

Abstract
Aims
The fibroblast growth factor (FGF) family plays an important role in cardiac growth and development. However, only FGF-16 RNA levels are reported to increase during the perinatal period and to be expressed preferentially in the myocardium, suggesting control at the transcriptional level and a role for FGF-16 in the postnatal heart. Beyond the identification of two TATA-like elements (TATA1 and TATA2) in the mouse FGF-16 promoter region and the preferential cardiac activity of TATA2, there is no report of Fgf-16 gene regulation. Assessment of promoter sequences, however, reveals putative nuclear factor-kappaB (NF-κB) elements, suggesting that Fgf-16 is regulated via NF-κB activation and thereby implicated in a number of cardiac events. Thus, the Fgf-16 gene was investigated as a target for NF-κB activation in cardiac cells.
Methods and results
Assessments of Fgf-16 promoter activity were made using truncated and transfected hybrid genes with NF-κB inhibitors and/or β-adrenergic stimulation via isoproterenol (IsP) treatment (a known NF-κB activator) in culture, and on endogenous mouse and human Fgf-16 genes in situ. The mouse Fgf-16 promoter region was stimulated in response to IsP treatment, but this response was lost with NF-κB inhibitor pretreatment. Deletion analysis revealed IsP responsiveness linked to sequences between TATA2 and TATA1 and, more specifically, a NF-κB element upstream and adjacent to TATA1 that associates with NF-κB p50/p65 subunits in chromatin. Finally, TATA1 and the proximal NF-κB element are conserved in the human genome and responsive to IsP.
Conclusion
The mouse and human Fgf-16 gene is a target for NF-κB activation in the postnatal heart.
Keywords: FGF-16, Gene regulation, Nuclear factor-kappaB, Isoproterenol, Mouse and human heart
1. Introduction
Members of the fibroblast growth factor (FGF) family play important roles in cardiac growth, development, and disease. FGF-16 is expressed in the epicardium and endocardium during embryonic development, where it stimulates cardiomyocyte growth in a paracrine manner.1–3 FGF-16 levels also increase preferentially in the mouse and rat myocardium during the perinatal period,3–5 however, its role in the postnatal heart is unclear.5 Furthermore, no substance, factor, or condition has been reported to modulate either rodent or human Fgf-16 gene expression.6
Analysis of the mouse (m) Fgf-16 upstream flanking DNA,6 identified multiple putative nuclear factor-kappaB (NF-κB) responsive DNA elements. NF-κB can function as a hetero- or homodimer of subunits, and act as an activator or repressor.7 The p50, p52, p65/RelA, and RelB NF-κB subunits have been identified in cardiomyocytes.7,8 Until recently, NF-κB dimers were considered to exist in an inactive form, associated with an inhibitory subunit (IκB), in the cytoplasm. Translocation to the nucleus and interaction with target genes required activation to release IκB, thereby unmasking a nuclear localization signal.9 There is now evidence, however, that NF-κB can be detected in the nucleus of non-stimulated cells, raising the possibility that the basal activity or repression of some genes is regulated by NF-κB.7,10
The activation of NF-κB in the heart is also dependent on the type of stimulus, its intensity, and interactions with different protein complexes. In this way, NF-κB is involved in numerous cardiac processes not only development, but also contraction, dilation, angina, atherosclerosis, inflammation, ischaemia, hypertrophy, and heart failure.7,11 A role for FGF-16 has been suggested under pathological conditions in the heart.3 Thus, we investigated the possibility that FGF-16 is a target for NF-κB in postnatal cardiac cells, through the use of tumour necrosis factor alpha (TNF-α) and β-adrenergic stimulation, which are known to activate NF-κB.12,13 We show that mouse Fgf-16 RNA expression is responsive to NF-κB activation and is linked to sequences located between two FGF-16 TATA/promoter regions. The downstream promoter region is conserved in the human genome, which is also responsive to NF-κB activation in transfected rat cardiomyocytes. The implications of these data are discussed in relation to regulation of Fgf-16 gene expression and possible functional roles for FGF-16 in the postnatal myocardium.
2. Methods
2.1 Cultures/gene transfer
All procedures involving animals, their tissues and cells conform to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996). Ventricular myocytes were isolated from 1-day-old rat hearts (36–40) using enzymatic digestion and Percoll gradient fractionation as previously described.6 Cells were transfected using calcium phosphate/DNA precipitation 24 h after plating as previously described,6,14 using 10 μg test plasmid per plate with 20 ng TKp.Rluc (encoding Renilla luciferase) to correct for variations in transfection efficiency.
For TNF-α treatment, cells were cultured in high glucose Dulbecco’s modified Eagle’s medium (DMEM) and antibiotics, without serum and the presence or absence of 25 ng/mL recombinant rat TNF-α (Biosource, Camarillo, CA, USA).13 For isoproterenol (IsP) treatment, myocytes were cultured in DMEM with 10% foetal bovine serum (FBS), in the presence or absence of 10 μM IsP (Sigma-Aldrich, Oakville, ON, Canada).15 Inhibition of NF-κB activation was done as reported.16,17 Briefly, cells were pre-incubated for 1 h with 100 μM pyrrolidine dithiocarbamate (PDTC) or 1.5 h with 10 μM N-(6-chloro-9H-β-carbolin-8-yl) nicotinamide (PS-1145) in 10% FBS-DMEM, before IsP addition for 6 h. Cells were then collected in 100 mM Tris, 0.1% Triton X-100 and assessed by a Dual Luciferase Assay System (Promega, Madison, WI, USA).
