

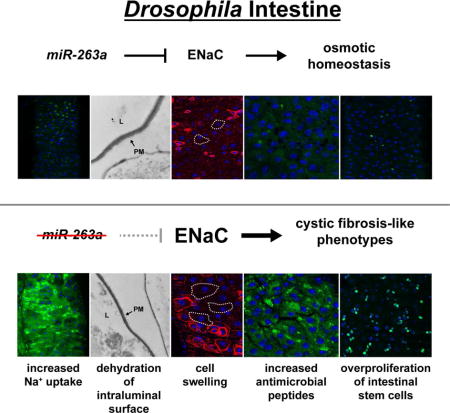
Summary
Proper regulation of osmotic balance and response to tissue damage is crucial in maintaining intestinal stem cell (ISC) homeostasis. We found that Drosophila miR-263a downregulates the expression of epithelial sodium channel (ENaC) subunits in enterocytes (ECs) to maintain osmotic and ISC homeostasis. In the absence of miR-263a, the intraluminal surface of the intestine displays dehydration-like phenotypes, Na+ levels are increased in ECs, stress pathways are activated in ECs and ISCs overproliferate. Furthermore, miR-263a mutants have increased bacterial load and expression of antimicrobial peptides. Strikingly, these phenotypes are reminiscent of the pathophysiology of cystic fibrosis (CF) in which loss-of-function mutations in the chloride channel CF transmembrane conductance regulator can elevate the activity of ENaC, suggesting that Drosophila could be used as a model for CF. Finally, we provide evidence that overexpression of miR-183, the human ortholog of miR-263a, can also directly target the expressions of all three subunits of human ENaC.
eTOC Blurb
Kim et al. demonstrate that miR-263a regulates the expression of the epithelial sodium channel ENaC to maintain osmotic and intestinal stem cell homeostasis. Furthermore, miR-263a mutants display phenotypes that are reminiscent of the pathophysiology of cystic fibrosis, suggesting that Drosophila could be used as a model for cystic fibrosis.
Introduction
The Drosophila intestinal system is an attractive model for studying signaling events that control stem cell homeostasis given its anatomical and functional similarities to human epithelial systems, including the intestine (Jiang and Edgar, 2011). The adult midgut is continuously damaged during feeding, as well as by chemicals and pathogens they encounter in the food, and thus needs to be constantly renewed. The renewal process requires tight regulation of the activities of multiple conserved signaling pathways in response to various types of intestinal epithelial injuries. These responses promote both intestinal stem cell (ISC) proliferation and enteroblast (EB) differentiation, expediting the rapid generation of new midgut epithelial cells to replace damaged ones (Biteau and Jasper, 2011; Buchon et al., 2010; Jiang and Edgar, 2011; Jiang et al., 2009; Osman et al., 2012; Zhou et al., 2013).
MicroRNAs (miRNAs) are small non-coding RNAs that post-transcriptionally regulate gene expression. In the past few years, miRNAs have been shown to play an important role in stem cell homeostasis by regulating differentiation and self-renewal (Gangaraju and Lin, 2009; Mathieu and Ruohola-Baker, 2013; Yi and Fuchs, 2012). Here, we found that a well-conserved miRNA, miR-263a, is necessary for maintaining ISC homeostasis. We show that deletion of miR-263a in the adult midgut ECs activates a stress response that, in turn, activates signaling pathways required for ISC proliferation, resulting in midgut hyperplasia. We identified well-conserved subunits of the epithelial sodium channel (ENaC) as biologically important targets of miR-263a and demonstrate that regulation of these subunits by miR-263a is critical for maintaining proper osmotic homeostasis in the midgut epithelium. Remarkably, many of the phenotypes of miR-263a mutants are reminiscent of the pathophysiology of cystic fibrosis (CF), an autosomal recessive disorder caused by mutations in the gene encoding the chloride channel CF transmembrane conductance regulator (CFTR) (Riordan, 2008). In CF patients, loss-of-function mutations in the CFTR can elevate the activity of ENaC through a mechanism that is not fully understood (Berdiev et al., 2009). ENaC is present at the apical plasma membrane in many epithelial tissues throughout the body to regulate sodium reabsorption, and control total body salt and water homeostasis (Garty and Palmer, 1997). The most common symptoms of CF are potential lethal blockages of distal small intestines, airway mucus obstruction, and chronic airway inflammation (Grubb and Gabriel, 1997; Mall et al., 2004), which are consistent with the model that upregulation in ENaC activity increases sodium and water reabsorption, ultimately leading to dehydration of the intraluminal surface and reduction in mucus transport (Bhalla and Hallows, 2008). Interestingly, we provide evidence that overexpression of miR-183, the human ortholog of miR-263a, can also directly target all three subunits of human ENaC to regulate its activity. Altogether, our findings describe the role of a miRNA in regulating ENaC levels and suggest that the Drosophila intestine could be used as a model for CF.
Results
miR-263a is Required for ISC Homeostasis
To identify miRNAs that regulate ISC homeostasis, we screened for miRNAs that alter the basal number of ISCs in the adult posterior midgut using publically available miRNA deletion mutants. From the screen, we identified miR-263a as necessary for maintaining ISC homeostasis. Using a homozygous viable null allele of miR-263a (miR-263aΔ) (Hilgers et al., 2010), we found that miR-263a mutants have an increased number of ISCs, as marked by Delta (Dl) expression (Figure 1A–1B). In addition, many of these ISCs were juxtaposed, suggesting that they undergo symmetric division (Figure 1B). This phenotype is similar to the symmetric division of ISCs seen in neuralized and βν integrin (βint-ν) mutants, where downregulation of Dl/Notch signaling leads to frequent ISC duplication expanding the pool of ISCs (Ohlstein and Spradling, 2007; Okumura et al., 2014).
Figure 1. Phenotypes of miR-263a mutant.
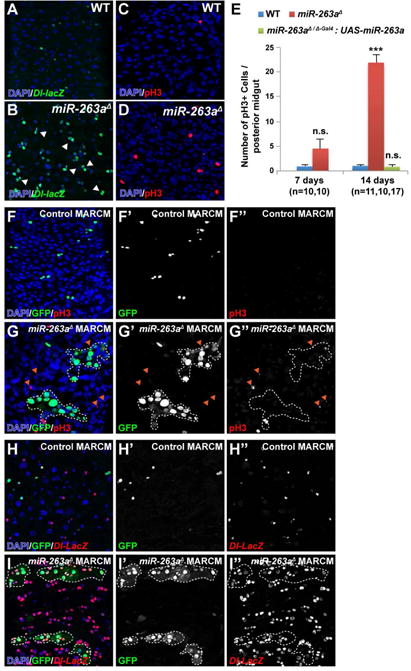
(A–B) The posterior midguts of 14 days old wild-type and miR-263a mutant stained with anti-β-gal to mark Dl (green). White arrowheads mark symmetrically divided ISCs (B). (C–D) The posterior midguts of wild-type and miR-263a mutant stained with anti-pH3 to mark mitotic ISCs (red). (E) The average number of pH3+ cells in the posterior midguts at 7 and 14 days old. “n” denotes the number of posterior midguts examined for each genotype. Error bars indicate SEM. ***P < 0.001 (two-tailed t-test). (F–I″) MARCM clones are labeled with GFP. White dotted lines outline the miR-263a mutant clones. (F–G″) miR-263a mutant clones can induce ISC proliferation non-cell-autonomously as indicated by the increased number of pH3+ cells (red) outside the mutant clones (orange arrowheads). (H–I″) Non-cell-autonomous increase in the number of ISCs, marked with Dl-LacZ (red), was observed outside the miR-263a mutant clones. See also Figure S1.
In order to determine where in the midgut epithelium miR-263a is expressed, we took advantage of a Gal4 knock-in allele of miR-263a (miR-263aΔ-Gal4), in which the miR-263a hairpin sequences are replaced by Gal4 (Hilgers et al., 2010), to drive the expression of a UAS-GFP transgene, thus marking all cells that express miR-263a. GFP expression was restricted to ECs, as evident by a lack of GFP expression in either ISCs or EBs, which were marked with esg-LacZ (Figure S1A-S1A″), or in enteroendocrine cells (EEs), which were marked with immunostaining of Prospero (Pros) (Figure S1B–S1B″).
To ask if the increased number of ISCs was due to an increase in proliferation, we stained the midguts using anti-phospho-Histone H3 (pH3), a marker for cells undergoing mitosis (Figure 1C–1D). In miR-263a mutants, the average number of pH3+ cells increased with age while the numbers of pH3+ cells were relatively constant in the control (Figure 1E and Figure S1C). Although miR-263a mutants had higher averages of pH3+ cells in both 7 and 14 days old guts compared to controls, only the 14 days old average was statistically different. Overexpression of a miR-263a in a miR-263a mutant background completely suppressed the increase in pH3+ cells (Figure 1E), indicating that the phenotype was due to the absence of functional miR-263a.
