

Abstract
Objective:
To investigate CSF biomarkers in presymptomatic and symptomatic mutation carriers with hereditary cerebral hemorrhage with amyloidosis–Dutch type (HCHWA-D), a model for sporadic cerebral amyloid angiopathy, and to determine the earliest deposited form of β-amyloid (Aβ).
Methods:
HCHWA-D mutation carriers and controls were enrolled in the cross-sectional EDAN (Early Diagnosis of Amyloid Angiopathy Network) study. The HCHWA-D group was divided into symptomatic carriers with a previous intracerebral hemorrhage and presymptomatic carriers. CSF concentrations of Aβ40, Aβ42, total tau, and phosphorylated tau181 proteins were compared to those of controls of a similar age. Correlations between CSF biomarkers, MRI markers, and age were investigated with multivariate linear regression analyses.
Results:
We included 10 symptomatic patients with HCHWA-D (mean age 55 ± 6 years), 5 presymptomatic HCHWA-D carriers (mean age 36 ± 13 years), 31 controls <50 years old (mean age 31 ± 7 years), and 50 controls ≥50 years old (mean age 61 ± 8 years). After correction for age, CSF Aβ40 and Aβ42 were significantly decreased in symptomatic carriers vs controls (median Aβ40 1,386 vs 3,867 ng/L, p < 0.001; median Aβ42 289 vs 839 ng/L, p < 0.001) and in presymptomatic carriers vs controls (median Aβ40 3,501 vs 4,684 ng/L, p = 0.011; median Aβ42 581 vs 1,058 ng/L, p < 0.001). Among mutation carriers, decreasing CSF Aβ40 was associated with higher lobar microbleed count (p = 0.010), increasing white matter hyperintensity volume (p = 0.008), and presence of cortical superficial siderosis (p = 0.02).
Conclusions:
Decreased levels of CSF Aβ40 and Aβ42 occur before HCHWA-D mutation carriers develop clinical symptoms, implicating vascular deposition of both Aβ species as early steps in cerebral amyloid angiopathy pathogenesis. CSF Aβ40 and Aβ42 may serve as preclinical biomarkers of cerebral amyloid angiopathy pathology.
Sporadic cerebral amyloid angiopathy (sCAA) is a major cause of intracerebral hemorrhage (ICH), especially in the elderly. sCAA is caused by vascular accumulation of β-amyloid (Aβ), but much remains unknown about its underlying pathophysiology.1 Although clinical criteria for the diagnosis exist (the Boston criteria based on CAA-related hemorrhagic lesions on neuroimaging),2 the definite diagnosis of sCAA can be established only postmortem or by brain biopsy, which hampers research on this disease.3 Recognizing CAA in an early stage could limit bleeding complications by avoiding anticoagulation therapy or thrombolysis4 and could provide insights into the preclinical pathophysiology of CAA, knowledge needed for designing future preventive trials.
Hereditary cerebral hemorrhage with amyloidosis–Dutch type (HCHWA-D) provides a unique opportunity to study the preclinical phase of CAA. The underlying pathology of amyloid deposition is likely similar to that of sCAA.5 In HCHWA-D, Aβ accumulation is caused by a point mutation at codon 693 of the amyloid precursor protein (APP) gene located on chromosome 21, producing symptomatic disease with essentially 100% clinical penetrance. HCHWA-D can therefore be diagnosed by genetic analysis in presymptomatic individuals.6
Direct measurement of Aβ may identify early stages of CAA development. While in Alzheimer disease senile plaques in the brain parenchyma consist mainly of Aβ42, vascular deposits contain mostly Aβ40.7,8 In Alzheimer disease, decreased concentrations of Aβ42 and elevated concentrations of total tau (t-tau) and phosphorylated tau (p-tau) have been detected in the CSF.9 In patients with sCAA with advanced vascular damage, decreased CSF Aβ42 and Aβ40 and mildly elevated t-tau and p-tau concentrations have been found.10,11
We aimed to find biomarkers of the earliest, potentially reversible phases of CAA. We investigated whether altered CSF levels of Aβ and tau species are detectable in presymptomatic and symptomatic hereditary CAA mutation carriers.
METHODS
Study design and participants.
Patients and controls participated in the larger EDAN (Early Diagnosis of Amyloid Angiopathy Network) study, a collaboration among the Leiden University Medical Center (LUMC), Massachusetts General Hospital in Boston, and Erasmus University Medical Center in Rotterdam. The general aim of the study is to identify early biomarkers of CAA. In the LUMC, a cross-sectional study was performed to detect early MRI markers in HCHWA-D mutation carriers. In addition to MRI, all LUMC participants were asked to undergo a lumbar puncture.
Inclusion criteria for the LUMC EDAN study were age ≥18 years; diagnosis of HCHWA-D, family history of HCHWA-D, or willing to participate as control; able and willing to provide written informed consent; no contraindications for MRI.
