

Key Clinical Message
Patients with antisynthetase‐positive interstitial lung disease (ILD) alone sometimes develop myositis during follow‐up, but myasthenia gravis (MG) overlapping on antisynthetase syndrome is unusual. A 56‐year‐old woman with ILD and anti‐EJ antibody treated for 8 years developed MG. Physicians should consider myositis and MG when patients develop muscle symptoms during follow‐up.
Keywords: Anti‐EJ antibody, antisynthetase syndrome, dermatomyositis, myasthenia gravis, polymyositis
Introduction
Antisynthetase syndrome (ASS) is a heterogeneous systemic autoimmune syndrome characterized by inflammatory myopathy, arthritis or arthralgias, interstitial lung disease (ILD), fever, Raynaud's phenomenon, and mechanic's hands 1, 2 associated with antisynthetase antibodies. There are three patterns of onset of ILD and myositis: simultaneous onset, myositis preceding ILD, and ILD preceding myositis. When physicians treat and follow patients with ASS‐positive ILD alone, it is well known that careful attention should be paid to the development of myositis during follow‐up. Here, we report a 56‐year‐old woman whose ILD with anti‐EJ antibody had been treated, but 8 years after the onset of ILD, myasthenia gravis (MG) developed. MG is an autoimmune disorder characterized by weakness and skeletal muscle fatigue, and it is sometimes associated with other autoimmune disorders. However, only two cases of polymyositis (PM) with positive antisynthetase antibody‐positive PM overlapping MG have been reported 3, 4. Both had anti‐Jo‐1 antibody, and there have been no reports of patients with antisynthetase antibody other than anti‐Jo‐1 antibody who had overlapping MG. Our case suggests that not only myositis but also overlapping of MG should be considered when antisynthetase antibody‐positive ILD patients develop muscle symptoms during their follow‐up.
Case Report
A 56‐year‐old woman with no past history or family history of note developed dyspnea on effort in November 2007 without muscle pain or arthralgia and presented to a local physician. Heliotrope rush and Gottron's papules were not found. Serum muscle enzymes were within normal range, and she was negative for anti‐Jo‐1 antibody, anti‐Scl‐70 antibody, anti‐dsDNA antibody, anti‐SS‐A/Ro antibody, anti‐SS‐B/La antibody, anti‐RNP antibody, and antinuclear cytoplasmic antibodies. Chest X‐ray showed reticular shadows and ground‐glass opacities (GGOs) in both lung fields, and her serum KL‐6 was elevated to 1460 U/mL. She was diagnosed as having idiopathic interstitial pneumonia, and prednisolone (PSL) 30 mg/day was started. As a result, her conditions improved, as shown by chest X‐rays, and the dose of PSL was tapered; however, she developed dyspnea and dry cough when the daily dose of PSL was tapered to 10 mg/day. Another chest X‐ray showed the development of new GGOs in both lung fields, and she was referred to Saitama Cardiovascular and Respiratory Center for further evaluation in August 2008.
On presentation, auscultation detected fine crackles in both chest fields. She did not have any muscle weakness. Mechanic's hands were found, but heliotrope rush and Gottron's papules, which are characteristic of PM/dermatomyositis (DM), were not found. Arterial blood gas analysis showed a pH of 7.46, PaCO2 of 39.2 Torr, and PaO2 of 78.5 Torr under ambient air, and her serum KL‐6 was 1982 U/mL. Serum antinuclear antibody was 160 titers (speckled pattern), and she was negative for anti‐Jo‐1 antibody, anti‐dsDNA antibody, anti‐SS‐A/Ro antibody, anti‐SS‐B/La antibody, anti‐RNP antibody, anti‐Scl‐70 antibody, and anti‐CCP antibody. Chest X‐ray showed reticular shadows in both lung fields, and chest computed tomography showed GGOs and reticular shadows predominantly in the lower lobes (Fig. 1A and B). Bronchoalveolar lavage fluid showed 48.6% macrophages, 39.3% lymphocytes, 5.6% neutrophils, and 6.5% eosinophils, but the specimen obtained via transbronchial lung biopsy was inadequate for evaluation. We suspected her