

Abstract
Matrix rigidity has important effects on cell behavior and is increased during liver fibrosis; however, its effect on primary hepatocyte function is unknown. We hypothesized that increased matrix rigidity in fibrotic livers would activate mechanotransduction in hepatocytes and lead to inhibition of hepatic-specific functions. To determine the physiologically relevant ranges of matrix stiffness at the cellular level, we performed detailed atomic force microscopy analysis across liver lobules from normal and fibrotic livers. We determined that normal liver matrix stiffness was around 150Pa and increased to 1–6kPa in areas near fibrillar collagen deposition in fibrotic livers. In vitro culture of primary hepatocytes on collagen matrix of tunable rigidity demonstrated that fibrotic levels of matrix stiffness had profound effects on cytoskeletal tension and significantly inhibited hepatocyte-specific functions. Normal liver stiffness maintained functional gene regulation by hepatocyte nuclear factor 4 alpha (HNF4α) whereas fibrotic matrix stiffness inhibited the HNF4α transcriptional network. Fibrotic levels of matrix stiffness activated mechanotransduction in primary hepatocytes through focal adhesion kinase (FAK). In addition, blockade of the Rho/Rho-associated protein kinase (ROCK) pathway rescued HNF4α expression from hepatocytes cultured on stiff matrix.
Conclusion
Fibrotic levels of matrix stiffness significantly inhibit hepatocyte-specific functions in part by inhibiting the HNF4α transcriptional network mediated through the Rho/ROCK pathway. Increased appreciation of the role of matrix rigidity in modulating hepatocyte function will advance our understanding of the mechanisms of hepatocyte dysfunction in liver cirrhosis and spur development of novel treatments for chronic liver disease.
Keywords: matrix stiffness, fibrosis, atomic force microscopy, mechanotransduction, Rho/ROCK pathway
It is increasingly evident that the rigidity of extracellular matrix has a profound effect on cell behavior, including cell function, proliferation, and motility (1, 2). The response of epithelial cells to matrix stiffness is mediated by an intricate mechanotransduction cascade in which cells sense the mechanical signals in their environment and activate intracellular signaling pathways. These pathways involve integrin clustering and activation of focal adhesion kinase (FAK), the Rho/Rho-associated protein kinase (ROCK) pathway, and the extracellular signal-regulated kinase (ERK) pathway (3–5). While these fundamental mechanosensing mechanisms are likely generalizable, how they uniquely regulate the cellular function of hepatocytes, a highly specialized epithelial cell, is unknown.
In the liver, fibrosis, and ultimately cirrhosis, is the final common pathway of chronic liver diseases due to a variety of causes that span viral infections (hepatitis B and C), chemical insults (e.g. alcohol), inflammatory conditions (non-alcoholic steatohepatitis, autoimmune hepatitis), and metabolic dyscrasias (alpha-1 antitrypsin deficiency, hemochromatosis, Wilson’s disease, etc.). The degree of liver fibrosis in humans has been shown to correlate well with liver tissue stiffness as determined by non-invasive diagnostic methods such as ultrasound-based transient and shear-wave elastography (6) and magnetic resonance elastography (7, 8). The significance of matrix rigidity in liver fibrosis has been established by experiments demonstrating that increased matrix stiffness was required to activate stellate cells and portal fibroblasts, the key cell types responsible for abnormal extracellular matrix deposition in the fibrogenic process (9, 10). In addition, hepatocellular carcinomas, the majority of which arise in the setting of liver fibrosis, show increased proliferation and resistance to chemotherapeutic agents when cultured on matrix with greater rigidity (11).
What remains unknown about the effect of increased matrix stiffness during the course of liver fibrosis is how it may alter the function of primary hepatocytes. This is an important question because the cause of liver failure in cirrhosis is not fully understood and there is evidence that hepatocytes removed from a cirrhotic microenvironment may regain lost function when replaced into a non-cirrhotic host liver (12). We hypothesize that increased matrix rigidity directly inhibits hepatic-specific functions and is a major mechanism of liver dysfunction in the setting of advanced liver fibrosis. There is mounting evidence that the process of liver fibrosis may be reversed by elimination of the injurious agent(s) and, potentially, by targeted anti-fibrotic therapies (13). If changes in matrix stiffness directly modulate primary hepatocyte functions, then such changes may be a likely mechanism to explain how liver function improves as fibrosis resolves. If proven, this mechanism would also provide a rationale for developing therapies that intercept the mechanotransduction signal cascade in primary hepatocytes in the setting of advanced cirrhosis with the goal of preserving liver function.
A major barrier to investigating the physiologically relevant effects of matrix stiffness on hepatocyte function is that normal versus pathological levels of matrix rigidity have not been fully defined at the cellular level. Transient and shear-wave elastography, expressing stiffness in elastic modulus (E), estimated normal human liver stiffness to be around 5kPa and grade 4 cirrhotic livers to be 15–20kPa (6). Magnetic resonance elastography, expressing stiffness in shear modulus (G′), determined normal human liver stiffness to be 2kPa and placed the matrix stiffness cutoff for grade 4 cirrhotic livers at >5kPa (7, 8). For all these non-invasive technologies, liver stiffness values are extrapolated from the response of tissues to generated shear waves and may not accurately reflect mechanical tissue stiffness. Mechanical measurements of bulk liver tissue from humans and rodents by rheometry suggest that true liver stiffness is likely much softer, ranging between 400–600Pa for normal liver and 1.2–1.6kPa for fibrotic liver (14, 15). Liver fibrosis, however, is not a homogenous process at the tissue level and bulk tissue measurements at best represent an average of what individual hepatocytes encounter at a cellular level. Even in cases of advanced fibrosis where there are bridging fibrotic bands, areas of relatively less collagen deposition remain, suggesting that changes in matrix rigidity may be specific to the local microenvironment.
Therefore, in order to determine whether physiologically relevant levels of matrix rigidity may modulate primary hepatocyte function, we first performed detailed microscale analyses across liver lobules using atomic force microscopy (AFM) to define the normal and pathological ranges of matrix stiffness that hepatocytes encounter within the liver. We then isolated primary hepatocytes from mice and cultured them on collagen matrices with tunable rigidity to determine the effect of these levels of matrix stiffness on hepatocyte function and mechanotransduction.
Material and Methods
Atomic Force Microscopy (AFM)
Measurement s on fresh liver tissues were carried out on samples embedded in 4% low melting point agarose and cut into 50 μm sections using a Leica VT1000S vibratome (Buffalo Grove, IL). Measurements on frozen liver tissues were performed on livers embedded in OCT compound (Sakura, Torrance, CA), snap frozen via direct immersion into liquid nitrogen, and cut into 50μm sections on a Leica CM1900-13 cryostat. During AFM analysis, samples were maintained in protease inhibitor cocktail (Roche Diagnostics, Indianapolis, IN) and propidium iodide (Sigma). Similar to methods previously described (16), measurements were conducted using a MFP3D-BIO inverted optical AFM (Asylum Research, Santa Barbara, CA) mounted on a Nikon TE200-U inverted fluorescence microscope (Melville, NY). Silicon nitride cantilevers (k = 0.06 N/m) modified with a 5μm diameter borosilicate glass spherical tip (Novascan Tech, Ames, IA) were used for indentation. For each session, cantilevers were calibrated using the thermal oscillation method. AFM force maps were performed on 90μm×90μm fields. Gradients were generated post-acquisition. Each experimental group included at least 3 different mice, with 3 sections from each mouse, and 2 gradients (one orthogonal and one parallel to the fibrosis) generated per section. Data analyses were done using the Hertz model in Igor Pro v. 6.22A (WaveMetrics, Lake Oswego, OR) and a Poisson’s ratio of 0.5.
