

Abstract
Objective
This study examined whether a history of traumatic brain injury (TBI) is associated with earlier onset of Alzheimer disease (AD), independent of apolipoprotein ε4 status (Apoe4) and gender.
Method
Participants with a clinical diagnosis of AD (n=7625) were obtained from the National Alzheimer’s Coordinating Center Uniform Data Set, and categorized based on self-reported lifetime TBI with loss of consciousness (LOC) (TBI+ vs TBI-) and presence of Apoe4. ANCOVAs, controlling for gender, race, and education were used to examine the association between history of TBI, presence of Apoe4, and an interaction of both risk factors on estimated age of AD onset.
Results
Estimated AD onset differed by TBI history and Apoe4 independently (p’s <.001). The TBI+ group had a mean age of onset 2.5 years earlier than the TBI- group. Likewise, Apoe4 carriers had a mean age of onset 2.3 years earlier than non-carriers. While the interaction was non-significant (p = .34), participants having both a history of TBI and Apoe4 had the earliest mean age of onset compared to those with a TBI history or Apoe4 alone (MDifference = 2.8 & 2.7 years, respectively). These results remained unchanged when stratified by gender.
Conclusions
History of self-reported TBI can be associated with an earlier onset of AD-related cognitive decline, regardless of Apoe4 status and gender. TBI may be related to an underlying neurodegenerative process in AD, but the implications of age at time of injury, severity, and repetitive injuries remain unclear.
Keywords: National Alzheimer’s Coordinating Center (NACC), Traumatic Brain Injury (TBI), Dementia, Alzheimer’s Disease (AD), Age of Onset
Mounting evidence suggests that a history of TBI is a risk factor for later cognitive consequences, including Alzheimer disease (AD), for some individuals. Although not all studies are supportive (Dams-O’Connor, 2013; Fratiglioni, Ahlbom, Viitanen, & Winblad, 1993; Helmes, Ostbye, & Steenhuis, 2011; Lindsay et al., 2002; Mehta et al., 1999; Xu et al., 2015), the increased likelihood for the later development of AD years or decades following TBI has ranged from 1.3 to a 9.9 - fold higher risk in case-control (Guo et al., 2000; Mortimer, French, Hutton, & Schuman, 1985; Plassman et al., 2000; van Duijn et al., 1992), meta-analytic (Fleminger, Oliver, Lovestone, Rabe-Hesketh, & Giora, 2003; Mortimer et al., 1991), and medical chart review (Barnes et al., 2014; Wange et al., 2012) studies. However, neurodegenerative dementias such as AD have an insidious onset, with support that this process develops over several decades. As such, the potential mechanism(s) underlying TBI as a risk factor remains poorly understood.
The neuropathology of AD involves the accumulation of tau-related neurofibrillary tangles (NFT) and amyloid-β (Aβ) plaques, although these pathologies appear to represent distinct yet synergistic processes (Nelson, Braak, & Markesbery, 2009). NFTs first appear in the mesial temporal lobe and then progress to involve the inferior frontal region before coursing superiorly and posteriorly to include the entire frontal, parietal, and occipital regions as well (Braak & Braak, 1991; Nelson et al., 2009; Thal, Attems, & Ewers, 2014). Aβ plaques, on the other hand, initially occur in the neocortex, but then progress to the mesial temporal lobe, followed by the basal ganglia, brainstem, and cerebellum (Dietmar R Thal, Rüb, Orantes, & Braak, 2002; Dietmar Rudolf Thal et al., 2014). TBI has a predilection for affecting frontal and temporal structures, and, even at milder severities, can produce white matter injury (Johnson et al., 2013), which recently has been linked to the presence of greater Aβ deposition in some individuals with moderate-to-severe TBI (Scott et al., 2016). Thus, TBI-related processes may contribute to the buildup of pathological burden in AD and related conditions, potentially increasing the risk for earlier onset of these conditions. Fueled by the media’s increased focus on sports-related concussions and the possible link with cognitive decline later in life, there has been greater attention to TBI and its potential association with AD, the most common type of dementia. However, research on later-life risk and the clinical trajectory for dementia following TBI remains in its infancy.
The first study that suggests TBI may be a risk factor for earlier expression of AD was published in 1987. Sullivan et al. performed a medical chart review to retrospectively diagnose AD in a convenience sample of 13 cases (Sullivan, Petitti, & Barbaccia, 1987). Six cases were also found to have a history of TBI of mixed severity. The authors observed the age of AD onset to be approximately 9 years earlier for those with a history of TBI. Gedye et al. extended this finding by publishing a 1989 medical chart review of 148 cases diagnosed with AD using standard clinical criteria (Gedye, Beattie, Tuokko, Horton, & Korsarek, 1989). The authors collected lifetime TBI information and rated severity according to duration of loss of consciousness (LOC) and presence of neurologic symptoms following TBI (i.e., headaches, dizziness, confusion). There were 35 cases with a history of TBI, and although mild TBI cases did not differ in terms of AD onset from those without TBI history, those with moderate and severe TBI were found to have a nearly 7 year earlier onset.
In contrast to the early reports indicating that a history of TBI may accelerate the expression of AD, the literature thereafter has been mixed. In 1991, Mortimer et al. published a meta-analysis of 11 case-control AD studies with a total of 1,113 participants, of whom 84 had a history of TBI with LOC, while 1,029 did not. Although the authors found TBI with LOC was linked to increased risk for AD, a history of TBI failed to show an association with earlier age of AD onset (Mortimer et al., 1991). A similar finding was reported in 1995 by Rasmussin et al. when comparing 20 AD cases with any history of TBI, regardless of LOC, and 48 non-injury cases (Rasmusson, Brandt, Martin, & Folstein, 1995). In 1999, Nemetz et al. performed a medical chart review among Rochester, MN residents with a documented history of TBI from 1935 to 1984 (Nemetz et al., 1999). TBI was defined as a head injury resulting in LOC, posttraumatic amnesia (PTA), neurological symptoms, or skull fracture. There were 1,283 TBI cases, of whom 31 were later diagnosed with AD. The authors concluded that those with a history of TBI developed AD approximately 8 years earlier than the general Rochester population.
These publications were soon followed by two articles in what remain perhaps some of the best-controlled studies examining the link between TBI and AD to date. In 2000, Plassman et al. used standard clinical evaluations to diagnose AD in 35 male World War II veterans, out of 1,811 who were hospitalized during their military service (approximately 40 years prior) due to a TBI or a non-TBI condition (Plassman et al., 2000). The authors classified TBI as mild (LOC or PTA <30 minutes), moderate (LOC or PTA between 30 minutes and 24 hours and/or skull fracture), and severe (extended LOC or posttraumatic amnesia >24 hours). Although it was concluded that moderate and severe TBI were associated with an increased likelihood for an AD diagnosis, when the authors compared TBI (n = 17) and non-TBI cases (n = 18), a history of TBI was not linked to an earlier age of AD onset. In addition, Guo et al. published one of the largest studies to date reporting on this association by examining 2,234 cases diagnosed with AD according to standard clinical criteria, of whom 398 had a history of TBI resulting in LOC or medical care, and 1,835 did not (Guo et al., 2000). The authors reported that while a history of TBI was associated with a greater likelihood of an AD diagnosis, age of AD onset was similar between those with and without a history of TBI.
