

Abstract
Acute inflammatory demyelinating polyneuropathy (AIDP) is an aggressive antibody- and T cell-mediated variant of Guillain-Barré Syndrome (GBS), a prominent and debilitating autoimmune disorder of the peripheral nervous system. Despite advancements in clinical management, treatment of patients with AIDP/GBS and its chronic variant CIDP remains palliative and relies on the use of non-specific immune-modulating therapies. Our laboratory has previously reported that therapeutic administration of statins safely attenuates the clinical severity of experimental autoimmune neuritis (EAN), a well-characterized animal model of AIDP/GBS, by restricting the migration of autoreactive leukocytes across peripheral nerve microvascular endoneurial endothelial cells (PNMECs) that form the blood-nerve barrier. Despite these advancements, the clinical application of systemically-administered statins for the management of inflammatory disorders remains controversial due to disappointingly inconclusive phase trials. Here, poly(lactic-co-glycolic) acid (PLGA) nanoparticles were evaluated as an alternative strategy by which to locally administer statins for the management of EAN. When tested in vitro, lovastatin-encapsulating PLGA nanoparticles elicited a marked increase in RhoB mRNA content in PNMECs, similar to cells treated with activated un-encapsulated lovastatin. Unilateral peri-neural administration of lovastatin-encapsulating PLGA nanoparticles, but not empty nanoparticles, to naïve Lewis rats similarly enhanced RhoB mRNA content in adjacent nerve and muscle tissue. When administered in this manner, serum levels of lovastatin were below the level of detection. Bilateral peri-neural administration of lovastatin-encapsulating PLGA nanoparticles to EAN-induced Lewis rats significantly attenuated EAN clinical severity while protecting against EAN-induced peripheral nerve morphological and functional deficits. This study provides the first proof-of-concept approach for the application of a nanoparticle-based local drug delivery platform for the management of inflammatory demyelinating diseases, including AIDP/GBS.
Keywords: Guillain-Barré Syndrome, nanoparticles, lovastatin, poly(lactic-co-glycolic) acid (PLGA)

In this issue
Statins attenuate disease severity in established animal models of inflammatory autoimmune disease. When applied clinically, however, phase trials evaluating statins for the treatment of inflammatory autoimmune disease have been disappointingly inconclusive. Nanotechnology-based drug delivery represents a relatively new and innovative approach by which to administer therapeutic compounds exhibiting low bioavailability, high systemic toxicity, and/or poor water solubility in a controlled and sustained manner. In this study, polymers of poly(lactic co-glycolic) acid (PLGA) were used to form injectable, biodegradable nanoparticles encapsulating lovastatin. A one-time, non-surgical, peri-neural administration of lovastatin-loaded nanoparticles significantly attenuated the clinical development of experimental autoimmune neuritis (EAN), a well-established animal model of AIDP/GBS (see graph). This study provides the first proof-of-concept approach for the application of a nanoparticle-based local drug delivery platform for the management of inflammatory demyelinating diseases, including AIDP/G BS.
INTRODUCTION
Acute inflammatory demyelinating polyneuropathy (AIDP) is a North American and European variant of Guillain-Barré Syndrome (GBS), a leading cause of acute flaccid paralysis in Western countries (Flachenecker 2006) with a reported annual stable incidence rate of 0.2–4.0 cases per 100,000 (McGrogan et al. 2009, Sejvar et al. 2011). In the U.S. alone, nearly 10,000 GBS cases are reported annually with an overall yearly socioeconomic burden exceeding $1.7 billion (Frenzen 2008). Characterized clinically by acute aggressive ascending paresis, this autoimmune peripheral nerve disorder can affect individuals of all age groups (5–89 years) with a unique male:female preferential ratio ~1.6:1 (Blum et al. 2013). Although rarely fatal, in some affected patients life-preserving mechanical ventilation and prolonged supportive clinical care is required. Despite its prevalence and overwhelming socioeconomic impact, the pathophysiology of AIDP/GBS remains unclear. Aberrant cell-mediated autoimmune responses directed against specific constituents of peripheral nerve myelin are considered causal in AIDP (Hartung et al. 2001, Hartung et al. 1988). Enhanced infiltration of inflammatory cells into peripheral nerves is strongly suggestive of a Th1 cell-mediated process (Willison 2005, Csurhes et al. 2005, McCombe & Csurhes 2010). Therapeutic strategies for the management of AIDP/GBS currently utilize non-specific therapies that target the immune system as a whole (Lindenbaum et al. 2001).