2.2 Hybrid FGF-16/luciferase genes
Generation of −4.7/+1.1, −4.7, −2.7, −1.2, −0.2 mFGF-16p.luc genes was described previously,6 but are renamed here −5771/−12, −5771/−1039, −3773/−1039, −2309/−1039, and −1268/−1039 mFGF-16p.luc based on assigning the adenosine residue in the methionine translation start site nucleotide position +1. The −1292/−30 mFGF-16p.luc gene was generated by polymerase chain reaction (PCR) amplification of the −1292/−30 mFGF-16 fragment (Table 1) and insertion directly into the promoterless reporter gene vector -p.luc. The −1292/−760 and −1292/−627 mFGF-16p.luc constructs were generated by the digestion of −1292/−30 mFGF-16p.luc construct with SacI and BglI, respectively, and the −747/−12 mFGF-16p.luc construct was generated by SacI/KpnI digestion of −5771/−12 mFGF-16p.luc construct, all followed by religation. The human (h) −737/−1 hFGF-16p.luc construct was obtained through PCR amplification of the −737/−1 hFGF-16 fragment (Table 1) from human placenta DNA (DNeasey Blood & Tissue Kit, Qiagen, ON, Canada), and insertion into -p.luc.
Table 1.
Primers used for PCR
| Primer name | Sequence |
|---|---|
| hFGF-16 exon 1/2 junction F | 5′-ACAGCCGCTTCGGAATCC-3′ |
| hFGF-16 exon 2 R | 5′-CCTCGCTCATTCATTCCTAGGT-3′ |
| −879 hFGF-16 F | 5′-CCCCGCCAACCTATACAGGTA-3′ |
| −794 hFGF-16 R | 5′-TCGGAGGCTGCGGAAAG-3′ |
| −310 hFGF-16 F | 5′-AAAGTGCCGTTTGCATCTGAT-3′ |
| −230 hFGF-16 R | 5′-TCCTGACCTTGCTCTCAGTCAA-3′ |
| Sequencing primers hFGF-16 | |
| −892 hFGF-16 F | 5′-CACTTCCTCGGTACCCCG-3′ |
| −385 hFGF-16 R | 5′-CACACCTCATTGGCATCCCCTC-3′ |
| −2035 hFGF-16 F | 5′-CCCCTCACCCAGCTTAAC-3′ |
| −1427 hFGF-16 R | 5′-CCCTGGCACTCGGCGTCCCG-3′ |
| −490 hFGF-16 R | 5′-AGAGGGAGTGAAGCTCG-3′ |
| −735 hFGF-16 F | 5′-CCAACTCCCGTCTCGAGGCACTTTCCA-3′ |
| −735 hFGF-16 R | 5′-CTGGGCCGATTGCGG-3′ |
| mB2m F | 5′-GCTATCCAGAAAACCCCTCAAA-3′ |
| mB2m R | 5′-GCGGGTGGAACTGTGTTACG-3′ |
| mFGF-16 exon 1/2 junction F | 5′-ACAGCCGCTTCGGAATTCT-3′ |
| mFGF-16 exon 2 R | 5′-CTCTCCTCGCTCATTCATTCCT-3′ |
| −1292/−30 mFGF-16 F | 5′-CCCCCGGTACCCGCCAGTTCCCACCCCTG-3′ |
| −1292/−30 mFGF-16 R | 5′-CCCCCGGTACCGGGCCGAGCCGGGGCC-3′ |
| hFGF-16 UTP F | 5′-ACCCCGCCAACCTATACA-3′ |
| hFGF-16 DTP F | 5′-GAGCAAGGTCAGGAGCACG-3′ |
| hFGF-16 exon 1/2 junction F | 5′-ACAGCCGCTTCGGAATCC-3′ |
| hFGF-16 exon 2 R | 5′-CCTCGCTCATTCATTCCT-3′ |
| hGAPDH F | 5′-CCATGGAGAAGGCTGGGG-3′ |
| hGAPDH R | 5′-CAAAGTTGTCATGGATGACC-3′ |
| ChIP assay | |
| −1233/−1051 F | 5′-AGGACAGAGCGAGGAAACAC-3′ |
| −1233/−1051 R | 5′-AGGGGTGGAGGCTAAGACAT-3′ |
| −659/−572 F | 5′-CAGCAGACCGACAGACAGAC-3′ |
| −659/−572 R | 5′-TGAAATAACGGAGCCCAGTA-3′ |
| −395/−314 F | 5′-ACCCACACACCAATCTCTGA-3′ |
| −395/−314 R | 5′-CACTCCCCTTCCATTGACAT-3′ |
| Untr 6 F | 5′-TCAGGCATGAACCACCATAC-3′ |
| Untr 6 R | 5′-AACATCCACACGTCCAGTGA-3′ |
2.3 DNA sequencing
Genomic DNA from human placenta and blood was isolated using the DNeasy Blood and Tissue Kit (Qiagen). Collection and manipulation of all human tissue conforms to the principles outlined in the Declaration of Helsinki for use of human tissue or subjects. Primers (Table 1) were designed to amplify regions of unassigned nucleotides (gaps) in the human genome database. The size and specificity of the amplicon was assessed by electrophoresis (1% agarose) and sequencing (Robarts Research Institute, ON, Canada), using forward and reverse primers designed within the known sequences flanking the gaps. The analysis was then repeated using forward and reverse primers from within the newly sequenced DNA fragment.
2.4 RNA isolation and reverse transcriptase–PCR
RNA was isolated using the RNeasy Plus Mini Kit (Qiagen) according to the manufacturer’s instructions and assessed for quality on an agarose gel. Total RNA from adult human hearts was obtained from commercial suppliers (female, age 73, Ambion, ON, Canada; pools of three males, ages 30–39 and three males/females, ages 20–42, Clontech, CA, USA). Total RNA (1 μg) was converted to cDNA using the QuantiTect Reverse Transcription Kit (Qiagen) according to the manufacturer’s instructions.
For PCR, 250 ng from an RT reaction or genomic DNA, or 100 ng plasmid DNA, as appropriate, was used with 1 μM each of forward and reverse primers (Table 1), 2 U Taq DNA polymerase and buffers included in the PCR amplifier kit (Qiagen), according to the manufacturer’s instructions. PCR reactions were normally for 30 cycles (denaturation 95°C/5 min, annealing 52–65°C/45 s, extension 72°C/1 min). Products were visualized by agarose gel electrophoresis and ethidium bromide staining.