In all participants, genetic testing was performed for the mutation in the APP gene.6 After DNA was isolated from blood samples, the APP mutation test was performed with APP PCR and sequencing.6 Because both mutation-positive and -negative participants were enrolled, at-risk individuals who did not wish to know their genetic status could still participate in the EDAN study without learning their mutation status.
HCHWA-D mutation carriers were divided into a symptomatic and a presymptomatic group. Symptomatic participants with HCHWA-D were defined as mutation carriers who had previously developed one or multiple clinical ICHs, of which at least one caused clinical symptoms and was confirmed by CT or MRI. Presymptomatic carriers were defined as participants without previous symptomatic ICH. EDAN controls were genetically negative controls and were mainly spouses or family members who did not carry the APP mutation.
CSF samples from additional controls of a similar age were obtained from the Radboud University Nijmegen Medical Center (RUNMC). These controls were 18- to 79-year-old individuals who visited the neurology outpatient clinic of the RUNMC for various reasons but had no neurologic diagnosis after the diagnostic workup. CSF samples were obtained as part of the clinical diagnostic workup. Samples were coded and used with the consent of the patients. Their CSF samples contained normal leukocyte and erythrocyte counts, normal glucose and lactate levels, normal total protein, and no oligoclonal immunoglobulin G bands. The data of the RUNMC controls >50 years of age have been previously published.10
We divided the controls into those <50 years and those ≥50 years of age. For both the EDAN participants and the additional RUNMC controls, demographic data were collected. EDAN participants underwent a neurologic examination by a neurologist (M.J.H.W. or G.M.T.), including the NIH Stroke Scale and modified Rankin Scale. Cognitive screening was performed by a neuropsychologist (S.v.R.), including a Mini-Mental State Examination.
Standard protocol approvals, registrations, and patient consents.
The ethics committee of the LUMC approved the study protocol, and written informed consent was obtained from all participants.
Imaging.
EDAN participants underwent 3T MRI before the lumbar puncture on an Achieva MRI scanner using a standard 32-channel head coil (Philips Medical Systems, Best, the Netherlands). Three-dimensional T1-weighted images with repetition time (TR)/echo time (TE) = 9.7/4.6 milliseconds, flip angle (FA) = 8°, and nominal voxel size (1.17 × 1.17 × 1.4 mm); T2-weighted images (TR/TE = 4,200/80 milliseconds, FA = 90°); fluid-attenuated inversion recovery images (TR/TE = 11,000/125 milliseconds, FA = 90°); and T2*-weighted images (TR/TE = 45/31 milliseconds, FA = 13°) were acquired.
Microbleeds were identified on T2* and defined as punctate, hypointense foci (<5 mm in diameter) involving the cortex, distinct from vascular flow voids.12 and cortical superficial siderosis (cSS), which was defined as linear gyriform hypointensities13 scored as absent or present. Enlarged perivascular spaces (EPVSs) were rated on T2-weighted sequences according to the STRIVE (Standards for Reporting Vascular Changes on Neuroimaging) recommendations14 in the centrum semiovale and basal ganglia and, in line with previous studies,15,16 dichotomized as low (<20) or high (>20). White matter hyperintensity was defined as areas of increased signal intensity within the white matter on both fluid-attenuated inversion recovery and T2-weighted images. White matter hyperintensity volume was measured semiautomatically as previously described.17
CSF analysis.
For the CSF study described here, we included all participants who consented to undergo a lumbar puncture under standardized conditions. CSF samples of all participants were obtained with the same protocol. We collected CSF in polypropylene tubes, transferred the samples to laboratories within 30 minutes at 4°C, centrifuged them (622g for 5 minutes at 4°C–8°C), and stored the samples in polypropylene aliquots at −80°C. CSF was analyzed for Aβ40, Aβ42, t-tau, and p-tau181. All analyses were performed in the RUNMC with an ELISA as described previously (all from Innogenetics NV, Gent, Belgium).18 We performed standard CSF analysis, including leukocyte count, erythrocyte count, total protein, and glucose. A leucocyte count ≤4/μL, red blood cells <1,500/μL, total protein <700 mg/L, and glucose <4.3 mmol/L were considered normal. CSF results from the RUNMC and EDAN controls did not differ significantly. Personnel performing the analyses were blinded to clinical diagnosis.
Statistical analyses.
We compared CSF concentrations and the Aβ40/Aβ42 ratios between patients with HCHWA-D and controls, as well as the presence of cSS and high number of EPVSs, using the Mann-Whitney test. We compared presymptomatic mutation carriers with controls <50 years old and symptomatic mutation carriers with controls ≥50 years old. To correct for the remaining age effect, we performed multivariate linear regression analyses for CSF values (square root transformed) with age as a covariate and a categorical variable with 3 levels (presymptomatic vs symptomatic vs controls) as a factor. Multivariate linear regression correcting for age was used to assess the correlation between CSF levels, microbleed count, and white matter hyperintensity volume in mutation carriers. Microbleed count was transferred logarithmically (in case of zero microbleeds, the log[microbleeds] was set at zero). To look at the correlation between age, Aβ40, and Aβ42, scatterplots with separate regression lines for mutation carriers and controls were plotted. Intercepts at the mean age of the study population and the slopes of the regression lines were compared with the use of a multivariate linear regression model.
