

Abstract
Objective
Current anticonvulsant screening programs are based on seizures evoked in normal animals. One‐third of epileptic patients do not respond to the anticonvulsants discovered with these models. We evaluated a tiered program based on chronic epilepsy and spontaneous seizures, with compounds advancing from high‐throughput in vitro models to low‐throughput in vivo models.
Methods
Epileptogenesis in organotypic hippocampal slice cultures was quantified by lactate production and lactate dehydrogenase release into culture media as rapid assays for seizure‐like activity and cell death, respectively. Compounds that reduced these biochemical measures were retested with in vitro electrophysiological confirmation (i.e., second stage). The third stage involved crossover testing in the kainate model of chronic epilepsy, with blinded analysis of spontaneous seizures after continuous electrographic recordings.
Results
We screened 407 compound‐concentration combinations. The cyclooxygenase inhibitor, celecoxib, had no effect on seizures evoked in normal brain tissue but demonstrated robust antiseizure activity in all tested models of chronic epilepsy.
Interpretation
The use of organotypic hippocampal cultures, where epileptogenesis occurs on a compressed time scale, and where seizure‐like activity and seizure‐induced cell death can be easily quantified with biomarker assays, allowed us to circumvent the throughput limitations of in vivo chronic epilepsy models. Ability to rapidly screen compounds in a chronic model of epilepsy allowed us to find an anticonvulsant that would be missed by screening in acute models.
Introduction
In the absence of sufficiently detailed insights into the pathophysiology of epilepsy,1 development of anticonvulsant therapies has relied on empirical drug screening efforts. Decades of screening and subsequent drug development have produced many new anticonvulsants, yet, epilepsy remains medically intractable in one‐third of patients.2 Recently, it has been proposed that this lack of progress is a consequence of the method by which compounds are screened.3, 4 To date, initial screening is based on acute application of convulsant conditions to normal brain tissue,2, 4 but this produces patterns of epileptic activity that are substantially different from spontaneous epileptiform activity in chronically epileptic brain tissue.5 Thus, we considered the hypothesis that screening for anticonvulsants in chronically epileptic tissue would uncover agents that may be uniquely effective in medically intractable epilepsy.6
Unfortunately, most epileptiform activity in chronically epileptic brain is comprised of brief interictal electrographic spikes. While not benign,7 spikes are not the target of anticonvulsants.8 Actual seizure activity in experimental epilepsy is rare, unpredictable, and develops slowly after an inciting brain injury.9 Morbidity and mortality are significant once seizure frequencies are sufficiently high for screening studies.10 At all seizure frequencies, clustering of seizures substantially increases the sampling required to discern anticonvulsant effects.11 Thus, in vivo models of chronic seizures have not been practical for drug screening. Accordingly, we developed a staged screening program in which initial evaluations are based on spontaneous seizures in a chronically epileptic in vitro preparation in which these practical shortcomings are obviated.
Organotypic hippocampal slice cultures12 preserve the key circuitry of the in vivo hippocampus.13 However, brain slice preparation involves massive traumatic axotomy at the cut surfaces, so that the slice can be considered a model of severe traumatic sheer injury and an in vitro extension of the undercut cortical model of posttraumatic epilepsy.14 As a consequence of the deafferentation, robust sprouting takes place,15, 16 so that connectivity between pyramidal cells increases from 3% in acute slices to 30% in organotypic slices.17, 18 In light of this abundant reciprocal excitation, it is not surprising that organotypic slice cultures are hyperexcitable19 and even spontaneously epileptic20, 21 (see Heinemann and Staley22 for review). We have characterized epileptogenesis in this preparation.23, 24 Increases in extracellular lactate accompanied seizures in human hippocampus,25, 26 and we found that lactate production was correlated with seizure‐like activity in organotypic cultures. Lactate dehydrogenase (LDH) is released into culture medium after plasma membrane loses its integrity,27, 28, 29 and its concentration is correlated with cell death in cultures. We then demonstrated that this preparation and lactate and LDH assays can be used to evaluate anticonvulsant efficacy, using phenytoin and manipulation of mTOR pathway as examples.30, 31 We now show the utility of this in vitro model of chronic epilepsy as a first, blinded, moderate throughput stage for drug screening. Repeat biochemical as well as electrophysiological confirmation in vitro comprised the second stage of screening. The final stage was comprised of double blind, crossover controlled, in vivo testing in the kainate model of severe chronic epilepsy9, 32 with seizure quantification by continuous electrographic monitoring.33
Materials and Methods
Organotypic hippocampal cultures
Hippocampal slices of 350 μm thickness were dissected from postnatal day 7–8 Sprague‐–Dawley male rat pups (Fig. 1A). Slices were placed onto poly‐D‐lysine (PDL)‐coated 6‐well tissue culture plates (Fig. 1B and C). Culture medium consisted of Neurobasal‐A/B27 supplemented with 0.5 mmol/L GlutaMAX and 30 μg/mL gentamicin (Life Technologies). Slices were maintained at 37°C in a humidified atmosphere that contained 5% CO2. After attachment, culture plates were placed on a rocking platform (Fig. 1D and E). All of the procedures conformed to the NIH guidelines (i.e., as described in The Guide for Care and Use of Laboratory Animals) and were approved by the Massachusetts General Hospital Subcommittee on Research Animal Care.
Figure 1.
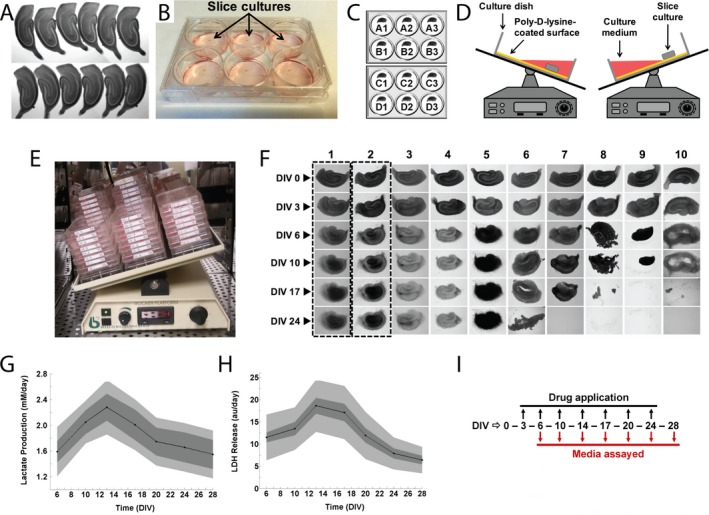
Screen design. (A) Twelve hippocampal slices derived from the same rat pup, imaged on day in vitro (DIV) 0 immediately after dissection. (B) Photograph of slice cultures in a poly‐D‐lysine‐coated 6‐well tissue culture plate with culture medium. (C) Twelve slices were cultured in four sets of triplicates, allowing for screening of four conditions per experiment. One of the four conditions tested per experiment was always a control (vehicle). (D) A single well of a 6‐well plate with a slice culture is shown in cross‐section on a rocking platform. Slice was offset from the center of the well allowing it to be submerged in culture medium and aerated in a continuously alternating manner during rocking. (E) Slices were cultured in 6‐well plates on a rocking platform in a humidified chamber with 5% CO 2 at 37°C. (F) Morphological progression of a control organotypic hippocampal slice culture over 24 days in vitro is shown in column 1. The toxic effects of various compounds (first application on DIV 3) on organotypic hippocampal slice cultures (columns 3–10) in comparison with a compound with a nontoxic effect (column 2) are also shown. (G) Lactate production and (H) Lactate dehydrogenase (LDH) release measured in spent culture media from control organotypic hippocampal slice cultures (n = 318 slices) as biomarkers of seizure‐like activity and ictal neuronal death, respectively. The lighter gray areas represent the overall population variation, whereas the darker gray areas represent the intertriplicate variation. Data are expressed as mean ± SD. (I) Chronic application experiments were carried out for 28 days in vitro with compounds added to culture media starting on DIV 3. Every 3–4 days, spent culture medium was collected and fresh culture medium with appropriate drug(s) was added to slice cultures. Collected spent culture medium was assayed for biomarkers of seizure activity and ictal neuronal death.
Compound application and assays
We organized cultures into four experimental groups of three slices each, all from the same animal (Fig. 1C).
Compounds were applied starting from day in vitro (DIV) 3, and reapplied with every biweekly medium change (Fig. 1I), except wash‐out experiments where compound application stopped on indicated DIV. To blind the experimenter, compounds and vehicle controls were labeled with randomly assigned numbers. Maps of compound/vehicle numbers and corresponding experiments were maintained by a different investigator. Culture supernatant (spent culture medium) was collected and relative concentrations of markers of seizure‐like activity and ictal neuronal death, lactate, and LDH, were measured using commercially available kits (Biovision, Milpitas, CA).
Morphology scoring
The natural progression of slice morphology over first 24 days in vitro is shown in the first column of Figure 1F. We noticed that some drugs significantly altered slice morphology, and that these changes correlated with the number of surviving neurons in the slice. We therefore developed a scoring metric for morphology that was used together with LDH measurements to screen drugs and compounds for toxicity. Slices that appeared lighter or same as controls were assigned a score of 0, slices that were smaller or darker than controls were assigned a score of 1, slices with significantly changed shape or with neuronal layers that were no longer distinguishable were assigned a score of 2, and slices which disintegrated or detached from substrate were assigned a score of 3 (Fig. 1F).
