

Abstract
Polyketide metabolites produced by modular type I polyketide synthases (PKS) acquire their chemical diversity through the variety of catalytic domains within modules of the pathway. Methyltransferases are among the least characterized of the catalytic domains common to PKS systems. We determined the domain boundaries and characterized the activity of a PKS C-methyltransferase (C-MT) from the curacin A biosynthetic pathway. The C-MT catalyzes S-adenosylmethionine-dependent methyl transfer to the α-position of β-ketoacyl substrates linked to acyl carrier protein (ACP) or a small-molecule analog, but does not act on β-hydroxyacyl substrates or malonyl-ACP. Key catalytic residues conserved in both bacterial and fungal PKS C-MTs were identified in a 2-Å crystal structure and validated biochemically. Analysis of the structure and the sequences bordering the C-MT provides insight into the positioning of this domain within complete PKS modules.
Graphical abstract
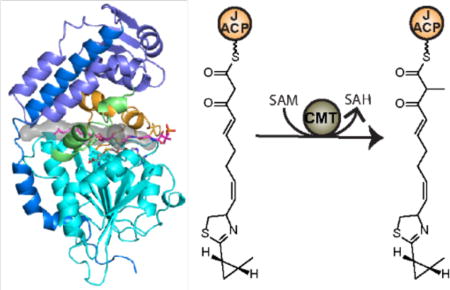
Introduction
Polyketides and their derivatives are among the most chemically and biologically diverse natural products, and their biosynthetic pathways contain a wealth of synthetic potential. Type I polyketide synthases (PKSs) are large multifunctional enzymes that generate complex polyketides with exquisite stereo and regioselectivity1–3. Typical type I PKSs contain acyltransferase (AT) and ketosynthase (KS) domains for the selection of acyl-CoA building blocks and extension of polyketide intermediates as well as an acyl carrier protein (ACP) domain to shuttle intermediates between domains. Additionally, PKSs may have one or more modification domains, such as a ketoreductase (KR), dehydratase (DH), enoylreductase (ER) and methyltransferase (MT), for further processing of the β-ketoacyl-ACP product of KS extension (Figure 1a). In bacteria, type I PKSs are commonly found as modular assembly lines, where each module catalyzes a single two-carbon extension and modification of the polyketide and then passes the intermediate to the next module for further elongation. In some cases, PKS modules lack embedded AT domains and rely on in trans ATs encoded in the biosynthetic gene cluster for the delivery of acyl-CoAs. Fungal type I PKSs typically occur as single modules that catalyze multiple rounds of chain elongation and modification1, 4.
Figure 1.
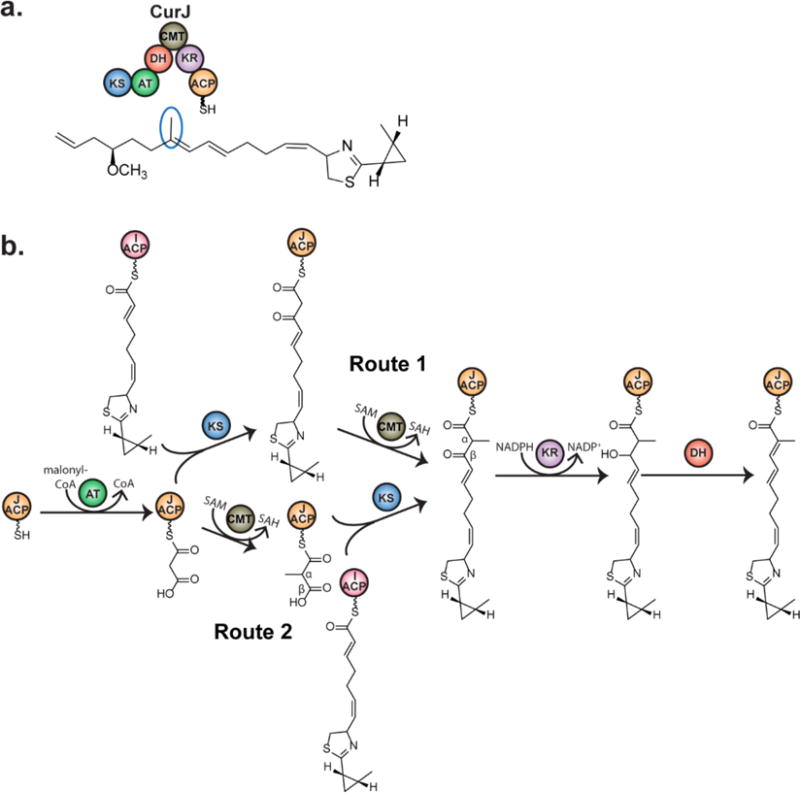
Introduction of α-methyl by CurJ C-MT. a) CurJ composition. Curacin A contains a single methyl branch derived from the C-MT in module CurJ, which contains ketosynthase (KS), acyltransferase (AT), dehydratase (DH), C-methyltransferase (CMT), ketoreductase (KR), and acyl carrier protein (ACP) domains. b) Potential routes for C-methylation in PKSs. Route 1-Methylation occurs on the β- ketoacyl intermediate after the KS condensation reaction. Route 2-Methylation occurs on malonyl-ACP prior to KS condensation.
Methyl groups can be introduced at the α-position of a polyketide intermediate through the incorporation of methylmalonyl-CoA extender units selected by the AT or through the action of embedded C-methyltransferase (C-MT) domains. S-adenosylmethionine (SAM)-dependent PKS C-MT domains were initially characterized in the lovastatin biosynthetic pathway of the fungus Aspergillus terreus and the mixed PKS / non-ribosomal peptide synthetase (NRPS) yersiniabactin pathway5–7 and subsequently annotated in multiple fungal PKSs as well as in bacterial cis-AT and trans-AT pathways8–16.
PKS C-MTs are members of the class I MT superfamily17, 18, identified by six sequence motifs in the SAM-binding core19. Despite structural conservation of the MT core, class I MTs lack conserved active site residues due to the intrinsic reactivity of SAM and the great variety of methyl acceptor substrates. Moreover, numerous loop and domain insertions to the MT core facilitate substrate binding or oligomer formation, resulting in an extremely divergent superfamily with very low overall sequence identity (generally below 20%). C-MTs are the only domains commonly found within PKS modules that have remained structurally uncharacterized. Beyond the easily recognized sequence motifs for SAM binding, the PKS C-MT domain boundaries are uncertain. However, a pseudo-methyltransferase (Ψ-MT) domain is present in the PKS homolog, the metazoan fatty acid synthase (mFAS)20, but the Ψ-MT lacks the SAM binding motifs and appears to be significantly shorter than the PKS C-MTs.