Next, we assessed the impact of miR-263a on overall midgut proliferation by generating miR-263a mutant clones using the MARCM approach (Lee and Luo, 2001). Clones of miR-263a mutant cells grew larger than control clones (Figure 1F–1I and Figure S1D–S1E), consistent with an increase in ISC proliferation. Furthermore, miR-263a mutant clones induced non-cell-autonomous ISC proliferation, as indicated by an increased number of pH3+ and Dl+ cells outside the mutant clones (Figure 1F–1I″). These results indicate that loss of miR-263a expression in the ECs can have a non-cell-autonomous effect on ISC homeostasis.
Surprisingly, the miR-263a mutation also had an effect on the overall morphology of the adult midgut. The posterior midguts of the miR-263a mutants were shorter in length and larger in width compared to control (Figure S1F–S1K). We further observed that shortening and thickening of the midgut was due to multi-layering of the epithelium (Figure S1L–S1N). In addition, we observed that multi-layering of epithelial cells was followed by delamination and anoikis of ECs, which is evident by the presence of large lysosomes (Figure S1O).
Absence of miR-263a Induces Stress and Developmental Signaling Pathways to Promote ISC Hyperproliferation
In the midgut the Jun N-terminal Kinase (JNK) pathway is activated in response to EC stress/damage to promote ISC proliferation and differentiation, leading to rapid regeneration of the epithelium to replace damaged ECs (Jiang et al., 2009). To test whether ECs in miR-263a mutants are stressed/damaged, we used a puckered (puc) LacZ reporter line (puc-LacZ) to monitor JNK pathway activity. Using the MARCM approach, we generated miR-263a mutant clones and observed high levels of JNK pathway activation in both mutant clones and neighboring cells (Figure 2A–2B″). This non-cell-autonomous activation of the JNK pathway is likely caused by ECs that are detaching from the basement membrane due to their larger and more proliferative neighboring miR-263a mutant clones. In a recent study, Notch-defective ISC tumors have been shown to displace surrounding ECs, competing with them for basement membrane space and causing their detachment, extrusion and apoptosis (Patel et al., 2015). In fact, the multi-layering phenotypes of the miR-263a mutant midgut (Figure S1M and S1N) is very similar to the EC detachment phenotype observed in the previous study. Further, this non-cell-autonomous activation of the JNK pathway likely stimulates the proliferation of neighboring ISCs (Figure 1I–I″), which may further expand the expression of puc-LacZ.
Figure 2. Absence of miR-263a activates stress and developmental signaling pathways.
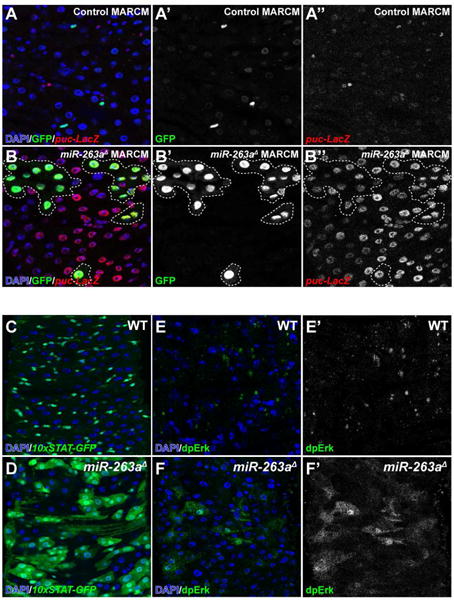
(A–B″) Activation of the JNK pathway in miR-263a mutants as visualized by the increased number of puc-LacZ expressing cells. White dotted lines outline miR-263a mutant clones. (C–D) Enhancement of JAK/STAT pathway activity in miR-263a mutants as visualized by the increased expression of 10xSTAT-GFP reporter. (E–F″) Enhancement of EGFR pathway activity in miR-263a mutants as visualized by the increased expression of dpERK. See also Figure S2.
Upon activation of the JNK pathway, damaged ECs and surrounding visceral muscle (VM) release ligands that activate JAK/STAT and EGFR/Ras/MAPK signaling pathways to promote ISC proliferation and differentiation (Biteau and Jasper, 2011; Buchon et al., 2010; Jiang and Edgar, 2011; Jiang et al., 2009; Osman et al., 2012; Zhou et al., 2013). Therefore, we examined the expression of these ligands in miR-263a mutant midguts using real-time quantitative PCR (qPCR). Indeed, we found that expression of the Unpaired cytokines (Upd, Upd2 and Upd3), which activate JAK/STAT signaling, and EGFR ligands, vein (vn), Spitz (Spi) and Keren (Krn), were significantly induced (Figure S2). Next, we examined whether induction of these ligands truly activate JAK/STAT and EGFR/Ras/MAPK signaling. To measure JAK/STAT pathway activity, we used a Stat92E reporter line driving the expression of GFP (10xSTAT-GFP). In control, the expression of 10xSTAT-GFP was restricted to ISCs and EBs (Figure 2C), while large patches of cells expressed the reporter in the miR-263a mutant (Figure 2D). Furthermore, the expression of 10xSTAT-GFP was also present in large misdifferentiated EC-like polyploid cells. To monitor EGFR/Ras/MAPK pathway activity, we examined the levels of phosphorylated ERK (dpERK) (Gabay et al., 1997). In control, dpERK signals were weakly detected in ISCs and EBs (Figure 2E–2E′). However, in miR-263a mutants, ectopic dpERK signals were mainly observed in large differentiated ECs (Figure 2F–2F′). Collectively, these results demonstrate that loss of miR-263a in the midgut activates the JNK pathway, which in turn increases the production of both JAK/STAT and EGFR/Ras/MAPK pathway ligands to promote hyperproliferation of ISCs.
Identification of Putative miR-263a Targets
To further understand the molecular function of miR-263a in maintaining ISC homeostasis, we searched for miR-263a target genes using TargetScan (Ruby et al., 2007), which predicted 158 targets. Since miR-263a expression is specific to ECs in the adult midgut, we cross-referenced the list of predicted miR-263a target genes with a list of genes that we identified to be specifically expressed in ECs, using the cell-type-specific gene expression profiling technique TADA (Southall et al., 2013) (Targeted DNA adenine methyltransferase identification using the RNA polymerase II-DAM fusion protein; (Doupé and Perrimon, personal communication)), thus restricting the list to 32 genes. Next, we examined the transcript levels of all 32 genes by qPCR and found that 5 genes had elevated transcript levels (>2-fold on average) in the miR-263a mutant midguts compared to the control (Table S1). Interestingly, one gene, Nach, had approximately 41-fold higher transcript level in the mutant compared to the control (Table S1 and Figure 3A). Furthermore, we raised an antibody against Nach and found that mutant midguts had significantly more Nach proteins while it is undetectable in the control (Figure 3B–3C). These striking increases in both the transcript and protein levels prompted us to further investigate whether Nach is a functionally important target of miR-263a in the adult midgut.
Figure 3. Regulation of ENaC by miR-263a.
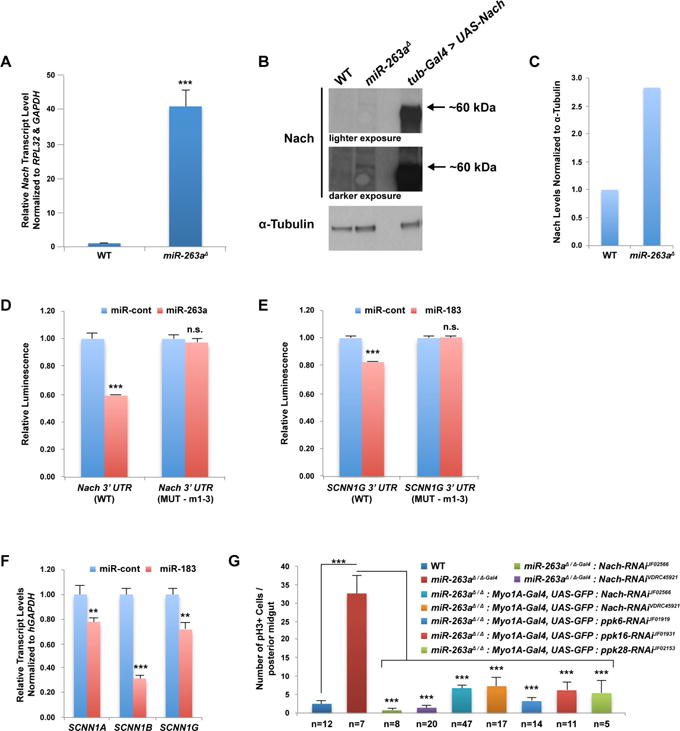
(A) Relative Nach transcript level in miR-263a mutant midguts. (B) Western blot analysis of Nach in miR-263a mutant midguts. α-Tubulin was used as loading control. (C) Quantitative analysis of the Western blot from (B). (D) Nach 3′ UTR luciferase reporter assay. (E) SCNN1G 3′ UTR luciferase reporter assay. (F) Relative transcript levels of human ENaC subunits after overexpression of miR-183 in CFBE41o- cells. (G) The average number of pH3+ cells in the posterior midguts at 14 days old. Depletion of Nach using two independent RNAi lines and two different Gal4 drivers in the miR-263a mutant background suppressed the increased number of pH3+ cells. The phenotype was also suppressed when other ENaC subunits were depleted in the mutant background. “n” denotes the number of posterior midguts examined for each genotype. Error bars indicate SEM. **P < 0.05 and ***P < 0.001 (two-tailed t-test). See also Figure S3 and Table S1.