In general, various agents that exhibit anti-inflammatory properties (Zhang et al. 2009, Li et al. 2011, Han et al. 2014, Xiao et al. 2014) or antagonists of inflammatory mediators (Archelos et al. 1993, Bao et al. 2003, Mao et al. 2010, Yuan et al. 2014) show therapeutic potential (Hahn 1996, Gold et al. 2000) at lessening experimental disease severity, consistent with a central role of aberrant inflammation in the pathogenesis of AIDP/GBS. Strategies which selectively target and disrupt autoreactive leukocyte trafficking into affected peripheral nerves, however, represent an attractive and promising new option for improved therapeutic management of patients with AIDP/GBS. One such experimental strategy previously evaluated by our laboratory involves systemic administration of statins, a class of generally well-tolerated cholesterol lowering agents. A short-term high-dose course of statins was found to markedly restrict the transendothelial trafficking of autoreactive leukocytes into peripheral nerves of Lewis rats and safely attenuate the development and progression of experimental autoimmune neuritis (EAN), a well-established model of AIDP/GBS (Sarkey et al. 2007).
In vitro, we have shown that statins significantly attenuate TNF-α mediated release of the leukocyte-recruiting chemokine (C-C motif) ligand 2 (CCL2) from cultured peripheral nerve microvascular endoneurial endothelial cells, the barrier-forming cells that line the vessels of the peripheral nerves (Langert et al. 2014, Langert et al. 2013b). Moreover, statins attenuate endothelial cell adhesion molecule expression, NFκB activation, and CD40 expression (Zapolska-Downar et al. 2004, Takeuchi et al. 2000, Wu et al. 2013, Lin et al. 2006, Stach et al. 2012). These findings are consistent with well-documented pleiotropic effects of statins (Cai et al. 2015, Goldstein 2007, Kim et al. 2007). While the precise mechanisms remain to be determined, it is well-established that statins can limit the trafficking of autoreactive leukocytes across activated endothelial barriers (Maher et al. 2009, Ifergan et al. 2006, Greenwood et al. 2003). In vivo, statins attenuate disease severity in both EAE and EAN animal models of inflammatory autoimmune disease (Stanislaus et al. 2002, Stanislaus et al. 2001, Sarkey et al. 2007, Greenwood et al. 2003). When applied clinically either as a monotherapy or as an add-on therapy to interferon-beta, however, phase trials evaluating statins (up to 80 mg daily) for the treatment of relapsing-remitting or progressive multiple sclerosis (MS) or clinically isolated syndrome (CIS) have been disappointingly inconclusive (Chataway et al. 2014, Pihl-Jensen et al. 2015, Sorensen et al. 2011). There is evidence to support a beneficial effect of statins in secondary progressive MS (Pihl-Jensen et al. 2015). Whether due to immunomodulation or neuroprotection, the potential benefit of statins for the management of autoimmune neuropathies warrants further study.
Nanotechnology-based drug delivery represents a relatively new and innovative approach by which to administer therapeutic compounds exhibiting low bioavailability, high systemic toxicity, and/or poor water solubility, including statins (Romana et al. 2014, Sirtori 2014, Wischke & Schwendeman 2008). Poly(lactic-co-glycolic) acid (PLGA) nanoparticles are ideal biodegradable candidates which efficiently encapsulate hydrophobic compounds for controlled and sustained release (Astete & Sabliov 2006, Kulkarni & Kompella 2014). Local administration of statin encapsulating PLGA nanoparticles accelerates healing in preclinical bone fracture models by increasing transcription of BMP-2 at the site of injury (Garrett et al. 2007, Ho et al. 2011).
Here, we report that a bilateral peri-neural injection of lovastatin-encapsulating biodegradable PLGA nanoparticles protects against the clinical progression of EAN and associated peripheral nerve functional, histological, and neuropathological deficits.