RESULTS
Between January 2013 and April 2014, 57 participants were enrolled in the EDAN study at the LUMC. Twenty-five of the 57 participants gave consent for a lumbar puncture. Of these, 5 were presymptomatic HCHWA-D mutation carriers, 11 were symptomatic mutation carriers, and 9 were controls. CSF samples of 73 additional controls from the RUNMC were collected.
Presymptomatic carriers had a mean age of 36 ± 13 years and were all women (table 1). None of them had neurologic or cognitive symptoms. One presymptomatic patient had 32 lobar microbleeds on 3T MRI and one slightly larger (5.4 × 3.5 mm), asymptomatic hemorrhage in the right occipital lobe. The other 4 participants had no microbleeds and only a little white matter hyperintensity volume (median 1 mL). Presymptomatic mutation carriers who did not undergo a lumbar puncture were slightly younger (mean age 33 years) and were more often men (M/F 3/4 vs 0/5) compared with presymptomatic carriers who did participate in the CSF substudy.
Table 1.
Clinical characteristics
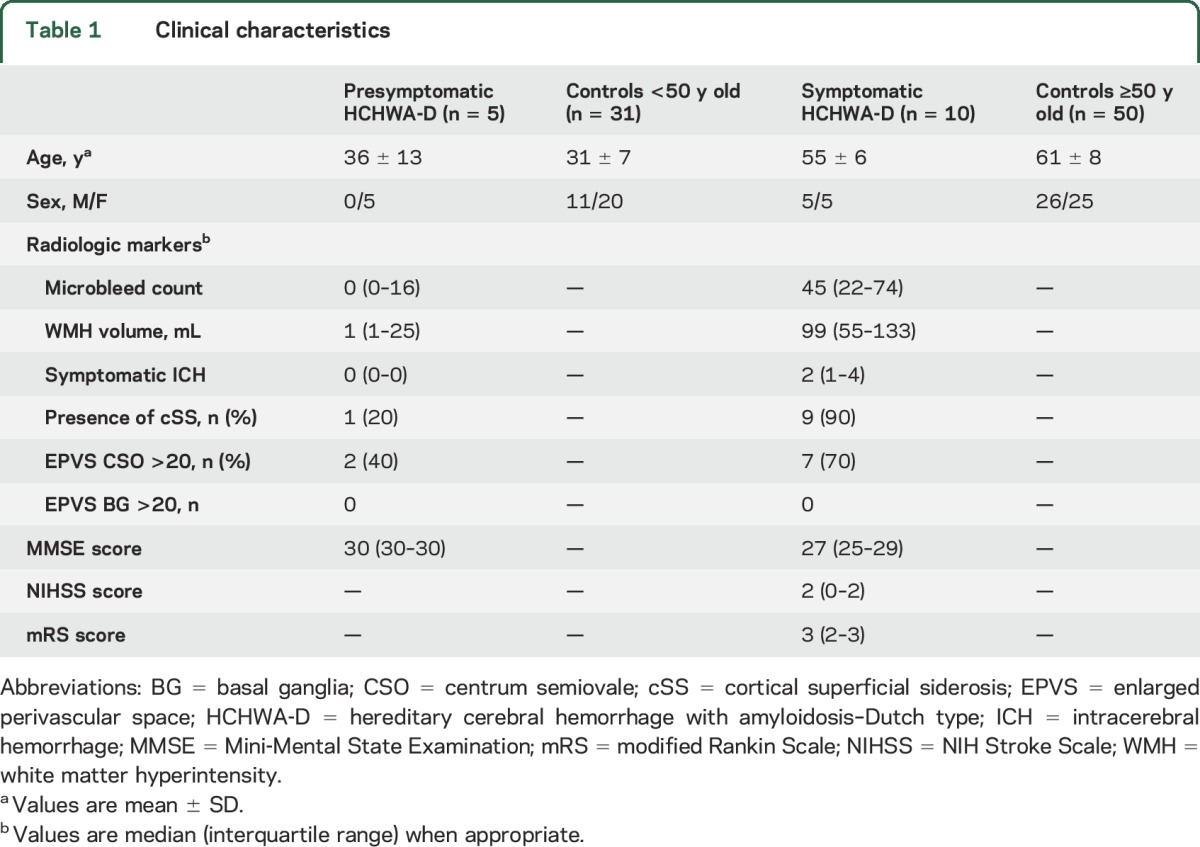
Symptomatic carriers had a mean age of 55 ± 6 years and an equal sex distribution. One symptomatic carrier was excluded from the analysis because of a high erythrocyte count. All symptomatic carriers had at least one symptomatic ICH confirmed by previous MRI (6 carriers had >1 ICH). They had a median NIH Stroke Scale score of 2, a median modified Rankin Scale score of 3, and a median Mini-Mental State Examination score of 27 (table 1). Symptomatic carriers who did not undergo lumbar puncture did not differ in age or sex distribution from symptomatic carriers who participated in the CSF study.