2.5 IsP treatment in vivo
Mice (6–8 weeks) were injected intraperitoneally (i.p.) with ketoprofen (5 mg/kg body weight). One hour later, IsP (80 mg/kg) or saline vehicle was administrated (i.p.). Following treatment, animals were euthanized and hearts excised and rapid frozen. For chromatin immunoprecipitation (ChIP) assay, IsP treatment was for 2 h and hearts were processed by Genpathway, Inc. (http://www.genpathway.com/) using NF-κB p65 (sc-109, Santa Cruz Biotechnology) and p50 (ab7971, Abcam) specific antibodies; antibodies were validated against known NF-κB sites in the NF-κB inhibitor (alpha) and interleukin 8 genes. Primer sequences for each NF-κB site assessed and for the untranscribed region of chromosome 6 (Untr 6) are provided in Table 1.
2.6 Real-time RT–PCR
Amplifications were performed in triplicate in 20 μL using the SYBR GREEN PCR Master Mix Kit (10 μL; A&B Applied Biosystems, Warrington, UK), with 50 ng of template cDNA and primers for FGF-16 or Beta-2 microglobulin (B2m; Table 1) in an ABI 7500 Real-Time PCR System; cycle conditions were: incubation 50°C/2 min, denaturation 95°C/10 min, then 40 cycles with denaturation 95°C/15 s and annealing/extension 60°C/1 min. B2m was assumed to be constitutively expressed, and used to normalize data. FGF-16 RNA levels were analysed using the comparative CT method. The average CT values were determined for both FGF-16 and B2m RNA in each sample from the treated and untreated (control) groups (n = 3 per group). The average delta CT value (ΔCT) was determined for each group by subtracting the average CT value for B2m from the average CT value for FGF-16. The ΔΔCT value was then determined by subtracting ΔCT value for untreated group from the ΔCT value for the treated group. The relative quantity (RQ) was determined for each group using the RQ Study feature within the 7500 Real Time PCR System Sequence Detection Software, version 1.3.1 (A&B Applied Biosystems). To correct for differences in RNA input and/or in efficiencies of reverse transcription and amplification reaction, the data were validated and the slope of the two standard curves (FGF-16 and B2m) were compared for accuracy. FGF-16 and B2m were amplified with a similar efficiency and correlation coefficients over a range of 104.
2.7 Statistical analysis
Analysis of variance (ANOVA) with a post hoc Dunnet’s test and the unpaired (Welch’s) t-test were used for multiple and single comparisons, respectively. Mean values were considered significantly different if P < 0.05 (*P < 0.05, **P < 0.01, ††P < 0.01, and ***P < 0.001 in figures).
3. Results
3.1 The mouse FGF-16 promoter region responds to NF-κB activation
Analysis of 6 kb of murine (m) Fgf-16 gene sequences upstream of the methionine (ATG) start codon revealed a series of six putative NF-κB DNA elements in the forward direction and one in the reverse. Four of these NF-κB DNA elements are located within a 1300 bp putative proximal promoter region containing two TATA-like sequences (TATA1 and TATA2) (Figure 1).6 The ~6 kb Fgf-16 fragment was inserted upstream of a luciferase reporter gene to generate −5771/−12 mFGF-16p.luc (Figure 1). This gene fragment (previously reported as −4.7/+1.1 FGF-16p) supports significant basal activity in neonatal rat cardiomyocytes and transgenic mouse hearts.6 To test for a response to NF-κB activation, neonatal rat cardiomyocytes transfected with −5771/−12 mFGF-16p.luc were treated with TNF-α and IsP. Both compounds are known activators of NF-κB in cardiac cells.12,13 Initially, the effect of TNF-α treatment was assessed at 1, 2, and 6 h. A significant increase in luciferase activity was observed at 2 and 6 h but not 1 h post-treatment (Figure 2A). The effect of TNF-α and IsP treatment for 24 h was then assessed, and highly significant stimulations of −5771/−12 mFGF-16p.luc activities were observed (Figure 2B).
Figure 1.

Schematic of DNA upstream of the mouse FGF-16 ATG start codon, indicating six putative NF-κB sites and two TATA-like elements (TATA1 and TATA2) identified by sequence analysis. The position of each site (first nucleotide) is indicated. A seventh putative NF-κB site but in the reverse/anti-sense direction was also identified at −700/−708. Regions of the mouse Fgf-16 gene assessed by hybrid luciferase reporter gene assay are also indicated.
Figure 2.

(A) Cardiomyocytes were transfected with −5771/−12 mFGF-16p.luc and treated without or with TNF-α for 1, 2, and 6 h as indicated, or (B) TNF-α or isoproterenol (IsP) for 24 h, or IsP for 6 h with or without pretreatment with NF-κB inhibitors (C) PDTC and (D) PS-1145 for 60 and 90 min, respectively. Results are presented relative to luciferase activity in untreated (Control) cultures, which has been arbitrarily set to 1.0. The activity levels for ‘Control’ samples expressed as mean ±standard error of the mean (SEM) for (A) 1, 2 and 6 h are 14.74 ±0.05; 12.82 ±0.06 and 19.73 ±0.03, respectively; for (B) TNF-α and IsP are 9.82 ±0.03 and 6.65 ±0.13, respectively; and for (C) 13.95 ±0.02 and (D) 11.84 ±0.01 (n = 3).
To confirm the IsP response was related to NF-κB activation, cardiomyocytes were pretreated with NF-κB inhibitors PDTC or PS-1145 and then in conjunction with IsP treatment; PDTC and PS-1145 target different points in the activation of NF-κB for inhibition, specifically, dissociation of NF-κB from IκB and the IκB kinase complex.16,17 A significant increase in −5771/−12 mFGF-16p.luc activity was observed 6 h post-IsP treatment. In contrast, no significant increase in expression was observed following pretreatment of cells with PDTC or PS-1145 before IsP treatment (Figure 2C). Furthermore, neither inhibitor had significant effects on reporter gene activity when used alone.