Despite the mixed findings in the literature, we were unable to find any additional studies published over the past 15 years regarding the association between a history of TBI and earlier onset of AD, even though a number of potentially confounding variables have yet to be explored. Various methodological factors may account for the mixed findings in the literature, including limited numbers of cases, sampling procedures, methods of data collection, different criteria for TBI classification, and methods of AD diagnosis. Additionally, other factors may play a role in the mixed findings, including gender, as some studies have reported a higher risk for AD only in males with a history of TBI (Fleminger et al., 2003; Guo et al., 2000; Mortimer et al., 2001), and presence of genetic susceptibility. The apolipoprotein E gene (Apoe) has 3 alleles (ε2, ε3, ε4) that encode a protein involved in lipid transport based on which combination of two alleles is present (ε2/ε2/, ε2/ε3, ε3/ε3, ε2/ε4, ε3/ε4, ε4/ε4); the Apoe ε4 allele (Apoe4) is well-known to play a role in the pathogenesis and earlier onset of AD (Blacker et al., 1997; Corder et al., 1993; Maiti et al., 2015; Naj et al., 2014). The primary goal of this study was to examine whether a history of TBI with LOC is associated with an earlier age of onset, independent of Apoe4 status and gender, in a large cohort of well-characterized individuals with a diagnosis of AD. These findings would support TBI as a risk factor for later cognitive decline and may suggest a possible role for TBI in a neurodegenerative process.
Method
Participants
Sociodemographic, clinical, and biomarker information was obtained for participants with a diagnosis of AD from the National Alzheimer’s Coordinating Center (NACC) Uniform Data Set (UDS) (Morris et al., 2006). The NACC has consolidated data from 34 past and present National Institute of Aging (NIA) - funded Alzheimer’s Disease Centers (ADC) throughout the U.S. from September, 2005. The NACC’s data collection procedures were approved by institutional review boards at each participating ADC. Selection criteria included participants 50 years and older with a diagnosis of possible or probable AD at the initial visit to an ADC between September, 2005 and December, 2013. ADC clinicians reviewed neurological exam results, medical history, neuropsychological test performance, laboratory and neuroimaging results, psychosocial background, and information from informants to determine a diagnosis of AD using standard NINCDS/ADRDA guidelines (McKhann et al., 1984). Age of onset was defined as 1) the ADC clinician’s estimate for age of cognitive decline based on participant/informant report for when cognitive abilities began declining (hereafter referred to as age of symptom onset) and 2) the participant’s age at the initial ADC visit when they were diagnosed with AD.
Measures
TBI information in the NACC database is based upon three questions answered during a medical history interview with participants/informants at the initial ADC visit. Participants were asked whether they had ever experienced a TBI resulting in <5 minutes loss of consciousness (mLOC), ≥5 mLOC, and if there was a chronic deficit as a result of the injury. Responses to all three questions were recorded as absent, recent/active (occurring within 1 year of visit or requiring ongoing treatment), remote/inactive (occurring >1 year of visit and either having recovered from the injury or no current treatment is underway), or unknown. The current study included two groups, one with a reported absence of TBI history with LOC (TBI-), and a second group that reported a TBI with any LOC, regardless of duration, and no chronic deficits, occurring more than one year prior to the initial visit (TBI+). Because our goal was to examine the link between TBI history and the development of AD, we attempted to limit potential confounding from cognitive decline directly related to a TBI by excluding participants who reported a recent TBI (defined as < 1 year prior to initial ADC visit; n = 55) or any history of TBI resulting in a chronic deficit (n = 14).
For the present study, age, gender, race, years of education, family history of dementia (at least 1 member), and number of Apoe4 alleles were requested due to their known link with AD. Since vascular factors and depression have been associated with increased risk for AD (Luchsinger et al., 2005; Modrego & Ferrández, 2004), we examined their potential influence from available data. At the initial visit, participants are asked questions via participant/informant interview if they ever had: 1) a history of heart attack, atrial fibrillation, angioplasty, cardiac bypass, pacemaker, congestive heart failure, stroke, transient ischemic attack, hypertension, hypercholesterolemia, and/or diabetes, 2) a history of depression occurring >2 years prior, and 3) a history of depression within 2 years of their visit. Vascular conditions are coded similar to the TBI information in the NACC database as absent, recent/active, remote/inactive, or unknown. Depression was coded only as absent or present and defined as consulting a clinician about depressed mood, being prescribed antidepressant medication, or receiving a mood disorder diagnosis (i.e., major depression, dysthymia, bipolar).
Statistical Analyses
Chi-square and independent t-tests were used to assess whether sociodemographic variables and number of Apoe4 alleles differed between TBI+ and TBI- groups. Since the two groups had similar proportions for Apoe4 status, this variable was collapsed into two categories, based on absence or presence of at least one Apoe4 allele. ANOVA was used to initially assess whether estimated age of symptom onset and age at diagnosis of AD significantly differed between those reporting a mild TBI (< 5 mLOC), more serious injuries (≥5 mLOC), and those without any history of TBI with LOC. Next, ANCOVA was used to assess whether estimated age of symptom onset and age at diagnosis of AD differed between the TBI+ and TBI- groups, after controlling for relevant factors. A two-way multivariate ANCOVA was conducted to examine the association between presence of Apoe4, history of TBI with LOC, and an interaction of both risk factors on estimated age of symptom onset in participants with AD. In order to examine potential gender-related effects, the two-way multivariate ANCOVA was performed, stratified by gender. Multivariate analyses were performed again only for participants diagnosed with probable AD to conservatively assess whether associations remained. Assumptions for all tests were reviewed, and unequal variances were observed for the primary analysis when examining age of symptom onset and age of diagnosis in AD. A Welch ANOVA was used to determine if unequal variances resulted in a Type I error. All analyses were conducted using IBM© SPSS Statistics V22 (IBM Corp, SPSS Statistics V22, Armonk, NY, 2013) with p < .05 as the level for significance; a Bonferroni correction of .01 (.05/5) was used to maintain a 5% error rate across analyses.
Results
Demographic and Sample Characteristics
Of the 7,625 AD cases in the NACC database who met inclusion criteria, 6,603 were diagnosed with probable AD. A total of 422 participants reported a history of mild TBI with < 5 mLOC, 149 a history of more serious TBI (≥ 5 mLOC), and 7,054 reported no such history, although an unknown proportion of the TBI- group may have a history of TBI without LOC. Demographic characteristics for participants with a history of TBI with any LOC (TBI+) and those without (TBI-) can be found in Table 1. Few participants were missing data on most variables, but 4% were missing vascular medical conditions, 3% were missing a history of depression, and nearly 30–40% were missing Apoe4 status. TBI+ and TBI- groups significantly differed in terms of gender, race, and education (p <.001). Specifically, TBI+ participants with AD were 38% female, 89% Caucasian, and had an average education of 14.5 (SD = 3.6). In contrast, TBI- participants were 59% female, 80% Caucasian, and had an average education of 13.8 (SD = 3.9). TBI+ and TBI- groups with AD did not differ by family history of dementia (p = .76), vascular medical conditions (p’s ≥ .13), or lifetime history of depression (p’s ≥ .07). Additionally, of the 4,744 AD participants with known Apoe4 status, the number of Apoe4 alleles did not differ (p = .28) between the TBI+ (n = 386; 67%) and TBI- (n = 4,312; 56%) groups.