In total, 31 participants were included in the control group <50 years old (mean age 31 ± 7 years) and 50 in the control group ≥50 years of age (mean age 61 ± 8 years). One EDAN control ≥50 years of age was excluded from the analyses because of a high total protein in the CSF.
CSF concentrations.
Median CSF concentrations are shown in table 2. Both CSF Aβ40 concentration (p < 0.001, figure 1A) and CSF Aβ42 concentration (p < 0.001, figure 1B) in symptomatic carriers were decreased compared with controls ≥50 years of age. Similarly, in presymptomatic mutation carriers, both Aβ40 (p = 0.005, figure 1A) and Aβ42 (p = 0.001, figure 1B) were decreased compared with controls <50 years. Aβ40 and Aβ42 levels were lower in symptomatic HCHWA-D mutation carriers compared with presymptomatic mutation carriers (p = 0.002 for Aβ40 and p = 0.003 for Aβ42, figure 1, A and B). After correction for the remaining age effect, both Aβ40 and Aβ42 remained decreased in symptomatic mutation carriers (p < 0.001 for Aβ40 and p < 0.001 for Aβ42) and presymptomatic mutation carriers (p = 0.011 for Aβ40 and p < 0.001 for Aβ42) compared with controls. Furthermore, the Aβ40/Aβ42 ratio was not significantly higher in presymptomatic compared to symptomatic HCHWA-D mutation carriers (6.2 vs 4.8; p = 0.18).
Table 2.
β-Amyloid and tau protein CSF levels
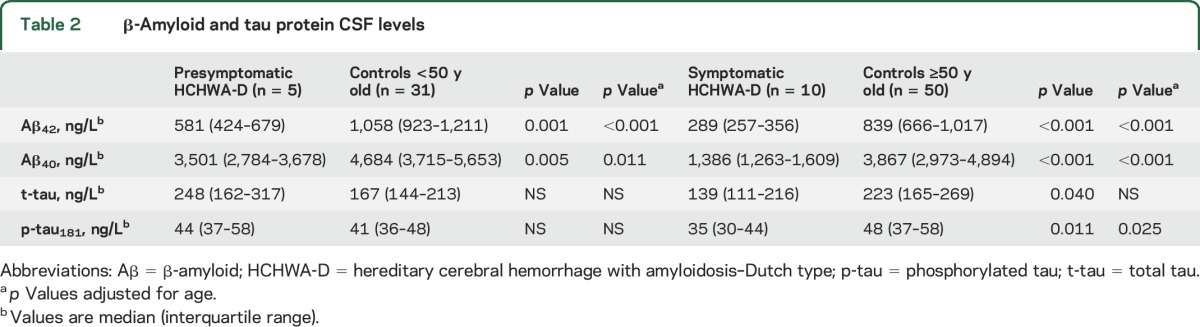
Figure 1. CSF measurements.

Scatterplots of CSF measurements of Aβ40 (A), Aβ42 (B), total tau (t-tau; C), and phosphorylated tau181 (p-tau181; D). Aβ = β-amyloid; HCHWA-D = hereditary cerebral hemorrhage with amyloidosis–Dutch type.
In symptomatic HCHWA-D mutation carriers, CSF t-tau (p = 0.040, figure 1C) and CSF p-tau181 (p = 0.011, figure 1D) concentrations were decreased compared with controls ≥50 years of age. After correction for age, this difference was not significant for t-tau (p = 0.061) but remained significant for p-tau181 (p = 0.025). In presymptomatic HCHWA-D carriers, there was no difference in the levels of tau protein (figure 1, C and D). The levels of p-tau181 and t-tau were not different between presymptomatic and symptomatic mutation carriers.
CSF concentrations and MRI markers.
In mutation carriers, both CSF Aβ40 and CSF Aβ42 concentrations decreased with increasing number of microbleeds (r = −0.77, p = 0.001 for Aβ40, figure 2A; and r = −0.60, p = 0.018 for Aβ42, figure 2B) and increasing white matter hyperintensity volume (r = −0.73, p = 0.002 for Aβ40, figure 2C; and r = −0.55, p = 0.034 for Aβ42, figure 2D). After correction for age, Aβ40 still correlated significantly with microbleed count (p = 0.010) and white matter hyperintensity volume (p = 0.008), but Aβ42 did not (microbleed count p = 0.64, white matter hyperintensity volume p = 0.47). The median Aβ40 and Aβ42 were lower when cSS was present (Aβ40 p = 0.02, Aβ42 p = 0.05) and when mutation carriers had a high number (>20) of EPVSs in the centrum semiovale (Aβ40 p = 0.10, Aβ42 p = 0.10). The number of ICH did not correlate with CSF concentrations.