3.2 TATA1-related promoter activity responds to NF-κB activation
To further localize the region of DNA that is responsive to IsP treatment, hybrid genes containing TATA1, TATA2, or both (Figure 1) were assessed in transfected cardiomyocytes (Figure 3A). Expression of genes containing TATA1, either alone (−747/−12 mFGF-16p.luc) or with TATA2 (−5771/−12 mFGF-16p.luc) were increased significantly 24 h posttreatment. The construct −5771/−1039 mFGF-16p.luc, containing the TATA2 but not TATA1 sequences was not responsive, suggesting that elements required for a response reside downstream of TATA2. To assess a requirement for NF-κB activation, the −747/−12 mFGF-16p.luc gene containing the most proximal promoter sequences (Figure 1) was tested for sensitivity to PDTC and PS-1145 pretreatments (Figure 3B). A significant increase in luciferase activity was observed following IsP treatment, but not following inhibition of NF-κB activation.
Figure 3.

(A) Hybrid mFGF-16p.luc genes with truncated regions of the upstream DNA flanking the ATG start codon containing either TATA1 or TATA2 alone, or TATA1 and TATA2 were used to transfect cardiomyocytes. Cultures were treated with or without IsP and luciferase activity measured at 24 h. (B) The −747/−12 mFGF-16p.luc gene containing TATA1 and putative NF-κB DNA elements (Figure 1) was tested for the effect of NF-κB inhibitors (PDTC and PS-1145) pretreatment on IsP responsiveness (6 h). Results for each hybrid gene in (A) and (B) are expressed relative to luciferase activity in untreated (Control) cultures, which has been arbitrarily set to 1.0. Control activity levels for −5771/−1039, −5771/−12, and −747/−12 mFGF-16p.luc are 16.20 ±0.03, 23.84 ±0.04, and 21.69 ±0.07, respectively, and are means ±SEM (n = 3–6).
3.3 The human FGF-16 sequences contain conserved TATA1 and NF-κB elements
The human, like the mouse, Fgf-16 gene is located on the X chromosome.19 On investigation, the available sequence upstream of the human Fgf-16 coding region was incomplete, containing two predicted gaps of 887 and 416 bp, both within 2 kb of the translation start site. Primers were designed to fill these gaps and sequence data were obtained (GenBank: GQ273509). The 887 bp gap was determined to be only 442 bp long, spanning positions −847 to −455, whereas the second gap was 449 bp long, spanning positions −1949 to −1468. The 5′-flanking regions of the human and murine Fgf-16 genes were then compared. TATA1 but not TATA2 was conserved in the human sequence. Alignment of 500 bp immediately upstream of the ATG start codon revealed 87% identity similarity, with several insertions in the alignment and 10% mismatched nucleotides. Importantly, the (forward) NF-κB binding site located approximately 65–70 bp upstream of TATA1 at nucleotides −375/−367 (5′-GGGGATGCC-3′) but not the (reverse) site at −700/−708 was conserved in both sequence and location in the human gene (Figure 4A).
Figure 4.

(A) Alignment of human (−383/−267) and mouse (−375/−259) Fgf-16 gene sequences. Positions of putative TATA1 and NF-κB DNA sequences are shown. Mismatched sequences are indicated (no vertical bar/lower case). (B) Schematic showing the relative positions of the upstream (UTP) and downstream TATA1 primer (DTP) as well as the exon 1 and exon 2 (Ex1/Ex2) primers, together with expected amplicon sizes after RT–PCR. Three human (h) heart RNA samples were assessed by RT–PCR using Ex1/Ex2 primers for 30 and 35 cycles. Results are visualized by electrophoresis and ethidium bromide staining. To assess whether the ‘TATA’ is likely part of the hFGF-16 promoter region, RNA was amplified (RT–PCR) using UTP vs. DTP partnered with an exon 2 (Ex2) primer for 20 cycles. A second round of PCR using Ex1/Ex2 primers (30 cycles) was used to detect FGF-16 RNA as a 100 bp product. GAPDH RNA was also assessed (±RT) as a control. The mobility of a 100 bp marker is shown. (C) Neonatal rat cardiomyocytes were transfected with −735/−1 hFGF-16p.luc or -p.luc, and treated without or with IsP for 6 h, and without or with PDTC or PS-1145 pretreatment. Results are expressed relative to luciferase activity in untreated (Control) cultures, which has been arbitrarily set to 1.0. Basal levels of activity for −735/−1 hFGF-16p.luc and -p.luc are 7.28 ±0.08 and 11.42 ±0.08, respectively, and are means ±SEM (n = 3).
The presence of FGF-16 in three adult human heart RNA samples was assessed by RT–PCR, using a primer spanning the exon 1–exon 2 boundary with an exon 2 primer (100 bp amplicon) for 30 and 35 cycles (Figure 4B). Two of these samples represent pools from three individuals and the third is from one individual. For a control, PCR was also done (±RT) using specific glyceraldehyde-3-phosphate dehydrogenase (GAPDH) primers. A 100 bp FGF-16 RNA-related product was detected in all three RNA samples.
The possibility that the TATA sequence at −314/−309 (5′-TATAAA-3′) in the human gene reflects the proximal promoter region in cardiac cells was examined. On the basis of an expectation of low levels of human cardiac FGF-16 expression,19 RNA was assessed by two rounds of PCR. In the first, the three human heart RNA samples were amplified with forward primers specific to sequences upstream (UTP) and downstream (DTP) of the putative TATA region, partnered with an exon 2 primer for 20 cycles, to generate appropriate products as indicated in the schematic representation (Figure 4B). A second round of PCR was done on these samples using the exon 1–exon 2 primer set as above (100 bp amplicon) for 30 cycles. For an additional negative control, −RT reactions for the UTP/exon 2 and GAPDH primer set were done. Products were assessed by (2%) agarose gel electrophoresis and ethidium bromide staining (Figure 4B). Increased levels of the 100 bp exon 1–exon 2 amplicon above those seen with exon 1–exon 2 primers after 30 cycles were detected in RT–PCR products generated downstream but not upstream of the TATA region, consistent with the conservation of TATA1 and this representing the proximal human Fgf-16 promoter region in cardiac cells.