C-MTs could potentially act on the unmodified α-carbon at any point during the PKS catalytic cycle, unlike KS, KR, DH and ER domains, which act in a specific sequence (Figure 1b). In an earlier study, it was proposed that PKS C-MT domains act after KS condensation and prior to subsequent reductive modifications based on the accumulation of a 2-methyl-1,3-diketo-containing shunt product in a mutagenized lovastatin producing fungus (Figure 1b, Route 1)6. Methylation of the β-ketoacyl intermediate via Route 1 was observed directly in two fungal PKSs14, 21. However, two bacterial PKS C-MTs that dimethylate the α-position of polyketide intermediates were shown to act exclusively via Route 2 in epothilone biosynthesis (Figure 1b, Route 2) and either on the β-ketoacyl intermediate of Route 1 or on malonyl-ACP preceding KS condensation in yersiniabactin biosynthesis22, 23. Thus it was unclear when monomethylation occurs in modular C-MTs. Recent work demonstrated C-MT activity via Route 1 in bacterial trans-AT and cis-AT pathways24, 25.
The biosynthetic pathway for the antimitotic polyketide curacin A (Figure 1a) produced by the marine cyanobacterium Moorea producens has provided a wealth of structural insight into conventional and unusual PKS biosynthetic capabilities8, 26–35. Modules CurG-CurM catalyze typical PKS extension reactions. Like many PKS pathways from Moorea species, a C-MT domain in CurJ installs a single α-methyl (Figure 1a). Here we report the crystal structure of CurJ C-MT at 2.1 Å resolution and demonstrate that CurJ C-MT follows Route 1 by methylating the α-position of the β-keto acyl substrate. The structure reveals key conserved features of PKS C-MT active sites and provides important clues about how C-MTs are positioned within PKS modules.
Results and Discussion
Identification of C-MT domain boundaries
C-MT domains within PKS modules have been annotated through identification of MT motifs (Supplementary Figure 1). However, all have a large unannotated region between the preceding catalytic domain and MT motif I, for example 228 amino acids following the DH domain of CurJ. Moreover C-MTs from cis-AT and trans-AT PKS pathways cluster separately in a phylogenetic tree (Supplementary Figure 2)15, and have distinct positions within PKS modules, typically preceding KR domains in cis-AT pathways and following KR domains in trans-AT pathways.
The C-MT N-terminal extension is less conserved than the MT core (for example, 30% identity for the CurJ N-terminal extension and 50% for the MT core compared to the homologous regions of the GphI C-MT from the myxobacterial gephyronic acid pathway15). However, the N-terminal extension is likely to have a defined structure, as it is highly conserved among cyanobacterial C-MTs, (for example, 78% identity to CrpA from Nostoc sp. ATCC 5378010). We identified approximate domain boundaries based on a multiple sequence alignment of C-MTs from cyanobacterial PKS pathways, created fifteen constructs encoding the CurJ C-MT with various N- and C-termini (Table S4), and tested twelve for production of soluble protein. Ultimately, CurJ residues 1269–1658, herein referred to as 1–390, yielded a pure, stable, recombinant C-MT domain that was monomeric in solution as determined by size exclusion chromatography (data not shown).
Methyltransferase Activity
Based on precedent for C-methylation in PKSs, we expected that CurJ C-MT would act on either malonyl-ACP via Route 2 or the β-ketoacyl intermediate formed after KS-condensation via Route 1 (Figure 1b)14, 21, 23–25. As native CurJ substrates were unavailable, acetoacetyl-ACP (1) was tested using an assay based on intact protein mass spectrometry (MS). Complete conversion of acetoacetyl-ACP (1) to α-methyl-acetoacetyl-ACP (2) (Figure 2a) was observed by LC-MS. However, no methylation occurred with malonyl-ACP (3) (Figure 2c), establishing that CurJ follows Route 1 and not Route 2.
Figure 2.
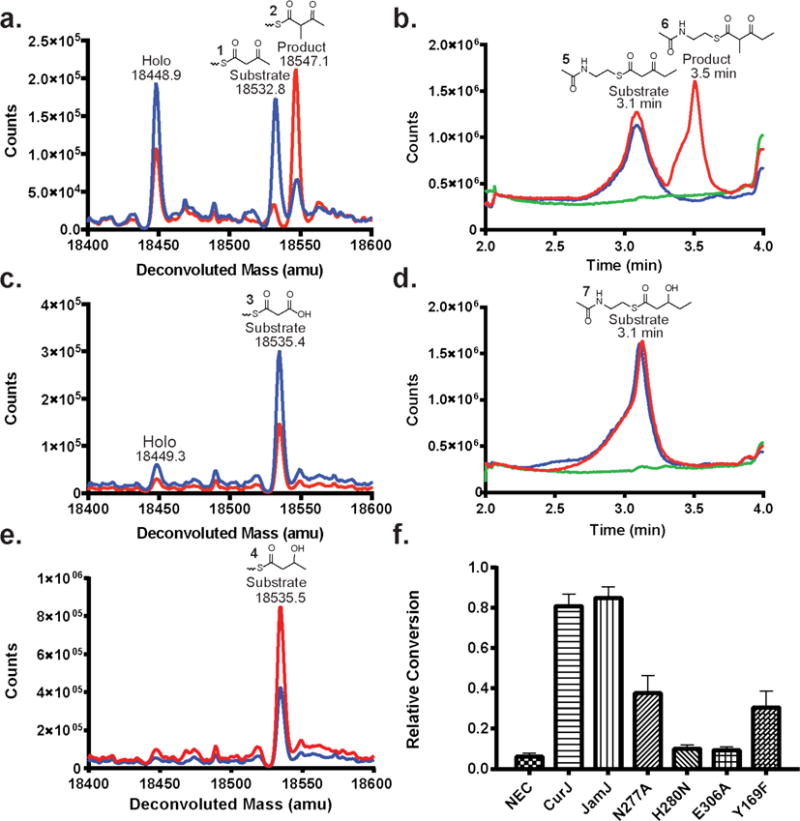
LC-MS analysis of C-MT activity. Ion counts are shown in red for reactions via Route 1 in a) acetoacetyl-ACP (1) and b) β-ketopentanoyl-NAC (5), via Route 2 in c) malonyl-ACP (3), and via Route 3 in d) β-hydroxypentanoyl-NAC (7) and e) D,L-β-hydroxybutyryl-ACP (4). Blue traces are no-enzyme control reactions; green traces are no-substrate control reactions. See Supplementary Table 1 for expected masses and Supplementary Figure 3 for mass spectra of total ion chromatographs of acyl-NACs. f) Relative methylation activities of wild type CurJ C-MT, JamJ C-MT, and CurJ C-MT site-directed mutants with acetoacetyl-ACP (1) in an MS assay based on phosphopantetheine ejection (Supplementary Figure 5). NEC = no enzyme control.
We also tested a potential methylation Route 3 where the C-MT acts after KR reduction of the β-ketoacyl intermediate, i.e. on the β-hydroxyacyl intermediate. Reactions performed with DL-β-hydroxybutyryl-ACP (4) to mimic the β-hydroxy intermediate failed to generate a methylated product (Figure 2e), indicating that the C-MT is specific for the β-ketoacyl intermediate. CurJ C-MT also methylated an N-acetyl cysteamine (NAC-) linked β-ketopentanoyl substrate (5) (Figure 2b) to (6), but not β-hydroxypentanoyl-NAC (7) (Figure 2d).