Table 1.
Demographics and Age of Alzheimer Disease Onset in Participants With and Without a History of Traumatic Brain Injury
| TBI+ (n=571) | TBI– (n=7054) | ||||||
|---|---|---|---|---|---|---|---|
| M | SD | Range | M | SD | Range | p | |
| Age of Onset | 68.3 | 10.3 | 41–102 | 70.9 | 9.5 | 37–103 | < .001* |
| Age of Diagnosis | 73.9 | 10.2 | 50–102 | 76.2 | 9.4 | 50–104 | < .001* |
| Education | 14.5 | 3.6 | 0–20 | 13.8 | 3.9 | 0–20 | < .001* |
| Female, n (%) | 216 | (38) | 4136 | (59) | < .001* | ||
| Caucasian, n (%) | 508 | (89) | 5634 | (80) | < .001* | ||
| Family History of Dementia, n (%) | 309 | (54) | 3770 | (53) | 0.76 | ||
| Heart Attack, n (%) | 570 | (7) | 7029 | (6) | 0.16 | ||
| Atrial Fibrillation, n (%) | 570 | (8) | 7022 | (7) | 0.36 | ||
| Angioplasty, n (%) | 571 | (8) | 7049 | (7) | 0.13 | ||
| Cardiac Bypass, n (%) | 571 | (5) | 7049 | (5) | 0.17 | ||
| Pacemaker, n (%) | 571 | (5) | 7053 | (4) | 0.50 | ||
| Congestive Heart Failure, n (%) | 571 | (3) | 7040 | (3) | 0.68 | ||
| Stroke, n (%) | 563 | (6) | 7026 | (6) | 0.64 | ||
| Transient Ischemic Attack, n (%) | 566 | (7) | 7001 | (6) | 0.52 | ||
| Hypertension, n (%) | 568 | (50) | 7036 | (54) | 0.22 | ||
| Hypercholesterolemia, n (%) | 566 | (51) | 6976 | (51) | 0.66 | ||
| Diabetes, n (%) | 571 | (12) | 7038 | (13) | 0.70 | ||
| Remote History of Depression, n (%) | 565 | (21) | 6917 | (18) | 0.07 | ||
| Recent History of Depression, n (%) | 567 | (41) | 6981 | (41) | 0.77 | ||
| APOE ε4 alleles, n (%) | 0.43 | ||||||
| 0 ε4 alleles | 158 | (41) | 1788 | (41) | |||
| 1 ε4 allele | 172 | (45) | 2033 | (47) | |||
| 2 ε4 alleles | 56 | (14) | 537 | (12) | |||
Note. * p < .05. n = 7,625. TBI+ = presence of a history of traumatic brain injury with any loss of consciousness and no chronic deficits occurring more than one year prior to the initial visit. TBI– = absence of a history of traumatic brain injury with loss of consciousness. APOE ε4+ = presence of at least one apolipoprotein E-e4 allele. APOE ε4– = absence of an apolipoprotein E-e4 allele.
TBI History on Age of Symptom Onset and Age of Diagnosis
Participants reporting a history of mild TBI with <5 mLOC (MOnset Age = 68.1; MAge of Diagnosis = 73.8) and more serious injuries (MOnset Age = 68.9; MAge of Diagnosis = 74.1) had a significantly earlier age of AD onset (F (2, 7619) = 14.8, p < .001; η2partial = .004) and diagnosis (F (2, 7619) = 16.8, p < .001; η2partial = .004) compared to those without a TBI history (MOnset Age = 70.9; MAge of Diagnosis = 76.2). However, no significant difference was observed between the TBI samples. Controlling for gender, race, and education, clinician-estimated age of symptom onset again differed (F (1, 7620) = 33.0, p <.001; η2partial = .004) between groups, with TBI+ (MAge = 68.3, SD = 10.3) participants having an average onset 2.6 years earlier than the TBI- group (MAge = 70.9, SD = 9.5). Likewise, AD was also diagnosed a mean of 2.3 years earlier (F (1, 7620) = 28.1, p <.001; η2partial = .004) in the TBI+ group (MAge = 73.9, SD = 10.2) relative to the TBI- sample (MAge = 76.2, SD = 9.4). Levene’s test was significant (p < .01) for both analyses, indicating that the TBI+ and TBI- groups had significantly unequal variances for estimated age of symptom onset and age of diagnosis in AD; as a result, a nonparametric Welch ANOVA was conducted to control for these discrepancies. Results from the two Welch ANOVAs showed that despite the unequal variances, the TBI+ group had a significantly earlier age of symptom onset (Welch’s F (1, 650.262) = 34.3, p <.001) and age of diagnosis (Welch’s F (1, 650.099) = 27.8, p <.001) than the TBI- group.
Age of Symptom Onset and Associations with Apoe4 and TBI History
When presence of Apoe4 and history of TBI were entered as independent variables in a multivariate ANCOVA model, controlling for gender, race, and education, there was a main effect for both Apoe4 and history of TBI. Specifically, Apoe4 carriers (MAge = 69.6, SD = 8.8) had a mean estimated age of symptom onset 2.3 years earlier (F (1, 4737) = 26.1, p <.001; η2partial = .005) than non-carriers (MAge = 71.9, SD = 10.3). Likewise, estimated age of symptom onset remained 2.5 years earlier (F (1, 4737) = 19.3, p <.001; η2partial = .004) in the TBI+ group (MAge = 68.2, SD = 10.6) compared to the TBI- group (MAge = 70.7, SD = 9.4). Interestingly, relative to those with a history of TBI or APOE ε4 alone and in the absence of both risk factors, individuals having both Apoe4 and a history of TBI had the earliest age of symptom onset (M Difference ≥2.7 years vs all others, Table 2); however, this interaction was non-significant (F (1, 4737) = .93, p = .34; η2partial < .001). Among participants diagnosed with probable AD, age of symptom onset and diagnosis remained significantly earlier for Apoe4 carriers (F (1, 4141) = 22.6, p < .001; η2partial = .004) and those with a history of TBI alone (F (1, 4141) = 21.4, p < .001; η2partial = .004).
Table 2.