Primary Hepatocyte Isolation and Culture
Hepatocytes were isolated from wild-type, 6–12 week-old C57BL/6 mice (Jackson Laboratory, Bar Harbor, ME). In some experiments, hepatocytes were isolated from Hnf4afl/fl:Alb Cre+ mice that lacked HNF4α expression in adult hepatocytes and Hnf4afl/fl:Alb Cre− wild-type littermates (kind gift from Dr. Frank J. Gonzalez) (17). All mice were cared for in accordance to the National Institutes of Health “Guide for the Care and Use of Laboratory Animals.” Hepatocyte isolation was performed by two-step perfusion using Liver Perfusion and Liver Digest Media (Life Technologies, Pleasanton, CA) followed by separation with 50% Percoll (GE Healthcare Life Sciences, Pittsburgh, PA) density gradient. Purity of live hepatocytes was routinely ≥90% by trypan blue exclusion. Hepatocytes were cultured in 5% fetal calf serum (Hyclone, Logan, UT) in DMEM supplemented with L-glutamine, antibiotics, insulin-transferrin-selenium, and HEPES (Mediatech, Manassas, VA). Inhibitors used and their final concentrations in cell culture were: 50μM Y-27632, 25μM FAK inhibitor-14, 0.5μM blebbistatin, or 20μM U-0126 (Santa Cruz Biotech, Dallas, TX). Unless otherwise noted, functional assays and gene expression analysis were performed 24h after hepatocyte plating.
Collagen-conjugated polyacrylamide gels
Polyacrylamide gels were made from 40% acrylamide and 2% bis acrylamide (Bio-Rad, Hercules, CA) where varying ratios of acrylamide and bis acrylamide were used to create gels of known reproducible stiffness (18). Gels were cast on glutaraldehyde-modified coverglasses and polymerization was activated by 1% ammonium persulfate (Sigma). The PA gel surfaces were then conjugated to acrylic acid N-hydroxy-succinimide ester activated by 365nm UV exposure and Irgacure catalyst (BASF Resins, Laramie, WY). Rat-tail collagen I (VWR, Brisbane, CA) was then added to the surface at 150ug/mL to allow coupling to the gel through side-chain primary amines.
Please refer to Supplemental Methods for additional methodological details.
Results
Normal liver matrix stiffness at the cellular level was around 150Pa and pathological matrix stiffness ranged between 1kPa to 6kPa in fibrotic livers
To determine the matrix rigidity of normal and fibrotic liver tissue, livers were harvested from untreated wild-type C57BL/6 mice and mice induced with mild or severe forms of fibrosis by chronic carbon tetrachloride (CCl4) injections or 1% 5-diethoxycarbonyl-1,4-dihydrocollidine (DDC) diet. These two models were chosen because CCl4 induced fibrosis by causing hepatocellular necrosis whereas DDC produced biliary fibrosis. To ensure homogenous experimental groups for comparison, liver sections were stained with hematoxylin and eosin (H&E) or Sirius Red and evaluated by an expert liver pathologist who was blinded to the identity of the samples. The liver pathologist was able to categorize the samples by pattern of fibrosis into groups that corresponded to the model of fibrosis induction. Samples graded as no fibrosis corresponded to no treatment (Figure 1A). Central-central fibrosis corresponded to the mild CCl4 protocol, whereas presence of central-portal and portal-portal fibrosis corresponded to the severe CCl4 protocol. DDC diet generated some mice with mild disease showing fibrotic expansion limited to portal zones and some mice with severe disease showing bridging fibrosis between portal tracts. To quantitatively evaluate the degree of fibrosis, the percent tissue area positive for fibrillar collagen was measured by Sirius Red fluorescence under polarized light (Figure 1B). This analysis revealed that there was significantly more fibrillar collagen deposition in mice treated with CCl4 or DDC compared to untreated mice. Furthermore, there was significantly more fibrillar collagen deposition in the severe forms of CCl4- or DDC-induced fibrosis than in their respective mild forms, corroborating the qualitative histological assessments.
Figure 1.

Increased pathological severity grade of liver fibrosis corresponds to increased quantified fibrillar collagen deposition. (A) Representative images of H&E and Sirius Red staining (brightfield & polarized light) demonstrate distinct patterns of collagen deposition depending on the model and severity of experimental liver fibrosis. Scale bars represent 200μm. (B) Quantification of fibrillar collagen deposition in Sirius Red-stained liver sections. *p<0.05 and **p<0.01 by one-way ANOVA and Tukey’s post-hoc test for pairwise comparison. There were 3 mice per group and 3 sections were quantified per mouse, n=3. Error bars represent SEM.
To determine normal liver matrix stiffness at the cellular level, AFM was used to obtain detailed measurements across liver lobules from untreated wild-type mice. We initially performed our AFM studies on fresh liver tissue and subsequently switched to frozen tissue after confirming that there were no differences in tissue extracellular matrix stiffness between the two conditions (Supplemental Figure 1A). Measurements were carried out spanning from periportal zones to pericentral zones as well as from periportal zones to adjacent periportal zones. We found that matrix rigidity ranged consistently around 150Pa in normal livers with minimal variation from portal to central zones or portal to portal zones (Figure 2B). When stiffness measurements in pericentral and periportal zones were extracted and directly compared with each other, we found that matrix stiffness in periportal zones had slightly greater variability and trended slightly higher compared to pericentral zones (Supplemental Figure 1B), potentially reflecting the fact that portal tracts normally have more extracellular matrix than areas around central veins.
Figure 2.

Liver fibrosis results in treatment and region specific increases in tissue matrix stiffness as determined by AFM. (A) Diagram depicting the directionality of AFM measurement acquisition either along fibrotic tracts or orthogonal to fibrotic tracts. (B) Quantitative AFM analysis of tissue matrix stiffness of a liver lobule from untreated normal liver. Measurements were taken either from central (location ratio 0) to portal zones (location ratio 1) or from portal to portal zones. Tissue matrix rigidity did not vary significantly within normal liver lobules and averaged around 150Pa. (C) AFM analysis orthogonal to fibrotic tracts demonstrates that tissue stiffening is region specific with the greatest increases in matrix rigidity in areas of greatest fibrosis. Measurements were carried out within a liver lobule starting remote from the fibrotic tract (location ratio 0) toward the fibrotic tract (location ratio 1). Graphs depict changes between all conditions (no treatment, black; CCl4, blue; DDC, red), between mild (orange) vs. severe (blue) CCl4-induced fibrosis, or between mild (purple) vs. severe (red) DDC-induced fibrosis. *p<0.01 between all conditions and **p<0.01 for the two treatment groups compared to non-treated mice within the region of comparison denoted by dotted line. (D) AFM analysis demonstrates significant increases in tissue stiffness along fibrotic tracts in both CCl4-induced and DDC-induced disease compared to non-treated normal liver. Measurements were carried out either from central-central zones or portal-portal zones depending on disease model and severity. *p<0.01 between all conditions across the entire fibrotic tract. Sample size = 3 mice for each condition and 3 lobules were examined per mouse. For “All Conditions” plots, severe and mild forms of disease were combined so that n=6 for CCl4 and DDC treatment. Statistical significance was calculated using one-way ANOVA and Tukey’s post-hoc test for pairwise comparison. Error bars represent SEM.
