in bringing remotely located recognition sites into proximity. Indeed, our experiments with bending vectors showed that IHF bends the linear CRISPR DNA (Figure 2). Supercoiled plasmids carrying a CRISPR DNA were shown to act as in vitro substrates for integration whereas no integration was observed when linearized CRISPR encompassing plasmids were used (22). In comparison to linear DNA, supercoiled plasmids are inherently compact and bent and therefore it is intrinsically possible to bring remotely located recognition sites into juxtaposition. However, in the case of linear DNA, we identify that IHF is indispensable and it may facilitate favourable conformation of DNA for integration (Figures 1–3). Further, since some transposases such as Tn10 prefer deformed target DNA (52), it is possible that the IHF mediated bent DNA conformation could become a substrate for Cas1–2 complex. In addition to this, the fact that the presence of IHF ensue reduced disintegration of protospacer implies that IHF induced DNA bending appears to stabilize the integration intermediate by modulating the integrase/excisionase activity of Cas1–2 complex (Supplementary Figure S6). This shows semblance to how IHF along with λ integrase promotes integration over excision (51,53).
The involvement of IHF in the protospacer acquisition is recently reported (34); however, the precise connection between IHF induced DNA bending and directional integration of the protospacer remains elusive. Further, while IHF binding to linear DNA was shown earlier, the extent to which it deforms the leader region is not clear (34). Our findings suggest that IHF bends the linear CRISPR DNA by ∼120°, which is likely to prompt reversal in the DNA direction (Figure 2). One possible consequence of this bending could be to bring the leader region in proximity to the first repeat. While pursuing this hypothesis, we discovered that in addition to the IHF binding site, the leader region also harbours binding site for Cas1–2 complex (referred as IAS) that is located just upstream of IBS (Figures 4 and 5). We also observed IAS to be highly conserved within the leader region among the type I-E organisms that harbour IHF (boxed in dotted line in Figure 3A). This presents an attractive proposition that the IHF induced DNA bending is likely to facilitate the proximity between the Cas1–2 complex and the leader-repeat junction. The higher order nucleoprotein complex (vide. Super-shifted band in Figures 3–5) that appears in the presence of Cas1–2 complex and IHF is also noted in the case of site-specific recombination catalyzed by λ integrase and IHF (53,54). However, in the absence of IHF, since CRISPR DNA is not bound by Cas1–2 complex, it is likely that IHF induced DNA bending precedes the loading of Cas1–2 complex onto the CRISPR DNA (Figure 3). Moreover, we observed no appreciable changes in the bending angle even in the presence of Cas1–2 complex suggesting that the loading of Cas1–2 complex doesn't introduce further DNA deformation (Supplementary Figure S11).
Cas1 is reported to have an intrinsic specificity towards the sequences spanning the leader-repeat junction (24). In the vast genome sequence, it is not infrequent for Cas1 to encounter such nucleotide preference and hence this is unlikely to be a principal specificity determinant. Therefore, the role of IHF could be attributed to biasing the preference of Cas1–2 complex towards shape-based recognition as exhibited by homing endonucleases (55). In this context, it is tempting to propose that Cas1–2 complex prefers a bipartite binding site that is complemented by a part of the leader region (IAS) and leader-repeat junction. This is akin to the distantly located low affinity core site and high affinity attachment site in the case of λ integrase (51). Proximity of these complementary sites—IAS and leader-repeat junction—mediated by the IHF induced DNA bending is aptly poised to regenerate the cognate binding site for Cas1–2 complex. The following observations appear to bolster this conjecture: First, the formation of higher order nucleoprotein complex requires IHF induced DNA bending—akin to ‘intasome’ in the case of bacteriophage λ integration—suggesting that the loading of Cas1–2 complex onto the CRISPR DNA is contingent upon the proximity of the aforementioned complementary sites (Figures 3–5). Therefore, in the absence of such proximity-induced regeneration of the cognate binding site, Cas1–2 complex is unlikely to facilitate the protospacer integration into the leader proximal end. Second, in line with the above, we could observe IHF binding onto linear CRISPR DNA in the absence of Cas1–2 complex and not vice versa (Figures 3–5). Third, in conjunction with the acquisition assay, we noted that the presence of IHF abolishes the non-specific nicking activity of Cas1–2 complex (Supplementary Figure S5). Given this, it is possible to reiterate that Cas1–2 complex loading onto the CRISPR DNA is governed by the IHF mediated regeneration of the distantly located bipartite binding site.