Methylation of the β-ketoacyl intermediate via Route 1 is the most thermodynamically favorable for the formation of the carbanion nucleophile. Among the potential substrates via Routes 1, 2 and 3, the β-ketoacyl intermediate has the lowest pKa ~10 due to the flanking carbonyls. By contrast, the pKa of the α-proton in malonyl-ACP is several units higher as a result of the carboxylate anion. Additionally, the β-hydroxy intermediate produced by the KR has the least acidic α-proton with a pKa estimated ~21, making it the least favorable substrate for methyl transfer.
Structure of CurJ C-MT
The crystal structure of selenomethionine-labeled CurJ C-MT was solved to 2.1 Å by single-wavelength anomalous diffraction (SAD) phasing (Figure 3 and Table 1). The CurJ C-MT has a class I MT core domain for SAM binding (residues 1–27, 172–306, 342–381), comprised of a seven-stranded β-sheet flanked by α-helices. A 32-residue insertion (307–341, including helix 13) between β-strand 8 and helix 14 of the MT core distinguishes the PKS C-MT from a minimal class I MT (Figure 3). The “core insertion” extends over the active site, and is conserved in PKS C-MTs from cis-AT (49% identity to GphI C-MT), trans-AT (44% to RhiB) and fungal PKS pathways (44% to SQTKS) (Supplementary Figure 1). The mFAS Ψ-MT has a homologous 26-residue insertion in the equivalent position (Supplementary Figure 4).
Figure 3.
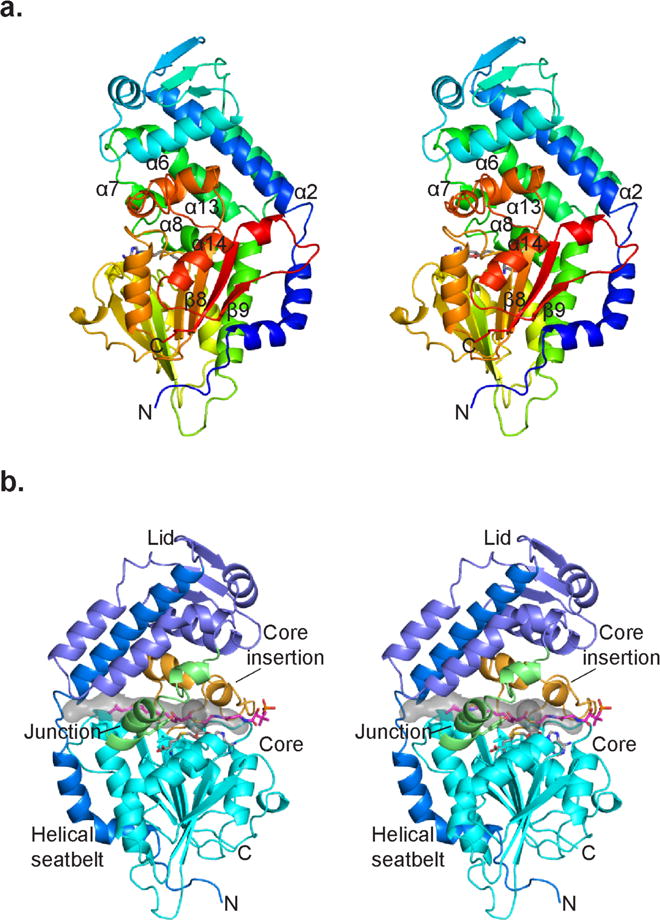
CurJ C-MT structure. a) CurJ C-MT colored as a rainbow from blue (N-terminus) to red (C-terminus), shown in stereo. SAH is shown in sticks with atomic colors (C, gray; O, red; N, blue; S, yellow). b) CurJ C-MT colored by structure region. Helical seatbelt, blue; lid, dark blue; lid-to-core junction green; core, cyan; core insertion, orange; SAH, sticks with gray C. The transparent gray surface represents the substrate tunnel, which is lined with hydrophobic residues (Ile35, Phe157, Leu168, Val174, Ala307, Trp313, Val314, Phe318, Leu338). The CurJ C-MT substrate and phosphopantetheine (sticks with magenta C) were modeled into the active site. The views in A and B are from opposite sides of the C-MT.
Table 1.
Crystallographic information
| Data Collection | SeMet | Wild type |
|---|---|---|
| Space group | P21 | P21 |
| Cell Dimensions | ||
| a,b,c (Å) | 48.4, 130.4, 62.8 | 46.1, 129.1, 63.1 |
| α, β, γ (°) | 90, 112.0, 90 | 90, 111.5, 90 |
| X-ray Source | APS 23ID-D | APS 23ID-D |
| Wavelength (Å) | 0.979 | 1.033 |
| dmin (Å) | 2.09 (2.16–2.09)a | 2.10 (2.18–2.10) |
| R-merge | 0.1703 (1.492) | 0.1508 (1.048) |
| Inner-shell R-merge | 0.053 (9.3 Å)b | 0.049 (6.2 Å)b |
| Avg I/σ (I) | 11.62 (2.55) | 9.3 (1.37) |
| Completeness (%) | 100 (100) | 100 (100) |
| Multiplicity | 10.8 (10.4) | 6.6 (6.6) |
| Total observations | 463,157 (44,285) | 263,797 (26,230) |
| CC1/2 | 0.998 (0.683) | 0.997 (0.703) |
| CC* | 1 (0.901) | 0.999 (0.909) |
| Refinement | ||
| Data range (Å) | 44.48–2.09 | 43.43–2.10 |
| Reflections used in refinement | 42,794 | 39,893 |
| Rwork/Rfree (%) | 16.9/21.6 | 18.5/25.2 |
| Number of non-hydrogen atoms | 6355 | 6220 |
| macromolecules | 6049 | 6040 |
| ligands | 40 | 19 |
| water | 328 | 214 |
| Amino acid residues | 765 | 765 |
| Deviation from ideality | ||
| bond lengths (Å) | 0.003 | 0.007 |
| bond angles (°) | 0.60 | 0.92 |
| Average B-factor | 46.3 | 51.9 |
| protein | 46.0 | 52.0 |
| ligands | 86.3 | 70.3 |
| solvent | 46.8 | 44.5 |
| Ramachandran plot | ||
| favored (%) | 98 | 96 |
| allowed (%) | 2.5 | 3.4 |
| outliers (%) | 0 | 0.4 |
| PDB | 5THY | 5THZ |
Values in parentheses pertain to the outermost shell of data.
dmin inner shell
Much of the unannotated N-terminal extension of CurJ C-MT forms a helical “lid” over the MT core. The large lid (residues 28–171) includes eight helices and a three-stranded β-sheet and is connected to the core by a “lid-to-core junction” containing helices 7 and 8 (Figure 3). Helices 1 and 2 (Figure 3) form a long amphipathic N-terminal helical “seatbelt” (residues 9–54) that wraps around the MT core and extends into the lid. Nearly all other C-MTs from bacterial cis-AT pathways contain a lid of similar length with an amphipathic sequence at the N-terminus (Supplementary Figure 1), suggesting that the seatbelt is present in nearly all of them. The seatbelt serves an architectural function of bringing together the N- and C-termini at the bottom of the core domain (15 Å separation). This is analogous to the mFAS Ψ-MT, which lacks helix 1 of the seatbelt and instead contributes a β-strand to the core β-sheet, bringing the domain termini within ~20 Å of one another (Supplementary Figure 4). The mFAS Ψ-MT lid is a truncated variant of the PKS C-MT lid.