MATERIALS AND METHODS
PLGA Nanoparticle Preparation and Characterization
Unless otherwise specified, analytical grade reagents were obtained from Sigma-Aldrich (St. Louis, MO). Nanoparticles were prepared using an oil-in-water single emulsion technique (McAlvin et al. 2013, McCall & Sirianni 2013). Briefly, 100 mg of PLGA (85:15 or 50:50, viscosity 0.26-0.54, Durect Corp., Birmingham, AL) was dissolved in dichloromethane (1 ml) and slowly added to ice-cold 1% (w/v) polyvinyl alcohol (PVA, 10 ml) while vigorously vortexing. The resultant suspension was emulsified by probe sonication and subsequently diluted with 100 ml ice-cold PVA. The organic solvent was allowed to evaporate with constant stirring for 3h at 23°C and the resulting PLGA nanoparticles were isolated by centrifugation (25,000g × 20 min @ 4°C) and washed 3x with deionized water. Washed nanoparticles were resuspended in 10 ml deionized water and 50 mg sucrose was added as a lyoprotectant (Howard et al. 2012). PLGA nanoparticles were lyophilized and stored at −80°C. Lovastatin-encapsulating PLGA nanoparticles were prepared in a similar manner by dissolving 10–50 mg lovastatin (Calbiochem/Millipore, Billerca, MA) in the initial organic phase. PLGA nanoparticle size was determined by dynamic light scattering using a Malvern NanoSight NS500 analyzer.
To determine percent drug loading and encapsulation efficiency, PLGA nanoparticles were dissolved in dimethyl sulfoxide (5 mg/ml) and encapsulated lovastatin resolved by HPLC (Hitachi LaChrom Elite, model 2200) using a Beckman Coulter Ultrasphere 5 μm C18 reverse-phase column (4.6 mm × 25 cm). An isocratic mobile phase of acetonitrile: 0.05N formic acid (70:30, v/v) was used to elute lovastatin from the column at a rate of 1 ml/min. Lovastatin was quantified (240 nm) using an external standard curve prepared in DMSO.
Static Dialysis of Lovastatin-Encapsulating PLGA Nanoparticles
Lovastatin-encapsulating PLGA nanoparticles (2.5–10 mg) were suspended in 1 ml phosphate-buffered saline (pH 7.4, 37°C), placed in 5cm × 10mm pre-soaked and rinsed Spectra/Por Biotech regenerated cellulose dialysis membrane tubing (Spectrum Laboratories, MWCO: 3.5–5 kDa), and the suspension dialyzed at 37°C by immersion in 50 ml PBS containing 0.05% NaN3 with continuous gentle stirring. At timed intervals, an aliquot of the dialysate was removed and replaced with an equal volume of PBS. Lovastatin present in the dialysate was quantified by HPLC as described above, with an external standard curve prepared in PBS.
Peripheral Nerve Microvascular Endoneurial Endothelial Cell Culture
Unless otherwise specified, cell-culture reagents were obtained from Life Technologies (Grand Island, NY). An immortalized cell line of peripheral nerve microvascular endoneurial endothelial cells (PNMECs) was prepared from purified rat primary PNMECs using a replication-deficient SV40 retrovirus encoding a temperature sensitive, non-SV40-origin binding mutant of the large T antigen and a selectable neomycin resistance gene as previously described (Langert et al. 2013a). Immortalized PNMEC cultures were maintained in Ham's F10 basal media supplemented with 10% FBS, 5.6 μg/ml amphotericin B, 100 U/ml penicillin, 100 μg/ml streptomycin, 50 μg/ml ECGS (BD Biosciences, San Jose, CA), and 0.4 μg/ml heparin (Sigma-Aldrich), under an atmosphere of 5% CO2/95% air.
In vitro Evaluation of PLGA Nanoparticle Function
Empty- or lovastatin-encapsulating PLGA nanoparticles were suspended in culture media and 40 μg added to confluent PNMEC cultures seeded on collagen-coated (6.0 μg/cm2, rat tail collagen type I, Millipore) 6-well tissue culture plates. After 24h, total RNA was extracted from PNMEC cultures with TRIzol reagent and 5 μg was reverse-transcribed using Super Script III First Strand Synthesis system (Life Technologies) as previously described (Von Zee et al. 2009). Rat-specific RhoA and RhoB cDNA sequences were amplified by real-time PCR on a Mini-Opticon PCR detection system using iQ SYBR Green Supermix (Bio-Rad, Hercules, CA) with the following primer pairs: RhoA: forward, 5'- TGAAAACTATGTGGCGGATATCG; reverse, 5'- TCTGCTTCTTCAGGTTTAACCGG; RhoB: forward, 5'- AGGACTACGATCGTTTACGGCC; reverse, 5'- CAGCCATTCTGGGATCCGTAG. For each sample, GAPDH (forward, 5'-TCCCTCAAGATTGTCAGCAA; reverse, 5'-AGATCCACAACGGATACATT) was used as a reference control. Optimized amplification steps of 94°C × 5 min followed by 94°C × 15s, annealing at 55°C × 30s, and elongation at 72°C × 60s were used. Reaction efficiencies were typically >90%. For each sample, the specificity of the real-time reaction product was determined by melt curve analysis.