Nach is a Functional Target of miR-263a in the Adult Posterior Midgut Epithelium
In Drosophila, Nach (also known as ppk4) is one of 31 family members (termed pickpocket (ppk) genes) that represent ion channels called the Degenerin/epithelial sodium channel (DEG/ENaC) (Zelle et al., 2013). Although the physiological functions of most family members are not known, some have been shown to act as non-voltage gated sodium channels (Bianchi and Driscoll, 2002; Garty and Palmer, 1997). The Nach 3′ untranslated region (3′ UTR) contains three potential miR-263a binding sites (Figure S3A). To determine whether miR-263a regulates the expression of Nach through these binding sites, we generated a luciferase reporter carrying the full length Nach 3′ UTR. Co-expression of the Nach 3′ UTR luciferase reporter with miR-263a in Drosophila S2R+ cells significantly reduced luciferase activity, while mutating the miR-263a seed sequences, the minimal sequence required for silencing of targets (Brennecke et al., 2005), completely relieved the suppression (Figure 3D). These results suggest that miR-263a can directly regulate the expression of Nach through these binding sites.
Drosophila encodes a second miRNA closely related in sequence to miR-263a called miR-263b. Interestingly, homozygous null mutants of miR-263b do not perturb ISC homeostasis (Figure S1C). One possibility is that slight differences in miR-263a and miR-263b seed sequences allow each miRNA to target a different set of genes. In fact, Nach is not a predicted target of miR-263b. Nevertheless, both miR-263a and miR-263b are members of a conserved family of miRNAs that includes mammalian miR-183, miR-96 and miR-182. The high degree of sequence conservation prompted us to ask if human miR-183, the miRNA most closely related in sequence to miR-263a, has analogous function in regulating the activity of ENaC. In humans, three genes termed sodium channel, non-voltage-gated 1 (SCNN1), alpha, beta, and gamma (SCNN1A, SCNN1B, and SCNN1G, respectively) encode components of the heteromultimeric ENaC (Canessa et al., 1994; Lingueglia et al., 1993; Waldmann et al., 1995). Based on ortholog prediction (Hu et al., 2011), Nach has two predicted orthologs, SCNN1G and SCNN1B; however, Nach is more closely related to SCNN1G. Strikingly, SCNN1G 3′ UTR contains three potential miR-183 binding sites (Figure S3B), and co-expression of the SCNN1G 3′ UTR luciferase reporter with miR-183, modestly, but significantly reduced luciferase activity, whereas mutating the miR-183 seed regions completely relieved the suppression (Figure 3E).
To further investigate the inhibitory potential of miR-183 on SCNN1G, we performed qPCR analyses to measure the endogenous transcript level of SCNN1G after overexpressing miR-183. We used immortalized human CF bronchial epithelial (CFBE41o-) cells, bearing the most frequent mutation in the CFTR gene (ΔF508), which has increased ENaC activity. Overexpression of miR-183 in CFBE41o- cells significantly reduced the endogenous SCNN1G transcript level (Figure 3F), likely through the three miR-183 binding sites we identified (Figure S3B). Strikingly, we also observed significant decreases in the endogenous transcript levels of SCNN1A and SCNN1B (Figure 3F). To investigate whether these are also due to direct interactions with miR-183, we searched for miR-183 binding sites and found multiple sites (Figure S3C–S3D). Co-expression of the 3′ UTR luciferase reporters with miR-183, also modestly, but significantly reduced the luciferase activities (Figure S3F). These results demonstrate that overexpression of miR-183 can repress ENaC activity by directly targeting all three ENaC subunits.
To investigate whether misregulation of Nach contributes to the overproliferation of ISCs in miR-263a mutants, we depleted Nach in the miR-263a mutant background. Depletion of Nach by RNAi suppressed the increase in pH3+ cells (Figure 3G). Further, to address whether Nach activity was required for proliferation of ISCs in general, we examined the consequence of depleting Nach activity on proliferation of ISCs in a gut regeneration model induced by feeding flies with dextran sodium sulfate (DSS) or bleomycin (Jiang et al., 2009; Ren et al., 2010). Knockdown of Nach did not suppress DSS or bleomycin-induced proliferation of ISCs (Figure S3H), indicating that Nach is dispensable for damage-induced ISC proliferation.
In mammals, overexpression of either SCNN1A or SCNN1B subunit alone is sufficient for ENaC activity (Canessa et al., 1994; Mall et al., 2004). Therefore, we asked whether overexpression of Nach alone is sufficient to disrupt ISC homeostasis by increasing ENaC activity. Overexpression of Nach in an otherwise wild-type background failed to increase the number of pH3+ cells (Figure S3I), suggesting that Nach may require additional subunits for full ENaC activity. This result led us to hypothesize that additional ENaC subunits may be misregulated in the miR-263a mutant and, therefore, began searching for other ENaC subunits in flies. Out of 12 ppk genes that are predicted orthologs of human SCNN1 genes, only 4 ppk genes (Nach/ppk4, ppk6, ppk16 and ppk28) are expressed in ECs (Figure S3J) (Doupé and Perrimon, personal communication), and all have elevated expression in miR-263a mutant midguts (Figure 3A and Figure S3K). Depleting individual ppk genes by RNAi in the miR-263a mutant background suppressed the increase in pH3+ cells (Figure 3G), suggesting that together with Nach, they may form a functional ENaC in flies. To determine whether this regulation is direct, we searched and found potential miR-263a binding sites in all 3 putative fly ENaC subunits (ppk6, ppk16, and ppk28) and performed 3′ UTR luciferase assays. We found that miR-263a was, in addition to Nach, only able to directly regulate the expression of ppk28 (Figure S3E and S3G). Collectively, these results suggest that miR-263a can directly and indirectly regulate the expressions of multiple candidate ENaC subunits.
Sodium Levels increase in miR-263a Mutant Midgut Epithelium
To examine whether misregulation of ENaC subunits in miR-263a mutants lead to Na+ imbalance in the midgut epithelium, we used a Na+-sensitive fluorescent indicator, Sodium Green, to observe the amount of Na+ in the midgut epithelium. Sodium Green is frequently used to provide spatial and temporal resolution of Na+ concentrations with sufficient selectivity in the presence of physiological concentrations of other monovalent cations (Amorino and Fox, 1995; Minta and Tsien, 1989). Overall, miR-263a mutants displayed higher amount of intracellular Na+ along the entire length of the midgut as indicated by the increased fluorescence, with three very distinct regions where the increase was most significant (Figure S4A–S4B). Since many of the miR-263a mutant phenotypes we observed are manifested in the posterior region of the midgut, we focused our analyses to this specific region. The posterior midguts of miR-263a mutants had approximately 4–5-fold higher amount of intracellular Na+ compared to control (Figure 4A–4B′ and Figure S4C–S4C′ & S4H), suggesting that they are allowing more Na+ across the epithelial membrane. Although disruption of the cell membrane in ECs undergoing delamination and anoikis may allow uptake of more Sodium Green and increase the fluorescent signal in the miR-263a mutants, only a small percentage of the ECs undergo delamination and anoikis, therefore, we do not believe that dying cells are the major contributors of increased Sodium Green fluorescence. Overexpression of miR-263a or depletion of Nach and other ENaC subunits in the miR-263a mutant background completely suppressed the phenotype (Figure 4C–4D′ and Figure S4D–S4G′ & S4H). Interestingly, while overexpression of Nach alone failed to increase the number of pH3+ cells, we observed an increase in the amount of Na+ uptake in the midgut epithelium (Figure 4E–4E′). Quantification of Sodium Green fluorescence revealed approximately 2-fold increase in the intracellular Na+ levels, which is modest compared to the intracellular Na+ of the miR-263a mutants (Figure S4H). This result suggests that overexpression of Nach may partially increase ENaC activity, but not enough to stimulate ISC overproliferation. Collectively, these results suggest that increased intracellular Na+ in the miR-263a mutant midgut epithelium is due to upregulation of ENaC activity as a result of misregulation of multiple ENaC subunits.
Figure 4. Increased Na+ in the miR-263a mutant posterior midgut epithelium.
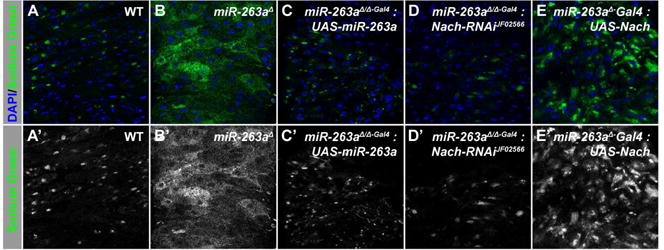
(A-A′) Na+ in the midgut epithelium of control midgut. (B-B′) Enhancement of Na+ in the miR-263a mutant midgut epithelium. (C–D′) Suppression of increased Na+ uptake by overexpressing miR-263a (C-C′) or depleting Nach (D-D′) in the mutant background. (E-E′) Overexpression of Nach partially increased the amount Na+ in the midgut epithelium. See also Figure S4.