Because patterns of fibrosis differed depending on the model of experimental fibrotic disease, we conducted a series of AFM measurements orthogonal to the fibrotic tract or along the fibrotic tract within a lobule (see diagram in Figure 2A). This analysis revealed that increased matrix stiffness within fibrotic livers was localized to the microenvironments around fibrotic tracts. Measurements orthogonal to fibrotic tracts showed that liver matrix was significantly stiffer in regions approaching fibrillar collagen deposition and returned to near normal rigidity in areas remote from it within the same lobule (Figure 2C). Although, in a few instances, non-fibrotic areas of CCl4- and DDC-treated livers showed a trend toward slightly greater matrix stiffness than normal livers, there were no statistically significant differences when considered in aggregate (Supplemental Figure 1C). Matrix stiffness was significantly greater in severe forms of CCl4- or DDC-induced disease compared to their respective mild forms, but only in areas with or near fibrotic tracts. Along the fibrotic tracts, the average matrix stiffness was around 1kPa in CCl4-treated mice and ranged 2–3kPa in DDC-treated mice, both significantly greater than the stiffness across a normal liver lobule (Figure 2D). There were also statistically significant increases in stiffness along fibrotic tracts in severe CCl4- or DDC-induced disease compared to mild disease, suggesting that the rigidity of fibrotic tracts increased as disease progressed. In general, fibrotic tracts in DDC-induced disease were stiffer than in CCl4-induced disease, peaking near 6kPa in the former compared to 2kPa in the latter. These results demonstrated that matrix stiffness was significantly increased along or near fibrotic tracts compared to normal liver tissue. Importantly, matrix rigidity was highly region-specific and varied between local microenvironments within fibrotic livers depending on the proximity to fibrotic tracts and the disease model.
Matrix stiffness has significant effects on primary hepatocyte cytoskeletal tension and function
Isolated primary hepatocytes were cultured on top of collagen-conjugated polyacrylamide gels ranging in stiffness from below physiologic (75Pa) to highly supra-physiologic levels (60kPa) to determine the effect on cell spreading. Increased cell spreading, as determined by increased cell area, indicated greater cytoskeletal tension (19). We found that primary hepatocyte cell area, and therefore cytoskeletal tension, increased logarithmically with increasing matrix stiffness (Figure 3A). As demonstrated on a linear graph (Figure 3A inset), hepatocytes responded to increasing matrix stiffness most dynamically at the lower ranges of rigidity between 75Pa and 1kPa. Interestingly, 1kPa was the lower limit within the range of pathological matrix stiffnesses detected in fibrotic livers near fibrotic tracts. At stiffnesses greater than 1kPa, hepatocyte cell size continued to increase but at a more gradual rate. Representative photos of hepatocytes on the collagen-PA gels demonstrated that at 140Pa, the matrix stiffness of normal liver, cells were rounded and their nuclei were obscured by the spherical contour of the cell (Figure 3B). As matrix stiffness increased to 1kPa, hepatocytes flattened and became spread so that their nuclei were more visible. Cell spreading and flattening continued to increase from 1kPa to 6kPa, the upper limit of pathological liver matrix rigidity in fibrotic livers. At the supra-physiological stiffness of 60kPa, hepatocytes had spread and flattened even more compared to 6kPa, although the change in cell area and morphology was less dramatic than at lower ranges of matrix stiffness. In summary, these results showed that physiological and pathological ranges of matrix stiffness had profound effects on hepatocyte cell morphology and cytoskeletal tension.
Figure 3.
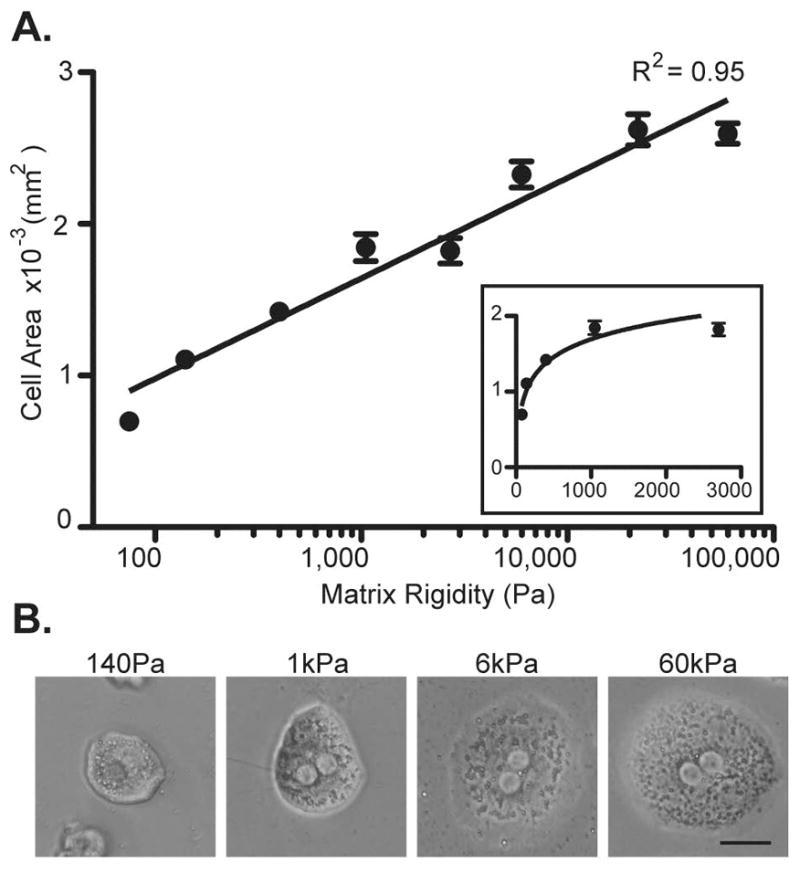
Primary hepatocyte cell area increases logarithmically with increasing matrix stiffness. (A) Primary hepatocytes were cultured on collagen-conjugated polyacrylamide gels at 8 different stiffnesses: 75, 140, 400, 1k, 2.7k, 6k, 22k, and 60k Pa. Images were taken after 24h of culture and the area of individual cells were determined by digital imaging analysis. Data represent the average of 2–5 independent experiments in which at least 20 cells were measured for each stiffness level per experiment. The best-fit curve follows a logarithmic function with an R2 = 0.95 as demonstrated by the semi-log plot. The inset linear plot shows that the most dynamic range for increasing cell size was between 75 and 1k Pa. Error bars represent SEM. (B) Representative phase-contrast images of single-cell primary hepatocytes cultured at 140, 1k, 6k, and 60k Pa for 24h. Scale bar = 25μm. Total magnification 80x.