Main Effects and Interaction for Traumatic Brain Injury and Apolipoprotein E-ε4 Status on Age of Symptom Onset of Alzheimer Disease
| n | M (SD) | F | p | |
|---|---|---|---|---|
| Main Effect for APOE Status | 19.27 | < .001* | ||
| APOE ε4 + | 2,798 | 69.6 (8.8) | ||
| APOE ε4 – | 1,946 | 71.9 (10.3) | ||
| Main Effect for TBI Status | 26.06 | < .001* | ||
| TBI + | 386 | 68.2 (10.6) | ||
| TBI – | 4,358 | 70.7 (9.4) | ||
| Interaction for TBI and APOE ε4 | 0.93 | 0.34 | ||
| TBI + APOE ε4 + | 228 | 67.1 (9.9) | ||
| TBI + APOE ε4 – | 158 | 69.9 (11.3) | ||
| TBI – APOE ε4 + | 2,570 | 69.8 (8.7) | ||
| TBI – APOE ε4 – | 1,788 | 72.1 (10.2) | ||
Note. * p < .05. n = 4,744. df = 1. TBI+ = presence of a history of traumatic brain injury with any loss of consciousness and no chronic deficits occurring more than one year prior to the initial visit. TBI− = absence of a history of traumatic brain injury with loss of consciousness. APOE ε4+ = presence of at least one apolipoprotein E-e4 allele. APOE ε4– = absence of an apolipoprotein E-e4 allele.
Age of Symptom Onset by Gender and Associations with Apoe4 and TBI History
When stratified by gender, the results remained relatively unchanged. Main effects for Apoe4 and history of TBI were found for both females and males. Estimated age of symptom onset was 2.6 years earlier for Female participants with Apoe4 (MAge = 69.9; F (1, 2636) = 10.4, p = .001; η2partial = .005) and 2.7 years earlier for females with a history of TBI (MAge = 68.4; F (1, 2636) = 10.2, p = .001; η2partial = .003), compared to those without (MAge = 72.6 and 71.1, respectively). Among male participants, estimated age of symptom onset was 1.8 and 2.1 years earlier, respectively, for Apoe4 carriers (MAge = 69.3; F (1, 2096) = 14.6, p < .001; η2partial = .005) and those with a TBI history (MAge = 68.1; F (1, 2096) = 8.9, p = .003; η2partial = .005), compared to participants without TBI (MAge = 71.1 and 70.2, respectively). Regardless of gender, there was no significant interaction between Apoe4 and a history of TBI (Females: F (1, 2636) = .94, p = .34; η2partial < .001); Males: F (1, 2096) = .44, p = .51; η2partial < .001). However, female and male participants having both risk factors continued to show the earliest onset of symptoms relative to those with a history of TBI or Apoe4 alone and in the absence of both risk factors (M Difference: females ≥3.0 years; males ≥2.4 years), as shown in Figures 1 and 2. Examination of participants with probable AD alone showed the same results (not shown).
Figure 1.
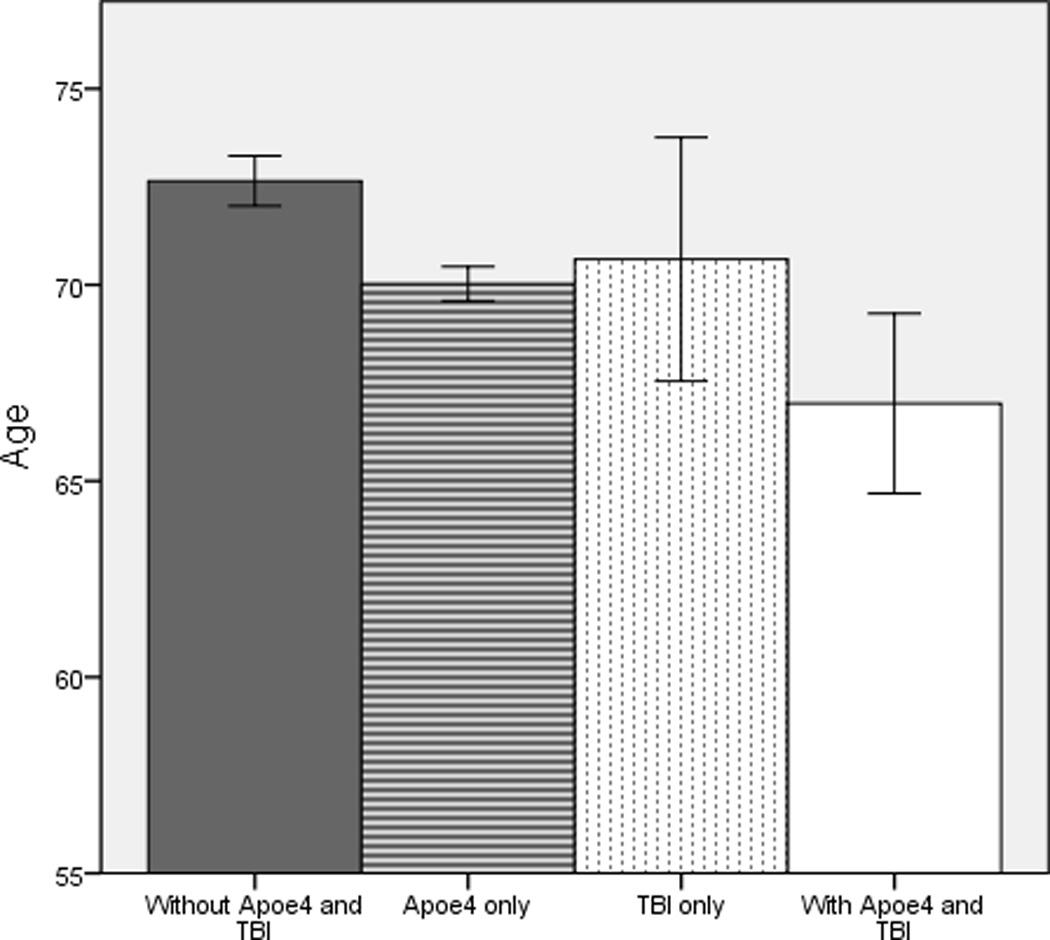
Mean age of symptom onset in female patients with Alzheimer disease with and without risk factors apolipoprotein E-ε4 and history of traumatic brain injury. Bars represent 95% confidence intervals for each group. TBI = traumatic brain injury with loss of consciousness. Apoe4 = apolipoprotein E-e4 allele.
Figure 2.