While type I-E system requires accessory factor for protospacer acquisition, it was shown in vitro that type II-A system exhibits robust polarized protospacer incorporation into linear CRISPR DNA in the absence of any host factor (56). Further, another study showed that substitution or deletion of leader region (−1 to −5 from repeat) bordering leader-repeat junction (termed as leader-anchoring sequence or LAS) in Streptococcus pyogenes (type II-A) induces an ectopic spacer incorporation at fifth repeat where the sequence derived from fourth spacer acts as LAS (57). In Sulfolobus solfataricus (type I-A), it was observed that CRISPR locus E alone exhibits ectopic spacer incorporation whereas polarized acquisition was observed in loci C and D (58). CRISPR locus E encompasses a deletion of −47 to −70 in the leader region (58,59), which could possibly disrupt the accessory factor/Cas1–2 binding site. This in turn may impair bipartite site formation and since ssoCas1 is shown to have intrinsic sequence specificity (24), it could favour integration at region that closely resembles that of leader-repeat junction thus tuning it towards ectopic acquisition. These studies lend credence to our hypothesis that the distance between IAS and leader-repeat junction (bipartite site for Cas1–2 binding) governs the requirement of accessory factor(s) for protospacer incorporation (see below).
In addition to the sequences bordering the leader-repeat junction, modification of the repeat sequences or structure in vivo is also reported to inhibit the protospacer integration (32,60,61). On the contrary, we noticed that the protospacer integration due to such modifications remains unaltered in vitro (Supplementary Figure S8). This suggests that the bipartite binding site of Cas1–2 complex is unlikely to extend deep into the CRISPR repeat region. Based on fragment analysis, under our experimental conditions, we deciphered that the integration of the protospacer occurs into the top strand and we found no integration into the bottom strand (Supplementary Figure S5). This allows us to infer that since the underlying leader region harbouring the IBS and IAS remains intact, in such scenario, the modification of repeat sequences or structure is not expected to inhibit the top strand invasion. On the other hand, we speculate that such modification could reduce the efficiency of bottom strand invasion—wherein the integrity of the repeat sequences or structure could play a leading role in determining the specificity towards repeat1-spacer1 junction—leading to unproductive full-site integration in agreement with spacer integration assay (32,60,61).
Based on our data and previous reports (13,22,24,32,34,56–58), we present an updated model for CRISPR adaptation (Figure 6). This model can be dichotomized based on the proximity between IAS and leader-repeat junction, which allows us to predict the requirement of accessory factor(s). In cases where IAS and leader-repeat junction are segregated, in order to bring them into proximity for Cas1–2 binding, accessory factor(s) may be required. As exemplified by type I-E, this role is adopted by IHF in E. coli. IHF binding to the leader region of the CRISPR locus (IBS) leads to DNA bending. This deformed conformation ensue proximity of the distantly located IAS and leader-repeat junction that leads to the regeneration of the cognate binding site for the Cas1–2 integrase complex. Subsequently, this allows the Cas1–2 complex to orient the 3′-OH end of the protospacer fragment suitably for nucleophilic attack on the leader-repeat junction thus leading to the first nick on the top strand. This is followed by the second nucleophilic attack on the bottom strand leading to the full integration of the protospacer. We analyzed the distribution of IHF in organisms possessing type I CRISPR systems (type I-E and non type I-E systems). Out of 76 organisms encompassing type I-E CRISPR system, we found that 56 of them possess IHF (about 73%) and its distribution is predominant among enteric bacteria (Supplementary Table S5). In the case of non type I-E, 104 out of 242 organisms (∼43%) carry IHF (Supplementary Table S5). Interestingly, wherever IBS is conserved in type I-E systems, we also noted a strong correlation for the existence of IAS suggesting that these two sites co-evolve to preserve the CRISPR adaptation active (Figure 3A). However, since few organisms that harbour type I-E system in our analysis lack IHF (27%), it is possible to envisage the participation of other DNA architectural proteins such as HU or other auxiliary Cas proteins to facilitate protospacer integration (62,63). It may be noted that HU is structurally similar to IHF; however, unlike IHF, it binds DNA non-specifically. Further inspection of the type I-E organisms that lack IHF showed that a few of them lack cas operon suggesting that they are non-functional similar to E. coli BL21. A few others co-exist with other CRISPR subtypes including other type I (non type I-E), type II and type III, which suggests that the acquisition machinery may be shared across subtypes. On the contrary, if the IAS and leader-repeat junction lie juxtaposed as observed in type II-A system (33,56,57), the requirement of accessory factor(s) may be precluded (Figure 6). Nevertheless, co-opting the host proteins during adaptation epitomizes just the tip of the iceberg of the functional diversity embodied in the CRISPR–Cas system.