To determine whether changes in hepatocyte morphology in response to matrix stiffness were associated with changes in function, we quantitatively measured albumin production, glycogen storage, and cytochrome P450 activity from isolated primary hepatocytes cultured on top of collagen-PA gels of varying stiffness (Figure 4A–C). We found that albumin production from hepatocytes cultured on top of stiffer matrix decreased significantly compared to those cultured on the physiologically normal stiffness of 140Pa. Interestingly, there appeared to be a threshold effect in which albumin production significantly decreased from 140Pa to 1kPa, with no further significant decreases detected at matrix stiffnesses greater than 1kPa compared to 1kPa. There was a trend toward decreased hepatocyte glycogen storage ability with increasing matrix stiffness. Intrinsic cytochrome P450 1A activity was low in hepatocytes plated on top of collagen-PA gels and not significantly affected by matrix rigidity. Hepatocyte proliferation increased on stiff compared to soft matrix and may reflect activation of aberrant proliferative signal transduction pathways (Supplemental Figure 2). These findings indicated that, in conjunction with changes in cytoskeletal tension, increasing matrix stiffness significantly inhibited albumin production and may also adversely affect other hepatocyte-specific functions.
Figure 4.
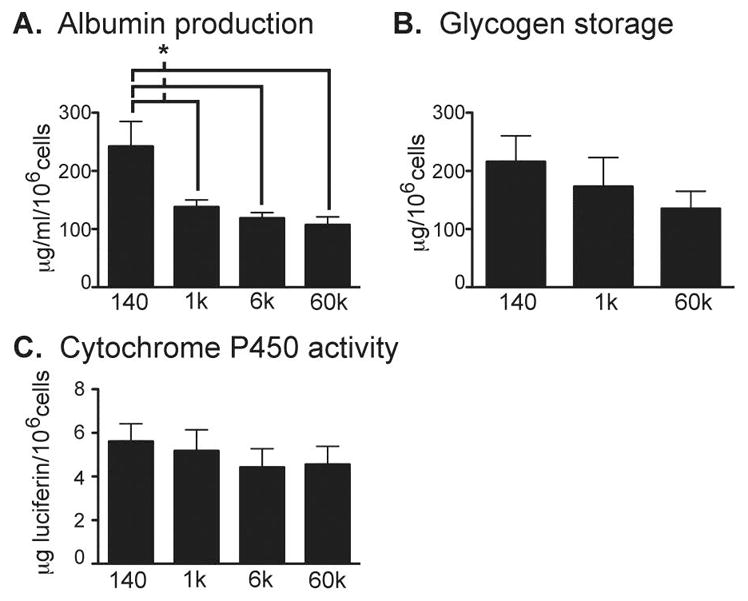
Matrix rigidity modulates various primary hepatocyte functions to differing degrees. (A) Albumin production, (B) glycogen storage, and (C) cytochrome P450 1A activity were measured quantitatively with correction for cell numbers after 24h of culture on matrices of varying stiffness. Data represent the average of 3 independent experiments. *p<0.05 by Student’s t-test. Error bars represent SEM.
Fibrotic ranges of matrix stiffness decreased the expression of functional genes within the hepatocyte nuclear factor 4 alpha (HNF4α regulatory network
We previously demonstrated that HNF4α was a key regulator for maintaining hepatocyte-specific functions in three-dimensional (3D) culture compared to two-dimensional (2D) monolayer culture (20). Since difference in substrate rigidity was one facet of the differences between 3D and 2D physical environments, we hypothesized that HNF4α and its downstream targets may be modulated by matrix rigidity. We co-stained liver tissue from untreated, CCl4-, and DDC-treated mice for HNF4α and fibronectin and found that the percentage of HNF4α positive cells was significantly decreased in fibrotic compared to normal livers (Figure 5A and B). Indeed, expression of Hnf4a itself and 3 important transcriptional functional targets (Baat, bile acid-CoA:amino acid N-acyltransferase; F7, factor VII; Gys2, glycogen synthase 2), was significantly decreased in hepatocytes cultured on stiffer matrices compared to normal liver matrix stiffness (Figure 5C). Similarly to the effect of increasing stiffness on albumin production, there was a threshold effect at the pathological stiffness level of 1kPa, at which there was a significant decrease in the expression of these functional genes. These findings indicated that pathological levels of liver matrix rigidity suppressed the expression of HNF4α and consequently the expression of several of its downstream functional targets.
Figure 5.

HNF4α expression is decreased in fibrotic livers in vivo and in hepatocytes cultured on stiff matrix in vitro. (A) HNF4α (red) was co-stained with fibronectin (green) to depict areas of fibrosis in livers of untreated, CCl4-, and DDC-treated mice. Images are representative of at least 3 mice per group and at least 3 sections evaluated per mouse. Scale bars represent 100μm. (B) The percentage of HNF4α+ cells per low-power field in the livers of untreated, CCl4-, and DDC-treated mice were quantified by digital imaging analysis. Sample size = 3 mice for each group with at least 3 sections evaluated per mouse. *p<0.01 by one-way ANOVA and Tukey’s post-hoc test for pairwise comparison. Error bars represent SEM. (C) Expression levels of hepatocyte nuclear factor 4 alpha (Hnf4a), bile acid-CoA:amino acid N-acyltransferase (Baat), factor VII (F7), and glycogen synthase 2 (Gys2) mRNA were significantly inhibited on matrices stiffer than 140Pa in hepatocytes after 24h culture as determined by quantitative real-time reverse transcription polymerase chain reaction (qRT-PCR). Data represent the average of 3 independent experiments. *p<0.05 and **p<0.005 by Student’s t-test. Error bars represent SEM.
To clarify the interaction between matrix rigidity and the HNF4α transcriptional regulatory network, we isolated primary hepatocytes from Hnf4afl/fl:Alb Cre+ mice, in which there was hepatocyte-specific deletion of HNF4α, and from wild-type littermates (Hnf4afl/fl:Alb Cre−). We cultured wild-type and HNF4α-deficient hepatocytes on collagen-PA gels of normal liver matrix stiffness (140Pa) to determine whether softer matrix maintained expression of certain functional genes through the maintained expression of HNF4α. We found that expression of the functional genes Baat, F7, and Gys2 was significantly decreased in HNF4α-deficient compared to wild-type hepatocytes on 140Pa matrix, indicating that maintained expression of HNF4α was critical for the expression of these genes on matrix of normal liver stiffness (Figure 6A). Conversely, HNF4α has been shown to actively suppress the transcription of mesenchymal genes such as Snail (Snai1) and vimentin (Vim) in hepatocytes (21); these genes were significantly increased in HNF4α-deficient compared to wild-type hepatocytes on 140Pa matrix. These results suggest that normal soft matrix supported hepatocyte-specific functions in part by maintaining the HNF4α transcriptional regulatory network, while pathological stiff matrix inhibited hepatocyte function because HNF4α expression was lost.
Figure 6.

HNF4α transcriptional regulation is maintained in primary hepatocytes cultured on normal liver matrix stiffness in vitro. (A) Primary hepatocytes were isolated from the Hnf4afl/fl:Alb Cre+ mice and wildtype littermates (Hnf4afl/fl:Alb Cre−) and cultured on 140Pa collagen-PA gels for 24h. Expression of gene targets positively regulated by HNF4α (Baat, F7, and Gys2) was decreased whereas targets negatively regulated by HNF4α (Snai1 and Vim) were increased in Hnf4afl/fl:Alb Cre+ hepatocytes, indicating that maintained expression of those functional genes on 140Pa matrix was dependent on continued HNF4α expression. Data represent the average of 5 independent experiments. (B) Primary hepatocytes were cultured on 1kPa matrix and transfected with either control plasmid or plasmid over-expressing HNF4α. Gene expression as determined by qRT-PCR was determined 24h after transfection. HNF4α-plasmid transfection achieved high over-expression of Hnf4a mRNA (>85-fold compared to control plasmid transfection) and resulted in decreased expression of Snai1. Data represent the average of 3 independent biological samples. *p<0.05 and **p<0.005 by Student’s t-test. Error bars represent SEM.