To test function a hybrid luciferase gene was generated (−735/−1 hFGF-16p.luc) directed by 735 bp of human DNA sequence encompassing the conserved TATA and NF-κB elements. Following transfection, neonatal rat cardiomyocytes were treated with IsP in the presence and absence of PDTC and PS-1145 pretreatments (Figure 4C). The activity of −735/−1 hFGF-16p.luc increased significantly with IsP treatment after 6 h in a PDTC/PS-1145-sensitive manner, and above promoterless-luciferase levels, suggesting that the proximal NF-κB element is sufficient for IsP responsiveness human and, by extension, mouse Fgf-16 promoter.
3.4 IsP exerts a rapid but transient effect on endogenous mFGF-16 RNA
Endogenous FGF-16 RNA expression was assessed in adult mice injected (i.p.) without or with 80 mg/kg IsP after 2 and 6 h by real-time (q) RT–PCR. Primers spanning exons 1 and 2 of the Fgf-16 gene, with a 100 bp amplicon, were used to specifically detect RNA (Figure 5). IsP injection resulted in a rapid but transient stimulation of Fgf-16 gene expression, with a significant increase in RNA levels evident by 2 h, but was lost by 6 h.
Figure 5.
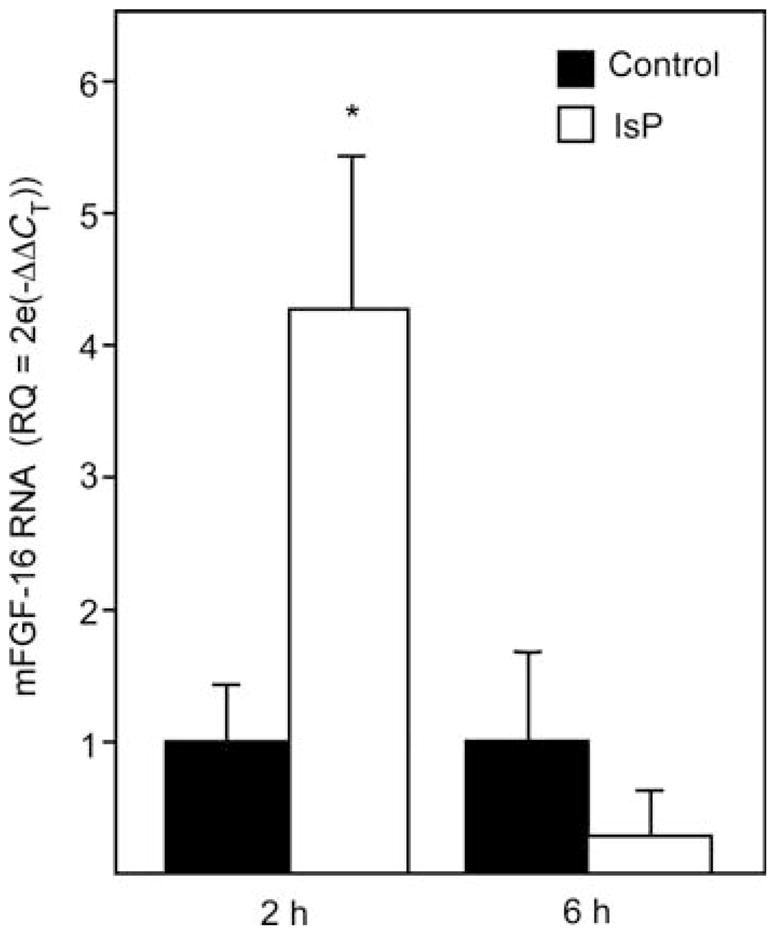
Mouse heart FGF-16 RNA levels are increased in response to IsP treatment in vivo. Mice were injected with IsP or vehicle (Control) and RNA assessed by real-time RT–PCR 2 and 6 h after injection using primer pairs from mFgf-16 exon 1 and 2 sequences, as well as the constitutively active B2m gene to normalize data. Comparative CT analysis for n = 3 independent injections per group is shown (mean ±standard deviation). RQ, relative quantitation. *P < 0.05 compared with control at the same time point.
3.5 NF-κB associates with the mFGF-16 promoter in situ
ChIP assay was done to assess the binding of NF-κB (p50 and p65) binding in situ, using isolated heart nuclei from adult mice injected with saline or IsP (80 mg/kg i.p., n = 3). Hearts were harvested 2 h after injection. PCR primers were designed to amplify DNA encompassing each of the three putative NF-κB DNA elements at −375/−367, −700/−708 (reverse) and −1005/−997 (Figure 1) as well as an untranscribed region on chromosome 6 (Untr6). The latter is used (by Genpathway, Inc.) as a measure of ‘background’ or non-specific association of protein-DNA detected by ChIP assay. In saline-injected hearts, all three putative sites showed significant association with p50 above ‘background’ levels after 2 h (Figure 6A). In contrast, only the most proximal site at −375/−367 showed any significant association with p65 after saline treatment (Figure 6B); the p65 subunit possesses the trans-activation domain. For IsP-treated hearts, however, there was a significant reduction in the levels of association of p50 and p65 at all three sites by 2 h posttreatment (Figure 6, white columns). As such, only the proximal site at −375/−367 was associated with both p50 and p65 NF-κB subunits.
Figure 6.