Reduced PM Thickness and EC Swelling Phenotypes in the miR-263a Mutant Midgut Epithelium
As an increase in ENaC activity can increase sodium and water reabsorption, ultimately leading to dehydration of the intraluminal surface and reduction in mucus transport (Bhalla and Hallows, 2008), we examined the midgut lumen and the features of the peritrophic matrix (PM), which plays a role analogous to that of mucous secretions in the vertebrate digestive tract, for signs of dehydration. Electron microscopy of posterior midgut sections revealed that the thickness of the PM in miR-263a mutants was significantly reduced (Figure 5A–5B and 5E) and the phenotype was completely suppressed by either depleting Nach or overexpressing miR-263a in the mutant background (Figure 5C–5E). Interestingly, either overexpression of miR-263a or depletion of Nach in the mutant background resulted in thicker PM as compared to the control (Figure 5E). Thus, reducing Nach levels by both miR-263a overexpression and Nach RNAi may further reduce sodium and water reabsorption, possibly hyperhydrating the PM and causing it to be thicker.
Figure 5. Increased ENaC activity disrupts intestinal homeostasis in miR-263a mutants.
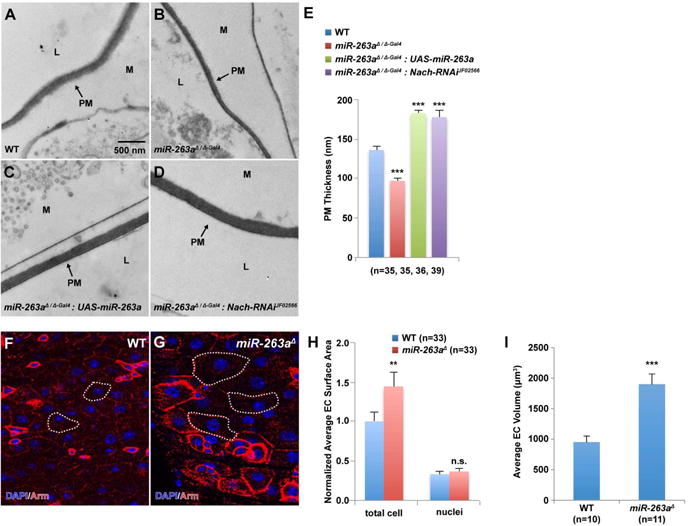
(A–D) EM cross-sections of posterior midguts. (B) miR-263a mutants show reduced PM thickness compared to wild-type (A). Overexpression of miR-263a (C) and depletion of Nach (D) in the mutant background. Arrows indicate the PM (peritrophic matrix), M (mucus), and L (lumen with digested food). (E) Quantitative measurements of the PM thickness. “n” denotes the number of PM thickness measurements for each genotype. (F–G) Enlarged ECs in the miR-263a mutants visualized by anti-Arm marking the cell membrane. White dotted lines outline the representative ECs. (H) Normalized quantitative measurements of the total EC and their respective nuclei surface area. “n” denotes the number of surface area measurements for each genotype. (I) Quantitative measurements of the total EC volume. “n” denotes the number of cell-volume measurements for each genotype. Error bars indicate SEM. **P < 0.05 and ***P < 0.001 (two-tailed t-test). See also Figure S5.
To investigate whether reduction in PM production was responsible for the reduced PM thickness in miR-263a mutants, we measured the transcript levels of drosocrystallin (dcy), an integral component of the PM (Kuraishi et al., 2011). Strikingly, dcy levels were approximately 16.5-fold higher in the miR-263a mutant midguts compared to the control (Figure S5A). In CF lung disease, excess mucus production and increased transcript levels of MUC5AC and MUC5B, major secreted gel-forming mucins in human airways (Davies et al., 1999; Hovenberg et al., 1996), are common (Hauber et al., 2004; Henderson et al., 2014), which results in mucus with concentrated mucins that collapses onto the underlying epithelium as a first step toward chronic infection and organ disease (Boucher, 2007; Chen et al., 2010; Matsui et al., 1998). Similarly, we found that transcripts of Hemolectin (Hml), a close ortholog of both the human MUC5AC and MUC5B (Hu et al., 2011), are significantly higher in the miR-263a mutant midguts (Figure S5B). Collectively, these results suggest that, similar to the CF airway, misregulation of ENaC in the miR-263a mutant midguts results in increased PM production, and that reduced PM thickness is not due to reduced PM production.
In C. elegans, gain-of-function mutations in the orthologs of ENaC (mec-4, mec-10, and deg-1) have been demonstrated to cause vacuolation, cell swelling and eventual cell lysis (Chalfie and Sulston, 1981; Chalfie and Wolinsky, 1990; Driscoll and Chalfie, 1991; Huang and Chalfie, 1994). Interestingly, we also frequently observed larger ECs in miR-263a mutant midguts suggesting that they may also be swollen. Staining of the midgut cell membrane, using antibodies against Armadillo (Arm), revealed that miR-263a mutant ECs have larger surface area compared to wild-type ECs (Figure 5F–5H). In addition, 3D reconstruction of 2D serial sections of ECs revealed that miR-263a mutant ECs have nearly twice the cell-volume compared to control ECs (Figure 5I). Next, although we saw a clear increase in the dcy and Hml transcript levels in miR-263a mutant midguts, we further investigated whether genetically disrupting the PM production/thickness was also sufficient to cause EC swelling and hyperproliferation of ISCs. Using a previously characterized loss-of-function allele of dcy (dcy1) (Kuraishi et al., 2011), we found that dcy mutant ECs were comparable in size to the control ECs, and that dcy mutants did not disrupt ISC homeostasis (data not shown). Collectively, these results suggest that EC swelling is not a direct consequence of reduce PM production/thickness, but rather, that increased expressions of ENaC subunits in the miR-263a mutant midgut epithelium leads to swelling of ECs through increased sodium and water reabsorption.
Increased Bacterial Load in miR-263a
Because structurally compromising the PM is also associated with increased susceptibility to bacterial infections (Kuraishi et al., 2011), we asked whether miR-263a mutants are more susceptible to bacterial infection. Since studies have shown that the Imd pathway regulates antimicrobial peptide (AMP) production in the gut and plays an important function in the resistance against bacterial infections (Liehl et al., 2006; Nehme et al., 2007; Ryu et al., 2006), we examined the activity of the Imd pathway by measuring the expression of the AMP genes Attacin A (AttA) and Diptericin (Dpt), and the NF-kB-like transcription factor Relish (Rel) in mutant midguts. qPCR analyses revealed that miR-263a mutants showed a stronger induction of all three genes (Figure 6A). Next, to directly visualize AMP expression in the midgut, we used a Drosomycin (Drs) reporter line driving the expression of GFP (Drs-GFP). As expected, miR-263a mutants expressed a higher level of Drs-GFP as compared to control (Figure 6B–C′).
Figure 6. Increased activation of the Imd pathway and susceptibility to bacterial infection in miR-263a mutants.
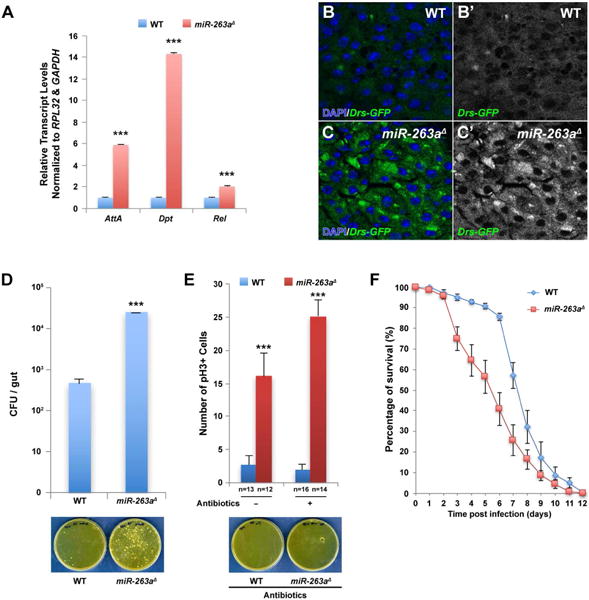
(A) Significant increase in the relative transcript levels of the AMPs was detected in miR-263a mutant midguts by qPCR. (B–C′) Expression of Drs-GFP in the posterior midgut. The expression of Drs-GFP is significantly lower in wild-type (B-B′) than in miR-263a mutants (C-C′). (D) Internal bacterial load significantly increased in the miR-263a mutants. Error bars indicate SEM. ***P < 0.001 (two-tailed t-test). Wild-type and miR-263a mutant flies homogenates were spread on the plate. The bacteria colonies were shown on the bottom panel. (E) The average number of pH3+ cells in the posterior midguts after antibiotics treatment for 16 days. Feeding antibiotics had no effect on the hyperproliferation of ISCs observed in the miR-263a mutant midguts. Bottom panel shows the bacteria colonies on medium plate spread with antibiotics fed wild-type and miR-263a mutant homogenate. Internal bacteria were absent in both wild-type and miR-263a mutant flies fed for 16 days with food containing antibiotics. “n” denotes the number of posterior midguts examined for each genotype. Error bars indicate SEM. ***P < 0.001 (two-tailed t-test). (F) Survival analysis of wild-type and miR-263a mutant flies upon oral infection with P. aeruginosa. The miR-263a mutant flies exhibited significant increase in lethality 3 days after infection. Error bars indicate SEM. See also Figure S6.