We then determined whether forced expression of HNF4α in hepatocytes cultured on stiff matrix was sufficient to restore expression of hepatic functional genes. We transfected primary hepatocytes plated on 1kPa matrix with either a control plasmid or plasmid expressing mouse HNF4α under the cytomegalovirus promoter. HNF4α plasmid transfection was able to achieve high over-expression of Hnf4a (>85-fold) compared to control plasmid. Forced over-expression of HNF4α significantly decreased the expression of Snai1 on stiff matrix, suggesting that the loss of HNF4α played an essential role in the initiation of the mesenchymal program in hepatocytes cultured on stiff matrix. On the other hand, forced expression of HNF4α did not increase expression of functional genes such as Baat, F7, and Gys2 on stiff matrix (Figure 6B). These findings suggest that HNF4α is necessary for the maintenance of hepatic functional genes on soft matrix, but not sufficient on its own to restore hepatic functional gene expression on stiff matrix. These results also suggest that increased matrix rigidity has other negative effects on the regulation of normal hepatocyte function in addition to the inhibition of HNF4α expression.
Fibrotic levels of matrix rigidity activated mechanotransduction in primary hepatocytes through FAK
In order to determine whether increased matrix stiffness in fibrotic liver disease led to initiation of the mechanotransduction cascade in vivo through activation of FAK, we performed immunostaining on liver tissue sections from untreated, CCl4-treated, and DDC-treated mice. Areas of fibrosis with increased extracellular matrix deposition in livers from CCl4- and DDC-treated mice were identified by phospho-FAKY397 and fibronectin co-staining. In addition, we performed phospho-FAKY397/HNF4α and activated β1-integrin/HNF4α co-staining to confirm that activation of the integrin-FAK pathway was taking place in hepatocytes. We found that hepatocytes near fibrotic tracts, and corresponding to areas of high matrix rigidity, showed significant expression of activated phospho-FAKY397 (Figure 7A). Interestingly, phospho-FAKY397 expression in hepatocytes was both membranous and cytoplasmic in livers from DDC-treated mice, whereas it was restricted to the membrane in hepatocytes of CCl4-treated mice. Activated β1-integrin was demonstrated on hepatocyte membranes in the livers of both CCl4- and DDC-treated mice, with higher levels of positive staining observed in DDC-treated mice. These findings showed that the mechanotransduction pathway through integrin and FAK was activated in hepatocytes near fibrotic tracts in vivo in fibrotic liver disease.
Figure 7.

Mechanotransduction is activated in primary hepatocytes by fibrotic levels of matrix stiffness in vivo and in vitro. (A) Activated phospho-FAKY397 (green) was expressed in hepatocytes near fibrotic tracts (highlighted by fibronectin staining in red) in the livers of CCl4- and DDC-treated mice. Expression of phospho-FAKY397 and activated β1-integrin (green) were localized to hepatocytes by HNF4α (red) co-staining to demonstrate activation of mechanotransduction within the hepatocytes of fibrotic livers. Images are representative of at least 3 mice per group and at least 3 sections evaluated per mouse. There was minimal background staining with secondary antibody-only controls (data not shown). (B) Isolated primary hepatocytes were cultured on 140, 1k, and 60k Pa matrices for 2h and cell lysates prepared for immunoblotting. Blot images are from one representative experiment. Densitometry graph shows the specific activation of phospho-FAKY397 relative to total FAK averaged over 5 independent experiments. *p<0.005 by Student’s t-test. Error bars represent SEM.
To determine whether activation of FAK in hepatocytes near fibrotic tracts was attributable to increased matrix stiffness, we cultured isolated primary hepatocytes on collagen-PA matrices and quantified the level of phospho-FAKY397 by immunoblotting. We found that phospho-FAKY397 was significantly increased in hepatocytes cultured on fibrotic (1kPa) and supra-physiological (60kPa) levels of matrix rigidity compared to normal liver stiffness (140Pa) (Figure 7B). Interestingly, a threshold phenomenon similar to albumin production and functional gene expression was observed. Phospho-FAKY397 was significantly greater in hepatocytes cultured on 1kPa compared to 140Pa and further increases in FAK activation were minimal between 1kPa and 60kPa. These results indicated that fibrotic levels of matrix rigidity at 1kPa were sufficient to activate FAK in primary hepatocytes.
Matrix rigidity modulated HNF4α expression in primary hepatocytes principally through the Rho/ROCK pathway
Mechanotransduction initiated by activated FAK is cross-regulated by the Rho/ROCK pathway, myosin contractility, and ERK (3–5). To determine whether aspects of this mechanotransduction circuit mediated the inhibition of the HNF4α network on stiff matrix, we cultured isolated primary hepatocytes on 1kPa matrix under control conditions or with small molecule inhibitors to FAK, ROCK, myosin contractility, or ERK kinase (MEK). Hnf4a mRNA expression in hepatocytes on stiff matrix was significantly increased by the inhibition of ROCK and there was a trend toward increased Hnf4a expression with FAK inhibition (Figure 8A). In contrast, neither inhibition of myosin contractility or MEK increased Hnf4a expression. Correlating with the effect on Hnf4a expression, inhibition of FAK also tended to increase the expression of Baat and decrease the expression of Vim. Importantly, inhibition of ROCK significantly increased the expression of Baat on stiff matrix and more profoundly decreased the expression of Vim compared to FAK inhibition. Albumin production was not affected (data not shown). To confirm that ROCK inhibition increased HNF4α expression on the protein level in hepatocytes cultured on stiff matrix, we performed immunoblotting for HNF4α from hepatocytes cultured on 140Pa matrix, 1kPa matrix, and 1kPa matrix with FAK or ROCK inhibition. We found that ROCK inhibition increased HNF4α protein expression in hepatocytes on 1kPa as early as 12h after plating to levels slightly higher than hepatocytes cultured on soft matrix (Figure 8B). Although protein expression of HNF4α decreased over time in all matrix conditions, ROCK inhibition in 1kPa matrix consistently maintained higher HNF4α protein expression compared to 1kPa alone. FAK inhibition on 1kPa matrix had minimal effect at earlier time points but increased HNF4α protein expression compared to 1kPa alone at the late time point. These findings indicated that stiff matrix inhibited the HNF4α transcriptional network primarily through signals downstream of the Rho/ROCK pathway and that signals directly downstream of activated FAK have a minor role.
Figure 8.

Inhibition of HNF4α expression in primary hepatocytes at fibrotic levels of matrix rigidity is mediated predominantly by the Rho/ROCK pathway. (A) Isolated primary hepatocytes were cultured on 1kPa matrices for 24h in the presence of control or inhibitors of FAK (FAKi-14), ROCK (Y-27632), myosin contractility (blebbistatin), or MEK (U-0126). Expression of Hnf4a and its target genes, Baat and Vim, were analyzed by qRT-PCR. Expression of Hnf4a and its positively regulated target, Baat, was significantly increased by blocking the Rho/ROCK pathway with Y-27632. Expression of Vim, a negatively regulated target of Hnf4a, was significantly inhibited by both FAK and ROCK inhibition. Data are representative of 3–5 independent experiments. *p<0.05 and **p<0.01 by Student’s t-test. Error bars represent SEM. (B) Primary hepatocytes were cultured on 140Pa matrix, 1kPa matrix, or 1kPa matrix in the presence of FAK or ROCK inhibitors for 12h, 24h, or 48h. HNF4α protein expression was determined by immunoblotting and quantified compared to the GAPDH housekeeping protein. Densitometry calculations were normalized to HNF4α expression in freshly isolated hepatocytes.