Knowledge of the C-MT domain boundaries within CurJ allowed us to locate a key structure element of the CurJ KR domain (Figure 4). An extended β-ribbon is an integral part of PKS KR domains, which are comprised of structural (KRS) and catalytic (KRC) sub-domains36. The extended β-ribbon contributes to the β-sheets of both KRS and KRC, but its two β-strands lie within inter-domain linker sequences, generally flanking the KRS sub-domain36–45. However, in CurJ the highly conserved first β-strand of the KR β-ribbon does not flank KRS, but is located between the DH and C-MT domains (amino acids 1254–1261, nine residues following the DH and eight residues preceding the C-MT, Figure 4). Interestingly, the first β-strand of the mFAS KR β-ribbon is located in the analogous location between DH and Ψ-MT domains. Together with the similarity of the C-MT and Ψ-MT structures and the similar positions of their N- and C-termini, this remarkable evolutionary conservation is a strong indication that the modifying region of CurJ (DH–C-MT–KR–ACP) and other PKS modules is organized similarly to the modifying region of mFAS (DH–Ψ–MT–KRS–ER–KRC–ACP), and that the structure of mFAS20 can guide modeling of the PKS module modifying region (Figure 4a).
Figure 4.
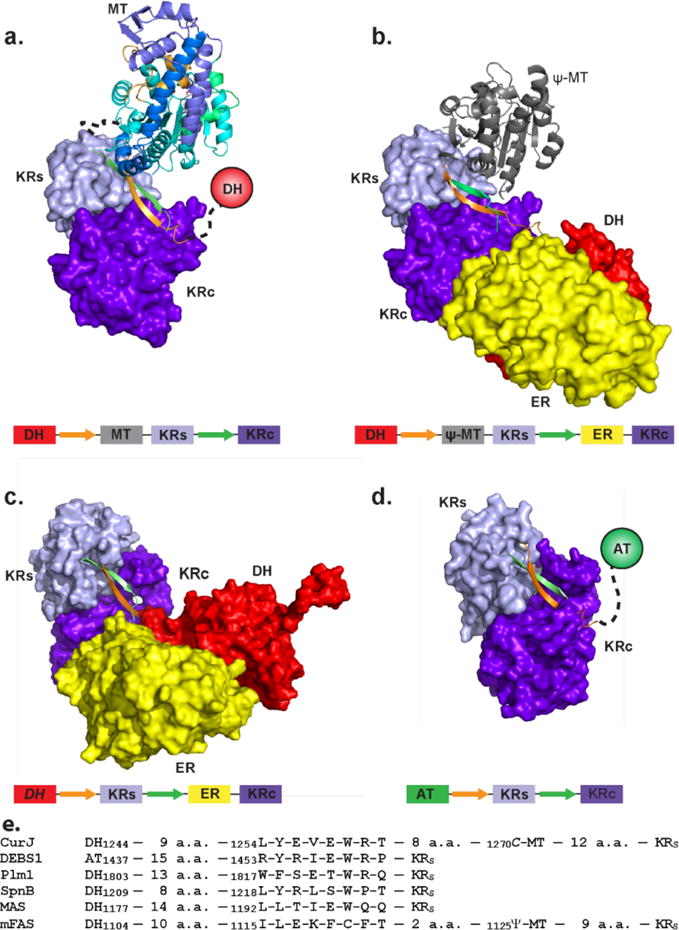
PKS and mFAS modifying regions. The β-ribbon that is both an essential part of the KR domain and also an inter-domain linker is orange (1st strand) and green (2nd strand). a) Model of CurJ C-MT and KR domains, arranged as in mFAS. The model is based on superpositions of the CurJ C-MT core with the mFAS Ψ-MT core and of a PKS KR with the mFAS KR20, 36. b) mFAS with DH, Ψ-MT, ER and KR domains20. c) MAS-like PKS with DH, ER and KR domains37. d) DEBS1 module 1 modifying region with KR domain only36. DH (red), ER (yellow), KRC (purple) and KRS (light blue) domains are represented as surfaces, the CurJ C-MT as a ribbon colored as in Fig. 3b, the mFAS Ψ-MT as a gray ribbon, and other domains as circles. Below each structure is a schematic of domain and β-ribbon strand arrangement colored as in the structure image. e) Sequence alignment and domain context of β-ribbon strand 1 in CurJ, DEBS module 136, phoslactomycin module 145, spinosyn module 239, MAS-like PKS37 and mFAS20.
Active Site Architecture
A hydrophobic tunnel between the MT core and the lid is long enough to accommodate the full CurJ C-MT substrate (Figure 3b), consistent with the observed activity on the hydrophobic β-ketoacyl substrate via Route 1, and the lack of activity with the negatively charged malonyl-ACP. S-adenosyl-L-homocysteine (SAH) was bound in the MT core below the tunnel, supported by clear electron density (Supplementary Figure 5). The conserved side chains of His280 and Glu306 form a hydrogen-bonded dyad 8 Å from the SAH (Figure 5). The His-Glu dyad is invariant in all bacterial and fungal PKS C-MTs, thus the imidazole may act as a catalytic base to deprotonate the substrate α-carbon and form a carbanion nucleophile to facilitate the MT reaction. The reactivity of the His-Glu dyad may be enhanced by the surrounding hydrophobic environment (conserved Ala281, Phe318, Trp324, Pro337, Leu338, Trp344). Additionally, conserved Tyr169 is ~4 Å from the presumed SAM sulfonium position, and may facilitate methyl transfer through a CH-O hydrogen bond with the SAM methyl46. The invariant Asn277 side chain points into the active site between SAH and the His-Glu dyad. Given its conservation and proximity to the SAM sulfonium, Asn277 could play a role in substrate positioning.
Figure 5.
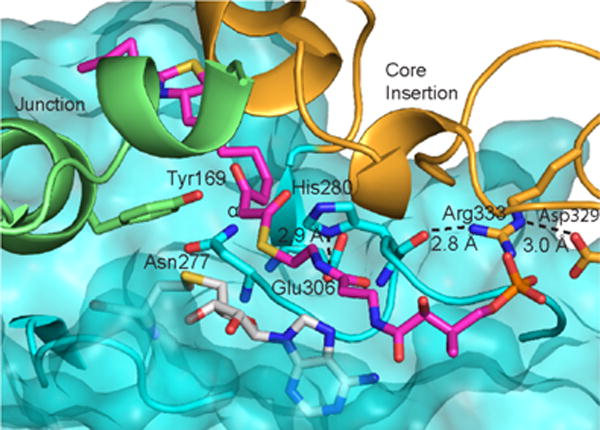
CurJ C-MT active site. Key amino acids, modeled substrate (magenta C), and SAH (white C) are shown in sticks. The MT core is represented as a transparent surface with ribbon. The junction and core insertion are shown as ribbons. C-MT structural regions are colored as in Fig. 3b.