Figure 6.

Model depicting the factor dependent and independent protospacer integration into CRISPR locus. Based on the proximity between IAS and leader-repeat junction, the requirement of accessory factor(s) may be predicted. In type I-E, where the IAS and leader-repeat junction are segregated, IHF is required to bring them into proximity (shown in the left panel). In this case, IHF (in cyan and blue ovals) binds to IBS (in grey) within the CRISPR leader (black) that leads to bending of the DNA. This brings IAS and leader-repeat1 (R1) junction into close proximity thereby regenerating the cognate binding site for the Cas1–2 integrase complex. This enables the loading of the protospacer-bound Cas1–2 complex that orients the 3′-OH end of protospacer for nucleophilic attack on the leader-repeat1 junction producing the half-site integration intermediate. Subsequent nucleophilic attack by 3′-OH on the other end of the protospacer generates the full-site integration. DNA repair by polymerases and ligases seals the nicks to generate an expanded CRISPR array. On the other hand, as evidenced by type II-A, the requirement of accessory factor(s) may be precluded if IAS and leader-repeat junction lie juxtaposed (shown in the right panel).
Acknowledgments
Vectors pdCas9-bacteria (Addgene #44249) and pgRNA-bacteria (Addgene #44251) were a kind gift from Stanley Qi; pOSIP-CT (Addgene #45981) was a kind gift from Drew Endy and Dr Keith Shearwin; pCas1–2[K] and pCSIR-T were a kind gift from F.J.M. Mojica; pBAD Strep TEV LIC cloning vector (p8R) (Addgene #37506), pET StrepII TEV LIC cloning vector (p1R) (Addgene #29664), pET StrepII TEV co-transformation cloning vector (p13SR) (Addgene #48328) and pET His6 Sumo TEV LIC cloning vector (p1S) (Addgene #29659) were a kind gift from Scott Gradia; pBend5 was a kind gift from Sankar Adhya; pKD46 (CGSC #7739) was a kind gift from Barry L. Wanner; E. coli IYB5101 was a kind gift from Udi Qimron; E. coli strains JW1702 (CGSC#: 9441) and JW0895 (CGSC#: 8917) were a kind gift from Hirotada Mori. We acknowledge the geniality of aforementioned scientists for sharing their plasmids and bacterial strains. We thank Payel Sarkar for technical assistance and all members of the MAB lab for their critical comments and suggestions. We acknowledge the Mass Spectrometry facility at C-CAMP, Bangalore for their services.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Department of Biotechnology (DBT) [BT/08/IYBA/2014-15, BT/406/NE/UEXCEL/2013, BT/PR5511/MED/29/631/2012 and BT/341/NE/TBP/2012]; Science and Engineering Research Board (SERB) [YSS/2014/000286]. The open access publication charge for this paper has been waived by Oxford University Press — NAR.
Conflict of interest statement. None declared.