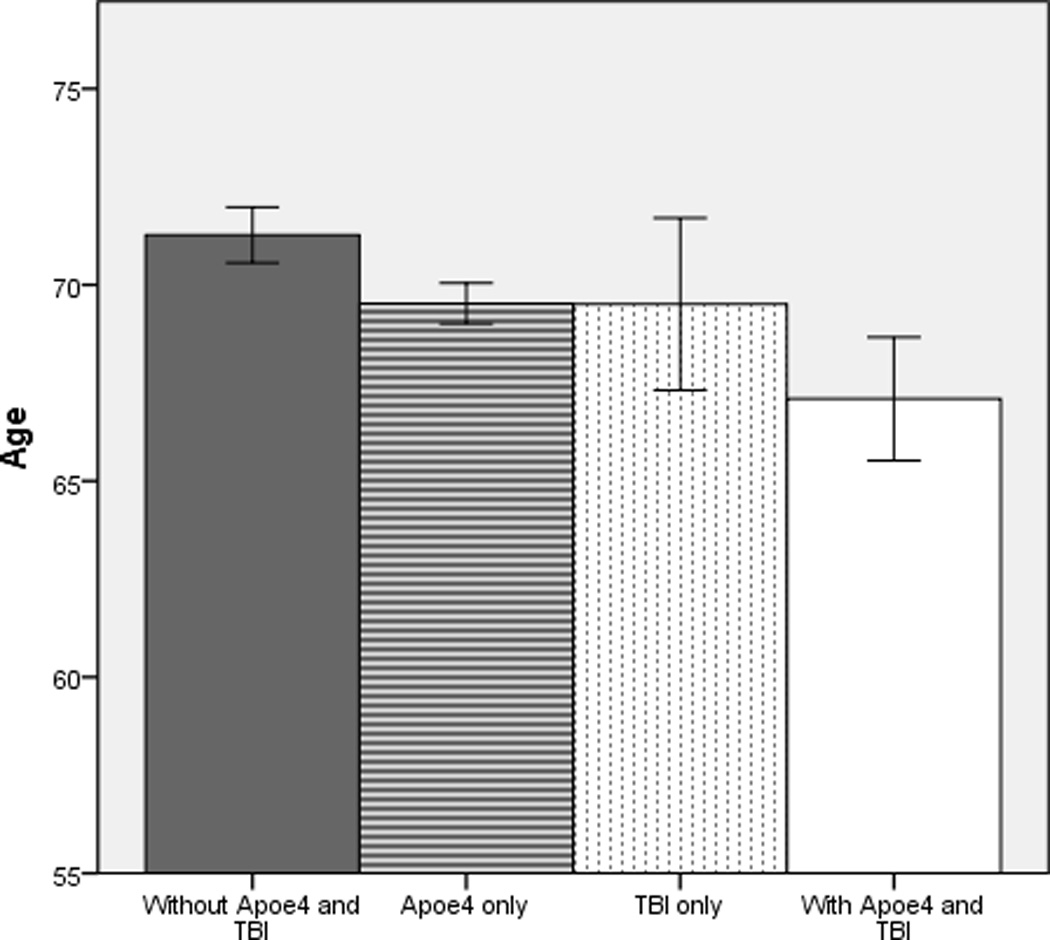
Mean age of symptom onset in male patients with Alzheimer disease with and without risk factors apolipoprotein E-ε4 and history of traumatic brain injury. Bars represent 95% confidence intervals for each group. TBI = traumatic brain injury with loss of consciousness. Apoe4 = apolipoprotein E-e4 allele.
Discussion
A history of TBI with LOC occurring more than one year prior to AD diagnosis was associated with an approximately 2.5 year earlier age of symptom onset and diagnosis of AD. In addition, this association remained even after controlling for potential effects related to gender and Apoe4 status, suggesting that, regardless of gender, a history of TBI is associated with an earlier expression of AD independent from well-known genetic risk factors. While these results differ from previous studies reporting no association between a history of TBI and earlier AD onset (Guo et al., 2000; Mortimer et al., 1991; Plassman et al., 2000; Rasmusson et al., 1995), the degree of this association (i.e., nearly 2.5 years earlier) also contrasts several smaller investigations reporting an earlier onset of approximately 7–9 years (Gedye et al., 1989; Nemetz et al., 1999; Sullivan et al., 1987) in those with a TBI history. As mentioned previously, this may reflect varying methodological differences across studies of less well-characterized participants, but also may be due, in part, to previous reports not taking Apoe4 status into account. However, three recent large investigations reported that all-cause dementia (Barnes et al., 2014), frontotemporal dementia (LoBue et al., 2016), and mild cognitive impairment (MCI) (LoBue et al., 2016) were diagnosed approximately 2 years earlier in participants with a history of TBI compared to those without, consistent with the findings related to AD in the present investigation. Therefore, it would appear that an emerging literature supports a history of TBI with LOC as being associated with an approximately 2 year earlier onset of neurodegenerative conditions, although additional questions remain.
The purpose of this investigation was to replicate and extend previous reports on the association between a TBI history and earlier onset of AD, but we also found an association with Apoe4. Our finding that Apoe4 was linked to a 2.3 year earlier onset of AD is consistent with results from a recent meta-analysis of 14 case-control datasets in the Alzheimer’s Disease Genetics Consortium (ADGC) that found AD onset to be reduced by 2.5 years in individuals with at least one Apoe4 allele (Naj et al., 2014). However, to our knowledge, the present study is the first to investigate a potential interaction between history of TBI and Apoe4 on age of symptom onset in AD. Despite both risk factors being independently linked to an earlier expression of AD, we did not observe a significant interaction between the two. This is consistent with previous reports that these two risk factors independently confer a heightened absolute risk for dementia rather than a “synergistic” association (Guo et al., 2000; O’Meara et al., 1997).
Biologically, TBI and Apoe4 are known risk factors for central nervous system impairment in some individuals. TBI involves acute neuronal/axonal damage that results in abnormal neurochemical cascades and inflammation (Johnson, Stewart, & Smith, 2012), whereas Apoe4 has been implicated in deficient neuronal maintenance and repair and reduced synaptic networks (Bu, 2009; Liu, Kanekiyo, Xu, & Bu, 2013). Some research furthermore suggests the mechanisms involved in TBI and Apoe4 may be linked to AD pathology. In an autopsy study, a subset of patients surviving a moderate-to-severe TBI for longer than a year showed presence of Aβ plaques that were more often fibrillary (Johnson, Stewart, & Smith, 2012), similar to plaque formations observed in AD, and unlike a predominantly diffuse pattern commonly present in normal aging (Price, & Morris, 1999). Fibrillary Aβ plaques have also been found to be more abundant and at a much earlier age in cognitively intact Apoe4 carriers relative to non-carriers (Kok et al., 2009; Fleisher et al., 2013). NFTs, on the other hand, have been reported to be more widespread in individuals with a single, moderate-to-severe TBI compared to age-matched controls (Johnson, Stewart, & Smith, 2012), while a link with Apoe4 is lacking. It would appear then that TBI and Apoe4 may involve similar biological processes to initiate and/or accelerate AD-like pathology, resulting in an earlier expression of AD. Although AD is the most common type of dementia, there is evidence that a history of TBI has a similar association with frontotemporal dementia (LoBue et al., 2015), indicating that the mechanisms involved with TBI may overlap other neurodegenerative disorders.
Several lines of evidence suggest gender-related effects can modify the association between TBI history or Apoe4 and AD. Two meta-analytic studies reported a history of TBI with LOC was associated with an increased risk for later developing AD, but when gender was accounted for, this association was only observed in males (Fleminger et al., 2003; Mortimer et al., 1991). In contrast, Apoe4 studies have shown that female Apoe4 carriers, relative to male carriers, have a greater risk for developing AD (Altmann, Tian, Henderson, & Greicius, 2014; Ungar, Altmann, & Greicius, 2014) and show more pronounced AD-like pathology in older adulthood (Corder et al., 2004). In the present study, however, gender-related effects were not observed. Both females and males had an approximately similar association for history of TBI and Apoe4 on age of onset independently. A significantly larger percentage of males were found to have a history of TBI with LOC than females, but this is generally consistent with overall TBI rates in the literature (Faul et al., 2010).