Remarkably, the SAH homocysteine and nearby amino acids are the least well ordered regions of the active site. The two slightly different crystal forms (Table 1) provided four independent views of the C-MT and revealed small differences in position for the SAH homocysteine, helix 8 in the lid-to-core junction, and MT motif I, which coordinates the SAM/SAH carboxylate (Figure 6, Supplementary Figure 5). For two of the four C-MT views, density indicated partial disorder for Tyr169 in helix 8 and for the SAH homocysteine, and multiple positions for Thr208 in motif I. In contrast, the SAH adenosine and His-Glu dyad are identically positioned and supported by strong density in all four views. The His280 position is stabilized by two hydrogen bonds within the 277–282 loop and by backbone hydrogen bonds with the conserved Arg333 side chain in the core insertion (Figure 5). These observations indicate that the substrate-binding region of the active site is flexible in absence of β-ketoacyl-ACP.
Figure 6.
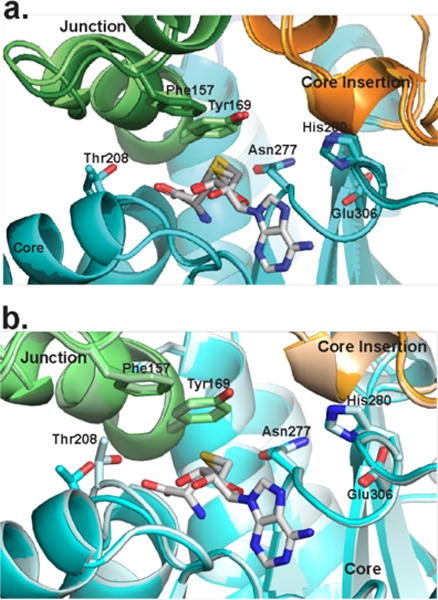
Movement surrounding the CurJ C-MT active site. Structural regions are colored as in Fig. 3b. a) Closely related SeMet and wild type CurJ C-MT crystal forms (light and dark colors) are different in the lid-to-core junction and SAH homocysteine position. b) The position of Thr208 in MT motif I differs in the two C-MT chains in the SeMet crystal form.
The CurJ C-MT core loops, the core insertion, and the lid-to-core junction surround the entrance to the active site, and amino acids in these regions are likely to interact with the ACP (Figure 3b, Figure 5). An analogous core insertion exists in the only other structurally characterized SAM-dependent enzyme with an ACP-linked substrate, cyclopropane synthase of mycolic acid biosynthesis47. As with cyclopropane synthase, the position and the highly conserved sequence of the PKS C-MT core insertion are consistent with a role in ACP recognition.
Activity of CurJ C-MT with active site substitutions
To probe the role of conserved amino acids in the active site, CurJ C-MT variants were produced and activity was evaluated using a phosphopantetheine ejection mass spectrometry assay (Figure 2f, Supplementary Figure 6)48. Substitutions in the active site His-Glu dyad abolished activity, consistent with their proposed role as a catalytic dyad. The melting temperatures of the wild type and the His variants were comparable (Supplementary Table 2), indicating that the reduced activity was not due to a decrease in the structural integrity of the protein. A Y169F variant had three-fold reduced activity, in agreement with a role in facilitating the methyltransfer reaction. The N277A variant reduced activity two fold suggesting that it plays a role in substrate recognition or positioning in the active site.
Substrate Modeling
His208, the strongest candidate for a catalytic base, is 8 Å from the SAM sulfonium on the opposite side of the active site. We modeled the full-length CurJ C-MT substrate into the active site tunnel, as attempts to soak small molecule NAC substrates (5, 7) yielded additional but uninterpretable density at the presumed substrate binding site between SAH and His280. The full length CurJ C-MT substrate was docked between the MT core and lid domain and manually edited to orient the α-carbon in line with the sulfonium (Figure 3b, Figure 5). While the tunnel can accommodate the full substrate, the sulfonium and His280 were too far apart for both to interact with the α-carbon of the β-ketoacyl intermediate (Figure 5). Additionally, hydrogen-bonding partners that could stabilize an enolate intermediate were not apparent. Thus, we propose that β-ketoacyl-ACP binding leads to small movements of the flexible active site elements (lid-to-core junction, core insertion and adjacent loops) and positions the substrate α-carbon, SAM methionine, Tyr169 and enolate-stabilizing hydrogen bonding partners closer to the His-Glu dyad and in optimal positions for methyl transfer. Due to the open active site in the crystal structures, we are unable to predict the stereochemical outcome of the reaction. Additionally, the acidity of the newly formed stereogenic proton precludes chemoenzymatic assignment. The chiral center in the CurJ C-MT product is eliminated by action of the CurJ DH, which catalyzes formation of a vinyl-methyl functionality in the curacin polyketide chain.
Similarities of CurJ C-MT to other PKS C-MTs
The CurJ C-MT structure is an excellent model for a pre-catalytic state of C-MTs embedded in PKS modules. These PKS MTs share several distinguishing features including the active site His-Glu dyad, the lid-to-core junction helices, and the MT core insertion (Supplementary Figure 1). Overall, C-MTs from trans-AT pathways and fungal iterative PKSs have lid domains approximately 30 residues shorter than the CurJ C-MT and others from cis-AT pathways (Figure 3, Supplementary Figure 1). Some PKS C-MTs produce gem-dimethyl products12, 23, 25, which the spacious C-MT active site could facilitate by substrate re-orientation between methyl-transfer reactions. Although the natural CurJ C-MT substrate is quite long, the domain was capable of methylating shorter β-keto acyl substrates, such as acetoacetyl-ACP. This is in contrast to the fungal LovB C-MT, which was selective for substrates of similar length to the native substrate and had no activity on an acetoacetyl analog21. Increased substrate specificity in iterative fungal C-MTs relative to bacterial type I PKS C-MTs can be rationalized, as fungal C-MTs have evolved to be active in specific rounds of the iterative catalytic cycle, whereas PKS C-MTs will encounter only one β-ketoacyl-ACP during the single round of catalysis within the module.
The biochemical and structural characterization of the CurJ C-MT is consistent with previous findings that monomethylating PKS C-MTs act on β-ketoacyl intermediates following KS mediated chain elongation, and further shows no activity with β-hydroxyacyl intermediates. Additionally, the structure provides new insights regarding the mechanism of α-methylation in both bacterial and fungal PKS C-MTs, including identification of a conserved His-Glu dyad and other active site elements that are flexible in absence of the methyl acceptor substrate. Similarities of the CurJ C-MT and mFAS Ψ-MT structures suggest that PKS C-MTs may share a similar position within a module as the Ψ-MT within the mFAS.