Discussion
Our study is the first to fully define the normal and pathological ranges of liver matrix rigidity on the cellular level across the microanatomical architecture of the liver lobule. Our results are also the first to show that matrix rigidity, as an isolated variable, is sufficient to modulate primary hepatocyte function through the effects of the Rho/ROCK pathway on the HNF4α transcriptional network. Previous studies have shown that changes in matrix composition (22), ligand density (23, 24), structure (25, 26), and cross-linking (27) altered hepatocyte function, proliferation, and response to growth factors. Dedifferentiation and redifferentiation of hepatocytes by altering matrix conformation correlated with HNF4α expression levels (28, 29). Although changes in those matrix variables also changed the substrate rigidity, those studies could not definitively determine whether matrix stiffness was primarily responsible for the observed effects. By culturing primary hepatocytes on collagen matrices of tunable rigidity, we showed that increased matrix stiffness in and of itself reduced hepatocyte-specific functions. We identified the physiologically relevant ranges of matrix stiffness by AFM and demonstrated that primary hepatocyte functions were preserved when cultured on matrix of normal liver stiffness, but were significantly reduced when cultured on matrix with the stiffness of fibrotic liver. Importantly, we showed that the most dynamic effects of matrix rigidity on primary hepatocyte morphology and function were in the relatively narrow range between 150Pa, the stiffness of normal liver, and 1kPa, the lower threshold of fibrotic liver stiffness.
Our findings demonstrated that reduced hepatocyte functions on stiff matrix were likely in part due to reduced expression of the master transcriptional regulatory factor, HNF4α. HNF4α is critical for normal liver development (30–32), maintaining mature liver functions (17, 33–35), and inducing differentiation of hepatocyte-like cells from stem cells and fibroblasts (36, 37). We confirmed previously published reports that HNF4α expression was decreased in fibrotic liver disease in vivo (38, 39). In addition, we showed that soft matrix equivalent to normal liver stiffness preserved the HNF4α transcriptional network, whereas stiff matrix equivalent to fibrotic liver inhibited it. Importantly, HNF4α was required to maintain functional gene expression on soft matrix. Although forced over-expression HNF4α was not sufficient to restore functional gene expression on stiff matrix, it had an essential role in suppressing the initiation of mesenchymal gene transcription. Others have shown that forced over-expression of HNF4α in vivo in cirrhotic rats restored hepatic functions (39). Our results might be limited by the in vitro nature of our experimental system in which primary hepatocytes were plated at single-cell density on a single layer of collagen in order to isolate the effect of matrix stiffness. These conditions as a starting point were suboptimal for maintenance of hepatic functions and might not be favorable for demonstrating HNF4’s ability to rescue function. Nevertheless, our findings provide a mechanistic link that increased matrix rigidity in cirrhotic livers can directly inhibit HNF4α expression through the Rho-ROCK pathway, leading to hepatocyte functional impairment.
The Rho/ROCK pathway is intricately connected to integrin-mediated FAK activation and ERK in a mechanoregulatory circuit (4, 5). Our results showed that FAK was activated in hepatocytes near fibrotic tracts in vivo and when cultured in vitro on matrix with the stiffness measured near fibrotic tracts. These findings indicated that pathological matrix stiffness found in fibrotic liver disease was sufficient to activate FAK-mediated mechanotransduction in hepatocytes. However, FAK inhibition had less effect on recovering the expression of HNF4α in hepatocytes on stiff matrix than compared to ROCK inhibition. In addition, inhibition of myosin contractility and ERK signaling did not increase HNF4α expression in the context of stiff matrix. Although the FAK and Rho/ROCK pathways have been closely linked in mechanotransduction, some studies have shown that matrix rigidity and cellular tension can regulate Rho/ROCK activation independently of FAK activation. Changes in cytoskeletal tension were demonstrated to directly regulate RhoA activation of ROCK (40). Stiff matrix could also destabilize microtubules, resulting in activation of guanine nucleotide exchange factors and subsequent downstream activation of Rho/ROCK (41). Finally, integrin-mediated mechanotransduction may be propagated through Src family tyrosine kinases that directly activate guanine nucleotide exchange factors through a pathway that does not rely on FAK activation (42) and also directly phosphorylate HNF4α to decrease its transcriptional activity (43). Our results indicate that while there may be redundant or compensatory mechanisms that activate Rho/ROCK when FAK activation is inhibited, signals downstream of ROCK are required to mediate inhibition of HNF4α expression in the context of stiff matrix.
Our results suggest that inhibition of Rho/ROCK signaling may help preserve hepatocyte function in cirrhotic liver disease by maintaining HNF4α expression. The ROCK inhibitor, Y-27632, has been shown to inhibit stellate cell activation and prevent development of liver fibrosis in rodent disease models (44–46). Unfortunately, systemic administration of Y-27632 caused severe hypotension and limited its clinical utility (47). Fasudil, a small molecule ROCK inhibitor that has been approved for clinical use in Japan and shown to ameliorate experimental lung fibrosis (48), may be further explored for treatment of liver fibrosis. On the other hand, FAK inhibition has also been shown to prevent experimental lung fibrosis (49) and toxicity studies in vivo showed that FAK inhibitor 14 does not have significant systemic effects (50). Given our results showing that FAK is activated by levels of matrix rigidity measured in fibrotic livers and detected in hepatocytes near fibrotic tracts in vivo, it will be of interest in future studies to determine whether FAK inhibition may have a therapeutic role in chronic fibrotic liver disease.
Supplementary Material
Acknowledgments
Financial Support: This work was supported by NIH K08-DK093708 to T.T.C., NHLBI T32 HL007544 to J.C.T., CIRM TR3-05542 to H.W. and V.M.W., and in part by NIH P30-DK026743 through the Liver Center at the University of California, San Francisco.