Although our results support a history of TBI with LOC as being a risk factor for later cognitive decline, the influence of TBI-related factors on this association remains unclear. Researchers have found TBI severity and age at injury influence risk for developing dementia. For example, Gardner and colleagues performed a 7 year medical chart review on patients over age 55 who presented to a hospital with a TBI of any severity (n = 52,000) or non-TBI trauma (n = 112,000) (Gardner et al., 2014). The authors reported that moderate-to-severe TBI was associated with a higher likelihood for developing dementia in persons aged 55 years and older, whereas a single, mild TBI was associated with increased risk among those older than 64 years. In addition, a “synergistic” link between Apoe4 and a history of TBI is even less clear, as this literature too has been mixed, with approximately 50% of studies reporting an absence of an additive effect on increasing risk for developing dementia (Lawrence, Comper, Hutchison, & Sharma, 2015). More severe TBI may be an important factor (Lawrence et al., 2015), but some Apoe4 carriers could also possess the Apoe ε2 (Apoe2) allele (genotype ε2/ ε4), which has been implicated as protective against neuronal injury (Miller et al., 2010) and from AD (Corder et al., 1994). It is possible that the presence of Apoe2 may offset Apoe4’s role in initiating and/or accelerating AD-like pathology, protecting against synergist mechanisms following TBI in some individuals, which could attenuate differences when comparing Apoe4 carriers to non-carriers. Interestingly, Apoe4 carriers with a history of TBI in the present study were observed to have an estimated onset of AD approximately 2.5 years earlier on average compared to those with each risk factor alone, and 5 years earlier than those with an absence of both risk factors; however, the interaction was non-significant. Given that TBI severity, age at injury, and potentially other related variables (e.g., Apoe2 and other genetic factors) could influence age of onset of AD differently, and may contribute to the mixed literature, further research is needed to determine their implications.
Limitations of the present investigation include the time since reported TBI and the manner in which TBI is classified within the NACC database. The definition for remote TBI (i.e., >1 year earlier) in NACC is relative, as injuries may have occurred as recently as nearly one year prior to the initial ADC visit but could have been decades earlier. Because the neurodegenerative process underlying AD occurs over years or decades, an unknown proportion of TBIs in this sample could have occurred once neurodegeneration had already begun and/or after symptom onset. In addition, while our classification of TBI with LOC is similar to previous investigations, NACC’s cutoff of <5 and ≥5 mLOC does not correspond to conventional criteria for TBI classification of severity, and further research is needed along these lines. In addition, the limited TBI data in the NACC database did not allow potentially important variables such as age at the time of injury, complications, or repetitive injuries to be examined that may play a role in the association between TBI and later cognitive decline. Further, TBI history as well as clinician-estimated age of onset were dependent on participant/informant reports; therefore, recall bias is possible and could limit accuracy in reporting a history of TBI and/or estimating symptom onset of AD. Last, NACC participants at ADCs are recruited via clinician referral or self-referral and often are predominantly Caucasian, well-educated, and express a commitment to involvement in longitudinal research. Even though NACC studies are not population-based and may not be generalizable to all individuals with AD, participants undergo extensive standard diagnostic evaluations and are well characterized.
These findings, derived from samples of carefully diagnosed AD cases obtained from a large, multicenter national database, suggest that a history of TBI with LOC may be associated with an earlier onset of AD-related cognitive decline, regardless of gender and Apoe4 status. The overlapping and synergist mechanisms underlying this apparent relationship remain unclear, but at a minimum, the results underscore the need for more thorough assessment of TBI history in individuals presenting for dementia evaluation later in life.
Acknowledgments
Funding for this study was provided in part by the NIH/NIA P3012300-19 Alzheimer’s Disease Center Grant at the University of Texas Southwestern Medical Center and the Texas Alzheimer’s Research Care and Consortium. The NACC database is funded by NIA/NIH Grant U01 AG016976. NACC data are contributed by the NIA funded: P30 AG019610 (PI Eric Reiman, MD), P30 AG013846 (PI Neil Kowall, MD), P50 AG008702 (PI Scott Small, MD), P50 AG025688 (PI Allan Levey, MD, PhD), P30 AG010133 (PI Andrew Saykin, PsyD), P50 AG005146 (PI Marilyn Albert, PhD), P50 AG005134 (PI Bradley Hyman, MD, PhD), P50 AG016574 (PI Ronald Petersen, MD, PhD), P50 AG005138 (PI Mary Sano, PhD), P30 AG008051 (PI Steven Ferris, PhD), P30 AG013854 (PI M. Marsel Mesulam, MD), P30 AG008017 (PI Jeffrey Kaye, MD), P30 AG010161 (PI David Bennett, MD), P30 AG010129 (PI Charles DeCarli, MD), P50 AG016573 (PI Frank LaFerla, PhD), P50 AG016570 (PI David Teplow, PhD), P50 AG005131 (PI Douglas Galasko, MD), P50 AG023501 (PI Bruce Miller, MD), P30 AG035982 (PI Russell Swerdlow, MD), P30 AG028383 (PI Linda Van Eldik, PhD), P30 AG010124 (PI John Trojanowski, MD, PhD), P50 AG005133 (PI Oscar Lopez, MD), P50 AG005142 (PI Helena Chui, MD), P30 AG012300 (PI Roger Rosenberg, MD), P50 AG005136 (PI Thomas Montine, MD, PhD), P50 AG033514 (PI Sanjay Asthana, MD, FRCP), and P50 AG005681 (PI John Morris, MD). The Alzheimer’s Disease Genetic Consortium (ADGC) is funded by NIA Grant U01 AG032984.
Footnotes
The authors disclose no financial interests.