Experimental Procedures
Construct Design, Protein Expression, and Purification
All PCR primers are listed in Supplementary Table 3. CurJ C-MT (CurJ residues 1269–1658) was amplified from cosmid pLM9 (GenBank accession code AY652953)8. JamJ C-MT (residues 1948–2337) and ACP (residues 3166–3302) were amplified from a mixture of jamacamide A cosmids9. C-MT and ACP constructs were inserted into pMCSG7 by ligation independent cloning (LIC) to create pMAS179 (CurJ C-MT), pMAS195 (JamJ C-MT), and pMAS197 (JamJ ACP). The QuikChange protocol (Stratagene) was used for site-directed mutagenesis. DNA sequencing at the UM DNA Sequencing Core verified all constructs and mutations. C-MT and ACP constructs were recombinantly expressed in Escherichia coli and purified using nickel affinity followed by gel filtration chromatography.
Protein Crystallization and Structure Determination
CurJ C-MT (native and SeMet) was crystallized by vapor diffusion from 2:2 μL ratio of protein stock (10 mg/mL of CurJ C-MT in buffer C with 1 mM SAM or SAH) and well solution (1.30–1.38 M sodium citrate, 4–9 mM glutathione (GSH)/glutathione disulfide (GSSG), 5% (v/v) acetone) at 20°C. Thin rod-like crystals appeared in a few hours and grew overnight. Crystals were harvested directly from the drop and flash cooled in liquid nitrogen. Single-wavelength anomalous diffraction was used to determine the CurJ C-MT structure. A fully refined SeMet structure was used to solve the structure of native CurJ C-MT crystals.
Enzyme Assays
Reaction mixtures contained 25 μM enzyme (CurJ C-MT, JamJ C-MT, or variants),100 μM JamJ ACP, 10 μM Streptomcyes verticillus phosphopantethinyl transferase (SVP)49, 1 mM MgCl2, 1 mM SAM, and 500 μM malonyl-CoA, acetoacetyl-CoA, or DL-β-hydroxybutyryl-CoA in 50 mM HEPES pH 7 (phosphopantetheine ejection) or 7.4 (intact protein), 150 mM NaCl (total volume 50 μL). Reactions were incubated at 30°C for 6 hr (phosphopantetheine ejection) or overnight (intact protein) and quenched with 1% (v/v) formic acid. JamJ ACP from the jamaicamide pathway of another Moorea producens strain9 (82% identical to CurJ ACP) was used as a surrogate due to the toxicity of CurJ ACP to E. coli35. Nearly identical levels of activity were observed for CurJ and JamJ C-MT (Figure 2f), indicating that the JamJ ACP is a suitable surrogate for CurJ ACP. NAC-linked substrates (5, 7) were synthesized as previously described50. For NAC-linked substrate reactions 25 μM C-MT was incubated overnight at 25 °C with 1 mM SAM and 500 μM 5 or 7 in 50 mM HEPES 7.4, 150 mM NaCl (50 μL). Reaction mixtures were quenched with 150 μL methanol. Portions of the reaction mixes for ACP-linked substrates (10 μL) and NAC-linked substrates (20 μL) were used for LC-MS analysis (Agilent 6520 Accurate Mass Q-TOF in the positive mode equipped with an Agilent 1290 HPLC system). Separation of samples for intact protein analysis and phosphopantetheine ejection was performed using reverse phase chromatography (Aeris widepore C4 column 3.6 μm, 50 × 2.10 mm) at a flow rate of 0.5 mL/min. Protein was eluted using a gradient of 5–100% B over 4 min (A- H2O with 0.2% (v/v) formic acid, B- acetonitrile with 0.2% (v/v) formic acid). Intact protein data were analyzed using the Agilent Mass Hunter Qualitative Analysis software with the maximum entropy deconvolution algorithm. Abundance of holo, acetoacetyl, and methylated acetoacetyl phosphopantetheine ejection masses were used to determine percent conversion of CurJ C-MT variants34. Data were normalized to exclude the spontaneous hydrolysis product (holo-ACP) of acetoacetyl or α-methyl-acetoacetyl-ACP. Three independent reactions were conducted and analyzed for each C-MT variant in the phosphopantetheine ejection assay. NAC-linked reaction mixes were separated using a Phenomenex Kinetix reverse-phase C18 column (40 mm × 2.1 mm, 2.6 μm) at a flow rate of 0.5 mL/min. Substrates were eluted using a gradient of 5–100% B over 4 min (A- H2O with 0.2% (v/v) formic acid, B- acetonitrile with 0.2% (v/v) formic acid).
Supplementary Material
Acknowledgments
This work was supported by NIH grants DK042303 to J.L.S., CA108874 to D.H.S., W.H.G. and J.L.S., and GM118101 to D.H.S. M.A.S. was supported by a predoctoral fellowship from the Cellular Biotechnology Training Program (T32GM008353) and Rackham Graduate School. GM/CA@APS is supported by the NIH National Institute of General Medical Sciences (AGM-12006) and National Cancer Institute (ACB-12002). The Advanced Photon Source is a US Department of Energy (DOE) Office of Science User Facility operated by Argonne National Laboratory under Contract No. DE-AC02-06CH11357.
Footnotes
Supporting Information
Supporting Information methods, tables, and figures. This material is available free of charge via the internet at http://pubs.acs.org.
Accession Codes
Atomic coordinates and structure factors have been deposited in the Protein Data Bank: 5THY (SeMet CurJ C-MT), 5 THZ (wild type CurJ C-MT).