REFERENCES
- 1.Barrangou R., Fremaux C., Deveau H., Richards M., Boyaval P., Moineau S., Romero D.A., Horvath P. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315:1709–1712. doi: 10.1126/science.1138140. [DOI] [PubMed] [Google Scholar]
- 2.Horvath P., Barrangou R. CRISPR/Cas, the immune system of bacteria and archaea. Science. 2010;327:167–170. doi: 10.1126/science.1179555. [DOI] [PubMed] [Google Scholar]
- 3.Fineran P.C., Charpentier E. Memory of viral infections by CRISPR–Cas adaptive immune systems: acquisition of new information. Virology. 2012;434:202–209. doi: 10.1016/j.virol.2012.10.003. [DOI] [PubMed] [Google Scholar]
- 4.Makarova K.S., Wolf Y.I., Alkhnbashi O.S., Costa F., Shah S.A., Saunders S.J., Barrangou R., Brouns S.J., Charpentier E., Haft D.H., et al. An updated evolutionary classification of CRISPR–Cas systems. Nat. Rev. Microbiol. 2015;13:722–736. doi: 10.1038/nrmicro3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marraffini L.A. CRISPR–Cas immunity in prokaryotes. Nature. 2015;526:55–61. doi: 10.1038/nature15386. [DOI] [PubMed] [Google Scholar]
- 6.Jansen R., Embden J.D., Gaastra W., Schouls L.M. Identification of genes that are associated with DNA repeats in prokaryotes. Mol. Microbiol. 2002;43:1565–1575. doi: 10.1046/j.1365-2958.2002.02839.x. [DOI] [PubMed] [Google Scholar]
- 7.Pougach K., Semenova E., Bogdanova E., Datsenko K.A., Djordjevic M., Wanner B.L., Severinov K. Transcription, processing and function of CRISPR cassettes in Escherichia coli. Mol. Microbiol. 2010;77:1367–1379. doi: 10.1111/j.1365-2958.2010.07265.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wright A.V., Nunez J.K., Doudna J.A. Biology and Applications of CRISPR systems: harnessing nature's toolbox for genome engineering. Cell. 2016;164:29–44. doi: 10.1016/j.cell.2015.12.035. [DOI] [PubMed] [Google Scholar]
- 9.Amitai G., Sorek R. CRISPR–Cas adaptation: insights into the mechanism of action. Nat. Rev. Microbiol. 2016;14:67–76. doi: 10.1038/nrmicro.2015.14. [DOI] [PubMed] [Google Scholar]
- 10.Levy A., Goren M.G., Yosef I., Auster O., Manor M., Amitai G., Edgar R., Qimron U., Sorek R. CRISPR adaptation biases explain preference for acquisition of foreign DNA. Nature. 2015;520:505–510. doi: 10.1038/nature14302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Datsenko K.A., Pougach K., Tikhonov A., Wanner B.L., Severinov K., Semenova E. Molecular memory of prior infections activates the CRISPR/Cas adaptive bacterial immunity system. Nat. Commun. 2012;3:945. doi: 10.1038/ncomms1937. [DOI] [PubMed] [Google Scholar]
- 12.Swarts D.C., Mosterd C., van Passel M.W., Brouns S.J. CRISPR interference directs strand specific spacer acquisition. PLoS One. 2012;7:e35888. doi: 10.1371/journal.pone.0035888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yosef I., Goren M.G., Qimron U. Proteins and DNA elements essential for the CRISPR adaptation process in Escherichia coli. Nucleic Acids Res. 2012;40:5569–5576. doi: 10.1093/nar/gks216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Diez-Villasenor C., Guzman N.M., Almendros C., Garcia-Martinez J., Mojica F.J. CRISPR-spacer integration reporter plasmids reveal distinct genuine acquisition specificities among CRISPR–Cas I-E variants of Escherichia coli. RNA Biol. 2013;10:792–802. doi: 10.4161/rna.24023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yosef I., Shitrit D., Goren M.G., Burstein D., Pupko T., Qimron U. DNA motifs determining the efficiency of adaptation into the Escherichia coli CRISPR array. Proc. Natl. Acad. Sci. U.S.A. 2013;110:14396–14401. doi: 10.1073/pnas.1300108110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deveau H., Barrangou R., Garneau J.E., Labonte J., Fremaux C., Boyaval P., Romero D.A., Horvath P., Moineau S. Phage response to CRISPR-encoded resistance in Streptococcus thermophilus. J. Bacteriol. 2008;190:1390–1400. doi: 10.1128/JB.01412-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mojica F.J., Diez-Villasenor C., Garcia-Martinez J., Almendros C. Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology. 2009;155:733–740. doi: 10.1099/mic.0.023960-0. [DOI] [PubMed] [Google Scholar]