Figure 2. CSF measurements and MRI markers.

Correlations between CSF Aβ40, Aβ42, microbleed (MB) count, and white matter hyperintensity (WMH) volume in presymptomatic mutation carriers (filled squares) and symptomatic mutation carriers (filled circles). After correction for age, Aβ40 remained a significant predictor for MB count (p = 0.010) and WMH volume (p = 0.008). Aβ = β-amyloid.
CSF concentrations and age.
Figure 3 shows correlations of age with CSF Aβ40 (figure 3A) and Aβ42 (figure 3B) for all participants. There was a yearly decrease in both Aβ40 and Aβ42. Age was a significant predictor for Aβ concentration in mutation carriers (Aβ40 p = 0.001, Aβ42 p < 0.001) but not in controls (Aβ40 p = 0.060, Aβ42 p = 0.062). At the mean age of the whole study population (49.5 years), the mean Aβ concentration of the mutation carriers was significantly lower (Aβ40 1,989 vs 4,373 ng/L, p < 0.001, figure 3A; and Aβ42 378 vs 939 ng/L, p < 0.001, figure 3B), but the slopes of the regression lines were not significantly different.
Figure 3. CSF markers and age.

Correlations between CSF β-amyloid (Aβ) with age in the combined presymptomatic (filled squares) and symptomatic (filled circles) mutation carriers and in the combined controls <50 years old (open squares) and controls ≥50 years old (open circles). Age vs Aβ40 (A): black line r = −0.64, p = 0.001; dotted line r = −0.34, p = 0.060. Age vs Aβ42 (B): black line r = −0.71, p < 0.001; dotted line r = −0.47 p = 0.062.
DISCUSSION
This study shows decreased CSF Aβ40 and Aβ42 concentrations in presymptomatic hereditary CAA mutation carriers, reflecting the earliest phase of CAA pathogenesis. Decreased Aβ concentrations can be demonstrated before imaging abnormalities associated with CAA-related pathology are found and continue to decrease in symptomatic patients with hereditary CAA. Thus, CSF Aβ concentrations may serve as biomarkers for early stages of CAA. Moreover, a high microbleeds count and white matter hyperintensity volume correlated with decreasing Aβ40 concentrations, and the presence of cSS was related to a lower Aβ40 and Aβ42 in mutation carriers.
Aβ is secreted by neurons and transported along the perivascular and glymphatic pathways to the subarachnoid space.19 The perivascular pathway transports Aβ via the interstitial fluid toward the cerebral vasculature. Because CAA is caused by Aβ accumulation in the vessel wall,20 the perivascular pathway is most likely involved in CAA pathophysiology.21
Two previous studies in patients with sporadic CAA also demonstrated decreased CSF Aβ40 and Aβ42 levels.10,11 However, in this study, the reduction in Aβ40 and Aβ42 was more pronounced, most likely because of the more severe phenotype of HCHWA-D.22 Reduction in both CSF Aβ40 and Aβ42 concentrations in HCHWA-D and sCAA differs from the pattern of low CSF Aβ42 and normal Aβ40 in patients with Alzheimer disease. Differences in CSF Aβ40 are presumably attributable to the observations that cerebrovascular Aβ deposits contain substantial amounts of Aβ407 and parenchymal senile plaques contain mainly Aβ42.8 Our data hinted at the possibility that presymptomatic patients had relatively greater reductions in Aβ42 (55% of control values) than Aβ40 (75% of control values; figure 1 and table 2). This observation (which will require larger numbers of participants to confirm) likely relates to reports that in the process of Aβ accumulation in the vessel wall, Aβ42 deposits earlier than Aβ40.23,24 Aβ42 is believed to seed in the vessels, followed by growth of these deposits by Aβ40 deposition, leading to vessel pathology.23,25–27
Because HCHWA-D is a dominantly inherited disorder, mutation carriers are heterozygous and produce both wild-type Aβ and mutant Dutch Aβ.28 The assays used in this study, however, measured total amounts of CSF Aβ. Future studies are needed to separately identify the amounts of wild-type and Dutch Aβ present in the CSF of patients with HCHWA-D.