Toxicity screening
We found that some drugs were toxic to organotypic hippocampal cultures at applied concentrations, as measured by increased LDH on DIV 6/7 or by morphology scoring. While morphology and LDH were well correlated (r 2 = 0.6, P < 0.001, n = 407 drug‐concentration pairs), several compounds or drugs increased LDH without a corresponding change in morphology, while others changed the morphology without a corresponding change in LDH (Fig. 2A). This may be due to different modes of cell death.27 We therefore adopted the following criteria for establishing toxicity of a given drug‐concentration pair: (1) morphology score >0, or (2) LDH on DIV 6/7 that was significantly higher than control. If either criterion was satisfied by a drug‐concentration pair, this pair was deemed toxic and excluded from further analysis.
Figure 2.
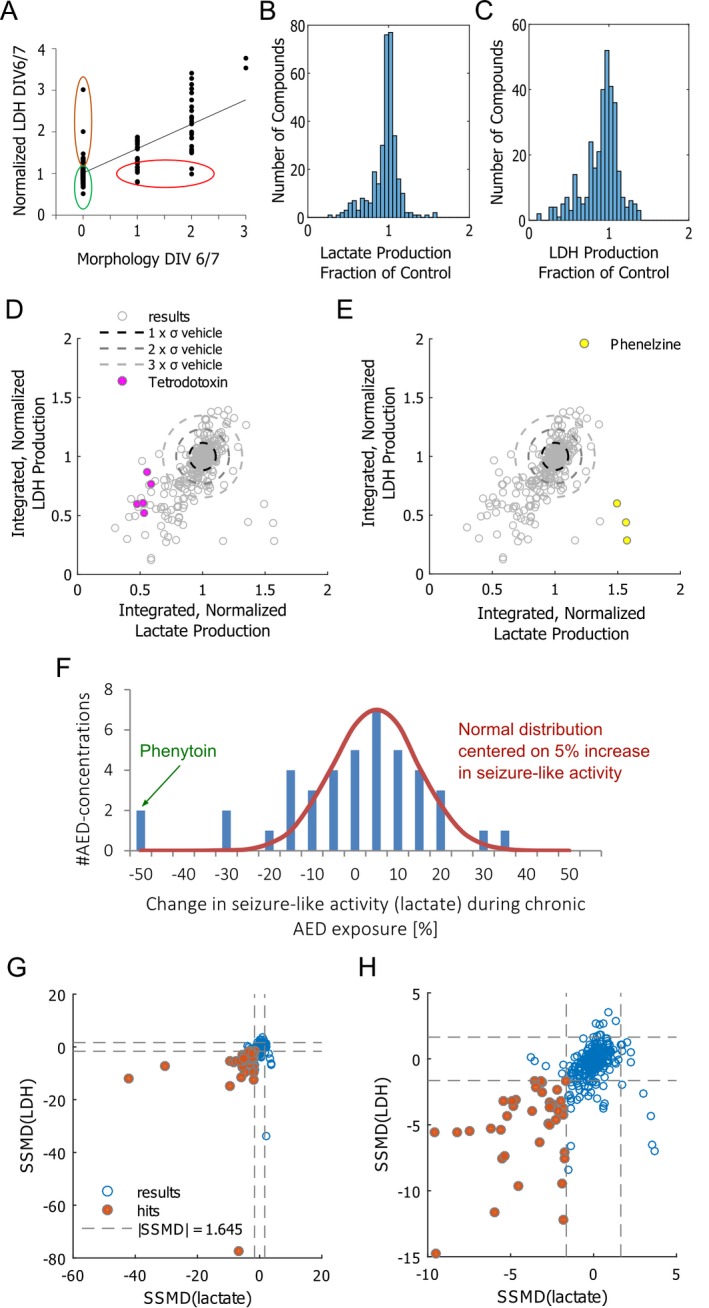
Drug effects. (A) Lactate dehydrogenase (LDH) values and morphology scores on day in vitro (DIV) 6/7 were well correlated (R2 = 0.6019, P < 0.001), suggesting that cell death led both to increased LDH release and changed culture morphology. However, in a few cases, applied compounds caused a significantly higher LDH release without accompanying change in morphology (brown outline). In other cases, differences in morphology were observed without a significant difference in LDH release (red outline). These compounds were also excluded from analysis. Compounds that were included in analysis are outlined in green. (B, C) Histograms of frequency of screened nontoxic compounds as fraction of controls for lactate production (B) and LDH release (C) (328 drug‐concentration pairs; n = 3 slices per pair). (D) Integrated lactate production (DIV 10–17), normalized to controls from the same pup versus integrated LDH production (DIV 1017), normalized to controls. σ = standard deviation of integrated lactate (horizontal axis) and LDH (vertical axis) of vehicle‐treated controls. Data are plotted for 328 drug‐concentration pairs. Effects of tetrodotoxin (D) and phenelzine (E) are highlighted for separate experiments (n = 3 each experiment) that were performed over 2–3 years to demonstrate reproducibility of the screen. (F) Histogram of effects of standard AEDs on seizure‐like activity (measured by lactate production) in organotypic hippocampal cultures. Normal distribution is included in the same plot for comparison. (G) Strictly standardized mean difference (SSMD) values of lactate and LDH production of drug‐treated cultures versus vehicle‐treated controls, evaluated between DIV 10–17. Significant results (SSMD < −1.645 for both lactate and LDH) are highlighted with brown color. (H) Same plot as (G), shown in more detail.
Data analysis and statistics
The strictly standardized mean difference (SSMD) metric was used to identify hits in the compound screen. SSMD was calculated according to the following formula:
, where is the mean of integrated lactate or LDH values for drug i, is the mean of integrated lactate or LDH for matched control, and and are drug and control variances, respectively. SSMD captures information about both the size of the effect (difference between drug and control means) and significance of the result, similar to Cohen's D.34 We used the following criteria: |SSMD| > 3 (very strong effect), 3 > |SSMD| > 2 (strong effect), 2 > |SSMD| > 1.645 (fairly strong effect), 1.645 > |SSMD| > 1 (moderate effect), 1 > |SSMD| > 0 (weak effect).35 P values for screen hits were calculated using Student's t‐test.
Acute recordings
Extracellular field potentials were recorded in the CA1 and/or CA3 pyramidal cell layer of intact hippocampi36 and organotypic hippocampal slices in a conventional submerged chamber using tungsten‐coated microelectrodes and ISO‐DAM8A amplifier (World Precision Instruments). Oxygenated (95% O2 and 5% CO2) artificial cerebrospinal fluid containing 126 mmol/L NaCl, 3.5 mmol/L KCl, 2 mmol/L CaCl2, 1.3 mmol/L MgCl2, 25 mmol/L NaHCO3, 1.2 mmol/L NaH2PO4, and 11 mmol/L glucose (pH 7.4), was continuously perfused at 33 ± 0.5°C. Flow rate was 2.5 mL/min. Before the actual recording, slices were allowed to stabilize in the recording chamber for 30–60 min. Electrical signals were digitized using an analog‐to‐digital converter (DigiData 1322A, Axon Instruments). pCLAMP 8.2 (Axon Instruments) and SigmaPlot 11.0 (Systat Software) programs were used for data acquisition and analysis. Recordings were sampled at 10 KHz and filtered from 1 Hz to 2 kHz. Ictal‐like epileptiform discharges were defined as hyper‐synchronous large amplitude (×3 baseline) and high‐frequency population spikes followed by sustained ictal‐tonic and intermittent ictal‐clonic after discharges, with the duration of the population spike and after‐discharge complex lasting more than 5–10 sec. Power spectrum analysis was performed from the electrical signals after applying a Hamming window function.
In vivo experiments
Kainate treatment and induction of status epilepticus
A description of the methods for and characteristics of the repeated, low‐dose kainate model have been published in detail elsewhere.9, 10, 37 Briefly, convulsive status epilepticus was induced in adult male Sprague–Dawley rats (180–200 g) with repeated (i.e., approximately every 30 min), low‐dose (7.5 mg/kg), intraperitoneal (IP) injections of kainic acid (Tocris Bioscience, Bristol, UK). During and after the kainate treatment, all rats were maintained in a climate‐controlled vivarium. All of the procedures conformed to the NIH guidelines (i.e., as described in The Guide for Care and Use of Laboratory Animals) and were approved by the University of Utah Animal Care and Use Committee (IACUC).
Surgical implantation of recording electrodes
Rats with kainate‐induced epilepsy were implanted with cortical electroencephalogram (EEG) electrodes. The head was held in a stereotaxic device, and the surgical site was clipped and washed with Betadine scrub and solution. The rat was anesthetized with 2% isoflurane and pretreated with 0.8 mL subcutaneous atropine (0.54 mg/kg) and 0.2 mL subcutaneous dexamethasone (4 mg/mL). After the incision and retraction of the skin, the dura was removed and six small holes were drilled through the skull, for implantation of support screws and electrodes. A bipolar stainless‐steel recording electrode pedestal was lowered onto the cortex of the right hemisphere (4.0 mm lateral, 8.0 mm caudal), and a ground electrode was placed on the cortex of the left hemisphere (2.0 mm lateral, 3.5 mm rostral). Dental acrylic fastened the electrode pedestal into place and covered and sealed the exposed skull surfaces. The incision was closed with 4–0 Dermalon sutures to the level of the dental cement covering the skull. Animals were then administered subcutaneous treatments of 3.0 mL lactated Ringer's, 0.1 mL marcaine (0.5%), 0.1 mL buprenorphine (0.3 mg/mL), and 0.2 mL penicillin (300,000 IU).