of having ASS, and serum antisynthetase antibodies other than anti‐Jo‐1 antibody were investigated by an immunoprecipitation method as reported elsewhere 1, and the result was positive for anti‐EJ antibody. Electromyographic findings were normal, but repetitive low‐frequency stimulation was not performed. Diagnostic criteria of PM/DM 5 were not satisfied. We diagnosed her as having ASS‐ILD and increased her PSL dose to 50 mg/day, which improved the lung opacities (Fig. 1C). The PSL was gradually tapered, but new lung opacities developed in July 2010 when the PSL dose was at 9 mg/day (Fig. 1D). We began cyclosporin A 150 mg/day and increased the PSL to 15 mg/day. The lung opacities improved again (Fig. 1E), but new lesions developed in January 2013 when the PSL was at 5 mg/day (Fig. 1F). The PSL was increased to 15 mg/day, and the cyclosporin A was changed to azathioprine, which was later stopped because of drug‐induced liver damage. Tacrolimus 2 mg/day was then introduced, and the PSL was increased to 30 mg/day, which improved the lung opacities, after which the PSL was again gradually tapered. From November 2015, the PSL was tapered from 20 to 17.5 mg/day, but then bilateral ptosis and diurnal variation in muscle weakness (morning‐time improvement and evening‐time worsening) developed in her proximal lower limbs in December 2015. She did not complain of muscle pain or dyspnea, but manual muscle testing showed muscle weakness (4/5) in her quadriceps. Findings of mechanic's hands; results of pulmonary function testing including vital capacity, forced expiratory volume in 1 sec, total lung capacity, and residual volume; and results of arterial blood gas analysis indicated no deterioration. Serum muscle enzymes including creatine kinase, aldolase, and myoglobin were not elevated. Her blood concentration of tacrolimus was low in January 2016 (2.9 ng/mL). Brain magnetic resonance imaging testing showed no abnormal findings, chest X‐ray and computed tomography showed no deterioration, and chest computed tomography showed no new pulmonary lesions or findings suggestive of thymic disorders. Systemic screening tests including a urinary test, gastrointestinal endoscopy, abdominal ultrasonography, and mammography did not show any findings suggestive of malignant diseases. Her muscle weakness partially and transiently improved immediately after intravenous administration of edrophonium chloride, and an electromyographic study revealed both waning of neuromuscular potentials on repetitive low‐frequency stimulation and myogenic changes (Fig. 2). Although her anti‐acetylcholine receptor antibody and anti‐MuSK (muscle‐specific tyrosine kinase) antibody were both negative, we diagnosed her as having seronegative MG (MG Foundation of America class II 6). Her HLA DR expression was DR8 and DR13. Pyridostigmine was started, and her muscle weakness and ptosis improved. Tacrolimus was increased to 3 mg/day in March 2016, which increased her blood concentration of tacrolimus to 6.0 ng/mL as measured 2 months later. She continues to be followed on an outpatient basis without deterioration of her ILD and no muscle symptoms.
Figure 1.
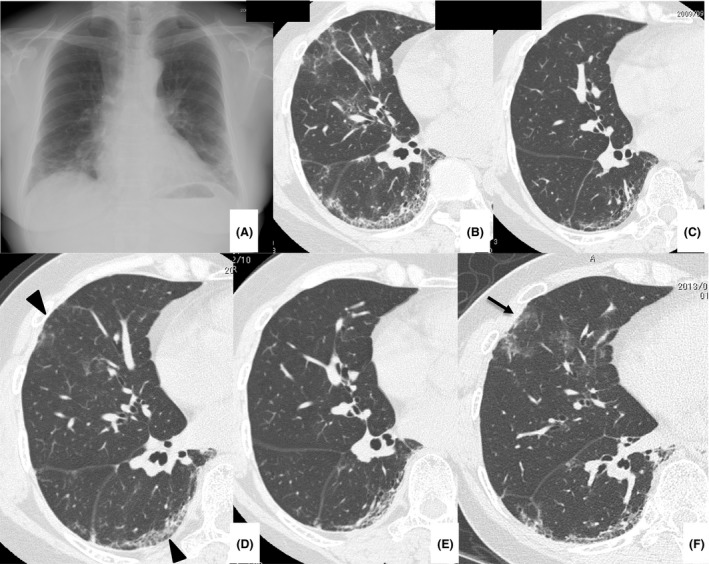
Chest X‐ray (A) and computed tomography (B) on presentation in November 2008. Chest X‐ray showed bilateral ground‐glass opacities (GGOs) and reticular shadows predominantly in the lower lung fields. Chest computed tomography showed bilateral GGOs, consolidation, and reticular shadows predominantly in the lower lobes. The volume of the lower lobes was reduced. Chest computed tomography performed in September 2009 (C) showed improvement of the GGOs; however, they increased in July 2010 (D, arrowheads). Computed tomography showed GGOs in the left lower lobe in October 2011 (E); however, they had increased in the left lower lobe in January 2013 (F, arrow).
Figure 2.

Electromyographic findings. An electromyographic study revealed waning of neuromuscular potentials on repetitive low‐frequency stimulation.