It has been hypothesized that Aβ-related pathology in Alzheimer disease starts decades before clinical symptoms appear. The Dominantly Inherited Alzheimer Network (DIAN) investigated CSF markers in carriers and noncarriers of autosomal dominant Alzheimer disease. Because the age at symptom onset is similar between generations, it can be estimated for presymptomatic mutation carriers.29 The DIAN studies showed that in mutation carriers Aβ42 starts to decline ≈25 years before the expected symptom onset.30 Although the age at onset in patients with HCHWA-D is not consistent within families, most mutation carriers develop their first ICH between 45 and 60 years of age.31 Because the group of presymptomatic HCHWA-D carriers had a mean age of 36 years, our results suggest that abnormal Aβ levels can be detected in the CSF at least 10 years before the disease symptoms manifest. The youngest presymptomatic mutation carrier with decreased Aβ concentrations was in the early 20s.
Increasing microbleed count, increasing white matter hyperintensity volume, and the presence of superficial siderosis were associated with decreasing CSF Aβ40 and Aβ42 levels. In the 4 presymptomatic carriers without microbleeds, with minimal white matter hyperintensity volume, and without indications for cSS, the CSF Aβ40 and Aβ42 levels were also decreased. This suggests that decreased CSF Aβ levels can be measured before radiologic markers of CAA can be found and may be good biomarkers for the earlier stages of CAA. Furthermore, Aβ40 was significantly associated with increasing microbleed count and increasing white matter hyperintensity volume independently of age.
We found significantly lower CSF p-tau181 concentrations only in symptomatic mutation carriers, after correction for age, albeit with complete overlap in the data points. Previous studies showed mildly increased levels of tau species in participants with sCAA compared with controls, although the levels were lower than in participants with Alzheimer disease.10,11 In Alzheimer disease, elevated CSF t-tau and p-tau181 concentrations are presumably related to the formation of neurofibrillary tangles, although elevated tau species also might represent a nonspecific axonal injury.32 The finding of elevated CSF tau levels in patients with sCAA might represent mixed CAA and Alzheimer pathology. In HCHWA-D, however, neuropathologic examination revealed no neurofibrillary tangles,33,34 consistent with the current CSF results. Taken together, these data suggest that HCHWA-D represents a pure form of CAA pathology without accompanying tau pathology.
The main limitation of this study is the relatively small sample size, which, however, reflects the limited number of available patients with HCHWA-D for in vivo examinations. Despite the small number of patients, we could still demonstrate significant differences in CSF Aβ biomarkers. Furthermore, the definition of presymptomatic and symptomatic patients, based solely on a symptomatic ICH, is debatable. Occasionally, patients with HCHWA-D experience cognitive decline before their first ICH.31 Additionally, small ICHs might occur without obvious clinical symptoms. In this study, only one presymptomatic mutation carrier showed multiple microbleeds and one slightly larger, asymptomatic hemorrhage. Still, none of the presymptomatic carriers demonstrated neurologic or cognitive symptoms. Lastly, our controls were derived from 2 different studies, the EDAN study and a cohort from RUNMC. However, all samples were handled the same and were analyzed in one laboratory using standardized techniques. Furthermore, CSF results did not differ significantly. For the RUNMC control group, no MRI scans were available. Therefore, we cannot rule out that some controls might have preclinical sCAA, although the controls were known to be without a clinical neurologic disorder. In addition, any undetected sCAA in the RUNMC controls would be expected to bias toward a null result for comparisons with participants with HCHWA-D.
In this hereditary form of CAA, both CSF Aβ40 and Aβ42 concentrations are markers of the earliest phase of CAA-related pathology before clinical or radiologic findings appear. This finding provides insight into the pathogenesis of CAA and provides important information for future trials aimed at preventing CAA-related ICH. Longitudinal studies in this unique hereditary CAA group will help to further unravel the dynamics of CSF biomarkers over time and their relationship to disease progression.
GLOSSARY
- Aβ
β-amyloid
- APP
amyloid precursor protein
- CAA
cerebral amyloid angiopathy
- cSS
cortical superficial siderosis
- DIAN
Dominantly Inherited Alzheimer Network
- EDAN
Early Diagnosis of Amyloid Angiopathy Network
- EPVS
enlarged perivascular space
- FA
flip angle
- HCHWA-D
hereditary cerebral hemorrhage with amyloidosis–Dutch type
- ICH
intracerebral hemorrhage
- LUMC
Leiden University Medical Center
- p-tau
phosphorylated tau
- RUNMC
Radboud University Nijmegen Medical Center
- sCAA
sporadic cerebral amyloid angiopathy
- STRIVE
Standards for Reporting Vascular Changes on Neuroimaging
- TE
echo time
- TR
repetition time
- t-tau
total tau
AUTHOR CONTRIBUTIONS
J.v.d.G, S.M.G., M.A.v.B, J.H., M.J.H.W., and G.M.T. contributed to conception and design of the study. E.S.v.E., M.M.V., R.Z., S.v.R., A.M.v.O., M.J.H.W., and G.M.T. contributed to acquisition of data. E.W.v.Z. provided statistical advice, and E.S.v.E. and E.W.v.Z. did the analyses. E.S.v.E., M.M.V., M.J.H.W., and G.M.T. contributed to drafting a significant portion of the manuscript and figures.