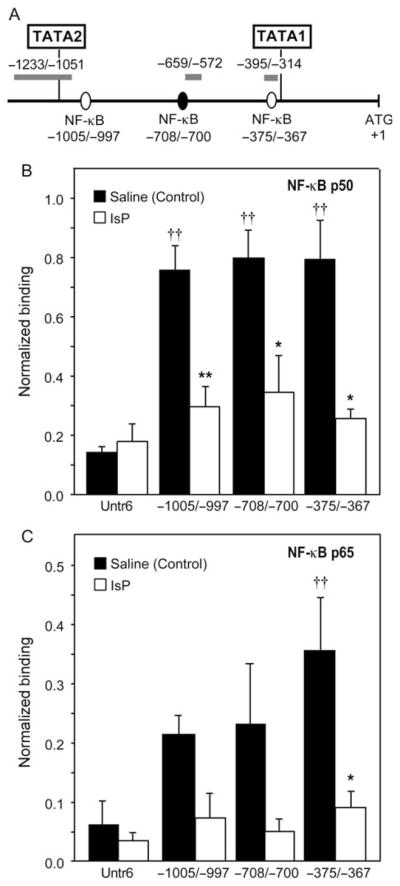
NF-κB associates with the Fgf-16 promoter region containing TATA1 sequences in mouse heart chromatin in situ. (A) Schematic showing the sequences upstream of the mouse Fgf-16 ATG start codon, indicating that the three putative NF-κB sites assessed by ChIP, and the relative position of the amplicons generated by PCR used for detection. Mouse heart nuclei harvested 2 h after saline (Control) or IsP treatments were assessed for (B) NF-κB p50 and (C) NF-κB p65 by ChIP assay. For each sample, the binding events data were normalized to the amount of input chromatin used for the immunoprecipitation reaction at each putative NF-κB site and the control untranscribed region (Untr6). Association of NF-κB subunits to each site is compared to that determined at Untr6 (††, P < 0.01, ANOVA with post-test), as well as the effect of IsP treatment on association at each site (*P < 0.05 and **P < 0.01, unpaired two-tailed t-test).
4. Discussion
The function of FGF-16 in postnatal cardiac cells is poorly understood. It has been suggested, however, that FGF-16 might play a role in the heart under pathological conditions, such as pressure overload and myocardial infarction.3 Here, we have used known NF-κB stimulators and inhibitors to investigate Fgf-16 gene expression as a target of NF-κB activation and, thus, cellular stress. More specifically, we show that endogenous mouse FGF-16 RNA levels are increased transiently through β-adrenergic (IsP) and cytokine (TNF-α) stimulation, and a mechanism linked to NF-κB activation. The mouse Fgf-16 gene upstream flanking region contains multiple NF-κB binding sites. Through deletion analysis, we observed that NF-κB stimulation of FGF-16 promoter activity is mediated by sequences located between two TATA sequences, the TATA1 at −314/−309 and TATA2 at −1126/−1121. The TATA1 sequence was identified but not pursued previously in cardiac cells,6 but is sufficient to support IsP responsiveness in conjunction with, it appears, the adjacent NF-κB element at −375/−367. This element alone showed significant association with both NF-κB p50 and p65 subunits in mouse heart chromatin in situ; a heterodimeric complex of these subunits is the most common and associated with transcriptional activation. Finally, this downstream putative promoter region, including both TATA1 and proximal NF-κB elements, are conserved in the human genome and is responsive to β-adrenergic stimulation after gene transfer, further indicating that the proximal NF-κB site in both the human and mouse Fgf-16 genes is sufficient to confer IsP responsiveness.
Our previous analysis of the murine FGF-16 5′-flanking region suggested cardiac-specific control via an upstream promoter region containing the TATA2 sequence 5′-TGTAAA-3′ at nucleotides −1126/−1121 but not TATA1 sequences, and was dependent on the binding of MEF-2.6 Our present analysis of the human Fgf-16 gene and flanking sequences did not reveal an analogous promoter region or, indeed, the MEF-2 site, although other putative MEF-2 sites can be found elsewhere in the human sequences. Instead, the downstream sequences containing TATA1 was highly conserved between the human and mouse genes, as was the most proximal NF-κB binding site. Although the −735/−1 hFGF-16p.luc was not expressed efficiently, this downstream promoter region of both the mouse (−747/−12) and human (−735/−1) Fgf-16 genes were increased significantly above background levels in response to adrenergic/IsP stimulation in a PDTC/PS-1145-sensitive manner in transfected cardiomyocytes. Although there is strong evidence for FGF-16 RNA expression in the postnatal rodent myocardium,2–5 similar data to support expression in the human heart have been lacking. For example, although FGF-16 RNA was observed in samples of human aorta, left atrium, and ventricle, there was a failure in the same study to detect human FGF-16 RNA in ‘adult heart’ or ‘foetal heart’.19 Our assessment of three independent RNA sources indicates a low level of Fgf-16 gene expression in the adult human heart, which is consistent with our promoter analysis. By extension, however, the promoter could be stimulated and thus relative FGF-16 expression may increase under certain conditions. Furthermore, the detection of amplifiable transcript corresponding to sequences downstream but not upstream of the TATA sequence, identified on the basis of similarity to TATA1 in the mouse, is consistent with the use of a conserved promoter region. However, the absence of TATA2 (and previously identified MEF-2)6 sequences in the human Fgf-16 gene indicates that while some expression and regulation (presumably via TATA1) will be conserved, mouse and human patterns of FGF-16 gene expression may not be identical. This would account for the apparent more readily detectable levels of FGF-16 RNA in rodent vs. human heart samples. In addition, TATA1 as opposed to TATA2 is likely to also play a role in FGF-16 expression outside the heart. Consistent with this expectation, both −747/−12 mFGF-16p.luc and −735/−1 hFGF-16p.luc genes are expressed at low levels in rat glial (C6) and skeletal myoblast (C2C12) cell lines after gene transfer (data not shown). Thus, although we cannot conclude that FGF-16 levels are regulated by adrenergic stimulation (or TNF-α) in the human heart or that this production would be cardiac specific, the RNA expression and transfection data strongly support a role for FGF-16 production at this site.