List of Abbreviations
- FAK
focal adhesion kinase
- ROCK
Rho-associated protein kinase
- ERK
extracellular signal-regulated kinase
- AFM
atomic force microscopy
- CCl4
carbon tetrachloride
- DDC
5-diethoxycarbonyl-1,4-dihydrocollidine
- qRT-PCR
quantitative reverse-transcriptase real-time PCR
- HNF4α
hepatocyte nuclear factor 4 alpha
- Baat
bile acid-CoA:amino acid N-acyltransferase
- F7
factor VII
- Gys2
glycogen synthase 2
- MEK
ERK kinase
References
- 1.Wells RG. The role of matrix stiffness in regulating cell behavior. Hepatology. 2008;47:1394–1400. doi: 10.1002/hep.22193. [DOI] [PubMed] [Google Scholar]
- 2.Yu H, Mouw JK, Weaver VM. Forcing form and function: biomechanical regulation of tumor evolution. Trends Cell Biol. 2010;21:47–56. doi: 10.1016/j.tcb.2010.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lessey EC, Guilluy C, Burridge K. From mechanical force to RhoA activation. Biochemistry. 2012;51:7420–7432. doi: 10.1021/bi300758e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, Reinhart-King CA, et al. Tensional homeostasis and the malignant phenotype. Cancer Cell. 2005;8:241–254. doi: 10.1016/j.ccr.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 5.Provenzano PP, Inman DR, Eliceiri KW, Keely PJ. Matrix density-induced mechanoregulation of breast cell phenotype, signaling and gene expression through a FAK-ERK linkage. Oncogene. 2009;28:4326–4343. doi: 10.1038/onc.2009.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ferraioli G, Tinelli C, Dal Bello B, Zicchetti M, Filice G, Filice C Liver Fibrosis Study G. Accuracy of real-time shear wave elastography for assessing liver fibrosis in chronic hepatitis C: a pilot study. Hepatology. 2012;56:2125–2133. doi: 10.1002/hep.25936. [DOI] [PubMed] [Google Scholar]
- 7.Yin M, Talwalkar JA, Glaser KJ, Manduca A, Grimm RC, Rossman PJ, Fidler JL, et al. Assessment of hepatic fibrosis with magnetic resonance elastography. Clin Gastroenterol Hepatol. 2007;5:1207–1213. e1202. doi: 10.1016/j.cgh.2007.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Venkatesh SK, Ehman RL. Magnetic resonance elastography of liver. Magn Reson Imaging Clin N Am. 2014;22:433–446. doi: 10.1016/j.mric.2014.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li Z, Dranoff JA, Chan EP, Uemura M, Sevigny J, Wells RG. Transforming growth factor-beta and substrate stiffness regulate portal fibroblast activation in culture. Hepatology. 2007;46:1246–1256. doi: 10.1002/hep.21792. [DOI] [PubMed] [Google Scholar]
- 10.Olsen AL, Bloomer SA, Chan EP, Gaca MD, Georges PC, Sackey B, Uemura M, et al. Hepatic stellate cells require a stiff environment for myofibroblastic differentiation. Am J Physiol Gastrointest Liver Physiol. 2011;301:G110–118. doi: 10.1152/ajpgi.00412.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schrader J, Gordon-Walker TT, Aucott RL, van Deemter M, Quaas A, Walsh S, Benten D, et al. Matrix stiffness modulates proliferation, chemotherapeutic response, and dormancy in hepatocellular carcinoma cells. Hepatology. 2011;53:1192–1205. doi: 10.1002/hep.24108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu L, Yannam GR, Nishikawa T, Yamamoto T, Basma H, Ito R, Nagaya M, et al. The microenvironment in hepatocyte regeneration and function in rats with advanced cirrhosis. Hepatology. 2012;55:1529–1539. doi: 10.1002/hep.24815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ramachandran P, Iredale JP. Reversibility of liver fibrosis. Ann Hepatol. 2009;8:283–291. [PubMed] [Google Scholar]
- 14.Georges PC, Hui JJ, Gombos Z, McCormick ME, Wang AY, Uemura M, Mick R, et al. Increased stiffness of the rat liver precedes matrix deposition: implications for fibrosis. Am J Physiol Gastrointest Liver Physiol. 2007;293:G1147–1154. doi: 10.1152/ajpgi.00032.2007. [DOI] [PubMed] [Google Scholar]
- 15.Yeh WC, Li PC, Jeng YM, Hsu HC, Kuo PL, Li ML, Yang PM, et al. Elastic modulus measurements of human liver and correlation with pathology. Ultrasound Med Biol. 2002;28:467–474. doi: 10.1016/s0301-5629(02)00489-1. [DOI] [PubMed] [Google Scholar]
- 16.Lopez JI, Kang I, You WK, McDonald DM, Weaver VM. In situ force mapping of mammary gland transformation. Integr Biol (Camb) 2011;3:910–921. doi: 10.1039/c1ib00043h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hayhurst GP, Lee YH, Lambert G, Ward JM, Gonzalez FJ. Hepatocyte nuclear factor 4alpha (nuclear receptor 2A1) is essential for maintenance of hepatic gene expression and lipid homeostasis. Mol Cell Biol. 2001;21:1393–1403. doi: 10.1128/MCB.21.4.1393-1403.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Przybyla L, Lakins JN, Sunyer R, Trepat X, Weaver VM. Monitoring developmental force distributions in reconstituted embryonic epithelia. Methods. 2015 doi: 10.1016/j.ymeth.2015.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tan JL, Tien J, Pirone DM, Gray DS, Bhadriraju K, Chen CS. Cells lying on a bed of microneedles: an approach to isolate mechanical force. Proc Natl Acad Sci U S A. 2003;100:1484–1489. doi: 10.1073/pnas.0235407100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chang TT, Hughes-Fulford M. Molecular mechanisms underlying the enhanced functions of three-dimensional hepatocyte aggregates. Biomaterials. 2014;35:2162–2171. doi: 10.1016/j.biomaterials.2013.11.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Santangelo L, Marchetti A, Cicchini C, Conigliaro A, Conti B, Mancone C, Bonzo JA, et al. The stable repression of mesenchymal program is required for hepatocyte identity: a novel role for hepatocyte nuclear factor 4alpha. Hepatology. 2011;53:2063–2074. doi: 10.1002/hep.24280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bissell DM, Arenson DM, Maher JJ, Roll FJ. Support of cultured hepatocytes by a laminin-rich gel. Evidence for a functionally significant subendothelial matrix in normal rat liver. J Clin Invest. 1987;79:801–812. doi: 10.1172/JCI112887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bhadriraju K, Hansen LK. Extracellular matrix- and cytoskeleton-dependent changes in cell shape and stiffness. Exp Cell Res. 2002;278:92–100. doi: 10.1006/excr.2002.5557. [DOI] [PubMed] [Google Scholar]
- 24.Mooney D, Hansen L, Vacanti J, Langer R, Farmer S, Ingber D. Switching from differentiation to growth in hepatocytes: control by extracellular matrix. J Cell Physiol. 1992;151:497–505. doi: 10.1002/jcp.1041510308. [DOI] [PubMed] [Google Scholar]
- 25.Fassett J, Tobolt D, Hansen LK. Type I collagen structure regulates cell morphology and EGF signaling in primary rat hepatocytes through cAMP-dependent protein kinase A. Mol Biol Cell. 2006;17:345–356. doi: 10.1091/mbc.E05-09-0871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Godoy P, Hengstler JG, Ilkavets I, Meyer C, Bachmann A, Muller A, Tuschl G, et al. Extracellular matrix modulates sensitivity of hepatocytes to fibroblastoid dedifferentiation and transforming growth factor beta-induced apoptosis. Hepatology. 2009;49:2031–2043. doi: 10.1002/hep.22880. [DOI] [PubMed] [Google Scholar]