- 18.Zetsche B., Gootenberg J.S., Abudayyeh O.O., Slaymaker I.M., Makarova K.S., Essletzbichler P., Volz S.E., Joung J., van der Oost J., Regev A., et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR–Cas system. Cell. 2015;163:759–771. doi: 10.1016/j.cell.2015.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sternberg S.H., Richter H., Charpentier E., Qimron U. Adaptation in CRISPR–Cas systems. Mol. Cell. 2016;61:797–808. doi: 10.1016/j.molcel.2016.01.030. [DOI] [PubMed] [Google Scholar]
- 20.Semenova E., Savitskaya E., Musharova O., Strotskaya A., Vorontsova D., Datsenko K.A., Logacheva M.D., Severinov K. Highly efficient primed spacer acquisition from targets destroyed by the Escherichia coli type I-E CRISPR–Cas interfering complex. Proc. Natl. Acad. Sci. U.S.A. 2016;113:7626–7631. doi: 10.1073/pnas.1602639113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nunez J.K., Harrington L.B., Kranzusch P.J., Engelman A.N., Doudna J.A. Foreign DNA capture during CRISPR–Cas adaptive immunity. Nature. 2015;527:535–538. doi: 10.1038/nature15760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nunez J.K., Lee A.S., Engelman A., Doudna J.A. Integrase-mediated spacer acquisition during CRISPR–Cas adaptive immunity. Nature. 2015;519:193–198. doi: 10.1038/nature14237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang J., Li J., Zhao H., Sheng G., Wang M., Yin M., Wang Y. Structural and mechanistic basis of PAM-dependent spacer acquisition in CRISPR–Cas systems. Cell. 2015;163:840–853. doi: 10.1016/j.cell.2015.10.008. [DOI] [PubMed] [Google Scholar]
- 24.Rollie C., Schneider S., Brinkmann A.S., Bolt E.L., White M.F. Intrinsic sequence specificity of the Cas1 integrase directs new spacer acquisition. eLife. 2015;4:e08716. doi: 10.7554/eLife.08716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li M., Wang R., Zhao D., Xiang H. Adaptation of the haloarcula hispanica CRISPR–Cas system to a purified virus strictly requires a priming process. Nucleic Acids Res. 2014;42:2483–2492. doi: 10.1093/nar/gkt1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heler R., Samai P., Modell J.W., Weiner C., Goldberg G.W., Bikard D., Marraffini L.A. Cas9 specifies functional viral targets during CRISPR–Cas adaptation. Nature. 2015;519:199–202. doi: 10.1038/nature14245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vorontsova D., Datsenko K.A., Medvedeva S., Bondy-Denomy J., Savitskaya E.E., Pougach K., Logacheva M., Wiedenheft B., Davidson A.R., Severinov K., et al. Foreign DNA acquisition by the I-F CRISPR–Cas system requires all components of the interference machinery. Nucleic Acids Res. 2015;43:10848–10860. doi: 10.1093/nar/gkv1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wei Y., Terns R.M., Terns M.P. Cas9 function and host genome sampling in Type II-A CRISPR–Cas adaptation. Genes Dev. 2015;29:356–361. doi: 10.1101/gad.257550.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Westra E.R., Pul U., Heidrich N., Jore M.M., Lundgren M., Stratmann T., Wurm R., Raine A., Mescher M., Van Heereveld L., et al. H-NS-mediated repression of CRISPR-based immunity in Escherichia coli K12 can be relieved by the transcription activator LeuO. Mol. Microbiol. 2010;77:1380–1393. doi: 10.1111/j.1365-2958.2010.07315.x. [DOI] [PubMed] [Google Scholar]