Chronic in vivo recordings
Rats with kainate‐induced epilepsy and implanted electrodes were placed in custom‐built Plexiglass recording chambers equipped with swivel commutators, and connected to spring‐covered cables (Plastics One, Roanoke, VA) to their skull caps. Electrographic signals were amplified with EEG100C amplifiers (high‐pass at 1 Hz; low‐pass at 100 Hz; notch filter at 60 Hz), digitized at 500 Hz with an MP150 digital‐analog converter, and acquired with AcqKnowledge software (BioPac Systems Inc.; Santa Barbara, California). Electrographic recordings were obtained continuously during vehicle and drug treatments. Data were stored on hard‐drives for offline analysis using AcqKnowledge software.
Repeated‐measures, crossover protocol for drug testing
Long‐term, continuous recording sessions were used to assess the effect of Celecoxib‐containing food versus control food on spontaneous recurrent electrographic seizures. A repeated‐measure, crossover protocol was used with 100 mg/kg/day Celecoxib in custom‐made food (Bio‐serv).38 The 100 mg/kg/day dose of Celecoxib would be equivalent to five injections of 20 mg/kg/day.39 Each trial involved eight treatments of Celecoxib and eight vehicle treatments administered on alternate days with two recovery days between the treatment days. The rationale for the repeated‐measure, crossover design was the high degree of statistical power inherent in this approach. For example, for α = 0.05, eight animals and six drug‐versus‐vehicle crossovers (i.e., a total of 48 drug‐versus‐vehicle tests per trial38) yields a power of 0.9.
Blinded seizure detection
EEG acquisition and analysis were performed at separate sites by different investigators. DClamp (sourceforge.net) was used to detect seizures using supervised algorithms.33 Detection parameters were set to increase sensitivity to the point that seizures were rarely missed in trial detections, but false‐positive detections were common. Human supervision consisted of review of all detected seizures for false‐positive detections and review of raw EEG for false negatives. In total, 532 true positives, 18578 false positives, and 94 false negatives were found. To blind the supervising investigator, the EEG files and channels were renamed using random numbers, and the map to the original names was managed by an investigator not involved in the analysis.
Results
Lactate and LDH in control cultures
We followed the natural progression of lactate and LDH production by epileptic control cultures from DIV 6 to DIV 28 (Fig. 1, n = 318). Lactate and LDH production reached their peaks between DIV 14 and 17 (Fig. 1G and H). Decrease in lactate and LDH production at later time points is likely a consequence of seizure‐induced neuron death, the peak of which occurs between DIV 14 and 17.30 We therefore used the period between DIV 10 and DIV 17 (the time between the onset of spontaneous seizure‐like activity and significant neuron death) to compare lactate and LDH production in vehicle‐treated and drug‐treated cultures. We found that variability between cultures dissected from the same animal was lower than variability between cultures dissected from different animals (Fig. 1G and H). To control for interanimal differences, all experimental assays were compared to vehicle‐treated cultures from the same animal.
Compound screen results
We screened 140 compounds, including combinations of drugs (Table 1). Different concentrations were used for each drug, and a total of 407 drug‐concentration pairs were screened in triplicate (n = 3 slices). Of these, 79 were found to be toxic (Table 2) and excluded from further analysis (Fig. 2A). We integrated the lactate and LDH production of the remaining 328 drug‐concentration pairs between DIV 10 and DIV 17 (n = 3), and normalized it by control (vehicle‐treated cultures from the same pup, n = 3) lactate and LDH production over the same period. Histograms of lactate and LDH production are shown in Figure 2B and C, and a scatter plot of lactate versus LDH is shown in Figure 2D. Values that are lower than unity indicate that a drug lowered lactate and/or LDH production to a level below that of a vehicle‐treated control. Some of the low lactate and LDH values were less than the mean standard deviation lower than the mean of controls, suggesting that significant reduction of seizure‐like.
Table 1.
List of screened drugs and compounds
| Anticonvulsants | Cholinergic | Glutamate | Dopamine | Chloride | Antiinflammatory |
| Carbamazepine | Amantadine HCl | ACPD | Clomipramine | Acetazolamide | Carprofen |
| Diazepam | Atropine sulfate | CPPG | Dopamine HCl | Bumetanide | Celecoxib |
| Ethosuximide | Carbachol | DCG IV | Haloperidol | Chlorothiazide | Celecoxib + |
| Felbamate | Diphenhydramine | Dextromethorphan | CLP257* | Phenytoin | |
| Gabapentin | Edrophonium | HBr | Serotonin | D4.2* | Deracoxib |
| Levetiracetam (keppra) | Nicotine bitartrate | dL APV | Ketanserin | Furosemide | Diclofenac |
| Phenacemide | Kynurenic acid | Tartrate | IS50699* | Firocoxib | |
| Phenobarbital | Monoamine NT | L‐AP4 | Methysergide | Mannitol | Indomethacin |
| Phenobarbital + | modulators | LY341495 | Zolmitriptan | Ketoprofen | |
| Bumetanide | Amitriptyline | MPEP | Sodium | Ketorolac | |
| Phenytoin | Amoxapine | Opioid | Bretylium Tosylate | LOXBlock‐1* | |
| Pregabalin | Atomoxetine | GABA | Nalbuphine HCl | Digoxin | Meclofenamate |
| Retigabine | Bupropion | Baclofen | Naloxone | Lidocaine HCl | Meloxicam |
| Topiramate | Clonidine | CGP55845 | Tramadol | Tetrodotoxin (TTX) | Piroxicam |
| Valproic acid | Desipramine | Diazepam + CLP257 | TG4‐155* | TTX + Fluoxetine | Rofecoxib |
| Doxepin | Diazepam + IS50699 | TTX + Nortriptyline | Sulindac | ||
| Intracellular signaling | Duloxetine | Flurothyl | Cannabinoid | Zileuton | |
| AG 1024 | Fluoxetine | Gabazine | Rimonobant | Calcium | |
| Akt 1,2 kinase inhibitor | Imipramine | Taurine | Flunarizine | Antioxidant | |
| API‐2 (Triciribine) | Maprotiline | Purine | Verapamil | Acetylcysteine | |
| DFMO | Methylphenidate HCl | Microtubule | Allopurinol | Alpha‐tocopherol | |
| Gefitinib (Iressa) | Modafinil | Colchicine | Caffeine | Potassium | GCEE* |
| K‐252a | Nortriptyline | Vinblastine | Glipizide | ||
| Lyothyronine | Phenelzine | Vincristine | Steroids | Growth factor | |
| PDMP* | Phenelzine + TTX | Dexamethasone | Blinded | Neuregulin‐1* | |
| Picropodophyllotoxin | Propranolol | Antiproliferative | Estradiol | LacCer* | |
| Rapamycin | Reserpine | Carboplatin | Letrozole | LDN‐209884(3‐1)* | Cell death |
| Sildenafil Citrate | Selegiline | Cycloheximide | Lovastatin | LDN‐212320(10‐10)* | Caspase‐3 inhibitor* |
| TGFbeta RI inhibitor* | Sertraline | Etoposide | Megestrol | LDN‐72457(5‐1)* | Erythropoietin |
| TrkB Fc chimera | Venlafaxine | AG 1478 | Tamoxifen citrate | Minocycline |
Drugs marked with an asterisk were obtained from collaborators, while the rest of the drugs were primarily obtained from the NINDS custom compound collection of known bioactives and FDA‐approved drugs.
Table 2.
List of drug‐concentration pairs excluded from analysis due to early toxicity
| Drug name | Concentration | Drug name | Concentration | Drug name | Concentration |
|---|---|---|---|---|---|
| Akt 1/2 kinase inhibitor | 10 μmol/L | Dextromethorphan HBr | 10 μmol/L | Maprotiline | 10 μmol/L |
| Akt 1/2 kinase inhibitor | 5 μmol/L | DFMO | 40 μmol/L | Megestrol acetate | 10 μmol/L |
| Akt 1/2 kinase inhibitor | 2 μmol/L | Diazepam + IS50699 | 1 + 10 μmol/L | Minocycline | 500 μmol/L |
| Alpha‐tocopherol | 10 μmol/L | Duloxetine | 10 μmol/L | Minocycline | 100 μmol/L |
| Alpha‐tocopherol | 1 μmol/L | Erythropoietin | 100 IU/mL | Nortriptyline | 10 μmol/L |
| Alpha‐tocopherol | 0.1 μmol/L | Erythropoietin | 10 IU/mL | Nortriptyline + TTX | 10 μmol/L + 1 μmol/L |
| Amitriptyline | 10 μmol/L | Erythropoietin | 1 IU/mL | Nortriptyline + TTX | 1 μmol/L + 1 μmol/L |
| Amoxapine | 10 μmol/L | Felbamate | 1 μmol/L | PDMP | 50 μmol/L |
| API‐2 (Triciribine) | 10 μmol/L | Flunarizine | 10 μmol/L | PDMP | 25 μmol/L |
| API‐2 (Triciribine) | 5 μmol/L | Fluoxetine | 10 μmol/L | Phenelzine + TTX | 1 μmol/L + 1 μmol/L |
| API‐2 (Triciribine) | 2 μmol/L | Fluoxetine | 2 μmol/L | Phenelzine + TTX | 0.1 μmol/L + 1 μmol/L |
| API‐2 (Triciribine) | 1 μmol/L | Fluoxetine + TTX | 10 μmol/L + 1 μmol/L | Picropodophyllotoxin | 10 μmol/L |
| Bupropion | 10 μmol/L | Gefitinib (Iressa) | 10 μmol/L | Picropodophyllotoxin | 5 μmol/L |
| Caspase‐3 inhibitor | 10 μmol/L | Gefitinib (Iressa) | 1 μmol/L | Picropodophyllotoxin | 2 μmol/L |
| CGP55845 | 1 μmol/L | Gefitinib (Iressa) | 0.1 μmol/L | Picropodophyllotoxin | 1 μmol/L |
| CGP55845 | 0.1 μmol/L | IS50699 | 10 μmol/L | Piroxicam | 10 μmol/L |
| CGP55845 | 0.01 μmol/L | IS50699 | 10 μmol/L | Rimonabant | 10 μmol/L |
| Clomipramine | 10 μmol/L | K‐252a | 1 μmol/L | Sertraline | 10 μmol/L |
| CLP257 | 100 μmol/L | K‐252a | 0.3 μmol/L | Sertraline | 2 μmol/L |
| CLP257 | 100 μmol/L | Ketanserin tartrate | 1 μmol/L | Tamoxifen citrate | 10 μmol/L |
| Colchicine | 1 μmol/L | Ketanserin tartrate | 0.1 μmol/L | TrkB Fc chimera | 1 μg/mL |
| Colchicine | 0.1 μmol/L | Ketanserin tartrate | 0.01 μmol/L | Verapamil | 100 μmol/L |
| Colchicine | 0.1 μmol/L | LacCer | 20 μg/mL | Verapamil | 10 μmol/L |
| D4.2 | 10 μmol/L | LacCer | 10 μg/mL | Vinblastine | 0.1 μmol/L |
| DCG IV | 10 μmol/L | LDN‐212320(10‐10) | 10 μmol/L | Vincristine | 0.1 μmol/L |
| DCG IV | 1 μmol/L | Lidocaine HCl | 10 μmol/L | ||
| Desipramine | 10 μmol/L | Lovastatin | 10 μmol/L |
We examined the long‐term reproducibility of our drug screening methodology by comparing values obtained over a period of 2–3 years (Fig. 2D and E). Lactate and LDH values for tetrodotoxin and phenelzine were found to reliably cluster into the same regions of lactate versus LDH scatter plot, demonstrating the stability and reproducibility of our results.