References
- Altmann A, Tian L, Henderson VW, Greicius MD. Sex modifies the APOE-related risk of developing Alzheimer disease. Annals of Neurology. 2014;75:563–573. doi: 10.1002/ana.24135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes DE, Kaup A, Kirby KA, Byers AL, Diaz-Arrastia R, Yaffe K. Traumatic brain injury and risk of dementia in older veterans. Neurology. 2014;83:312–319. doi: 10.1212/WNL.0000000000000616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blacker D, Haines JL, Rodes L, Terwedow H, Go RCP, Harrell LE, Albert MS. ApoE-4 and age at onset of Alzheimer’s disease the NIMH genetics initiative. Neurology. 1997;48:139–147. doi: 10.1212/wnl.48.1.139. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathologica. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- Bu G. Apolipoprotein E and its receptors in Alzheimer’s disease: Pathways, pathogenesis and therapy. Nature Reviews Neuroscience. 2009;10:333–344. doi: 10.1038/nrn2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small G, Pericak-Vance MA. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261:921–923. doi: 10.1126/science.8346443. Retrieved from http://www.jstor.org/stable/2882127. [DOI] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Risch NI, Strittmatterl WI, Schmechel DE. Protective effect of apolipoprotein E type 2 allele for late onset. Nature Genetics. 1994;7:180–184. doi: 10.1038/ng0694-180. [DOI] [PubMed] [Google Scholar]
- Corder EH, Ghebremedhin E, Taylor MG, Thal DR, OHM TG, Braak H. The biphasic relationship between regional brain senile plaque and neurofibrillary tangle distributions: Modification by age, sex, and APOE polymorphism. Annals of the New York Academy of Sciences. 2004;1019:24–28. doi: 10.1196/annals.1297.005. [DOI] [PubMed] [Google Scholar]
- Dams-O’Connor K, Gibbons LE, Bowen JD, McCurry SM, Larson EB, Crane PK. Risk for late-life re-injury, dementia and death among individuals with traumatic brain injury: A population-based study. Journal of Neurology, Neurosurgery & Psychiatry. 2013;84:177–182. doi: 10.1136/jnnp-2012-303938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faul M, Xu L, Wald MM, Coronado VG. Traumatic brain injury in the United States: Emergency department visits, hospitalizations, and deaths. Atlanta (GA): Centers for Disease Control and Prevention. National Center for Injury Prevention and Control. 2010;2:1–9. Retrieved from http://www.cdc.gov/TraumaticBrainInjury. [Google Scholar]
- Fleisher AS, Chen K, Liu X, Ayutyanont N, Roontiva A, Thiyyagura P, Sperling RA. Apolipoprotein E ε4 and age effects on florbetapir positron emission tomography in healthy aging and Alzheimer disease. Neurobiology of Aging. 2013;34:1–12. doi: 10.1016/j.neurobiolaging.2012.04.017. [DOI] [PubMed] [Google Scholar]
- Fleminger S, Oliver DL, Lovestone S, Rabe-Hesketh S, Giora A. Head injury as a risk factor for Alzheimer’s disease: The evidence 10 years on; A partial replication. Journal of Neurology, Neurosurgery & Psychiatry. 2003;74:857–862. doi: 10.1136/jnnp.74.7.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fratiglioni L, Ahlbom A, Viitanen M, Winblad B. Risk factors for late-onset Alzheimer’s disease: A population-based, case-control study. Annals of Neurology. 1993;33:258–266. doi: 10.1002/ana.410330306. [DOI] [PubMed] [Google Scholar]
- Gardner RC, Burke JF, Nettiksimmons J, Kaup A, Barnes DE, Yaffe K. Dementia risk after traumatic brain injury vs nonbrain trauma: The role of age and severity. JAMA Neurology. 2014;71:1490–1497. doi: 10.1001/jamaneurol.2014.2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gedye A, Beattie BL, Tuokko H, Horton A, Korsarek E. Severe head injury hastens age of onset of Alzheimer’s disease. Journal of the American Geriatrics Society. 1989;37:970–973. doi: 10.1111/j.1532-5415.1989.tb07283.x. [DOI] [PubMed] [Google Scholar]
- Guo Z, Cupples LA, Kurz A, Auerbach SH, Volicer L, Chui H, Johnson K. Head injury and the risk of AD in the MIRAGE study. Neurology. 2000;54:1316–1323. doi: 10.1212/wnl.54.6.1316. [DOI] [PubMed] [Google Scholar]
- Helmes E, Ostbye T, Steenhuis RE. Incremental contribution of reported previous head injury to the prediction of diagnosis and cognitive functioning in older adults. Brain Injury. 2011;25:338–347. doi: 10.3109/02699052.2011.556104. [DOI] [PubMed] [Google Scholar]
- Johnson VE, Stewart W, Smith DH. Widespread tau and amyloid-beta pathology many years after a single traumatic brain injury in humans. Brain Pathology. 2012;22:142–149. doi: 10.1111/j.1750-3639.2011.00513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson VE, Stewart W, Smith DH. Axonal pathology in traumatic brain injury. Experimental Neurology. 2013;246:35–43. doi: 10.1016/j.expneurol.2012.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kok E, Haikonen S, Luoto T, Huhtala H, Goebeler S, Haapasalo H, Karhunen PJ. Apolipoprotein E-dependent accumulation of Alzheimer disease-related lesions begins in middle age. Annals of Neurology. 2009;65:650–657. doi: 10.1002/ana.21696. [DOI] [PubMed] [Google Scholar]
- Lawrence DW, Comper P, Hutchison MG, Sharma B. The role of apolipoprotein E episilon (ε)-4 allele on outcome following traumatic brain injury: A systematic review. Brain Injury. 2015;29:1018–1031. doi: 10.3109/02699052.2015.1005131. [DOI] [PubMed] [Google Scholar]
- Lindsay J, Laurin D, Verreault R, Hébert R, Helliwell B, Hill GB, McDowell I. Risk factors for Alzheimer’s disease: A prospective analysis from the Canadian Study of Health and Aging. American Journal of Epidemiology. 2002;156:445–453. doi: 10.1093/aje/kwf074. [DOI] [PubMed] [Google Scholar]
- Liu CC, Kanekiyo T, Xu H, Bu G. Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nature Reviews Neurology. 2013;9:106–118. doi: 10.1038/nrneurol.2012.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LoBue C, Wilmoth K, Cullum CM, Rossetti HC, Lacritz LH, Hynan LS, Womack KB. Traumatic brain injury history is associated with earlier age of onset of frontotemporal dementia. Journal of Neurology, Neurosurgery & Psychiatry. 2016;87:817–820. doi: 10.1136/jnnp-2015-311438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LoBue C, Denney D, Hynan LS, Rossetti HC, Lacritz LH, Hart J, Cullum CM. Self-reported traumatic brain injury and mild cognitive impairment: Increased risk and earlier age of diagnosis. Journal of Alzheimer’s Disease. 2016;51:727–736. doi: 10.3233/JAD-150895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luchsinger JA, Reitz C, Honig LS, Tang M, Shea S, Mayeux R. Aggregation of vascular risk factors and risk of incident Alzheimer disease. Neurology. 2005;65:545–551. doi: 10.1212/01.wnl.0000172914.08967.dc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiti TK, Konar S, Bir S, Kalakoti P, Bollam P, Nanda A. Role of apolipoprotein E polymorphism as a prognostic marker in traumatic brain injury and neurodegenerative disease: a critical review. Neurosurgical Focus. 2015;39:E3. doi: 10.3171/2015.8.FOCUS15329. [DOI] [PubMed] [Google Scholar]