Discussion
We present a patient who developed MG during treatment of ILD with anti‐EJ antibody‐positive ASS for 8 years. ASS is a systemic autoimmune syndrome with antisynthetase antibodies. These antibodies are classified as myositis‐specific antibodies, and to date, eight types of autoantibodies specifically directed against the aminoacyl‐tRNA‐synthetase enzymes have been identified 7. These antibodies are detected mainly in PM/DM: Anti‐Jo‐1 (anti‐histidyl‐tRNA synthetase) antibody has a prevalence of 15–25% in patients with PM/DM 8, 9, 10, 11, anti‐PL‐7 (anti‐threonyl‐tRNA synthetase) and anti‐EJ antibody (anti‐glycyl‐tRNA synthetase) are identified in 5–10% of cases, and the others are found with even less frequency 12. Patients with ASS show different clinical features according to their autoantibodies; however, strategies are often worked out by using those of PM/DM as a reference.
Myasthenia gravis can be found in association with other autoimmune disorders such as systemic lupus erythematosus and rheumatoid arthritis 13, 14; however, the association of MG with ASS or PM/DM is less frequent. Reports of overlap of MG and PM/DM were limited to only about 20 cases from 1976 to 2014 15. Furthermore, PM with antisynthetase antibody was reported only in two patients 3, 4, both of whom had anti‐Jo‐1 antibody. Our case is, to our knowledge, the first case of ASS with antisynthetase antibodies other than anti‐Jo‐1 antibody.
Hamaguchi et al. reported that 29% of patient with anti‐EJ antibody‐positive ILD alone develop myositis during follow‐up 1. The present patient with anti‐EJ antibody‐positive ILD alone developed MG 8 years after the onset of ILD. This suggests that not only myositis but also overlapping of MG should be considered when antisynthetase antibody‐positive patients with ILD develop muscle symptoms during follow‐up.
Although corticosteroids and immunosuppressants possibly delay the development of occult MG, proximal muscle weakness with diurnal variation, ptosis, and abnormal electromyographic findings were not found when the patient presented to our hospital in 2008, which suggests the secondary overlap of MG on ASS. There are no drugs that are known to cause the development of MG. In patients with thymoma, the association of underlying abnormal immunity due to thymic disorders and the development of autoimmune disorders can be considered as a common cause of developing MG and PM/DM or ASS; however, thymic disorders were not found by computed tomography in our patient. The pathogenetic mechanisms of MG overlapping on ASS remain unknown. However, some reports suggest the existence of a shared pathophysiology between PM/DM and MG, which is especially emphasized in cases with the concomitant development of PM/DM and MG 16. For example, the susceptibility of a patient to generalized MG and PM seems to be related to human leukocyte antigen (HLA) DR 16, 17. Interestingly, the association of PM with HLA DR3 expression is claimed to be related to the coexistence of anti‐Jo‐1 antibody 18; however, the present patient did not express HLA DR3. Involvement of HLA in ASS has been unknown, especially in cases without muscle symptoms and elevated serum muscle enzymes, and future studies are needed to clarify a shared pathophysiology between ASS and MG as in the present case.
Myasthenia gravis developed in our patient during corticosteroid therapy with an added immunosuppressant, tacrolimus, which is a calcineurin inhibitor that is useful for both ASS‐ILD 19 and steroid‐resistant MG 20. The serum trough concentration in the present patient was low, and thus, the tacrolimus dose was increased. Other treatments for MG include intravenous immunoglobulin and plasma exchange, and these modalities can be considered depending on the patient's clinical course.
In conclusion, we report the case of a patient who developed MG 8 years after the onset of ASS‐ILD. Although rare, physicians should consider not only myositis but also MG when patients with ASS‐ILD develop muscle symptoms during the follow‐up of ASS.
Authorship
TI: is the guarantor of the paper, taking responsibility for the integrity of the work as a whole, from inception to published article. NK, EK, KO, YT, KK, and NT: aggregated the data, created the figures, and helped draft the discussion of the manuscript.
Conflicts of Interest
The authors have no conflicts of interest to declare.
Acknowledgment
We thank Kenzo Kaji and Kazuhiko Takehara of the Department of Dermatology, Kanazawa University Graduate School of Medical Science, for analyzing the antisynthetase antibodies.