Our screen included 42 drug‐concentration pairs (n = 3 per drug‐concentration pair) of standard antiepileptic drugs (AEDs), including acetazolamide (10 μmol/L), carbamazepine (10–70 μmol/L), diazepam (1 μmol/L), ethosuximide (10–1000 μmol/L), felbamate (0.01–1 μmol/L), gabapentin (1–50 μmol/L), levetiracetam (10–250 μmol/L), phenacemide (0.01–1 μmol/L), phenobarbital (100 μm), phenytoin (100 μmol/L), pregabalin (0.01–1 μmol/L), topiramate (10–100 μmol/L), and valproate (600 μmol/L). A histogram of the effects of these drugs on lactate production, a marker of seizure‐like activity in organotypics, is shown in Figure 2F. Data for individual drug‐concentration pairs are included in Table S1. Phenytoin was most effective, but seizure activity recrudesces after 3 weeks of exposure, that is, the slice cultures become refractory to phenytoin.30
We used the strictly standardized mean difference (SSMD) metric to make a robust determination of the significance of screen results, and to identify “hits”. SSMD takes both effect size and data variance into account, thus identifying hits that have the strongest and most statistically significant effect. We used a cutoff of SSMD <−1.645 to identify hits with a fairly strong35 reduction of both lactate and LDH production (Fig. 2G, H, and Table 3). Results for all screened compounds are listed in the Table S1.
Table 3.
Hits discovered with the screen (hits are highlighted in Figs 2F, G)
| Drug name | Concentration | Integrated, normalized lactate production | Integrated, normalized LDH production | p lactate | p LDH | SSMDlactate | SSMDLDH |
|---|---|---|---|---|---|---|---|
| Celecoxib + Phenytoin | 10 + 100 μmol/L | 0.41 | 0.49 | 2.133E‐07 | 3.355E‐05 | −42.04 | −11.83 |
| Celecoxib + Phenytoin | 10 + 100 μmol/L | 0.43 | 0.64 | 7.663E‐07 | 2.267E‐04 | −30.52 | −7.29 |
| Celecoxib | 10 μmol/L | 0.59 | 0.70 | 0.0001 | 0.0006 | −9.60 | −5.57 |
| Celecoxib + Phenytoin | 10 + 100 μmol/L | 0.30 | 0.39 | 0.0001 | 1.374E‐05 | −9.51 | −14.81 |
| Celecoxib | 10 μmol/L | 0.72 | 0.75 | 0.0001 | 0.0006 | −8.25 | −5.60 |
| API‐2 (Triciribine) | 0.5 μmol/L | 0.40 | 0.30 | 0.0002 | 0.0007 | −7.47 | −5.45 |
| Cycloheximide | 1 μmol/L | 0.54 | 0.34 | 0.0003 | 1.879E‐08 | −6.85 | −77.17 |
| TTX | 1 μmol/L | 0.53 | 0.61 | 0.0004 | 0.0008 | −6.17 | −5.27 |
| Taurine | 100 μmol/L | 0.73 | 0.32 | 0.0005 | 3.627E‐05 | −5.97 | −11.60 |
| Celecoxib | 10 μmol/L | 0.69 | 0.58 | 0.0006 | 0.0008 | −5.62 | −5.34 |
| LDN‐212320 (10‐10) | 3 μmol/L | 0.57 | 0.76 | 0.0007 | 0.0002 | −5.54 | −7.55 |
| Akt 1/2 kinase inhibitor | 1 μmol/L | 0.75 | 0.54 | 0.0007 | 0.0055 | −5.42 | −3.15 |
| Akt 1/2 kinase inhibitor | 0.5 μmol/L | 0.85 | 0.59 | 0.0008 | 0.0002 | −5.33 | −7.39 |
| Phenytoin | 100 μmol/L | 0.48 | 0.57 | 4.460E‐05 | 0.0001 | −5.23 | −4.34 |
| Doxepin | 10 μmol/L | 0.62 | 0.80 | 0.0011 | 0.0050 | −4.90 | −3.22 |
| Rapamycin (DIV3–28) | 20 nmol/L | 0.59 | 0.14 | 0.0011 | 0.0034 | −4.84 | −3.60 |
| TTX | 1 μmol/L | 0.48 | 0.60 | 0.0013 | 0.0060 | −4.66 | −3.07 |
| Celecoxib | 10 μmol/L | 0.58 | 0.59 | 0.0014 | 0.0001 | −4.56 | −9.66 |
| API‐2 (Triciribine) | 0.2 μmol/L | 0.70 | 0.49 | 0.0031 | 0.0024 | −3.70 | −3.96 |
| Topiramate | 100 μmol/L | 0.82 | 0.79 | 0.0036 | 0.0423 | −3.53 | −1.70 |
| Kynurenic acid | 3 mmol/L | 0.51 | 0.65 | 0.0004 | 0.0050 | −3.51 | −2.16 |
| TTX + Fluoxetine | 1 + 1 μmol/L | 0.63 | 0.55 | 0.0049 | 0.0004 | −3.24 | −6.32 |
| Phenobarbital + Bumetanide | 100 + 10 μmol/L | 0.86 | 0.81 | 0.0052 | 0.0420 | −3.20 | −1.70 |
| TTX | 1 μmol/L | 0.58 | 0.76 | 0.0058 | 0.0114 | −3.10 | −2.56 |
| TTX | 1 μmol/L | 0.53 | 0.52 | 0.0059 | 0.0370 | −3.09 | −1.78 |
| Rapamycin (DIV 3–14) | 20 nmol/L | 0.67 | 0.41 | 0.0088 | 0.0010 | −2.76 | −4.92 |
| Vinblastine | 0.01 μmol/L | 0.66 | 0.55 | 0.0096 | 0.0031 | −2.69 | −3.70 |
| Neuregulin‐1 | 10 ng/mL | 0.80 | 0.76 | 0.0096 | 0.0010 | −2.69 | −5.02 |
| LDN‐72457 (5‐1) | 10 μmol/L | 0.70 | 0.75 | 0.0103 | 0.0049 | −2.64 | −3.25 |
| Picropodophyllotoxin | 0.5 μmol/L | 0.71 | 0.28 | 0.0110 | 0.0038 | −2.59 | −3.50 |
| Rapamycin (DIV3–14) | 20 nmol/L | 0.59 | 0.12 | 0.0135 | 0.0032 | −2.44 | −3.67 |
| Acetylcysteine | 50 μmol/L | 0.85 | 0.64 | 0.0135 | 0.0038 | −2.43 | −3.49 |
| Furosemide | 1 mmol/L | 0.71 | 0.53 | 0.0143 | 0.0038 | −2.40 | −3.49 |
| Neuregulin‐1 | 100 ng/mL | 0.81 | 0.76 | 0.0167 | 0.0013 | −2.28 | −4.61 |
| Phenytoin | 100 μmol/L | 0.46 | 0.50 | 0.0044 | 0.0033 | −2.22 | −2.35 |
| Celecoxib | 10 μmol/L | 0.67 | 0.57 | 0.0213 | 0.0023 | −2.12 | −3.98 |
| Colchicine | 0.01 μmol/L | 0.81 | 0.48 | 0.0268 | 0.0051 | −1.97 | −3.22 |
| Akt 1/2 kinase inhibitor | 1 μmol/L | 0.79 | 0.29 | 0.0315 | 0.0001 | −1.87 | −9.43 |
| GCEE | 1 mmol/L | 0.73 | 0.47 | 0.0325 | 2.938E‐05 | −1.85 | −12.23 |
| Neuregulin‐1 | 1 ng/mL | 0.88 | 0.83 | 0.0334 | 0.0018 | −1.84 | −4.29 |
| Rapamycin (DIV 3 > ) | 20 nmol/L | 0.76 | 0.46 | 0.0339 | 0.0028 | −1.83 | −3.80 |
| Diazepam + CLP257 | 1 + 100 μmol/L | 0.69 | 0.35 | 0.0386 | 0.0002 | −1.75 | −7.11 |
| Carbamazepine | 50 μmol/L | 0.84 | 0.56 | 0.0415 | 0.0002 | −1.71 | −7.56 |
| Kynurenic acid | 3 mmol/L | 0.54 | 0.60 | 0.0151 | 0.0165 | −1.68 | −1.65 |
Compounds are organized based on increasing lactate SSMD. P values were calculated relative to vehicle‐treated controls from the same pup. Repeated drug‐concentration pairs represent replicated experiments in different animals.