To determine whether increased activation of the Imd pathway is a direct result of increased bacterial accumulation in the miR-263a mutant midgut, we quantified internal bacterial loads by measuring colony forming units (CFUs). Strikingly, the internal bacterial load in miR-263a mutants was significantly higher compared to the controls (Figure 6D). The observed increase in bacterial load in miR-263a mutants prompted us to investigate whether an inflammatory response to bacteria accumulation was causing ISC overproliferation. To test this hypothesis, we cultured control and miR-263a mutant flies in food containing antibiotics, which eliminated all internal bacteria (Figure 6E). Treatment with antibiotics did not suppress ISC overproliferation in the miR-263a mutants (Figure 6E), indicating that bacterial accumulation is not the primary cause of ISC proliferation.
Next, we investigated whether miR-263a mutants are more susceptible to bacterial infection using Pseudomonas aeruginosa (P. aeruginosa), a major pathogen in the cystic fibrosis lung (Ohman and Chakrabarty, 1982; Palmer et al., 2005). Wild-type flies orally infected with P. aeruginosa causes both acute and chronic infection and ultimately succumb to the infection (Mulcahy et al., 2011). After 72 hours of oral infection, miR-263a mutants exhibited a higher susceptibility than wild-type (Figure 6F), indicating that structurally compromised PM leads to increased bacterial infection. Together, our results show that structurally compromised PM in the miR-263a mutants allow for more bacterial infection and have elevated levels of the AMPs due to activation of the Imd pathway. This increased susceptibility to bacterial infection is likely an indirect result from structurally compromised PM, and is not the cause of ISC overproliferation.
miR-263a Mutants Disrupt Intestinal pH Homeostasis
In Drosophila adults, the anterior midgut region is neutral in pH, and is followed by an acidic zone (pH<4) called the copper-cell region. The posterior midgut is mildly alkaline (pH=7–9) and is followed by the hindgut, which is slightly acidic (pH=5) (Dubreuil, 2004; Shanbhag and Tripathi, 2009) (Figure S6A). To assess how structurally compromising the PM influences intestinal physiology, we tested whether intestinal pH homeostasis was perturbed in the miR-263a mutants. The intestinal pH was monitored colorimetrically by feeding animals phenol red as previously described (Shanbhag and Tripathi, 2009). In control midguts, only the posterior midgut is dark red, reflecting its high pH (Figure S6A). However, in the miR-263a mutant, almost the entire length of the midgut is alkaline, as revealed by expansion of the dark red zone (Figure S6B). Alkalization of the mutant midgut is suppressed by overexpressing miR-263a or by knocking down Nach (Figure S6C–S6D). These results indicate that the absence of miR-263a and subsequent increase in Nach activity disrupts intestinal pH homeostasis, which may provide a more favorable environment for bacterial survival.
Discussion
In this study, we describe a mechanism by which a miRNA regulates ENaC to maintain ISC and osmotic homeostasis in the Drosophila midgut epithelium. Specifically, we identified an evolutionarily conserved miRNA, miR-263a, that directly and indirectly regulates the expression of ENaC subunits (Figure 7). Strikingly, the phenotypes of miR-263a mutants are reminiscent of the pathophysiology of CF.
Figure 7. Model of regulation of ENaC by miR-263a.
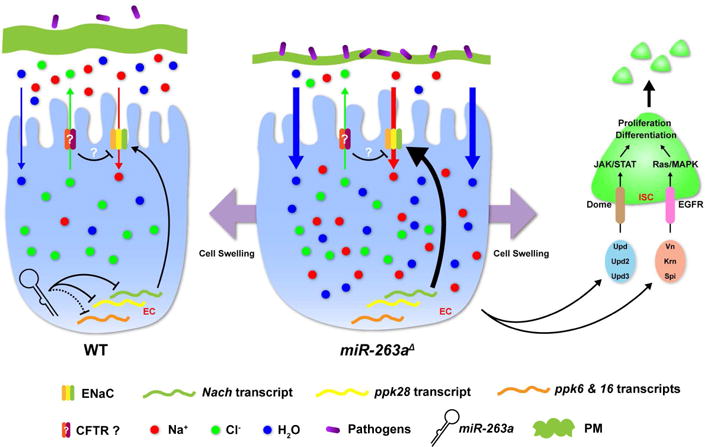
A model of miR-263a maintaining ISC and osmotic homeostasis in the midgut epithelium by directly and indirectly regulating the expression of ENaC subunits. See also Figure S7.
In CF, two different models have been proposed regarding the role of hydration and salt concentration in normal airway defense (Guggino, 2001). The hydration model proposes that increased absorption of fluid by the epithelium leads to dehydrated mucus and impaired mucociliary clearance that contributes to the establishment of an environment that promotes colonization of the lungs by bacteria. In contrast, the salt model proposes that the salt content of airway fluid in CF is too high and thus prevents salt-sensitive defensin molecules in the airway surface liquid (ASL) from killing bacteria, leading to increased susceptibility to lung infections. In Drosophila, the phenotypes associated with perturbation of ENaC are consistent with the hydration model, as misregulation of ENaC subunits in miR-263a mutants result in increased sodium reabsorption across the midgut epithelium. Further, we observed a dehydration-like phenotype of the PM, which is analogous to mucous secretions in the vertebrate digestive tract. Consistent with the PM providing protection against abrasive food particles and pathogens, miR-263a mutants appear more susceptible to bacterial infections as they succumb to P. aeruginosa infection faster than the controls. In addition, we observed increased bacterial load and AMP levels, and disruption of the intestinal pH in miR-263a mutants. Interestingly, ECs in miR-263a mutants appear swollen, which is likely due to increased water reabsorption through osmosis. Finally, we observed an activation of stressed pathways characteristic of damaged ECs, which correlates with increased proliferation of ISCs.
Consistent with previous reports that cell swelling can activate the JNK pathway (Berl et al., 1997; Huangfu et al., 2006; Sinning et al., 1997), JNK signaling is activated in miR-263a mutants that have large ECs. In addition, the JAK/STAT and EGFR pathways that regulate ISC proliferation (Biteau and Jasper, 2011; Buchon et al., 2010; Jiang and Edgar, 2011; Jiang et al., 2009; Osman et al., 2012; Zhou et al., 2013) are hyperactivated. Similarly, in CF airway and small intestine epithelia, cells in the airway epithelium and submucosal glands are more proliferative than cells in non-CF airways (Leigh et al., 1995). In addition, all CF mouse models in which CFTR has been deleted, goblet cell hyperplasia was observed in the small intestine (Gallagher and Gottlieb, 2001).
Although the existence of Drosophila CFTR is yet to be determined, given its phenotypic similarities to the pathophysiology of CF, miR-263a mutants may provide a cost-effective and high-throughput animal model for identifying potential therapeutic that can specifically target ENaC in vivo, as the Drosophila gut is amenable to large-scale small molecule screens (Markstein et al., 2014). In addition, miR-183 might itself be a potential therapeutic agent for regulating ENaC activity in CF based on our data that overexpression of miR-183 can directly target the expression of all 3 ENaC subunits in CFBE41o- cells (Figure S7). Thus, possibly, a combinational therapy for CF using the CFTR potentiator, Ivacaftor (aka Kalydeco, (McPhail and Clancy, 2013), which improves the transport of chloride through the mutated CFTR, together with overexpression of miR-183, could be imagined.
Materials and Methods
Drosophila stocks and genetics
The following Drosophila stocks were used in this study: miR-263aΔ, miR-263aΔ-Gal4, and miR-263bΔ (Hilgers et al., 2010); bft24 (Hardiman et al., 2002); UAS-miR-263a (Bejarano et al., 2012); puc-LacZ (pucE69) (Martin-Blanco et al., 1998); 10XSTAT-GFP (Bach et al., 2007); Drs-GFP; esg-LacZ (esgk00606); Dl-LacZ (Dl05151) (Bloomington Stock 11651); Myo1A-Gal4 (Jiang et al., 2009); hsFlp, Tub-Gal4, UAS-GFP/FM7; Tub-Gal80, FRT40A/CyO (Karpowicz et al., 2010); Nach RNAi (JF02566) (Bloomington Stock 27262); Nach RNAi (VDRC45921) (Vienna Drosophila Resource Center Stock 45921); ppk6 RNAi (JF01919) (Bloomington Stock 25880); ppk16 RNAi (JF01931) (Bloomington Stock 25890); and ppk28 RNAi (JF02153) (Bloomington Stock 31878). A FRT site (FRT40A) was introduced by recombination onto the chromosome arm carrying the miR-263aΔ. All stocks were maintained and crossed at 25°C. For MARCM clones, 2–3 days old adult flies were heat shocked for 1 h at 37°C to induce clones and kept a t 25°C for 14–21 days until dissection. The genotypes used were: (1) hsFlp, Tub-Gal4, UAS-GFP/FM7; Tub-Gal80, FRT40A/CyO; Dl-LacZ, (2) hsFlp, Tub-Gal4, UAS-GFP/FM7; Tub-Gal80, FRT40A, miR-263aΔ /CyO; Dl-LacZ, (3) hsFlp, Tub-Gal4, UAS-GFP/FM7; Tub-Gal80, FRT40A/CyO; puc-LacZ, and (4) hsFlp, Tub-Gal4, UAS-GFP/FM7; Tub-Gal80, FRT40A, miR-263aΔ/CyO; puc-LacZ.