- Mayeux R, Ottman R, Maestre G, Ngai C, Tang MX, Ginsberg H, Shelanski M. Synergistic effects of traumatic head injury and apolipoprotein-epsilon4 in patients with Alzheimer’s disease. Neurology. 1995;45:555–557. doi: 10.1212/wnl.45.3.555. [DOI] [PubMed] [Google Scholar]
- McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease Report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology. 1984;34:939–939. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- Mehta KM, Ott A, Kalmijn S, Slooter AJC, Van Duijn CM, Hofman A, Breteler MMB. Head trauma and risk of dementia and Alzheimer’s disease: The Rotterdam Study. Neurology. 1999;53:1959–1959. doi: 10.1212/wnl.53.9.1959. [DOI] [PubMed] [Google Scholar]
- Miller MA, Conley Y, Scanlon JM, Ren D, Ilyas Kamboh M, Niyonkuru C, Wagner AK. APOE genetic associations with seizure development after severe traumatic brain injury. Brain Injury. 2010;24:1468–1477. doi: 10.3109/02699052.2010.520299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modrego Pedro J, Ferrández Jaime Depression in patients with mild cognitive impairment increases the risk of developing dementia of Alzheimer type: a prospective cohort study. Archives of Neurology. 2004;61:1290–1293. doi: 10.1001/archneur.61.8.1290. [DOI] [PubMed] [Google Scholar]
- Morris JC, Weintraub S, Chui HC, Cummings J, DeCarli C, Ferris S, Beekly D. The Uniform Data Set (UDS): Clinical and cognitive variables and descriptive data from Alzheimer Disease Centers. Alzheimer Disease & Associated Disorders. 2006;20:210–216. doi: 10.1097/01.wad.0000213865.09806.92. [DOI] [PubMed] [Google Scholar]
- Mortimer JA, French LR, Hutton JT, Schuman LM. Head injury as a risk factor for Alzheimer’s disease. Neurology. 1985;35:264–264. doi: 10.1212/wnl.35.2.264. [DOI] [PubMed] [Google Scholar]
- Mortimer JA, Van Duijn CM, Chandra V, Fratiglioni L, Graves AB, Heyman A, Shalat SL. Head trauma as a risk factor for Alzheimer’s disease: A collaborative re-analysis of case-control studies. International Journal of Epidemiology. 1991;20:S28–S35. doi: 10.1093/ije/20.supplement_2.s28. [DOI] [PubMed] [Google Scholar]
- Naj AC, Jun G, Reitz C, Kunkle BW, Perry W, Park Y, Kauwe JS. Age-at-Onset in Late Onset Alzheimer Disease is Modified by Multiple Genetic Loci. JAMA Neurology. 2014;71:1394. doi: 10.1001/jamaneurol.2014.1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson PT, Braak H, Markesbery WR. Neuropathology and cognitive impairment in Alzheimer disease: a complex but coherent relationship. Journal of Neuropathology and Experimental Neurology. 2009;68:1–14. doi: 10.1097/NEN.0b013e3181919a48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemetz PN, Leibson C, Naessens JM, Beard M, Kokmen E, Annegers JF, Kurland LT. Traumatic brain injury and time to onset of Alzheimer’s disease: A population-based study. American Journal of Epidemiology. 1999;149:32–40. doi: 10.1093/oxfordjournals.aje.a009724. Retrieved from http://aje.oxfordjournals.org/content/149/1/32.short#cited-by. [DOI] [PubMed] [Google Scholar]
- O’Meara ES, Kukull WA, Sheppard L, Bowen JD, McCormick WC, Teri L, Larson EB. Head injury and risk of Alzheimer’s disease by apolipoprotein E genotype. American Journal of Epidemiology. 1997;146:373–384. doi: 10.1093/oxfordjournals.aje.a009290. Retrieved from http://aje.oxfordjournals.org/ [DOI] [PubMed] [Google Scholar]
- Plassman BL, Havlik RJ, Steffens DC, Helms MJ, Newman TN, Drosdick D, Guralnik JM. Documented head injury in early adulthood and risk of Alzheimer’s disease and other dementias. Neurology. 2000;55:1158–1166. doi: 10.1212/wnl.55.8.1158. [DOI] [PubMed] [Google Scholar]
- Price JL, Morris JC. Tangles and plaques in nondemented aging and “preclinical” Alzheimer’s disease. Annals of Neurology. 1999;45:358–368. doi: 10.1002/1531-8249(199903)45:3<358::aid-ana12>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- Rasmusson DX, Brandt J, Martin DB, Folstein MF. Head injury as a risk factor in Alzheimer’s disease. Brain Injury. 1995;9:213–219. doi: 10.3109/02699059509008194. [DOI] [PubMed] [Google Scholar]
- Scott G, Ramlackhansingh AF, Edison P, Hellyer P, Cole J, Veronese M, Heckemann RA. Amyloid pathology and axonal injury after brain trauma. Neurology. 2016;86:821–828. doi: 10.1212/WNL.0000000000002413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan P, Petitti D, Barbaccia J. Head trauma and age of onset of dementia of the Alzheimer type. The Journal of the American Medical Association. 1987;257:2289–2290. doi: 10.1001/jama.1987.03390170045014. [DOI] [PubMed] [Google Scholar]
- Sundström A, Nilsson LG, Cruts M, Adolfsson R, Van Broeckhoven C, Nyberg L. Increased risk of dementia following mild head injury for carriers but not for non-carriers of the APOE ε4 allele. International Psychogeriatrics. 2007;19:159–165. doi: 10.1017/S1041610206003498. [DOI] [PubMed] [Google Scholar]
- Thal DR, Rüb U, Orantes M, Braak H. Phases of Aβ-deposition in the human brain and its relevance for the development of AD. Neurology. 2002;58:1791–1800. doi: 10.1212/wnl.58.12.1791. [DOI] [PubMed] [Google Scholar]
- Thal DR, Attems J, Ewers M. Spreading of amyloid, tau, and microvascular pathology in Alzheimer’s disease: findings from neuropathological and neuroimaging studies. Journal of Alzheimer’s Disease. 2014;42:S421–S429. doi: 10.3233/JAD-141461. [DOI] [PubMed] [Google Scholar]
- Ungar L, Altmann A, Greicius MD. Apolipoprotein E, gender, and Alzheimer’s disease: An overlooked, but potent and promising interaction. Brain Imaging and Behavior. 2014;8:262–273. doi: 10.1007/s11682-013-9272-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Duijn CM, Tanja TA, Haaxma R, Schulte W, Saan RJ, Lameris AJ, Hofman A. Head trauma and the risk of Alzheimer’s disease. American Journal of Epidemiology. 1992;135:775–782. doi: 10.1093/oxfordjournals.aje.a116364. Retrieved from http://aje.oxfordjournals.org/content/135/7/775.short. [DOI] [PubMed] [Google Scholar]
- Wang HK, Lin SH, Sung PS, Wu MH, Hung KW, Wang LC, Tsai KJ. Population based study on patients with traumatic brain injury suggests increased risk of dementia. Journal of Neurology, Neurosurgery & Psychiatry. 2012;83:1080–1085. doi: 10.1136/jnnp-2012-302633. [DOI] [PubMed] [Google Scholar]
- Xu W, Tan L, Wang HF, Jiang T, Tan MS, Tan L, Yu JT. Meta-analysis of modifiable risk factors for Alzheimer’s disease. Journal of Neurology, Neurosurgery & Psychiatry. 2015;86:1299–1306. doi: 10.1136/jnnp-2015-310548. [DOI] [PubMed] [Google Scholar]