- 27.Semler EJ, Ranucci CS, Moghe PV. Mechanochemical manipulation of hepatocyte aggregation can selectively induce or repress liver-specific function. Biotechnol Bioeng. 2000;69:359–369. doi: 10.1002/1097-0290(20000820)69:4<359::aid-bit2>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 28.Runge D, Runge DM, Daskalakis N, Lubecki KA, Bowen WC, Michalopoulos GK. Matrix-mediated changes in the expression of HNF-4alpha isoforms and in DNA-binding activity of ARP-1 in primary cultures of rat hepatocytes. Biochem Biophys Res Commun. 1999;259:651–655. doi: 10.1006/bbrc.1999.0848. [DOI] [PubMed] [Google Scholar]
- 29.Runge D, Runge DM, Drenning SD, Bowen WC, Jr, Grandis JR, Michalopoulos GK. Growth and differentiation of rat hepatocytes: changes in transcription factors HNF-3, HNF-4, STAT-3, and STAT-5. Biochem Biophys Res Commun. 1998;250:762–768. doi: 10.1006/bbrc.1998.9384. [DOI] [PubMed] [Google Scholar]
- 30.Chen WS, Manova K, Weinstein DC, Duncan SA, Plump AS, Prezioso VR, Bachvarova RF, et al. Disruption of the HNF-4 gene, expressed in visceral endoderm, leads to cell death in embryonic ectoderm and impaired gastrulation of mouse embryos. Genes Dev. 1994;8:2466–2477. doi: 10.1101/gad.8.20.2466. [DOI] [PubMed] [Google Scholar]
- 31.Li J, Ning G, Duncan SA. Mammalian hepatocyte differentiation requires the transcription factor HNF-4alpha. Genes Dev. 2000;14:464–474. [PMC free article] [PubMed] [Google Scholar]
- 32.Parviz F, Matullo C, Garrison WD, Savatski L, Adamson JW, Ning G, Kaestner KH, et al. Hepatocyte nuclear factor 4alpha controls the development of a hepatic epithelium and liver morphogenesis. Nat Genet. 2003;34:292–296. doi: 10.1038/ng1175. [DOI] [PubMed] [Google Scholar]
- 33.Inoue Y, Yu AM, Inoue J, Gonzalez FJ. Hepatocyte nuclear factor 4alpha is a central regulator of bile acid conjugation. J Biol Chem. 2004;279:2480–2489. doi: 10.1074/jbc.M311015200. [DOI] [PubMed] [Google Scholar]
- 34.Stoffel M, Duncan SA. The maturity-onset diabetes of the young (MODY1) transcription factor HNF4alpha regulates expression of genes required for glucose transport and metabolism. Proc Natl Acad Sci U S A. 1997;94:13209–13214. doi: 10.1073/pnas.94.24.13209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lu H, Gonzalez FJ, Klaassen C. Alterations in hepatic mRNA expression of phase II enzymes and xenobiotic transporters after targeted disruption of hepatocyte nuclear factor 4 alpha. Toxicol Sci. 2010;118:380–390. doi: 10.1093/toxsci/kfq280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sekiya S, Suzuki A. Direct conversion of mouse fibroblasts to hepatocyte-like cells by defined factors. Nature. 2011;475:390–393. doi: 10.1038/nature10263. [DOI] [PubMed] [Google Scholar]
- 37.Takayama K, Inamura M, Kawabata K, Katayama K, Higuchi M, Tashiro K, Nonaka A, et al. Efficient generation of functional hepatocytes from human embryonic stem cells and induced pluripotent stem cells by HNF4alpha transduction. Mol Ther. 2012;20:127–137. doi: 10.1038/mt.2011.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yue HY, Yin C, Hou JL, Zeng X, Chen YX, Zhong W, Hu PF, et al. Hepatocyte nuclear factor 4alpha attenuates hepatic fibrosis in rats. Gut. 2010;59:236–246. doi: 10.1136/gut.2008.174904. [DOI] [PubMed] [Google Scholar]
- 39.Nishikawa T, Bell A, Brooks JM, Setoyama K, Melis M, Han B, Fukumitsu K, et al. Resetting the transcription factor network reverses terminal chronic hepatic failure. J Clin Invest. 2015;125:1533–1544. doi: 10.1172/JCI73137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bhadriraju K, Yang M, Alom Ruiz S, Pirone D, Tan J, Chen CS. Activation of ROCK by RhoA is regulated by cell adhesion, shape, and cytoskeletal tension. Exp Cell Res. 2007;313:3616–3623. doi: 10.1016/j.yexcr.2007.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Heck JN, Ponik SM, Garcia-Mendoza MG, Pehlke CA, Inman DR, Eliceiri KW, Keely PJ. Microtubules regulate GEF-H1 in response to extracellular matrix stiffness. Mol Biol Cell. 2012;23:2583–2592. doi: 10.1091/mbc.E11-10-0876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guilluy C, Swaminathan V, Garcia-Mata R, O’Brien ET, Superfine R, Burridge K. The Rho GEFs LARG and GEF-H1 regulate the mechanical response to force on integrins. Nat Cell Biol. 2011;13:722–727. doi: 10.1038/ncb2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chellappa K, Jankova L, Schnabl JM, Pan S, Brelivet Y, Fung CL, Chan C, et al. Src tyrosine kinase phosphorylation of nuclear receptor HNF4alpha correlates with isoform-specific loss of HNF4alpha in human colon cancer. Proc Natl Acad Sci U S A. 2012;109:2302–2307. doi: 10.1073/pnas.1106799109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Murata T, Arii S, Mori A, Imamura M. Therapeutic significance of Y-27632, a Rho-kinase inhibitor, on the established liver fibrosis. J Surg Res. 2003;114:64–71. doi: 10.1016/s0022-4804(03)00202-6. [DOI] [PubMed] [Google Scholar]
- 45.Murata T, Arii S, Nakamura T, Mori A, Kaido T, Furuyama H, Furumoto K, et al. Inhibitory effect of Y-27632, a ROCK inhibitor, on progression of rat liver fibrosis in association with inactivation of hepatic stellate cells. J Hepatol. 2001;35:474–481. doi: 10.1016/s0168-8278(01)00169-6. [DOI] [PubMed] [Google Scholar]
- 46.Tada S, Iwamoto H, Nakamuta M, Sugimoto R, Enjoji M, Nakashima Y, Nawata H. A selective ROCK inhibitor, Y27632, prevents dimethylnitrosamine-induced hepatic fibrosis in rats. J Hepatol. 2001;34:529–536. doi: 10.1016/s0168-8278(00)00059-3. [DOI] [PubMed] [Google Scholar]
- 47.Hennenberg M, Biecker E, Trebicka J, Jochem K, Zhou Q, Schmidt M, Jakobs KH, et al. Defective RhoA/Rho-kinase signaling contributes to vascular hypocontractility and vasodilation in cirrhotic rats. Gastroenterology. 2006;130:838–854. doi: 10.1053/j.gastro.2005.11.029. [DOI] [PubMed] [Google Scholar]
- 48.Zhou Y, Huang X, Hecker L, Kurundkar D, Kurundkar A, Liu H, Jin TH, et al. Inhibition of mechanosensitive signaling in myofibroblasts ameliorates experimental pulmonary fibrosis. J Clin Invest. 2013;123:1096–1108. doi: 10.1172/JCI66700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lagares D, Busnadiego O, Garcia-Fernandez RA, Kapoor M, Liu S, Carter DE, Abraham D, et al. Inhibition of focal adhesion kinase prevents experimental lung fibrosis and myofibroblast formation. Arthritis Rheum. 2012;64:1653–1664. doi: 10.1002/art.33482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Golubovskaya V, Curtin L, Groman A, Sexton S, Cance WG. In vivo toxicity, metabolism and pharmacokinetic properties of FAK inhibitor 14 or Y15 (1, 2, 4, 5-benzenetetramine tetrahydrochloride) Arch Toxicol. 2015;89:1095–1101. doi: 10.1007/s00204-014-1290-y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.