- 30.Ivancic-Bace I., Cass S.D., Wearne S.J., Bolt E.L. Different genome stability proteins underpin primed and naive adaptation in E. coli CRISPR–Cas immunity. Nucleic Acids Res. 2015;43:10821–10830. doi: 10.1093/nar/gkv1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Babu M., Beloglazova N., Flick R., Graham C., Skarina T., Nocek B., Gagarinova A., Pogoutse O., Brown G., Binkowski A., et al. A dual function of the CRISPR–Cas system in bacterial antivirus immunity and DNA repair. Mol. Microbiol. 2011;79:484–502. doi: 10.1111/j.1365-2958.2010.07465.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Arslan Z., Hermanns V., Wurm R., Wagner R., Pul U. Detection and characterization of spacer integration intermediates in type I-E CRISPR–Cas system. Nucleic Acids Res. 2014;42:7884–7893. doi: 10.1093/nar/gku510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wei Y., Chesne M.T., Terns R.M., Terns M.P. Sequences spanning the leader-repeat junction mediate CRISPR adaptation to phage in Streptococcus thermophilus. Nucleic Acids Res. 2015;43:1749–1758. doi: 10.1093/nar/gku1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nunez J.K., Bai L., Harrington L.B., Hinder T.L., Doudna J.A. CRISPR immunological memory requires a host factor for specificity. Mol. Cell. 2016;62:824–833. doi: 10.1016/j.molcel.2016.04.027. [DOI] [PubMed] [Google Scholar]
- 35.Fujita T., Fujii H. Efficient isolation of specific genomic regions and identification of associated proteins by engineered DNA-binding molecule-mediated chromatin immunoprecipitation (enChIP) using CRISPR. Biochem. Biophys. Res. Commun. 2013;439:132–136. doi: 10.1016/j.bbrc.2013.08.013. [DOI] [PubMed] [Google Scholar]
- 36.Datsenko K.A., Wanner B.L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U.S.A. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baba T., Ara T., Hasegawa M., Takai Y., Okumura Y., Baba M., Datsenko K.A., Tomita M., Wanner B.L., Mori H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2006;2:1–11. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Qi L.S., Larson M.H., Gilbert L.A., Doudna J.A., Weissman J.S., Arkin A.P., Lim W.A. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell. 2013;152:1173–1183. doi: 10.1016/j.cell.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.St-Pierre F., Cui L., Priest D.G., Endy D., Dodd I.B., Shearwin K.E. One-step cloning and chromosomal integration of DNA. ACS Synth. Biol. 2013;2:537–541. doi: 10.1021/sb400021j. [DOI] [PubMed] [Google Scholar]
- 40.Zwieb C., Adhya S. Plasmid vectors for the analysis of protein-induced DNA bending. Methods Mol. Biol. 2009;543:547–562. doi: 10.1007/978-1-60327-015-1_32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Waldminghaus T., Skarstad K. ChIP on Chip: surprising results are often artifacts. BMC Genomics. 2010;11:414. doi: 10.1186/1471-2164-11-414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Papapanagiotou I., Streeter S.D., Cary P.D., Kneale G.G. DNA structural deformations in the interaction of the controller protein C.AhdI with its operator sequence. Nucleic Acids Res. 2007;35:2643–2650. doi: 10.1093/nar/gkm129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Altschul S.F., Madden T.L., Schaffer A.A., Zhang J., Zhang Z., Miller W., Lipman D.J. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Swinger K.K., Rice P.A. IHF and HU: flexible architects of bent DNA. Curr. Opin. Struct. Biol. 2004;14:28–35. doi: 10.1016/j.sbi.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 45.Alkhnbashi O.S., Shah S.A., Garrett R.A., Saunders S.J., Costa F., Backofen R. Characterizing leader sequences of CRISPR loci. Bioinformatics. 2016;32:i576–i585. doi: 10.1093/bioinformatics/btw454. [DOI] [PubMed] [Google Scholar]
- 46.Crooks G.E., Hon G., Chandonia J.M., Brenner S.E. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Friedman D.I. Integration host factor: a protein for all reasons. Cell. 1988;55:545–554. doi: 10.1016/0092-8674(88)90213-9. [DOI] [PubMed] [Google Scholar]
- 48.Chalmers R., Guhathakurta A., Benjamin H., Kleckner N. IHF modulation of Tn10 transposition: sensory transduction of supercoiling status via a proposed protein/DNA molecular spring. Cell. 1998;93:897–908. doi: 10.1016/s0092-8674(00)81449-x. [DOI] [PubMed] [Google Scholar]
- 49.Rice P.A., Yang S., Mizuuchi K., Nash H.A. Crystal structure of an IHF-DNA complex: a protein-induced DNA U-turn. Cell. 1996;87:1295–1306. doi: 10.1016/s0092-8674(00)81824-3. [DOI] [PubMed] [Google Scholar]