Celecoxib effect on lactate and LDH production
We found that celecoxib, an antiinflammatory drug applied at the concentration of 10 μmol/L, was a strong inhibitor of lactate and LDH production in five separate experiments (n = 3 cultures, each experiment, −9.6 < SSMDlactate <−2.1, −9.7 SSMDLDH <−4). Lower concentration, 1 μmol/L, was less effective, while 100 μmol/L celecoxib was toxic to organotypic cultures (Figure S1). In contrast, other antiinflammatory drugs that we have tested did not have as strong effect on lactate and LDH production (Fig. 3A).
Figure 3.
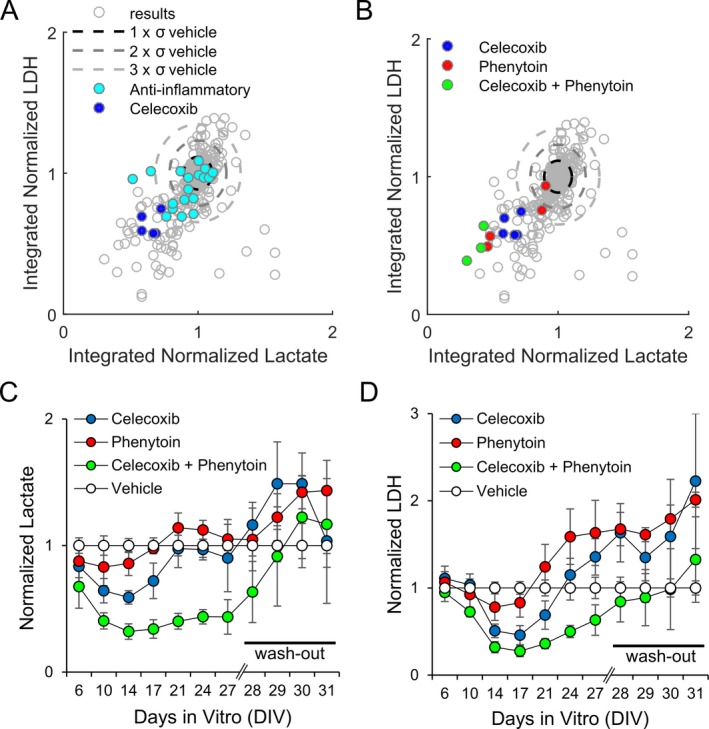
Anticonvulsive and neuroprotective effects of celecoxib, phenytoin, and celecoxib plus phenytoin. (A) The effects of celecoxib (n = 5 screens; three slices per screen) and 13 other screened antiinflammatory drugs (n = 18 screens; three slices per screen) in comparison with all screened nontoxic compounds. (B) The effects of celecoxib (10 μmol/L; n = 5 screens), phenytoin (100 μmol/L; n = 4 screens), and a combination of celecoxib (10 μmol/L) plus phenytoin (100 μmol/L) (n = 3 screens) in comparison with all screened nontoxic compounds. n = 3 slices per screen. (C, D) Lactate production (C) and lactate dehydrogenase (LDH) release (D), normalized to controls, for celecoxib (10 μmol/L), phenytoin (100 μmol/L), and celecoxib (10 μmol/L) plus phenytoin (100 μmol/L), two separate experiments with three slices per experiment per compound including control (n = 6), throughout a 4‐week chronic application phase followed by a 4‐day wash‐out phase.
We have previously found that anticonvulsant phenytoin is effective at suppressing ictal‐like activity and seizure‐induced cell death in this model of epilepsy, although the effect was temporary.30 Since phenytoin and celecoxib are likely reducing seizures through different mechanisms, we examined whether celecoxib + phenytoin (C+P) combination would have a stronger and/or longer lasting effect on lactate and LDH production than celecoxib or phenytoin alone. We found that this was the case, with C+P combination reducing both lactate and LDH production beyond that of celecoxib or phenytoin alone by DIV 14 (Fig. 3C and D, lactate P < 0.001 versus celecoxib or phenytoin alone, LDH P < 0.001 versus phenytoin alone, and P = 0.019 versus celecoxib alone, values from ANOVA with Bonferroni post hoc analysis, n = 6 each condition). Phenytoin and celecoxib were no longer effective in reducing lactate release from cultures by DIV 21, but effectiveness of C+P combination persisted until DIV 27 (C+P vs. vehicle, lactate P < 0.001). However, reduction in lactate release was no longer observed after wash‐out of all drugs on DIV 28. We examined the effects of drug wash‐out by comparing lactate values on DIV 27 (just before wash‐out) and DIV 30 (3 days after wash‐out). LDH values were compared between DIV 27 and DIV 31, since seizure‐induced cell death peak occurs slightly after the seizure peak in this model. We found that lactate and LDH release significantly increased in celecoxib‐treated cultures after celecoxib wash‐out (P < 0.001 lactate, P = 0.024 LDH, paired t‐test), and in C+P‐treated cultures after C+P wash‐out (P < 0.001 lactate, P = 0.007 LDH, paired t‐test), suggesting that celecoxib has a strong anticonvulsant, but not an antiepileptic, effect.
Lack of celecoxib effect in acute seizure model
Recurrent seizures were induced in intact hippocampus preparation by perfusion of low‐Mg artificial cerebrospinal fluid (aCSF). The extracellular field potential was monitored in the CA3 area in a recording chamber perfused with low‐Mg aCSF (Fig. 4A). The first appearance of seizure‐like activity was observed 25.2 ± 5.0 min after exposure to low‐Mg aCSF. After initial 3–10 seizures, seizure‐like activities were recorded over 1 h as baseline and then 10 μmol/L celecoxib was bath applied for another hour. Compared with baseline data before application, seizure parameters such as total number, percent change, and mean duration of seizures were not changed significantly by celecoxib (Fig. 4B) (P = 0.86, 0.8, and 0.25, respectively, n = 5). Interseizure interval tends to gradually decrease in this preparation over time,36 but celecoxib did not revert this trend (Fig. 4B). All these results indicate that celecoxib has no effect on acute epileptiform activity induced by low‐Mg in intact hippocampi from naïve animals.
Figure 4.
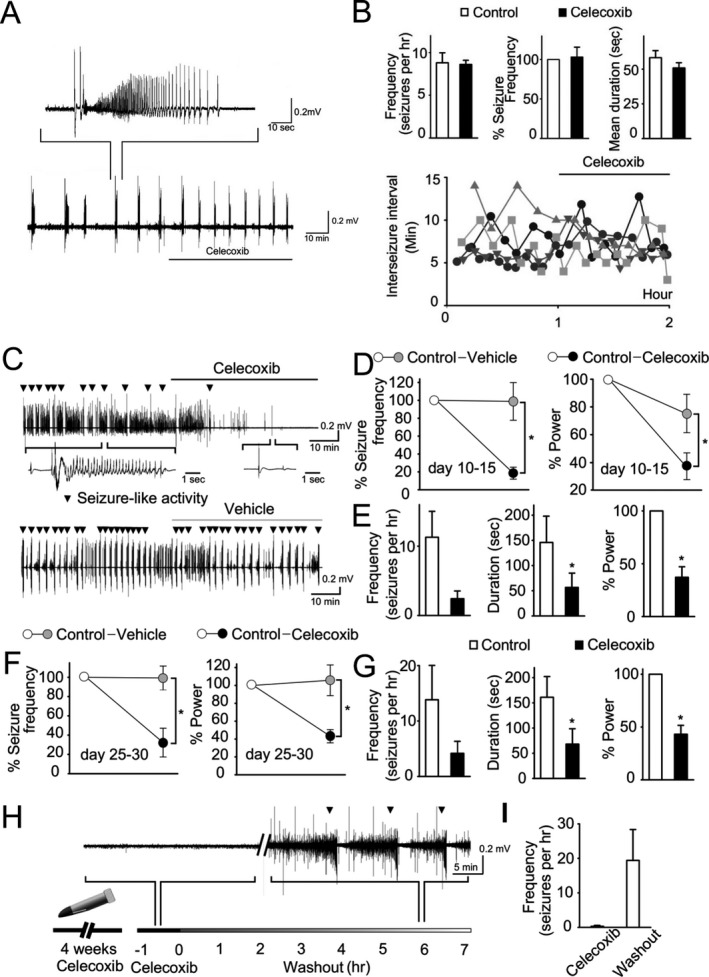
Celecoxib effects in vitro. (A) Application of celecoxib to intact hippocampal preparation in low‐Mg aCSF. A representative trace of field potential from intact hippocampal preparation treated with 10 μmol/L celecoxib with induced seizure‐like activity is shown. (B) Quantification of frequency and mean duration of seizure‐like activity showed that application of celecoxib did not alter those parameters in the intact hippocampal preparation. Different symbols and shading represent results from individual hippocampi. (C) Representative traces of field potential from organotypic hippocampal cultures treated with 10 μmol/L celecoxib or vehicle (DMSO). Inverted triangle indicates seizure‐like activity. (D, F) Frequency of seizure and power during bath application of celecoxib (black circle) or vehicle (gray circle) in younger slices (D) day in vitro (DIV 10–15; n = 8 slices per group) and older slices (F) (DIV 20–25; n = 8 for celecoxib and n = 6 for vehicle). (E, G) Comparison of frequency, total duration of seizure, and power, before and during exposure of celecoxib in younger slices (E) and older slices (G). (H, I) Hippocampal slices (n = 7) were cultured in the presence of 10 μmol/L celecoxib for 4 weeks and then washed out by artificial CSF for 7 h. Representative recording demonstrates the reappearance of seizure‐like activity after wash‐out. Error bars represent SEM, *P < 0.05 (t‐test). Statistical values were determined by one‐way analysis of variance or paired t‐test.