Generation of UAS-Nach transgenic fly
To construct UAS-Nach plasmid, Nach genomic fragment was amplified using specific primers (F, 5′-CGGAATTCATGGGCCACAGGAGGAGCTGAAGC-3′; R, 5′-GCTCTAGATTACGATTTGTGAATTATGAAAACTGAT-3′). Amplified fragment was digested with EcoRI and XbaI, and was ligated into the linearized pWALIUM10-moe vector. Transgenic fly was established by injecting UAS-Nach plasmid into flies carrying attP docking site, attP2, located on the third chromosome.
Immunostaining of the midgut
Prior to dissection, flies were fed on 5% sucrose for ~3 hr to remove food from the midgut. Female guts were dissected in 1xPBS and fixed in 4% paraformaldehyde diluted with 1xPBS for 30 minutes. Samples were washed with 1xPBS, blocked for 30 minutes in 1xPBS, 5% Donkey Serum and 0.1% Triton X-100. Samples were incubated overnight at 4°C using the following antibodies: mouse anti-Armadillo (1:50), mouse anti-Delta (1:50), mouse anti-Prospero (1:100) (Developmental Studies Hybridoma Bank), rabbit anti-β-galactosidase (1:1,000; Cappel), mouse anti-β-galactosidase (1:1,000; Promega), rabbit anti-phospho-Histone H3 (1:10,000; Millipore), and rabbit anti-P-p44/42 MAPK (dpERK) (1:200; Cell Signaling). Primary antibodies were detected using anti-mouse or anti-rabbit secondary antibodies conjugated to Alexa-Fluor 488 and 594 (1:1000; Invitrogen). Alexa-Fluor 488-conjugated phalloidin (Molecular Probe, 1:100) was used to stain F-actin. Fluorescent images were acquired with a Leica TCS SP2 AOBS.
Quantification of EC size
miR-263a mutant clones were generated using the MARCM approach as described above and immunostained using anti-Armadillo to label all cell membranes. The surface area of the miR-263a mutant ECs (GFP+) and their respective nuclei were measured using ImageJ (NIH). The surface area of the wild-type ECs (GFP−), right next to the mutant ECs (GFP+), was used as control. To analyze the cell-volume, serial sections (~19–32 sections per individual EC) of individual ECs in the MARCM clones were taken using confocal. Total cell-volume was calculated as follows: cell-volume (μm3) = total cell surface area (μm2) * thickness (μm) / sections * number of sections
miR-183 overexpression in CFBE41o- cells
Immortalized CFBE41o- cells were obtained from Dr. D. Gruenert (California Pacific Medical Center Research Institute, San Francisco, CA). CFBE41o- cells were grown in MEM (Minimum Essential Medium w/Earle’s salt; Life Technologies) supplemented with 10% FBS (Life Technologies), 2 mM L-Glutamine (Life Technologies), and 1% Penicillin/Streptomycin (Life Technologies) in 75-cm2 flasks and maintained in a 37°C humidified incubat or containing 5% CO2. Cells were routinely grown on flasks coated with an extracellular matrix containing fibronectin/vitrogen/BSA. miR-183 overexpression experiment was performed in 6-well plate. The day before the transfection, each well was seeded with 300,000 cells. Next day, either 1.0 μg of pcDNA3.1-miR-183 or empty pcDNA3.1 (control) plasmid was transfected using Effectene Transfection Reagent (Qiagen). After 72 hr, total RNA was extracted using TRIzol Reagent (Invitrogen). The pcDNA3.1-miR-183 plasmid was constructed by amplifying 399 nt fragment from HEK293 genomic DNA using specific primers (F, 5′-CTAGCTAGCAAGGTCATCTTGGGCTGATG-3′; R, 5′-CCGCTCGAGTCTCTGGGGACACACTGGAC-3′). Amplified fragment was digested with NheI and XhoI, and was ligated into the linearized pcDNA3.1 vector.
Nach antibody
Nach antibody was generated in rabbit against a peptide containing amino acids 516–535 (CPKANDTHSKEQKSVFIIHKS) and affinity-purified at YenZym Antibodies.
Western blots
Lysates prepared from dissected adult female midguts were separated by SDS-PAGE, blotted onto nitrocellulose membrane, and subjected to Western analysis using antibodies against Nach (1:500) and α-Tubulin (1:1000; Sigma). Blots were subsequently incubated with HRP-conjugated goat secondary antibody (Amersham), and processed for chemiluminescence (Pierce). For quantification of band intensity, the raw images were analyzed using ImageJ (NIH).
Sodium Green assay
Adult female flies, 14–16 days old, were fed 2 μM of cell-permeant Sodium Green tetraacetate Indicator (Invitrogen) diluted in 5% sucrose for 3–4 hr and dissected and fixed in 4% paraformaldehyde diluted with 1xPBS for 30 minutes. After a brief wash with 1xPBS, samples were mounted and images were taken immediately using a Leica TCS SP2 AOBS. For quantification of Sodium Green fluorescence, the raw images were analyzed using ImageJ (NIH).
Electron Microscopy
Dissected midguts were fixed in 0.1 M sodium cacodylate buffer (pH 7.4) containing 2.5% glutaraldehyde, 2% paraformaldehyde for overnight. The fixed samples were washed 3 times in distilled water, fixed again with 1% osmium tetroxide (OsO4) and 1.5% potassium ferrocyanide (KFeCN6) for 1 hr, and washed 3 times in distilled water. Then, the samples were washed in 1% maleate buffer, incubated in 1% aqueous uranyl acetate in 1% maleate buffer for 1 hr followed by 2 washes in 1% maleate buffer and subsequent dehydration in grades of alcohol. The samples were put in propylene oxide for 1 hr and embedded with a solution of 50% propylene oxide and 50% TAAB Epon overnight. Next day, samples were embedded in fresh TAAB Epon and polymerized for 2 days at 60°C. Ultrathin sections (about 60 nm) were cut on a Reichert Ultracut-S microtome, picked up on to copper grids, and then stained with lead citrate. The sections were examined in a JEOL 1200EX Transmission electron microscope, and images were recorded with an AMT 2k CCD camera.
Internal Bacterial Load
Age-matched cohorts of 10 control and 10 miR-263a mutant flies were co-housed in the same vial and were transferred to a new vial every other days. Three vials were used for each age. To culture the internal commensal bacteria, 4 flies were washed with 70% ethanol for 2 minutes and rinsed with sterile water twice to remove the external bacteria. Four flies were homogenized in 400 μl of MRS medium and serial dilutions were plated on the MRS plates. Plates were incubated at 30°C for 48–72 hr before counting colony-forming units.
Bacterial infection assays
P. aeruginosa PA14 was grown on LB medium overnight. Following morning, 200 μl of overnight culture was added to 10 ml of LB and cultured for another 6–8 hours to reach OD600 = 1.5. Bacteria/sucrose feeding solution was prepared by mixing 1 ml of the bacteria solution to 4 ml of sucrose solution to reach 4% sucrose final concentration. Bacterial infection assay was performed by placing twenty 1–3 days old female flies into individual vials containing Kimwipes containing bacteria/sucrose feeding solution at 25°C. Seven vials of each geno type were used. The number of dead flies were recorded every 24 hours. Flies were transferred to new vials with freshly prepared bacteria/sucrose solution after 3 days.
Feeding Assays
DSS and bleomycin feeding assays were performed as previously described (Ren et al., 2010). Briefly, 11–13 days old female flies were either fed 5% DSS or 25 μg/ml of bleomycin (Sigma) dissolved in 5% sucrose solution for 3 days at 25°C. For antibiotics feeding assays, standard laboratory fly food containing 30 μg/ml Kanamycin, 100 μg/ml Ampicillin, and 34 μg/ml Chloramphenicol was fed to newly eclosed female flies. Flies were transferred to fresh antibiotics food every 1–2 days. After 16 days, midguts were dissected and examined.
Gut pH Assay
The gut pH was assessed by feeding 14–16 days old female flies 0.2% phenol red dissolved in 5% sucrose solution for 3 hours at 25°C. The colors of the guts were photographed immediately after dissection with Zeiss Axioskop 2 compound microscope.