The association of NF-κB-related subunits p50 and p65 with an apparently conserved NF-κB DNA element immediately upstream of TATA1 in situ, and requirement of these sequences for significant promoter activity is consistent with a role for FGF-16 production in the mouse heart and involvement of NF-κB activation. NF-κB cellular availability increases after IsP treatment,20–22 and the sensitivity of the Fgf-16 IsP response to PDTC and PS-1145 indicates the involvement of NF-κB activation. At first glance, the decrease in the association of NF-κB subunits at −375/−367 suggested by ChIP analysis appears inconsistent with the increase in FGF-16 RNA levels and promoter activity at 2 h. However, assessments of RNA and reporter gene activity are measurements of accumulation and as such consequences of IsP treatment. Thus, while together they suggest that the effect is, at least in part, transcriptional, the pattern of transcription over the 2 h of treatment through this assessment is not known. Although accumulation may reflect a steady increase in RNA levels over the 2 h, it is also possible that an initial spike of activity occurred in the first 30 min of treatment followed by a decrease to basal levels (or lower), perhaps due to the expected loss of IsP effectiveness as a drug; in the dog IsP has been reported to peak 5–15 min after administration.23 In a similar manner, the apparent decrease in the levels of NF-κB association does not rule out increases during the 2 h of IsP treatment. In terms of binding seen in the absence of IsP stimulation, it is possible that procedures involved in handling mice and tissue isolation induce sufficient stress to stimulate NF-κB activity. Even in the case of IsP treatment, signs of administration are reported in the first minute. IsP treatment presumably results in a level or duration of stimulation beyond these ‘background’ levels as reflected by the significant PDTC/PS-1145 sensitive increase in mFGF-16 RNA levels observed at 2 h. However, although our data support a p50–p65 association with the NF-κB site at −375/−367 in vivo, we cannot rule out a contribution from alternative mechanisms. It is possible that p50, which appears to associate with multiple sites, can also interact with other NF-κB subunits (e.g. c-Rel or RelB) or even as a homodimer. The p50 subunit, like p52, lacks a trans-activation domain, and p50/p50 homodimers can function as transcriptional repressors, as they able to bind competitively to κB sites, thereby preventing heterodimer binding and trans-activation.7,8 NF-κB has also been linked with gene repression under certain circumstances, raising the possibility of a mechanism of derepression contributing to the response observed.7,11,24
As indicated, NF-κB is associated with several cardiac pathologies.7,11 Thus, the possibility that FGF-16 RNA expression is a NF-κB target is intriguing. The relevance of a transient increase in FGF-16 via β-adrenergic stimulation may lie in its ability to alter the threshold level of activity for other key signalling factors, either through simple receptor competition and/or through cross-talk and modification of the downstream signalling environment. Such action could conceivably be permissive or restrictive to the action of other factors. Previously, we showed that FGF-16 applied to neonatal cardiomyocytes could compete with FGF-2 for the occupation of FGF receptors; in addition, we observed potential cross-talk with IGF-1 receptor.5 In both cases, FGF-16 interfered with growth factor activity through the downstream signalling modulator protein kinase C. The ability of FGF-16 to alter the effects of other growth factors, perhaps even contributing to setting threshold levels on processes such as stress/inflammatory response, cell survival, and/or hypertrophy. The study of these pathophysiological processes in mice lacking a functional Fgf-16 gene may be an important step towards understanding the function of this evolutionarily conserved growth factor.
Acknowledgments
The authors thank Ms Yan Jin for her ‘footprint’, and Dr Cindy Ellison and Mrs Ionela Gheorghiu for help with PCR.
Funding
This work was supported by a grant from the Canadian Institutes of Health Research (MOP-62742). A.G.S. is the recipient of a Manitoba Health Research Council and Manitoba Institute of Child Health Studentship.
Footnotes
Conflict of interest: none declared.
References
- 1.Lavine KJ, Yu K, White AC, Zhang X, Smith C, Partanen J, et al. Endocardial and epicardial derived FGF signals regulate myocardial proliferation and differentiation in vivo. Dev Cell. 2005;8:85–95. doi: 10.1016/j.devcel.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 2.Lu SY, Sheikh F, Sheppard PC, Fresnoza A, Duckworth ML, Detillieux KA, et al. FGF-16 is required for embryonic heart development. Biochem Biophys Res Commun. 2008;373:270–274. doi: 10.1016/j.bbrc.2008.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hotta Y, Sasaki S, Konishi M, Kinoshita H, Kuwahara K, Nakao K, et al. FGF-16 is required for cardiomyocyte proliferation in the mouse embryonic heart. Dev Dyn. 2008;237:2947–2954. doi: 10.1002/dvdy.21726. [DOI] [PubMed] [Google Scholar]
- 4.Miyake A, Konishi M, Martin FH, Hernday NA, Ozaki K, Yamamoto S, et al. Structure and expression of a novel member, FGF-16, on the fibroblast growth factor family. Biochem Biophys Res Commun. 1998;243:148–152. doi: 10.1006/bbrc.1998.8073. [DOI] [PubMed] [Google Scholar]