Electrographic effects of celecoxib in chronic seizure models
Organotypic hippocampal cultures
We monitored extracellular field potentials in areas CA1 and CA3 simultaneously in organotypic hippocampal slice cultures at two time points, early (DIV 10–15) and late (DIV 25–20). Extracellular field potentials were recorded with celecoxib, vehicle, or neither. The acute effect of celecoxib was observed 10–20 min after the start of continuous application (Fig. 4C). Celecoxib‐treated slices were compared to vehicle‐treated slices to control for any temporal variance in epileptiform activity. In early slice cultures, 10 μmol/L celecoxib significantly reduced frequency of seizure‐like activity and the power of the field potential (n = 8, P = 0.01 and 0.047 for frequency and power reductions, respectively) (Fig. 4D). Celecoxib (10 μmol/L) also reduced seizure frequency, total duration of seizures, and total power when compared to baseline activity (prior to celecoxib in the same preparation; n = 8, P = 0.17, 0.02. 0.001, respectively). These data indicate that celecoxib exerts an acute anticonvulsant effect in chronically epileptic brain tissue in vitro (Fig. 4E), and that the inhibition of lactate production by celecoxib (Fig. 3) is due to an effect of celecoxib on seizures.
We also tested celecoxib effects on epileptiform activity recorded extracellularly in older slices with more advanced epilepsy30, 40 at 25–30 days of culture (Fig. 4F and G). Differences in seizure frequency and power between celecoxib and vehicle‐treated groups were significant (n = 6–8, P = 0.003 and 0.01, respectively). Frequency and total duration of seizure‐like activity decreased with celecoxib treatment (P = 0.08 and 0.01, respectively). This indicates an age‐independent effect of celecoxib treatment.
Next, we tested the effects of long‐term application of celecoxib followed by wash‐out to differentiate antiepileptogenic versus reversible anticonvulsant effects of celecoxib. Slices were incubated with 10 μmol/L celecoxib for 4 weeks and were then transferred to the recording chamber. Extracellular field potentials were recorded while applying 10 μmol/L celecoxib for 2 h to establish the baseline, and then aCSF only for 7 h to wash celecoxib out (Fig. 4H). Seizure‐like activity was not observed in most of slices during celecoxib application but reappeared after wash‐out (Fig. 4I) (n = 7, P = 0.05, comparison of seizure frequency in celecoxib and after wash‐out). This observation suggests that celecoxib functions as an anticonvulsant rather than an antiepileptogenic drug in organotypic hippocampal culture model of epilepsy.
Rats with kainate‐induced epilepsy
Rats were continuously monitored with traditional wired EEG beginning 2 months after epileptogenic kainate administration Rats were treated with either drug‐containing food or food without the drug on day 1 and then observed through day 3.32, 38 On day 4, the rats were crossed over to the other treatment (i.e., placebo if the first treatment was celecoxib), and observed through day 6. Continuous electrographic recordings through the entire 6‐day cycle were analyzed for seizures (Fig. 5B).
Figure 5.
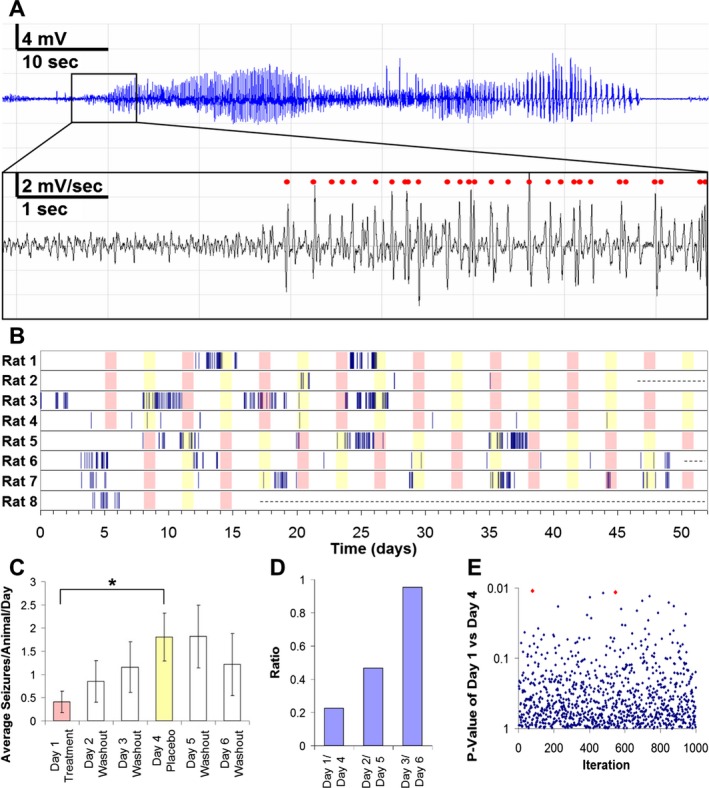
Celecoxib effects in vivo. (A) Raw electroencephalogram (EEG) recording of a seizure (top). First derivative of EEG recording (bottom); spikes marked automatically by dClamp illustrated with red dots. (B) Raster plot of seizure activity in rats (n = 8). Celecoxib treatment illustrated in pink; placebo treatment illustrated in yellow; rats removed from experiment illustrated with dashed line. (C) Average number of seizures per rat per day decreases following celecoxib treatment and increases following placebo treatment; data expressed as mean ± SEM (paired samples t‐test, *P = 0.0116). (D) Ratio of average number of seizures per rat per day showing effect of celecoxib treatment compared to placebo treatment. (E) Randomization of the order of interseizure intervals for each rat over total time course of experiment demonstrated two cases of false significance (P ≤ 0.0116, marked in red) for 1000 iterations.
Celecoxib treatment caused a significant reduction in seizure frequency compared to placebo (Fig. 5C and D; P = 0.0116, paired t‐test). Of note, seizures were clustered in this crossover study (Fig. 5B), so that the seizure intervals did not follow a random (Poisson) distribution. To exclude the possibility of a spuriously significant effect of celecoxib due to clustering, we randomly assigned the same clustered distribution of seizure intervals to test how often a significant result could be obtained by chance. Consistent with the paired samples t‐test, we found that only two randomizations out of 1000 produced a significant difference with P ≤ 0.0116 by chance (Fig. 5E). Thus, the in vivo testing in the kainate model of chronic epilepsy supported the findings of the biochemical and electrophysiological in vitro screening.
Discussion
Existing anticonvulsant drugs are ineffective in approximately 30% of patients. This may be due to an anticonvulsant drug discovery pipeline that relies on chemically or electrically induced seizures in otherwise normal brain tissue.4, 6 However, chronically epileptic tissue is significantly different from normal tissue,41 and may respond differently to drugs. We developed a three‐stage pipeline for discovering new anticonvulsants that relies on chronic epilepsy models at each stage. The organotypic hippocampal culture in vitro model of epilepsy was used in the first stage along with biomarkers of seizures to achieve the highest experimental throughput. Electrographic screening in organotypic hippocampal cultures was then used in the second stage to verify biomarker results. Upon electrographic confirmation in vitro, in vivo experiments were conducted in the kainate model of chronic epilepsy with continuous electrographic monitoring.
The use of organotypic hippocampal cultures, where epileptogenesis occurs on a compressed time scale,23, 24 and where seizures and seizure‐induced cell death can be easily quantified with biomarker assays,30, 31 allowed us to circumvent the throughput limitations of in vivo chronic epilepsy models.4 Thus, we were able to screen 140 compounds and combinations of compounds, in over 400 separate drug‐concentration experiments, and identify over 40 hits.
We discovered that celecoxib, a selective COX2 inhibitor, was surprisingly effective at inhibiting biomarkers of seizures and seizure‐related cell death. We verified these results in a second stage of screening using electrographic seizure quantification in organotypic cultures, and in the final stage of screening in vivo in the kainate model. Interestingly, celecoxib was not effective in attenuating acutely induced seizures in the intact hippocampal preparation, demonstrating that an anticonvulsant discovery pipeline that relies on induced seizures in normal brain tissue would likely miss the anticonvulsant effects of this compound.