References
- 1.Cox RJ. Polyketides, proteins and genes in fungi: programmed nano-machines begin to reveal their secrets. Org Biomol Chem. 2007;5:2010–2026. doi: 10.1039/b704420h. [DOI] [PubMed] [Google Scholar]
- 2.Keatinge-Clay AT. The structures of type I polyketide synthases. Nat Prod Rep. 2012;29:1050–1073. doi: 10.1039/c2np20019h. [DOI] [PubMed] [Google Scholar]
- 3.Smith S, Tsai SC. The type I fatty acid and polyketide synthases: a tale of two megasynthases. Nat Prod Rep. 2007;24:1041–1072. doi: 10.1039/b603600g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Crawford JM, Townsend CA. New insights into the formation of fungal aromatic polyketides. Nat Rev Microbiol. 2010;8:879–889. doi: 10.1038/nrmicro2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gehring AM, DeMoll E, Fetherston JD, Mori I, Mayhew GF, Blattner FR, Walsh CT, Perry RD. Iron acquisition in plague: modular logic in enzymatic biogenesis of yersiniabactin by Yersinia pestis. Chem Biol. 1998;5:573–586. doi: 10.1016/s1074-5521(98)90115-6. [DOI] [PubMed] [Google Scholar]
- 6.Hendrickson L, Davis CR, Roach C, Nguyen DK, Aldrich T, McAda PC, Reeves CD. Lovastatin biosynthesis in Aspergillus terreus: characterization of blocked mutants, enzyme activities and a multifunctional polyketide synthase gene. Chem Biol. 1999;6:429–439. doi: 10.1016/s1074-5521(99)80061-1. [DOI] [PubMed] [Google Scholar]
- 7.Kennedy J, Auclair K, Kendrew SG, Park C, Vederas JC, Hutchinson CR. Modulation of polyketide synthase activity by accessory proteins during lovastatin biosynthesis. Science. 1999;284:1368–1372. doi: 10.1126/science.284.5418.1368. [DOI] [PubMed] [Google Scholar]
- 8.Chang Z, Sitachitta N, Rossi JV, Roberts MA, Flatt PM, Jia J, Sherman DH, Gerwick WH. Biosynthetic pathway and gene cluster analysis of curacin A, an antitubulin natural product from the tropical marine cyanobacterium Lyngbya majuscula. J Nat Prod. 2004;67:1356–1367. doi: 10.1021/np0499261. [DOI] [PubMed] [Google Scholar]
- 9.Edwards DJ, Marquez BL, Nogle LM, McPhail K, Goeger DE, Roberts MA, Gerwick WH. Structure and biosynthesis of the jamaicamides, new mixed polyketide-peptide neurotoxins from the marine cyanobacterium Lyngbya majuscula. Chem Biol. 2004;11:817–833. doi: 10.1016/j.chembiol.2004.03.030. [DOI] [PubMed] [Google Scholar]
- 10.Magarvey NA, Beck ZQ, Golakoti T, Ding Y, Huber U, Hemscheidt TK, Abelson D, Moore RE, Sherman DH. Biosynthetic characterization and chemoenzymatic assembly of the cryptophycins. Potent anticancer agents from cyanobionts. ACS Chem Biol. 2006;1:766–779. doi: 10.1021/cb6004307. [DOI] [PubMed] [Google Scholar]
- 11.Ramaswamy AV, Sorrels CM, Gerwick WH. Cloning and biochemical characterization of the hectochlorin biosynthetic gene cluster from the marine cyanobacterium Lyngbya majuscula. J Nat Prod. 2007;70:1977–1986. doi: 10.1021/np0704250. [DOI] [PubMed] [Google Scholar]
- 12.Sudek S, Lopanik NB, Waggoner LE, Hildebrand M, Anderson C, Liu H, Patel A, Sherman DH, Haygood MG. Identification of the putative bryostatin polyketide synthase gene cluster from “Candidatus Endobugula sertula”, the uncultivated microbial symbiont of the marine bryozoan Bugula neritina. J Nat Prod. 2007;70:67–74. doi: 10.1021/np060361d. [DOI] [PubMed] [Google Scholar]
- 13.Tillett D, Dittmann E, Erhard M, von Dohren H, Borner T, Neilan BA. Structural organization of microcystin biosynthesis in Microcystis aeruginosa PCC7806: an integrated peptide-polyketide synthetase system. Chem Biol. 2000;7:753–764. doi: 10.1016/s1074-5521(00)00021-1. [DOI] [PubMed] [Google Scholar]
- 14.Winter JM, Sato M, Sugimoto S, Chiou G, Garg NK, Tang Y, Watanabe K. Identification and characterization of the chaetoviridin and chaetomugilin gene cluster in Chaetomium globosum reveal dual functions of an iterative highly-reducing polyketide synthase. J Am Chem Soc. 2012;134:17900–17903. doi: 10.1021/ja3090498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Young J, Stevens DC, Carmichael R, Tan J, Rachid S, Boddy CN, Muller R, Taylor RE. Elucidation of gephyronic acid biosynthetic pathway revealed unexpected SAM-dependent methylations. J Nat Prod. 2013;76:2269–2276. doi: 10.1021/np400629v. [DOI] [PubMed] [Google Scholar]
- 16.Tang L, Shah S, Chung L, Carney J, Katz L, Khosla C, Julien B. Cloning and heterologous expression of the epothilone gene cluster. Science. 2000;287:640–642. doi: 10.1126/science.287.5453.640. [DOI] [PubMed] [Google Scholar]
- 17.Ansari MZ, Sharma J, Gokhale RS, Mohanty D. In silico analysis of methyltransferase domains involved in biosynthesis of secondary metabolites. BMC Bioinformatics. 2008;9:454. doi: 10.1186/1471-2105-9-454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liscombe DK, Louie GV, Noel JP. Architectures, mechanisms and molecular evolution of natural product methyltransferases. Nat Prod Rep. 2012;29:1238–1250. doi: 10.1039/c2np20029e. [DOI] [PubMed] [Google Scholar]
- 19.Kozbial PZ, Mushegian AR. Natural history of S-adenosylmethionine-binding proteins. BMC Struct Biol. 2005;5:19. doi: 10.1186/1472-6807-5-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maier T, Leibundgut M, Ban N. The crystal structure of a mammalian fatty acid synthase. Science. 2008;321:1315–1322. doi: 10.1126/science.1161269. [DOI] [PubMed] [Google Scholar]
- 21.Cacho RA, Thuss J, Xu W, Sanichar R, Gao Z, Nguyen A, Vederas JC, Tang Y. Understanding Programming of Fungal Iterative Polyketide Synthases: The Biochemical Basis for Regioselectivity by the Methyltransferase Domain in the Lovastatin Megasynthase. J Am Chem Soc. 2015;137:15688–15691. doi: 10.1021/jacs.5b11814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miller DA, Luo L, Hillson N, Keating TA, Walsh CT. Yersiniabactin synthetase: a four-protein assembly line producing the nonribosomal peptide/polyketide hybrid siderophore of Yersinia pestis. Chem Biol. 2002;9:333–344. doi: 10.1016/s1074-5521(02)00115-1. [DOI] [PubMed] [Google Scholar]
- 23.Poust S, Phelan RM, Deng K, Katz L, Petzold CJ, Keasling JD. Divergent mechanistic routes for the formation of gem-dimethyl groups in the biosynthesis of complex polyketides. Angew Chem Int Ed Engl. 2015;54:2370–2373. doi: 10.1002/anie.201410124. [DOI] [PubMed] [Google Scholar]
- 24.Stevens DC, Wagner DT, Manion HR, Alexander BK, Keatinge-Clay AT. Methyltransferases excised from trans-AT polyketide synthases operate on N-acetylcysteamine-bound substrates. J Antibiot (Tokyo) 2016 doi: 10.1038/ja.2016.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wagner DT, Stevens DC, Mehaffey MR, Manion HR, Taylor RE, Brodbelt JS, Keatinge-Clay AT. α-Methylation Follows Condensation in the Gephyronic Acid Modular Polyketide Synthase. Chem Commun (Camb) 2016 doi: 10.1039/c6cc04418b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gerwick WH, Proteau PJ, Nagle DG, Hamel E, Blokin A, Slate DL. Structure of curacin A, a novel antimitotic, antiproliferative, and brine shrimp toxic natural product from the marine cyanobacterium Lyngbya majuscula. J Org Chem. 1994;59:1243–1245. [Google Scholar]