Supplementary Material
Highlights.
miR-263a regulates the expression of ENaC to maintain osmotic and ISC homeostasis
Loss of miR-263a leads to cystic fibrosis-like phenotypes in the Drosophila gut
Overexpression of human miR-183 can target the expression of human ENaC subunits
Acknowledgments
We thank Drs. S. Cohen, P. Watnick, and Rolf Bodmer for flies. We thank Dr. D. Gruenert for providing the CFBE41o- cells. We thank the Transgenic RNAi Resource Project, Vienna Drosophila Resource Center, and the Bloomington Drosophila Stock Center for flies, the Developmental Studies Hybridoma Bank for monoclonal antibodies, and the Drosophila RNAi Screening Center (Harvard Medical School) for plate-reader equipment. We thank D. Doupé for sharing unpublished results. We also thank M.-L. Samson, L. Rabinow, S. Mohr, R. Viswanatha, L. Perkins, R. Boucher, and members of Perrimon lab for discussion and critical comments on the manuscript. This work was supported in part by the STARR foundation and by the National Institute of Health (N.P.). R-J.H. is supported by the Jane Coffin Childs foundation. N.P. is a Howard Hughes Medical Institute investigator.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
K.K. and R-J.H. conceived, designed, and performed the experiments; K.K., R-J.H., and N.P. analyzed the data and wrote the paper.
References
- Amorino GP, Fox MH. Intracellular Na+ measurements using sodium green tetraacetate with flow cytometry. Cytometry. 1995;21:248–256. doi: 10.1002/cyto.990210305. [DOI] [PubMed] [Google Scholar]
- Bach EA, Ekas LA, Ayala-Camargo A, Flaherty MS, Lee H, Perrimon N, Baeg GH. GFP reporters detect the activation of the Drosophila JAK/STAT pathway in vivo. Gene Expr Patterns. 2007;7:323–331. doi: 10.1016/j.modgep.2006.08.003. [DOI] [PubMed] [Google Scholar]
- Bejarano F, Bortolamiol-Becet D, Dai Q, Sun K, Saj A, Chou YT, Raleigh DR, Kim K, Ni JQ, Duan H, et al. A genome-wide transgenic resource for conditional expression of Drosophila microRNAs. Development. 2012;139:2821–2831. doi: 10.1242/dev.079939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berdiev BK, Qadri YJ, Benos DJ. Assessment of the CFTR and ENaC association. Mol Biosyst. 2009;5:123–127. doi: 10.1039/b810471a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berl T, Siriwardana G, Ao L, Butterfield LM, Heasley LE. Multiple mitogen-activated protein kinases are regulated by hyperosmolality in mouse IMCD cells. Am J Physiol. 1997;272:F305–311. doi: 10.1152/ajprenal.1997.272.3.F305. [DOI] [PubMed] [Google Scholar]
- Bhalla V, Hallows KR. Mechanisms of ENaC regulation and clinical implications. J Am Soc Nephrol. 2008;19:1845–1854. doi: 10.1681/ASN.2008020225. [DOI] [PubMed] [Google Scholar]
- Bianchi L, Driscoll M. Protons at the gate: DEG/ENaC ion channels help us feel and remember. Neuron. 2002;34:337–340. doi: 10.1016/s0896-6273(02)00687-6. [DOI] [PubMed] [Google Scholar]
- Biteau B, Jasper H. EGF signaling regulates the proliferation of intestinal stem cells in Drosophila. Development. 2011;138:1045–1055. doi: 10.1242/dev.056671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucher RC. Cystic fibrosis: a disease of vulnerability to airway surface dehydration. Trends Mol Med. 2007;13:231–240. doi: 10.1016/j.molmed.2007.05.001. [DOI] [PubMed] [Google Scholar]
- Brennecke J, Stark A, Russell RB, Cohen SM. Principles of microRNA-target recognition. PLoS Biol. 2005;3:e85. doi: 10.1371/journal.pbio.0030085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchon N, Broderick NA, Kuraishi T, Lemaitre B. Drosophila EGFR pathway coordinates stem cell proliferation and gut remodeling following infection. BMC biology. 2010;8:152. doi: 10.1186/1741-7007-8-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canessa CM, Schild L, Buell G, Thorens B, Gautschi I, Horisberger JD, Rossier BC. Amiloride-sensitive epithelial Na+ channel is made of three homologous subunits. Nature. 1994;367:463–467. doi: 10.1038/367463a0. [DOI] [PubMed] [Google Scholar]
- Chalfie M, Sulston J. Developmental genetics of the mechanosensory neurons of Caenorhabditis elegans. Dev Biol. 1981;82:358–370. doi: 10.1016/0012-1606(81)90459-0. [DOI] [PubMed] [Google Scholar]
- Chalfie M, Wolinsky E. The identification and suppression of inherited neurodegeneration in Caenorhabditis elegans. Nature. 1990;345:410–416. doi: 10.1038/345410a0. [DOI] [PubMed] [Google Scholar]
- Chen EY, Yang N, Quinton PM, Chin WC. A new role for bicarbonate in mucus formation. Am J Physiol Lung Cell Mol Physiol. 2010;299:L542–549. doi: 10.1152/ajplung.00180.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies JR, Svitacheva N, Lannefors L, Kornfalt R, Carlstedt I. Identification of MUC5B, MUC5AC and small amounts of MUC2 mucins in cystic fibrosis airway secretions. Biochem J. 1999;344(Pt 2):321–330. [PMC free article] [PubMed] [Google Scholar]
- Driscoll M, Chalfie M. The mec-4 gene is a member of a family of Caenorhabditis elegans genes that can mutate to induce neuronal degeneration. Nature. 1991;349:588–593. doi: 10.1038/349588a0. [DOI] [PubMed] [Google Scholar]
- Dubreuil RR. Copper cells and stomach acid secretion in the Drosophila midgut. Int J Biochem Cell Biol. 2004;36:745–752. doi: 10.1016/j.biocel.2003.07.004. [DOI] [PubMed] [Google Scholar]
- Gabay L, Seger R, Shilo BZ. MAP kinase in situ activation atlas during Drosophila embryogenesis. Development. 1997;124:3535–3541. doi: 10.1242/dev.124.18.3535. [DOI] [PubMed] [Google Scholar]
- Gallagher AM, Gottlieb RA. Proliferation, not apoptosis, alters epithelial cell migration in small intestine of CFTR null mice. Am J Physiol Gastrointest Liver Physiol. 2001;281:G681–687. doi: 10.1152/ajpgi.2001.281.3.G681. [DOI] [PubMed] [Google Scholar]
- Gangaraju VK, Lin H. MicroRNAs: key regulators of stem cells. Nature reviews Molecular cell biology. 2009;10:116–125. doi: 10.1038/nrm2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garty H, Palmer LG. Epithelial sodium channels: function, structure, and regulation. Physiol Rev. 1997;77:359–396. doi: 10.1152/physrev.1997.77.2.359. [DOI] [PubMed] [Google Scholar]
- Grubb BR, Gabriel SE. Intestinal physiology and pathology in gene-targeted mouse models of cystic fibrosis. Am J Physiol. 1997;273:G258–266. doi: 10.1152/ajpgi.1997.273.2.G258. [DOI] [PubMed] [Google Scholar]
- Guggino WB. Cystic fibrosis salt/fluid controversy: in the thick of it. Nat Med. 2001;7:888–889. doi: 10.1038/90914. [DOI] [PubMed] [Google Scholar]
- Hardiman KE, Brewster R, Khan SM, Deo M, Bodmer R. The bereft gene, a potential target of the neural selector gene cut, contributes to bristle morphogenesis. Genetics. 2002;161:231–247. doi: 10.1093/genetics/161.1.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauber HP, Tsicopoulos A, Wallaert B, Griffin S, McElvaney NG, Daigneault P, Mueller Z, Olivenstein R, Holroyd KJ, Levitt RC, et al. Expression of HCLCA1 in cystic fibrosis lungs is associated with mucus overproduction. Eur Respir J. 2004;23:846–850. doi: 10.1183/09031936.04.00096504. [DOI] [PubMed] [Google Scholar]
- Henderson AG, Ehre C, Button B, Abdullah LH, Cai LH, Leigh MW, DeMaria GC, Matsui H, Donaldson SH, Davis CW, et al. Cystic fibrosis airway secretions exhibit mucin hyperconcentration and increased osmotic pressure. J Clin Invest. 2014;124:3047–3060. doi: 10.1172/JCI73469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilgers V, Bushati N, Cohen SM. Drosophila microRNAs 263a/b confer robustness during development by protecting nascent sense organs from apoptosis. PLoS Biol. 2010;8:e1000396. doi: 10.1371/journal.pbio.1000396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hovenberg HW, Davies JR, Carlstedt I. Different mucins are produced by the surface epithelium and the submucosa in human trachea: identification of MUC5AC as a major mucin from the goblet cells. Biochem J. 1996;318(Pt 1):319–324. doi: 10.1042/bj3180319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Flockhart I, Vinayagam A, Bergwitz C, Berger B, Perrimon N, Mohr SE. An integrative approach to ortholog prediction for disease-focused and other functional studies. BMC Bioinformatics. 2011;12:357. doi: 10.1186/1471-2105-12-357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang M, Chalfie M. Gene interactions affecting mechanosensory transduction in Caenorhabditis elegans. Nature. 1994;367:467–470. doi: 10.1038/367467a0. [DOI] [PubMed] [Google Scholar]