- 50.Leong J.M., Nunes-Duby S., Lesser C.F., Youderian P., Susskind M.M., Landy A. The phi 80 and P22 attachment sites. Primary structure and interaction with Escherichia coli integration host factor. J. Biol. Chem. 1985;260:4468–4477. [PubMed] [Google Scholar]
- 51.Moitoso de Vargas L, Kim S., Landy A. DNA looping generated by DNA bending protein IHF and the two domains of lambda integrase. Science. 1989;244:1457–1461. doi: 10.1126/science.2544029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pribil P.A., Haniford D.B. Target DNA bending is an important specificity determinant in target site selection in Tn10 transposition. J. Mol. Biol. 2003;330:247–259. doi: 10.1016/s0022-2836(03)00588-6. [DOI] [PubMed] [Google Scholar]
- 53.Segall A.M., Nash H.A. Architectural flexibility in lambda site-specific recombination: three alternate conformations channel the attL site into three distinct pathways. Genes Cells. 1996;1:453–463. doi: 10.1046/j.1365-2443.1996.d01-254.x. [DOI] [PubMed] [Google Scholar]
- 54.Kim S., Landy A. Lambda Int protein bridges between higher order complexes at two distant chromosomal loci attL and attR. Science. 1992;256:198–203. doi: 10.1126/science.1533056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lambert A.R., Hallinan J.P., Shen B.W., Chik J.K., Bolduc J.M., Kulshina N., Robins L.I., Kaiser B.K., Jarjour J., Havens K., et al. Indirect DNA sequence recognition and its impact on nuclease cleavage activity. Structure. 2016;24:862–873. doi: 10.1016/j.str.2016.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wright A.V., Doudna J.A. Protecting genome integrity during CRISPR immune adaptation. Nat. Struct. Mol. Biol. 2016;23:876–883. doi: 10.1038/nsmb.3289. [DOI] [PubMed] [Google Scholar]
- 57.McGinn J., Marraffini L.A. CRISPR–Cas systems optimize their immune response by specifying the site of spacer integration. Mol. Cell. 2016;64:616–623. doi: 10.1016/j.molcel.2016.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Erdmann S., Garrett R.A. Selective and hyperactive uptake of foreign DNA by adaptive immune systems of an archaeon via two distinct mechanisms. Mol. Microbiol. 2012;85:1044–1056. doi: 10.1111/j.1365-2958.2012.08171.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Garrett R.A., Shah S.A., Erdmann S., Liu G., Mousaei M., Leon-Sobrino C., Peng W., Gudbergsdottir S., Deng L., Vestergaard G., et al. CRISPR–Cas adaptive immune systems of the sulfolobales: unravelling their complexity and diversity. Life. 2015;5:783–817. doi: 10.3390/life5010783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Goren M.G., Doron S., Globus R., Amitai G., Sorek R., Qimron U. Repeat size determination by two molecular rulers in the type I-E CRISPR array. Cell Rep. 2016;16:2811–2818. doi: 10.1016/j.celrep.2016.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang R., Li M., Gong L., Hu S., Xiang H. DNA motifs determining the accuracy of repeat duplication during CRISPR adaptation in Haloarcula hispanica. Nucleic Acids Res. 2016;44:4266–4277. doi: 10.1093/nar/gkw260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wei Y., Terns M.P. CRISPR Outsourcing: Commissioning IHF for Site-Specific Integration of Foreign DNA at the CRISPR Array. Mol. Cell. 2016;62:803–804. doi: 10.1016/j.molcel.2016.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dillon S.C., Dorman C.J. Bacterial nucleoid-associated proteins, nucleoid structure and gene expression. Nat. Rev. Microbiol. 2010;8:185–195. doi: 10.1038/nrmicro2261. [DOI] [PubMed] [Google Scholar]
- 64.Lange S.J., Alkhnbashi O.S., Rose D., Will S., Backofen R. CRISPRmap: an automated classification of repeat conservation in prokaryotic adaptive immune systems. Nucleic Acids Res. 2013;41:8034–8044. doi: 10.1093/nar/gkt606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Waterhouse A.M., Procter J.B., Martin D.M., Clamp M., Barton G.J. Jalview Version 2–a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25:1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]