Several other compounds with similar COX2 inhibitory efficacy exhibited much more modest anticonvulsant effects in the organotypic model (Fig. 3A). Celecoxib also enhances the inhibitory potassium M current and reduces excitatory L‐type calcium currents in muscle cells.42 However, neither retigabine nor verapamil applied separately had significant activity in this model, suggesting that either these effects are synergistic or that other mechanisms underlie the anticonvulsant effects of celecoxib. Furthermore, the unique efficacy of celecoxib in chronic epilepsy is not readily explained by these particular off‐target effects, because compounds with similar agonist/antagonist profiles are also effective in acute models of epilepsy,43, 44 whereas celecoxib was not (Fig. 4B). Other non‐COX2 targets of celecoxib that have been identified in nonneuronal models45 may be relevant to its efficacy in chronic epilepsy. An interesting possible mechanism is suggested by the finding that the effects of COX2 antagonists can be substrate‐specific,46, 47 although a celecoxib‐specific substrate has not yet been discovered.
Celecoxib has previously been tested in several epilepsy models with quite mixed results.48, 49, 50, 51, 52 The lack of efficacy of other COX2 inhibitors has further dampened enthusiasm for this anticonvulsant strategy.53 However, our data clarify that celecoxib is only efficacious in chronic epilepsy models, and a review of the prior mixed results also demonstrates that it is effective in in vivo chronic epilepsy models,48, 49, 50 but not acutely evoked seizures.48, 51, 52 We suggest that this may be a signature of a compound that would be effective for the one‐third of epilepsy patients who do not respond to currently available anticonvulsants, all of which were discovered by screening against acutely evoked seizures.
Identification of drugs that prevent or modify epileptogenesis and the progression of epilepsy is an important goal in epilepsy research.2, 54 The investigation of drug effects on epileptogenesis is relatively straightforward in the first stage of our pipeline. However, there are important caveats. Most anticonvulsants had modest effects (<1.65 standard deviations) on biomarkers of seizure activity and cell death in the in vitro screen (Fig. 2F). Phenytoin had the most robust effects of the tested anticonvulsants, but resistance eventually developed (Fig. 3C and previous results30), as occurs after human brain injury.55 Thus, many drugs with robust anticonvulsant but not antiepileptogenic effects would be missed in this screen. This suggests that the current screen is an adjunct to, not a replacement of, current anticonvulsant screening methods. The second caveat is that while the lead compound celecoxib inhibited epilepsy for as long as the duration of the experiment without development of resistance (Figs. 3C and 5), this effect washed out immediately when drug was removed (Fig. 4H and I). Thus, the in vitro data indicate that this drug is not antiepileptogenic (i.e., it does not prevent the development of epilepsy). Rather the in vitro data indicate that it is an anticonvulsant that is effective in conditions where other anticonvulsants fail; that is, during the development of epilepsy after brain trauma.56
The only discrepancy between the three stages of evaluation regards the apparent antiepileptogenic effect of celecoxib. Chronic, continuous application of 10 μmol/L celecoxib in vitro provided no evidence of an antiepileptogenic effect upon wash‐out (Fig. 4H). However, a potential antiepileptogenic effect was observed in vivo following single doses administered PO at 6‐day intervals (Fig. 5B). Whether this is an effect of dosing interval or in vivo metabolism to an antiepileptogenic metabolite will require additional testing.
In the first‐stage in vitro screening, the compressed time course of epileptogenesis and ease of monitoring biochemical biomarkers of seizures during drug application and wash‐out allowed us to determine that celecoxib has an anticonvulsant (but not antiepileptogenic effect) in this model in <5 weeks. In contrast, antiepileptogenic screening in animal models of chronic epilepsy can take several months.57, 58 The second stage of screening was also accomplished in a matter of weeks. The final, in vivo stage of screening was by far the most labor‐intensive, requiring at least 2 months to develop chronic epilepsy with an adequate seizure rate after kainate treatment,9 2 months to execute the crossover trial, and several months to analyze the electrographic data with blind procedures. The speed of the first stage can be further enhanced by parallelization, so that this staged analysis could also be applied to medicinal chemistry optimization of lead compounds.
Author Contributions
Y.B., Y.S., K‐I.P., F.E.D, and K.J.S. designed experiments, analyzed data, and wrote the paper. Y.B., Y.S., K‐I.P, B.R., and W.P. conducted experiments. K.L. and W.S. analyzed data. All authors reviewed the manuscript.
Conflicts of Interest
Nothing to report.
Supporting information
Figure S1. Lactate and LDH in cultures after different concentrations of celecoxib were applied starting from DIV 3. Results are presented in cumulative form (DIV 6, DIV 6 + 11, DIV 6 + 11 + 14, etc.). Note toxicity of 100 μmol/L celecoxib indicated by higher LDH release on DIV 6 (3 days after celecoxib application). Each point is an average of results from n = 3 cultures.
{kind=link}
Table S1. Results of the screen, organized in alphabetical order. Each drug‐concentration pair has been applied to three slice cultures, and compared to three vehicle‐treated slice cultures. Duplicate drug‐concentration pairs represent repeated experiments.
Acknowledgments
This work was supported by NINDS grants NS072258, NS086364, and NS077908.
References
- 1. Staley K. Molecular mechanisms of epilepsy. Nat Neurosci 2015;18:367–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Löscher W, Schmidt D. Modern antiepileptic drug development has failed to deliver: ways out of the current dilemma. Epilepsia 2011;52:657–678. [DOI] [PubMed] [Google Scholar]
- 3. French JA, White HS, Klitgaard H, et al. Development of new treatment approaches for epilepsy: unmet needs and opportunities. Epilepsia 2013;54(Suppl 4):3–12. [DOI] [PubMed] [Google Scholar]
- 4. Wilcox KS, Dixon‐Salazar T, Sills GJ, et al. Issues related to development of new antiseizure treatments. Epilepsia 2013;54(Suppl 4):24–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sabolek HR, Swiercz WB, Lillis KP, et al. A candidate mechanism underlying the variance of interictal spike propagation. J Neurosci 2012;32:3009–3021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Galanopoulou AS, Kokaia M, Loeb JA, et al. Epilepsy therapy development: technical and methodologic issues in studies with animal models. Epilepsia 2013;54(Suppl 4):13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Holmes GL. What is more harmful, seizures or epileptic EEG abnormalities? Is there any clinical data? Epileptic Disord 2014;16(Suppl 1):12–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gotman J. Relationships between interictal spiking and seizures: human and experimental evidence. Can J Neurol Sci 1991;18(4 Suppl):573–576. [DOI] [PubMed] [Google Scholar]
- 9. Williams PA, White AM, Clark S, et al. Development of spontaneous recurrent seizures after kainate‐induced status epilepticus. J Neurosci 2009;29:2103–2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hellier JL, Patrylo PR, Buckmaster PS, Dudek FE. Recurrent spontaneous motor seizures after repeated low‐dose systemic treatment with kainate: assessment of a rat model of temporal lobe epilepsy. Epilepsy Res 1998;31:73–84. [DOI] [PubMed] [Google Scholar]
- 11. Kadam SD, White AM, Staley KJ, Dudek FE. Continuous electroencephalographic monitoring with radio‐telemetry in a rat model of perinatal hypoxia‐ischemia reveals progressive post‐stroke epilepsy. J Neurosci 2010;30:404–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gähwiler BH, Capogna M, Debanne D, et al. Organotypic slice cultures: a technique has come of age. Trends Neurosci 1997;20:471–477.[cited 2012 Jul 10] [DOI] [PubMed] [Google Scholar]
- 13. De Simoni A, Griesinger CB, Edwards FA. Development of rat CA1 neurones in acute versus organotypic slices: role of experience in synaptic morphology and activity. J Physiol 2003;550(Pt 1):135–147.[cited 2012 Jul 10] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Prince DA, Parada I, Graber K. Traumatic Brain Injury and Posttraumatic Epilepsy [Internet] In JL Noebels, M Avoli, MA Rogawski, et al., editors. Jasper's Basic Mechanisms of the Epilepsies. Bethesda (MD): National Center for Biotechnology Information (US); 2012. [cited 2016 Jan 26] Available at: http://www.ncbi.nlm.nih.gov/books/NBK98142/. [PubMed] [Google Scholar]
- 15. Sakaguchi T, Okada M, Kawasaki K. Sprouting of CA3 pyramidal neurons to the dentate gyrus in rat hippocampal organotypic cultures. Neurosci Res 1994;20:157–164. [DOI] [PubMed] [Google Scholar]
- 16. Gutiérrez R, Heinemann U. Synaptic reorganization in explanted cultures of rat hippocampus. Brain Res 1999;815:304–316. [DOI] [PubMed] [Google Scholar]
- 17. MacVicar BA, Dudek FE. Local synaptic circuits in rat hippocampus: interactions between pyramidal cells. Brain Res 1980;184:220–223. [DOI] [PubMed] [Google Scholar]