- 27.Blokhin AV, Yoo H, Geralds RS, Nagle DG, Gerwick WH, Hamel E. Characterization of the Interaction of the Marine Cyanobacterial Natural Prouduct Curacin A with the Colchicine Site of Tubulin and Initial Structure-Activity Studies iwth Analogues. Mol Pharmacol. 1995 [PubMed] [Google Scholar]
- 28.Akey DL, Razelun JR, Tehranisa J, Sherman DH, Gerwick WH, Smith JL. Crystal structures of dehydratase domains from the curacin polyketide biosynthetic pathway. Structure. 2010;18:94–105. doi: 10.1016/j.str.2009.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gehret JJ, Gu L, Gerwick WH, Wipf P, Sherman DH, Smith JL. Terminal alkene formation by the thioesterase of curacin A biosynthesis: structure of a decarboxylating thioesterase. J Biol Chem. 2011;286:14445–14454. doi: 10.1074/jbc.M110.214635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McCarthy JG, Eisman EB, Kulkarni S, Gerwick L, Gerwick WH, Wipf P, Sherman DH, Smith JL. Structural basis of functional group activation by sulfotransferases in complex metabolic pathways. ACS Chem Biol. 2012;7:1994–2003. doi: 10.1021/cb300385m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gu L, Geders TW, Wang B, Gerwick WH, Hakansson K, Smith JL, Sherman DH. GNAT-like strategy for polyketide chain initiation. Science. 2007;318:970–974. doi: 10.1126/science.1148790. [DOI] [PubMed] [Google Scholar]
- 32.Gu L, Wang B, Kulkarni A, Geders TW, Grindberg RV, Gerwick L, Hakansson K, Wipf P, Smith JL, Gerwick WH, Sherman DH. Metamorphic enzyme assembly in polyketide diversification. Nature. 2009;459:731–735. doi: 10.1038/nature07870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Khare D, Hale WA, Tripathi A, Gu L, Sherman DH, Gerwick WH, Hakansson K, Smith JL. Structural Basis for Cyclopropanation by a Unique Enoyl-Acyl Carrier Protein Reductase. Structure. 2015;23:2213–2223. doi: 10.1016/j.str.2015.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Khare D, Wang B, Gu L, Razelun J, Sherman DH, Gerwick WH, Hakansson K, Smith JL. Conformational switch triggered by alpha-ketoglutarate in a halogenase of curacin A biosynthesis. Proc Natl Acad Sci U S A. 2010;107:14099–14104. doi: 10.1073/pnas.1006738107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Whicher JR, Smaga SS, Hansen DA, Brown WC, Gerwick WH, Sherman DH, Smith JL. Cyanobacterial polyketide synthase docking domains: a tool for engineering natural product biosynthesis. Chem Biol. 2013;20:1340–1351. doi: 10.1016/j.chembiol.2013.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Keatinge-Clay AT, Stroud RM. The structure of a ketoreductase determines the organization of the beta-carbon processing enzymes of modular polyketide synthases. Structure. 2006;14:737–748. doi: 10.1016/j.str.2006.01.009. [DOI] [PubMed] [Google Scholar]
- 37.Herbst DA, Jakob RP, Zahringer F, Maier T. Mycocerosic acid synthase exemplifies the architecture of reducing polyketide synthases. Nature. 2016;531:533–537. doi: 10.1038/nature16993. [DOI] [PubMed] [Google Scholar]
- 38.Piasecki SK, Zheng J, Axelrod AJ, Detelich ME, Keatinge-Clay AT. Structural and functional studies of a trans-acyltransferase polyketide assembly line enzyme that catalyzes stereoselective alpha- and beta-ketoreduction. Proteins. 2014;82:2067–2077. doi: 10.1002/prot.24561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zheng J, Gay DC, Demeler B, White MA, Keatinge-Clay AT. Divergence of multimodular polyketide synthases revealed by a didomain structure. Nat Chem Biol. 2012;8:615–621. doi: 10.1038/nchembio.964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zeng J, Wagner DT, Zhang Z, Moretto L, Addison JD, Keatinge-Clay AT. Portability and structure of the four-helix bundle docking domains of trans-acyltransferase modular polyketide synthases. ACS Chem Biol. 2016 doi: 10.1021/acschembio.6b00345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zheng J, Taylor CA, Piasecki SK, Keatinge-Clay AT. Structural and functional analysis of A-type ketoreductases from the amphotericin modular polyketide synthase. Structure. 2010;18:913–922. doi: 10.1016/j.str.2010.04.015. [DOI] [PubMed] [Google Scholar]
- 42.Zheng J, Piasecki SK, Keatinge-Clay AT. Structural studies of an A2-type modular polyketide synthase ketoreductase reveal features controlling alpha-substituent stereochemistry. ACS Chem Biol. 2013;8:1964–1971. doi: 10.1021/cb400161g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zheng J, Fage CD, Demeler B, Hoffman DW, Keatinge-Clay AT. The missing linker: a dimerization motif located within polyketide synthase modules. ACS Chem Biol. 2013;8:1263–1270. doi: 10.1021/cb400047s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Keatinge-Clay AT. A tylosin ketoreductase reveals how chirality is determined in polyketides. Chem Biol. 2007;14:898–908. doi: 10.1016/j.chembiol.2007.07.009. [DOI] [PubMed] [Google Scholar]
- 45.Bonnett SA, Whicher JR, Papireddy K, Florova G, Smith JL, Reynolds KA. Structural and stereochemical analysis of a modular polyketide synthase ketoreductase domain required for the generation of a cis-alkene. Chem Biol. 2013;20:772–783. doi: 10.1016/j.chembiol.2013.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Horowitz S, Dirk LM, Yesselman JD, Nimtz JS, Adhikari U, Mehl RA, Scheiner S, Houtz RL, Al-Hashimi HM, Trievel RC. Conservation and functional importance of carbon-oxygen hydrogen bonding in AdoMet-dependent methyltransferases. J Am Chem Soc. 2013;135:15536–15548. doi: 10.1021/ja407140k. [DOI] [PubMed] [Google Scholar]
- 47.Huang CC, Smith CV, Glickman MS, Jacobs WR, Jr, Sacchettini JC. Crystal structures of mycolic acid cyclopropane synthases from Mycobacterium tuberculosis. J Biol Chem. 2002;277:11559–11569. doi: 10.1074/jbc.M111698200. [DOI] [PubMed] [Google Scholar]
- 48.Meluzzi D, Zheng WH, Hensler M, Nizet V, Dorrestein PC. Top-down mass spectrometry on low-resolution instruments: characterization of phosphopantetheinylated carrier domains in polyketide and non-ribosomal biosynthetic pathways. Bioorg Med Chem Lett. 2008;18:3107–3111. doi: 10.1016/j.bmcl.2007.10.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sanchez C, Du L, Edwards DJ, Toney MD, Shen B. Cloning and characterization of a phosphopantetheinyl transferase from Streptomyces verticillus ATCC15003, the producer of the hybrid peptide-polyketide antitumor drug bleomycin. Chem Biol. 2001;8:725–738. doi: 10.1016/s1074-5521(01)00047-3. [DOI] [PubMed] [Google Scholar]
- 50.Li Y, Dodge GJ, Fiers WD, Fecik RA, Smith JL, Aldrich CC. Functional Characterization of a Dehydratase Domain from the Pikromycin Polyketide Synthase. J Am Chem Soc. 2015;137:7003–7006. doi: 10.1021/jacs.5b02325. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.