STUDY FUNDING
Funding was provided by NIH R01 NS070834 (S.M.G.) This study was part of the CAVIA project (No. 733050202; M.M.V.), which has been made possible by ZonMW. The CAVIA project is part of Memorabel, the research and innovation program for dementia, as part of the Dutch national Deltaplan for Dementia (zonmw.nl/dementiaresearch). The CAVIA project is a consortium of RUNMC, LUMC, Erasmus University Medical Center, VU University Medical Center, ADX Neurosciences, Philips Healthcare, Stony Brook University, and Massachusetts General Hospital.
DISCLOSURE
E. van Etten reports no disclosures relevant to the manuscript. M. Verbeek was member of an advisory board for Roche. J. van der Grond, R. Zielman, S. van Rooden, E. van Zwet, A. van Opstal, J. Haan, S. Greenberg, M. van Buchem, and M. Wermer report no disclosures relevant to the manuscript. G. Terwindt reports independent support from NWO, ZonMW, the Dutch Heart Foundation, and the Dutch Brain Foundation. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Viswanathan A, Greenberg SM. Cerebral amyloid angiopathy in the elderly. Ann Neurol 2011;70:871–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Knudsen KA, Rosand J, Karluk D, Greenberg SM. Clinical diagnosis of cerebral amyloid angiopathy: validation of the Boston criteria. Neurology 2001;56:537–539. [DOI] [PubMed] [Google Scholar]
- 3.Greenberg SM. Cerebral amyloid angiopathy: prospects for clinical diagnosis and treatment. Neurology 1998;51:690–694. [DOI] [PubMed] [Google Scholar]
- 4.Charidimou A, Gang Q, Werring DJ. Sporadic cerebral amyloid angiopathy revisited: recent insights into pathophysiology and clinical spectrum. J Neurol Neurosurg Psychiatry 2012;83:124–137. [DOI] [PubMed] [Google Scholar]
- 5.Maat-Schieman ML, van Duinen SG, Bornebroek M, Haan J, Roos RA. Hereditary cerebral hemorrhage with amyloidosis-Dutch type (HCHWA-D), II: a review of histopathological aspects. Brain Pathol 1996;6:115–120. [DOI] [PubMed] [Google Scholar]
- 6.Bakker E, van Broeckhoven C, Haan J, et al. DNA diagnosis for hereditary cerebral hemorrhage with amyloidosis (Dutch type). Am J Hum Genet 1991;49:518–521. [PMC free article] [PubMed] [Google Scholar]
- 7.Herzig MC, Winkler DT, Burgermeister P, et al. Abeta is targeted to the vasculature in a mouse model of hereditary cerebral hemorrhage with amyloidosis. Nat Neurosci 2004;7:954–960. [DOI] [PubMed] [Google Scholar]
- 8.Smith EE, Greenberg SM. Beta-amyloid, blood vessels, and brain function. Stroke 2009;40:2601–2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Jong D, Kremer BP, Olde Rikkert MG, Verbeek MM. Current state and future directions of neurochemical biomarkers for Alzheimer's disease. Clin Chem Lab Med 2007;45:1421–1434. [DOI] [PubMed] [Google Scholar]
- 10.Verbeek MM, Kremer BP, Rikkert MO, Van Domburg PH, Skehan ME, Greenberg SM. Cerebrospinal fluid amyloid beta(40) is decreased in cerebral amyloid angiopathy. Ann Neurol 2009;66:245–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Renard D, Castelnovo G, Wacongne A, et al. Interest of CSF biomarker analysis in possible cerebral amyloid angiopathy cases defined by the modified Boston criteria. J Neurol 2012;259:2429–2433. [DOI] [PubMed] [Google Scholar]
- 12.Greenberg SM, Vernooij MW, Cordonnier C, et al. Cerebral microbleeds: a guide to detection and interpretation. Lancet Neurol 2009;8:165–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feldman HH, Maia LF, Mackenzie IR, Forster BB, Martzke J, Woolfenden A. Superficial siderosis: a potential diagnostic marker of cerebral amyloid angiopathy in Alzheimer disease. Stroke 2008;39:2894–2897. [DOI] [PubMed] [Google Scholar]
- 14.Wardlaw JM, Smith EE, Biessels GJ, et al. Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurol 2013;12:822–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhu YC, Tzourio C, Soumare A, Mazoyer B, Dufouil C, Chabriat H. Severity of dilated Virchow-Robin spaces is associated with age, blood pressure, and MRI markers of small vessel disease: a population-based study. Stroke 2010;41:2483–2490. [DOI] [PubMed] [Google Scholar]