- 18. Pavlidis P, Madison DV. Synaptic transmission in pair recordings from CA3 pyramidal cells in organotypic culture. J Neurophysiol 1999;81:2787–2797. [DOI] [PubMed] [Google Scholar]
- 19. Bausch SB, McNamara JO. Synaptic connections from multiple subfields contribute to granule cell hyperexcitability in hippocampal slice cultures. J Neurophysiol 2000;84:2918–2932.[cited 2012 Jul 10]. [DOI] [PubMed] [Google Scholar]
- 20. McBain CJ, Boden P, Hill RG. Rat hippocampal slices “in vitro” display spontaneous epileptiform activity following long‐term organotypic culture. J Neurosci Methods 1989;27:35–49.[cited 2012 Jul 10]. [DOI] [PubMed] [Google Scholar]
- 21. Gutnick MJ, Wolfson B, Baldino F. Synchronized neuronal activities in neocortical explant cultures. Exp Brain Res 1989;76:131–140. [DOI] [PubMed] [Google Scholar]
- 22. Heinemann U, Staley KJ. What is the clinical relevance of in vitro epileptiform activity? Adv Exp Med Biol 2014;813:25–41. [DOI] [PubMed] [Google Scholar]
- 23. Dyhrfjeld‐Johnsen J, Berdichevsky Y, Swiercz W, et al. Interictal spikes precede ictal discharges in an organotypic hippocampal slice culture model of epileptogenesis. J Clin Neurophysiol 2010;27:418–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lillis KP, Wang Z, Mail M, et al. Evolution of Network Synchronization during Early Epileptogenesis Parallels Synaptic Circuit Alterations. J Neurosci 2015;35:9920–9934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. During MJ, Fried I, Leone P, et al. Direct measurement of extracellular lactate in the human hippocampus during spontaneous seizures. J Neurochem 1994;62:2356–2361. [DOI] [PubMed] [Google Scholar]
- 26. Castillo M, Smith JK, Kwock L. Proton MR spectroscopy in patients with acute temporal lobe seizures. AJNR Am J Neuroradiol 2001;22:152–157. [PMC free article] [PubMed] [Google Scholar]
- 27. Bonfoco E, Krainc D, Ankarcrona M, et al. Apoptosis and necrosis: two distinct events induced, respectively, by mild and intense insults with N‐methyl‐D‐aspartate or nitric oxide/superoxide in cortical cell cultures. Proc Natl Acad Sci USA 1995;92:7162–7166.[cited 2012 Mar 26]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Decker T, Lohmann‐Matthes ML. A quick and simple method for the quantitation of lactate dehydrogenase release in measurements of cellular cytotoxicity and tumor necrosis factor (TNF) activity. J Immunol Methods 1988;115:61–69. [DOI] [PubMed] [Google Scholar]
- 29. Korzeniewski C, Callewaert DM. An enzyme‐release assay for natural cytotoxicity. J Immunol Methods 1983;64:313–320. [DOI] [PubMed] [Google Scholar]
- 30. Berdichevsky Y, Dzhala V, Mail M, Staley KJ. Interictal spikes, seizures and ictal cell death are not necessary for post‐traumatic epileptogenesis in vitro. Neurobiol Dis 2012;45:774–785.[cited 2012 Mar 11] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Berdichevsky Y, Dryer AM, Saponjian Y, et al. PI3K‐Akt signaling activates mTOR‐mediated epileptogenesis in organotypic hippocampal culture model of post‐traumatic epilepsy. J Neurosci 2013;33:9056–9067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ali A, Dua Y, Constance JE, et al. A once‐per‐day, drug‐in‐food protocol for prolonged administration of antiepileptic drugs in animal models. Epilepsia 2012;53:199–206. [DOI] [PubMed] [Google Scholar]
- 33. White AM, Williams PA, Ferraro DJ, et al. Efficient unsupervised algorithms for the detection of seizures in continuous EEG recordings from rats after brain injury. J Neurosci Methods 2006;152:255–266. [DOI] [PubMed] [Google Scholar]
- 34. Nakagawa S, Cuthill IC. Effect size, confidence interval and statistical significance: a practical guide for biologists. Biol Rev Camb Philos Soc 2007;82:591–605. [DOI] [PubMed] [Google Scholar]
- 35. Zhang XD. Illustration of SSMD, z score, SSMD*, z* score, and t statistic for hit selection in RNAi high‐throughput screens. J Biomol Screen 2011;16:775–785. [DOI] [PubMed] [Google Scholar]
- 36. Dzhala VI, Brumback AC, Staley KJ. Bumetanide enhances phenobarbital efficacy in a neonatal seizure model. Ann Neurol 2008;63:222–235. [DOI] [PubMed] [Google Scholar]
- 37. Hellier JL, Dudek FE. Chemoconvulsant Model of Chronic Spontaneous Seizures. Current Protocols in Neuroscience. 31:9.19:9.19.1–9.19.12. [DOI] [PubMed] [Google Scholar]
- 38. Grabenstatter HL, Clark S, Dudek FE. Anticonvulsant effects of carbamazepine on spontaneous seizures in rats with kainate‐induced epilepsy: comparison of intraperitoneal injections with drug‐in‐food protocols. Epilepsia 2007;48:2287–2295. [DOI] [PubMed] [Google Scholar]
- 39. Tegeder I, Niederberger E, Vetter G, et al. Effects of selective COX‐1 and ‐2 inhibition on formalin‐evoked nociceptive behaviour and prostaglandin E(2) release in the spinal cord. J Neurochem 2001;79:777–786. [DOI] [PubMed] [Google Scholar]
- 40. Dzhala V, Staley KJ. Acute and chronic efficacy of bumetanide in an in vitro model of posttraumatic epileptogenesis. CNS Neurosci Ther 2015;21:173–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Miyata H, Hori T, Vinters HV. Surgical pathology of epilepsy‐associated non‐neoplastic cerebral lesions: a brief introduction with special reference to hippocampal sclerosis and focal cortical dysplasia. Neuropathology 2013;33:442–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Brueggemann LI, Mackie AR, Mani BK, et al. Differential effects of selective cyclooxygenase‐2 inhibitors on vascular smooth muscle ion channels may account for differences in cardiovascular risk profiles. Mol Pharmacol 2009;76:1053–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hadar EJ, Yang Y, Sayin U, Rutecki PA. Suppression of pilocarpine‐induced ictal oscillations in the hippocampal slice. Epilepsy Res 2002;49:61–71. [DOI] [PubMed] [Google Scholar]
- 44. Qiu C, Johnson BN, Tallent MK. K+ M‐current regulates the transition to seizures in immature and adult hippocampus. Epilepsia 2007;48:2047–2058. [DOI] [PubMed] [Google Scholar]
- 45. Schönthal AH. Direct non‐cyclooxygenase‐2 targets of celecoxib and their potential relevance for cancer therapy. Br J Cancer 2007;97:1465–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hermanson DJ, Hartley ND, Gamble‐George J, et al. Substrate‐selective COX‐2 inhibition decreases anxiety via endocannabinoid activation. Nat Neurosci 2013;16:1291–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hermanson DJ, Gamble‐George JC, Marnett LJ, Patel S. Substrate‐selective COX‐2 inhibition as a novel strategy for therapeutic endocannabinoid augmentation. Trends Pharmacol Sci 2014;35:358–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gobbo OL, O'Mara SM. Post‐treatment, but not pre‐treatment, with the selective cyclooxygenase‐2 inhibitor celecoxib markedly enhances functional recovery from kainic acid‐induced neurodegeneration. Neuroscience 2004;125:317–327. [DOI] [PubMed] [Google Scholar]
- 49. Jung K‐H, Chu K, Lee S‐T, et al. Cyclooxygenase‐2 inhibitor, celecoxib, inhibits the altered hippocampal neurogenesis with attenuation of spontaneous recurrent seizures following pilocarpine‐induced status epilepticus. Neurobiol Dis 2006;23:237–246. [DOI] [PubMed] [Google Scholar]
- 50. Schlichtiger J, Pekcec A, Bartmann H, et al. Celecoxib treatment restores pharmacosensitivity in a rat model of pharmacoresistant epilepsy. Br J Pharmacol 2010;160:1062–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Baik EJ, Kim EJ, Lee SH, Moon C. Cyclooxygenase‐2 selective inhibitors aggravate kainic acid induced seizure and neuronal cell death in the hippocampus. Brain Res 1999;843:118–129. [DOI] [PubMed] [Google Scholar]
- 52. Fischborn SV, Soerensen J, Potschka H. Targeting the prostaglandin E2 EP1 receptor and cyclooxygenase‐2 in the amygdala kindling model in mice. Epilepsy Res 2010;91:57–65. [DOI] [PubMed] [Google Scholar]
- 53. Rojas A, Jiang J, Ganesh T, et al. Cyclooxygenase‐2 in epilepsy. Epilepsia 2014;55:17–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kovács R, Heinemann U. Models in research of pharmacoresistant epilepsy: present and future in development of antiepileptic drugs. Curr Med Chem 2014;21:689–703. [DOI] [PubMed] [Google Scholar]
- 55. Temkin NR, Dikmen SS, Wilensky AJ, et al. A randomized, double‐blind study of phenytoin for the prevention of post‐traumatic seizures. N Engl J Med 1990;323:497–502. [DOI] [PubMed] [Google Scholar]
- 56. Temkin NR. Preventing and treating posttraumatic seizures: the human experience. Epilepsia 2009;50(Suppl 2):10–13. [DOI] [PubMed] [Google Scholar]
- 57. Pitkänen A, Nehlig A, Brooks‐Kayal AR, et al. Issues related to development of antiepileptogenic therapies. Epilepsia 2013;54(Suppl 4):35–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Pitkänen A, Lukasiuk K, Dudek FE, Staley KJ. Epileptogenesis. Cold Spring Harb Perspect Med 2015;5:(10): a022822. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Lactate and LDH in cultures after different concentrations of celecoxib were applied starting from DIV 3. Results are presented in cumulative form (DIV 6, DIV 6 + 11, DIV 6 + 11 + 14, etc.). Note toxicity of 100 μmol/L celecoxib indicated by higher LDH release on DIV 6 (3 days after celecoxib application). Each point is an average of results from n = 3 cultures.
Table S1. Results of the screen, organized in alphabetical order. Each drug‐concentration pair has been applied to three slice cultures, and compared to three vehicle‐treated slice cultures. Duplicate drug‐concentration pairs represent repeated experiments.