References
- 1. Hamaguchi, Y. , Fujimoto M., Matsushita T., Kaji K., Komura K., Hasegawa M., et al. 2013. Common and distinct clinical features in adult patients with anti‐aminoacyl‐tRNA synthetase antibodies: heterogeneity within the syndrome. PLoS ONE 8:e60442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Katzap, E. , Barilla‐LaBarca M. L., and Marder G.. 2011. Antisynthetase syndrome. Curr. Rheumatol. Rep. 13:175–181. [DOI] [PubMed] [Google Scholar]
- 3. Bohan, A. , and Peter J. B.. 1975. Polymyositis and dermatomyositis (first of two parts). N. Engl. J. Med. 292:344–347. [DOI] [PubMed] [Google Scholar]
- 4. Diaco, M. , Ancarani F., Montalto M., Verrechia E., Evoli A., Servidei S., et al. 2004. Association of myasthenia gravis and antisynthetase syndrome: a case report. Int. J. Immunopathol. Pharmacol. 17:395–399. [DOI] [PubMed] [Google Scholar]
- 5. van de Warrenburg, B. P. , Hengstman G. J., Vos P. E., Boerman R. H., ter Laak H. J., and van Engelen B. G.. 2002. Concomitant dermatomyositis and myasthenia gravis presenting with respiratory insufficiency. Muscle Nerve 25:293–296. [DOI] [PubMed] [Google Scholar]
- 6. Fisher, A. , Swigris J. J., du Bois R. M., Lynch D. A., Downey G. P., Cosgrove G. P., et al. 2009. Anti‐synthetase syndrome in ANA and anti Jo‐1 negative patients presenting with idiopathic interstitial pneumonia. Respir. Med. 103:1719–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Targoff, I. N. 2008. Autoantibodies and their significance in myositis. Curr. Rheumatol. Rep. 10:333–340. [DOI] [PubMed] [Google Scholar]
- 8. Targoff, I. N. 2002. Laboratory testing in the diagnosis and management of idiopathic inflammatory myopathies. Rheum. Dis. Clin. North Am. 28:859–890. [DOI] [PubMed] [Google Scholar]
- 9. Brower, R. , Hengstman G. J., Vree Egberts W., Ehrfeld H., Bozic B., Ghirardello A., et al. 2001. Autoantibody profiles in the sera of European patients with myositis. Ann. Rheum. Dis. 60:116–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Selva‐O'Callaghan, A. , Labrador‐Horrillo M., Solans‐Laque R., Simeon‐Aznar C. P., Martínez‐Gómez X., and Vilardell‐Tarrés M.. 2006. Myositis‐specific and myositis‐associated antibodies in a series of eighty‐eight Mediterranean patients with idiopathic inflammatory myopathy. Arthritis Rheum. 55:791–798. [DOI] [PubMed] [Google Scholar]
- 11. Kalluri, M. , Sahn S. A., Oddis C. V., Gharib S. L., Christopher‐Stine L., Danoff S. K., et al. 2009. Clinical profile of anti‐PL‐12 autoantibody. Cohort study and review of literature. Chest 135:1550–1556. [DOI] [PubMed] [Google Scholar]
- 12. Paik, J. J. , Corse A. M., and Mammen A. L.. 2014. The co‐existence of myasthenia gravis in patients with myositis: a case series. Semin. Arthritis Rheum. 43:792–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Simpson, J. F. , Westerberg M. G., and Magee K. R.. 1996. Myasthenia gravis. An analysis of 295 cases. Acta Neurol. Scand. 42(Suppl. 23):1–27. [Google Scholar]
- 14. Osserman, K. E. , and Genkins G.. 1971. Studies in myasthenia gravis; review of a twenty‐year experience in over 1200 patients. Mt Sinai J. Med. 38:497–537. [PubMed] [Google Scholar]
- 15. Raschilas, F. , Mouthon L., André M. H., Azorin J., Couvelard A., and Guillevin L.. 1999. Concomitant polymyositis and myasthenia gravis reveal malignant thymoma. Ann. Med. Interne (Paris) 150:370–373. [PubMed] [Google Scholar]
- 16. Wilkes, M. R. , Sereika S. M., Fertig N., Lucas M. R., and Oddis C. V.. 2005. Treatment of antisynthetase‐associated interstitial lung disease with tacrolimus. Arthritis Rheum. 52:2439–2446. [DOI] [PubMed] [Google Scholar]
- 17. Cruz, J. L. , Wolff M. L., Vanderman A. J., and Brown J. N.. 2015. The emerging role of tacrolimus in myasthenia gravis. Ther. Adv. Neurol. Disord. 8:92–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jaretzki, A. 3rd , Barhon R. J., Ernstoff R. M., Kaminski H. J., Keesey J. C., Penn A. S., et al. 2000. Myasthenia gravis: recommendations for clinical research standards. Task Force of the Medical Scientific Advisory Board of the Myasthenia Gravis Foundation of America. Neurology 55:16–23. [DOI] [PubMed] [Google Scholar]
- 19. Ehrenstein, M. R. , Snaith M. L., and Isenberg D. A.. 1992. Idiopathic myositis: a rheumatological view. Ann. Rheum. Dis. 51:41–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Garlepp, M. J. 1993. Immunogenetics of inflammatory myopathies. Baillieres Clin. Neurol. 2:579–597. [PubMed] [Google Scholar]