- 16.Charidimou A, Boulouis G, Haley K, et al. White matter hyperintensity patterns in cerebral amyloid angiopathy and hypertensive arteriopathy. Neurology 2016;86:505–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Foster-Dingley JC, Moonen JE, van den Berg-Huijsmans AA, et al. Lower blood pressure and gray matter integrity loss in older persons. J Clin Hypertens 2015;17:630–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reijn TS, Rikkert MO, van Geel WJ, de Jong D, Verbeek MM. Diagnostic accuracy of ELISA and xMAP technology for analysis of amyloid beta(42) and tau proteins. Clin Chem 2007;53:859–865. [DOI] [PubMed] [Google Scholar]
- 19.Tarasoff-Conway JM, Carare RO, Osorio RS, et al. Clearance systems in the brain: implications for Alzheimer disease. Nat Rev Neurol 2015;11:457–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Attems J, Jellinger K, Thal DR, Van Nostrand W. Review: sporadic cerebral amyloid angiopathy. Neuropathol Appl Neurobiol 2011;37:75–93. [DOI] [PubMed] [Google Scholar]
- 21.Weller RO, Subash M, Preston SD, Mazanti I, Carare RO. Perivascular drainage of amyloid-beta peptides from the brain and its failure in cerebral amyloid angiopathy and Alzheimer's disease. Brain Pathol 2008;18:253–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang-Nunes SX, Maat-Schieman ML, van Duinen SG, Roos RA, Frosch MP, Greenberg SM. The cerebral beta-amyloid angiopathies: hereditary and sporadic. Brain Pathol 2006;16:30–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shinkai Y, Yoshimura M, Ito Y, et al. Amyloid beta-proteins 1-40 and 1-42(43) in the soluble fraction of extra- and intracranial blood vessels. Ann Neurol 1995;38:421–428. [DOI] [PubMed] [Google Scholar]
- 24.Natte R, Yamaguchi H, Maat-Schieman ML, et al. Ultrastructural evidence of early non-fibrillar Abeta42 in the capillary basement membrane of patients with hereditary cerebral hemorrhage with amyloidosis, Dutch type. Acta Neuropathol 1999;98:577–582. [DOI] [PubMed] [Google Scholar]
- 25.Alonzo NC, Hyman BT, Rebeck GW, Greenberg SM. Progression of cerebral amyloid angiopathy: accumulation of amyloid-beta40 in affected vessels. J Neuropathol Exp Neurol 1998;57:353–359. [DOI] [PubMed] [Google Scholar]
- 26.Maat-Schieman ML, van Duinen SG, Rozemuller AJ, Haan J, Roos RA. Association of vascular amyloid beta and cells of the mononuclear phagocyte system in hereditary cerebral hemorrhage with amyloidosis (Dutch) and Alzheimer disease. J Neuropathol Exp Neurol 1997;56:273–284. [DOI] [PubMed] [Google Scholar]
- 27.Vinters HV, Secor DL, Read SL, et al. Microvasculature in brain biopsy specimens from patients with Alzheimer's disease: an immunohistochemical and ultrastructural study. Ultrastruct Pathol 1994;18:333–348. [DOI] [PubMed] [Google Scholar]
- 28.Herzig MC, Van Nostrand WE, Jucker M. Mechanism of cerebral beta-amyloid angiopathy: murine and cellular models. Brain Pathol 2006;16:40–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lopera F, Ardilla A, Martinez A, et al. Clinical features of early-onset Alzheimer disease in a large kindred with an E280A presenilin-1 mutation. JAMA 1997;277:793–799. [PubMed] [Google Scholar]
- 30.Bateman RJ, Xiong C, Benzinger TL, et al. Clinical and biomarker changes in dominantly inherited Alzheimer's disease. N Engl J Med 2012;367:795–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bornebroek M, Haan J, Maat-Schieman ML, Van Duinen SG, Roos RA. Hereditary cerebral hemorrhage with amyloidosis-Dutch type (HCHWA-D), I: a review of clinical, radiologic and genetic aspects. Brain Pathol 1996;6:111–114. [DOI] [PubMed] [Google Scholar]
- 32.Blennow K, Vanmechelen E, Hampel H. CSF total tau, Abeta42 and phosphorylated tau protein as biomarkers for Alzheimer's disease. Mol Neurobiol 2001;24:87–97. [DOI] [PubMed] [Google Scholar]
- 33.van Duinen SG, Castano EM, Prelli F, Bots GT, Luyendijk W, Frangione B. Hereditary cerebral hemorrhage with amyloidosis in patients of Dutch origin is related to Alzheimer disease. Proc Natl Acad Sci USA 1987;84:5991–5994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tagliavini F, Ghiso J, Timmers WF, Giaccone G, Bugiani O, Frangione B. Coexistence of Alzheimer's amyloid precursor protein and amyloid protein in cerebral vessel walls. Lab Invest 1990;62:761–767. [PubMed] [Google Scholar]