- Huangfu WC, Omori E, Akira S, Matsumoto K, Ninomiya-Tsuji J. Osmotic stress activates the TAK1-JNK pathway while blocking TAK1-mediated NF-kappaB activation: TAO2 regulates TAK1 pathways. J Biol Chem. 2006;281:28802–28810. doi: 10.1074/jbc.M603627200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Edgar BA. Intestinal stem cells in the adult Drosophila midgut. Experimental cell research. 2011;317:2780–2788. doi: 10.1016/j.yexcr.2011.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Patel PH, Kohlmaier A, Grenley MO, McEwen DG, Edgar BA. Cytokine/Jak/Stat signaling mediates regeneration and homeostasis in the Drosophila midgut. Cell. 2009;137:1343–1355. doi: 10.1016/j.cell.2009.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpowicz P, Perez J, Perrimon N. The Hippo tumor suppressor pathway regulates intestinal stem cell regeneration. Development. 2010;137:4135–4145. doi: 10.1242/dev.060483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuraishi T, Binggeli O, Opota O, Buchon N, Lemaitre B. Genetic evidence for a protective role of the peritrophic matrix against intestinal bacterial infection in Drosophila melanogaster. Proc Natl Acad Sci U S A. 2011;108:15966–15971. doi: 10.1073/pnas.1105994108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee T, Luo L. Mosaic analysis with a repressible cell marker (MARCM) for Drosophila neural development. Trends Neurosci. 2001;24:251–254. doi: 10.1016/s0166-2236(00)01791-4. [DOI] [PubMed] [Google Scholar]
- Leigh MW, Kylander JE, Yankaskas JR, Boucher RC. Cell proliferation in bronchial epithelium and submucosal glands of cystic fibrosis patients. Am J Respir Cell Mol Biol. 1995;12:605–612. doi: 10.1165/ajrcmb.12.6.7766425. [DOI] [PubMed] [Google Scholar]
- Liehl P, Blight M, Vodovar N, Boccard F, Lemaitre B. Prevalence of local immune response against oral infection in a Drosophila/Pseudomonas infection model. PLoS Pathog. 2006;2:e56. doi: 10.1371/journal.ppat.0020056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingueglia E, Voilley N, Waldmann R, Lazdunski M, Barbry P. Expression cloning of an epithelial amiloride-sensitive Na+ channel. A new channel type with homologies to Caenorhabditis elegans degenerins. FEBS Lett. 1993;318:95–99. doi: 10.1016/0014-5793(93)81336-x. [DOI] [PubMed] [Google Scholar]
- Mall M, Grubb BR, Harkema JR, O’Neal WK, Boucher RC. Increased airway epithelial Na+ absorption produces cystic fibrosis-like lung disease in mice. Nat Med. 2004;10:487–493. doi: 10.1038/nm1028. [DOI] [PubMed] [Google Scholar]
- Markstein M, Dettorre S, Cho J, Neumuller RA, Craig-Muller S, Perrimon N. Systematic screen of chemotherapeutics in Drosophila stem cell tumors. Proc Natl Acad Sci U S A. 2014;111:4530–4535. doi: 10.1073/pnas.1401160111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Blanco E, Gampel A, Ring J, Virdee K, Kirov N, Tolkovsky AM, Martinez-Arias A. puckered encodes a phosphatase that mediates a feedback loop regulating JNK activity during dorsal closure in Drosophila. Genes Dev. 1998;12:557–570. doi: 10.1101/gad.12.4.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathieu J, Ruohola-Baker H. Regulation of stem cell populations by microRNAs. Adv Exp Med Biol. 2013;786:329–351. doi: 10.1007/978-94-007-6621-1_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui H, Grubb BR, Tarran R, Randell SH, Gatzy JT, Davis CW, Boucher RC. Evidence for periciliary liquid layer depletion, not abnormal ion composition, in the pathogenesis of cystic fibrosis airways disease. Cell. 1998;95:1005–1015. doi: 10.1016/s0092-8674(00)81724-9. [DOI] [PubMed] [Google Scholar]
- McPhail GL, Clancy JP. Ivacaftor: the first therapy acting on the primary cause of cystic fibrosis. Drugs Today (Barc) 2013;49:253–260. doi: 10.1358/dot.2013.49.4.1940984. [DOI] [PubMed] [Google Scholar]
- Minta A, Tsien RY. Fluorescent indicators for cytosolic sodium. J Biol Chem. 1989;264:19449–19457. [PubMed] [Google Scholar]
- Mulcahy H, Sibley CD, Surette MG, Lewenza S. Drosophila melanogaster as an animal model for the study of Pseudomonas aeruginosa biofilm infections in vivo. PLoS Pathog. 2011;7:e1002299. doi: 10.1371/journal.ppat.1002299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nehme NT, Liegeois S, Kele B, Giammarinaro P, Pradel E, Hoffmann JA, Ewbank JJ, Ferrandon D. A model of bacterial intestinal infections in Drosophila melanogaster. PLoS Pathog. 2007;3:e173. doi: 10.1371/journal.ppat.0030173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlstein B, Spradling A. Multipotent Drosophila intestinal stem cells specify daughter cell fates by differential notch signaling. Science. 2007;315:988–992. doi: 10.1126/science.1136606. [DOI] [PubMed] [Google Scholar]
- Ohman DE, Chakrabarty AM. Utilization of human respiratory secretions by mucoid Pseudomonas aeruginosa of cystic fibrosis origin. Infect Immun. 1982;37:662–669. doi: 10.1128/iai.37.2.662-669.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okumura T, Takeda K, Taniguchi K, Adachi-Yamada T. betanu integrin inhibits chronic and high level activation of JNK to repress senescence phenotypes in Drosophila adult midgut. PLoS One. 2014;9:e89387. doi: 10.1371/journal.pone.0089387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osman D, Buchon N, Chakrabarti S, Huang YT, Su WC, Poidevin M, Tsai YC, Lemaitre B. Autocrine and paracrine unpaired signaling regulate intestinal stem cell maintenance and division. Journal of cell science. 2012;125:5944–5949. doi: 10.1242/jcs.113100. [DOI] [PubMed] [Google Scholar]
- Palmer KL, Mashburn LM, Singh PK, Whiteley M. Cystic fibrosis sputum supports growth and cues key aspects of Pseudomonas aeruginosa physiology. J Bacteriol. 2005;187:5267–5277. doi: 10.1128/JB.187.15.5267-5277.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel PH, Dutta D, Edgar BA. Niche appropriation by Drosophila intestinal stem cell tumours. Nat Cell Biol. 2015;17:1182–1192. doi: 10.1038/ncb3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren F, Wang B, Yue T, Yun EY, Ip YT, Jiang J. Hippo signaling regulates Drosophila intestine stem cell proliferation through multiple pathways. Proc Natl Acad Sci U S A. 2010;107:21064–21069. doi: 10.1073/pnas.1012759107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riordan JR. CFTR function and prospects for therapy. Annu Rev Biochem. 2008;77:701–726. doi: 10.1146/annurev.biochem.75.103004.142532. [DOI] [PubMed] [Google Scholar]
- Ruby JG, Stark A, Johnston WK, Kellis M, Bartel DP, Lai EC. Evolution, biogenesis, expression, and target predictions of a substantially expanded set of Drosophila microRNAs. Genome Res. 2007;17:1850–1864. doi: 10.1101/gr.6597907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu JH, Ha EM, Oh CT, Seol JH, Brey PT, Jin I, Lee DG, Kim J, Lee D, Lee WJ. An essential complementary role of NF-kappaB pathway to microbicidal oxidants in Drosophila gut immunity. EMBO J. 2006;25:3693–3701. doi: 10.1038/sj.emboj.7601233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanbhag S, Tripathi S. Epithelial ultrastructure and cellular mechanisms of acid and base transport in the Drosophila midgut. J Exp Biol. 2009;212:1731–1744. doi: 10.1242/jeb.029306. [DOI] [PubMed] [Google Scholar]
- Sinning R, Schliess F, Kubitz R, Haussinger D. Osmosignalling in C6 glioma cells. FEBS Lett. 1997;400:163–167. doi: 10.1016/s0014-5793(96)01376-2. [DOI] [PubMed] [Google Scholar]
- Southall TD, Gold KS, Egger B, Davidson CM, Caygill EE, Marshall OJ, Brand AH. Cell-type-specific profiling of gene expression and chromatin binding without cell isolation: assaying RNA Pol II occupancy in neural stem cells. Dev Cell. 2013;26:101–112. doi: 10.1016/j.devcel.2013.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldmann R, Champigny G, Bassilana F, Voilley N, Lazdunski M. Molecular cloning and functional expression of a novel amiloride-sensitive Na+ channel. J Biol Chem. 1995;270:27411–27414. doi: 10.1074/jbc.270.46.27411. [DOI] [PubMed] [Google Scholar]
- Yi R, Fuchs E. A miR image of stem cells and their lineages. Current topics in developmental biology. 2012;99:175–199. doi: 10.1016/B978-0-12-387038-4.00007-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelle KM, Lu B, Pyfrom SC, Ben-Shahar Y. The genetic architecture of degenerin/epithelial sodium channels in Drosophila. G3 (Bethesda) 2013;3:441–450. doi: 10.1534/g3.112.005272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou F, Rasmussen A, Lee S, Agaisse H. The UPD3 cytokine couples environmental challenge and intestinal stem cell division through modulation of JAK/STAT signaling in the stem cell microenvironment. Dev Biol. 2013;373:383–393. doi: 10.1016/j.ydbio.2012.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.