- 5.Lu SY, Sontag DP, Detillieux KA, Cattini PA. FGF-16 is released from neonatal cardiac myocytes and alters growth-related signaling: a possible role in postnatal development. Am J Physiol Cell Physiol. 2008;294:C1242–C1249. doi: 10.1152/ajpcell.00529.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sofronescu AG, Jin Y, Cattini PA. A myocyte enhancer factor 2 (MEF2) site located in a hypersensitive region of the FGF-16 gene locus is required for preferential promoter activity in neonatal cardiac myocytes. DNA Cell Biol. 2008;27:173–182. doi: 10.1089/dna.2007.0689. [DOI] [PubMed] [Google Scholar]
- 7.Shaw J, Zhang T, Rzeszutek M, Yurkova N, Baetz D, Davie JR, et al. Transcriptional silencing of the death gene BNIP3 by cooperative action of NF-kappaB and histone deacetylase 1 in ventricular myocytes. Circ Res. 2006;99:1347–1354. doi: 10.1161/01.RES.0000251744.06138.50. [DOI] [PubMed] [Google Scholar]
- 8.Haudek SB, Bryant DD, Giroir BP. Differential regulation of myocardial NF kappa B following acute or chronic TNF-alpha exposure. J Mol Cell Cardiol. 2001;33:1263–1271. doi: 10.1006/jmcc.2001.1388. [DOI] [PubMed] [Google Scholar]
- 9.Hall G, Hasday JD, Rogers TB. Regulating the regulator: NF-kappaB signaling in heart. J Mol Cell Cardiol. 2006;41:580–591. doi: 10.1016/j.yjmcc.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 10.Ashburner BP, Westerheide SD, Baldwin AS., Jr The p65 (RelA) subunit of NF-kappaB interacts with the histone deacetylase (HDAC) corepressors HDAC1 and HDAC2 to negatively regulate gene expression. Mol Cell Biol. 2001;21:7065–7077. doi: 10.1128/MCB.21.20.7065-7077.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Valen G, Yan ZQ, Hansson GK. Nuclear factor kappa-B and the heart. J Am Coll Cardiol. 2001;38:307–314. doi: 10.1016/s0735-1097(01)01377-8. [DOI] [PubMed] [Google Scholar]
- 12.Chandrasekar B, Marelli-Berg FM, Tone M, Bysani S, Prabhu SD, Murray DR. Beta-adrenergic stimulation induces interleukin-18 expression via beta2-AR, PI3K, Akt, IKK, and NF-kappaB. Biochem Biophys Res Commun. 2004;319:304–311. doi: 10.1016/j.bbrc.2004.04.185. [DOI] [PubMed] [Google Scholar]
- 13.Condorelli G, Morisco C, Latronico MV, Claudio PP, Dent P, Tsichlis P, et al. TNF-alpha signal transduction in rat neonatal cardiac myocytes: definition of pathways generating from the TNF-alpha receptor. FASEB J. 2002;16:1732–1737. doi: 10.1096/fj.02-0419com. [DOI] [PubMed] [Google Scholar]
- 14.Doble BW, Chen Y, Bosc DG, Litchfield DW, Kardami E. Fibroblast growth factor-2 decreases metabolic coupling and stimulates phosphorylation as well as masking of connexin43 epitopes in cardiac myocytes. Circ Res. 1996;79:647–658. doi: 10.1161/01.res.79.4.647. [DOI] [PubMed] [Google Scholar]
- 15.Jimenez SK, Sheikh F, Jin Y, Detillieux KA, Dhaliwal J, Kardami E, et al. Transcriptional regulation of FGF-2 gene expression in cardiac myocytes. Cardiovasc Res. 2004;62:548–557. doi: 10.1016/j.cardiores.2004.01.032. [DOI] [PubMed] [Google Scholar]
- 16.Schreck R, Meier B, Mannel DN, Droge W, Baeuerle PA. Dithiocarbamates as potent inhibitors of nuclear factor kappa B activation in intact cells. J Exp Med. 1992;175:1181–1194. doi: 10.1084/jem.175.5.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Catley MC, Sukkar MB, Chung KF, Jaffee B, Liao SM, Coyle AJ, et al. Validation of the anti-inflammatory properties of small-molecule IkappaB Kinase (IKK)-2 inhibitors by comparison with adenoviral-mediated delivery of dominant-negative IKK1 and IKK2 in human airways smooth muscle. Mol Pharmacol. 2006;70:697–705. doi: 10.1124/mol.106.023150. [DOI] [PubMed] [Google Scholar]
- 18.Itoh N, Ornitz DM. Evolution of the Fgf and Fgfr gene families. Trends Genet. 2004;20:563–569. doi: 10.1016/j.tig.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 19.Antoine M, Wirz W, Tag CG, Gressner AM, Wycislo M, Müller R, et al. Fibroblast growth factor 16 and 18 are expressed in human cardiovascular tissues and induce on endothelial cells migration but not proliferation. Biochem Biophys Res Commun. 2006;346:224–233. doi: 10.1016/j.bbrc.2006.05.105. [DOI] [PubMed] [Google Scholar]
- 20.Freund C, Schmidt-Ullrich R, Baurand A, Dunger S, Schneider W, Loser P, et al. Requirement of nuclear factor-kappaB in angiotensin II- and isoproterenol-induced cardiac hypertrophy in vivo. Circulation. 2005;111:2319–2325. doi: 10.1161/01.CIR.0000164237.58200.5A. [DOI] [PubMed] [Google Scholar]
- 21.Takemoto Y, Yoshiyama M, Takeuchi K, Omura T, Komatsu R, Izumi Y, et al. Increased JNK, AP-1 and NF-kappa B DNA binding activities in isoproterenol-induced cardiac remodeling. J Mol Cell Cardiol. 1999;31:2017–2030. doi: 10.1006/jmcc.1999.1033. [DOI] [PubMed] [Google Scholar]
- 22.Rodriguez MS, Wright J, Thompson J, Thomas D, Baleux F, Virelizier JL, et al. Identification of lysine residues required for signal-induced ubiquitination and degradation of I kappa B-alpha in vivo. Oncogene. 1996;12:2425–2435. [PubMed] [Google Scholar]
- 23.Minatoya H, Lands AM, Portmann GA. Absorption and elimination profile of isoproterenol in anesthetized dogs. J Pharm Sci. 1965;54:968–972. doi: 10.1002/jps.2600540704. [DOI] [PubMed] [Google Scholar]
- 24.Zhong H, May MJ, Jimi E, Ghosh S. The phosphorylation status of nuclear NF-kappa B determines its association with CBP/p300 or HDAC-1. Mol Cell. 2002;9:625–636. doi: 10.1016/s1097-2765(02)00477-x. [DOI] [PubMed] [Google Scholar]