In Vivo Evaluation of PLGA Nanoparticle Function
This study was conducted using protocols approved by the Edward Hines Jr. VA Hospital Institutional Animal Care and Use Committee in accordance with the principles of laboratory animal care (NIH Publication No. 86-23, 1985). All animals were housed in pairs, allowed access to standard rat chow and water ad libitum, and maintained on a 10 h/14 h light/dark cycle. Naïve adult male Lewis rats (initial body weight 250 g; Envigo, Indianapolis, IN) were anesthetized with ketamine (100 mg/kg)-xylazine (5 mg/kg) prior to PLGA nanoparticle administration. Although AIDP/GBS develops slightly more frequently in males more than females, the use of male rats in this study was chosen based on our previously published findings (Sarkey et al. 2007).
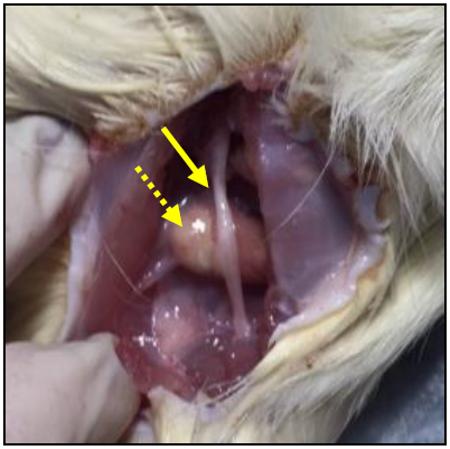
Empty or lovastatin-encapsulating PLGA nanoparticles were suspended in 1% sodium carboxymethylcellulose and 0.1% Tween 80 in sterile PBS at a concentration of 80 mg/ml. PLGA nanoparticles (250 μl) were administered bilaterally using a 20-gauge needle inserted posteromedial to the greater trochanter to a depth of 16 mm, pointing in a posterolateral direction. In some experiments, empty PLGA nanoparticles were injected on left side and lovastatin-encapsulating PLGA nanoparticles injected on the right side. In these studies, animals were sacrificed 7 days after PLGA nanoparticle injection, trunk blood was collected, and sciatic nerves and adjacent muscle were collected from each leg. PLGA nanoparticles administered in this manner were visible upon exposure as a localized globular deposit (dashed arrow) adjacent to the nerve fiber bundle (solid arrow). RNA was extracted from sciatic nerve and adjacent muscle tissue with TRIzol reagent, and RT-PCR analysis of RhoA and RhoB mRNA content was performed as described above.
Experimental Autoimmune Neuritis
Five days after administering PLGA nanoparticles, rats were actively induced with EAN as previously described (Calik et al. 2015, Sarkey et al. 2007). Rats were anesthetized with ketamine (100 mg/kg)–xylazine (5 mg/kg) and 100 μl of a freshly prepared fine-particle emulsion (1:1 v/v) containing of 100 μg of a synthetic neuritogenic P2 peptide (53–78, Peptide Synthesis Core Facility of the Simpson Querrey Institute, Northwestern University) suspended in sterile saline and incomplete Freund's adjuvant (Sigma-Aldrich) supplemented with 5 mg/ml heat-inactivated Mycobacteria tuberculosis (strain H37Ra; Difco Laboratories, Detroit, MI, USA) was injected into the left hind footpad. Rats were scored daily for EAN by investigators blinded to group assignment. The severity of clinical signs was scored as follows: 0 = no symptoms; 1 = flaccid tail; 2 = abnormal gait; 3 = mild paraparesis; 4 = severe paraparesis; 5 = paraplegia. Intermediate clinical signs were scored in increments of 0.5.
Peripheral Nerve Conduction Studies
Evoked compound muscle action potential (CMAP) amplitudes were recorded and sciatic nerve conduction velocities (NCV) were calculated as previously described (Sarkey et al. 2007). Prior to EAN induction (baseline) and again at peak of disease (Day 15–17), sciatic nerves of anesthetized rats were stimulated at the sciatic notch or at the ankle (tibial nerve) with unipolar pin electrodes using supramaximal stimuli (25 mA, 0.05-ms duration) at high frequency (1 Hz) for the direct measurement of motor nerve (M-wave) responses. Unipolar pin recording electrodes were used to record the evoked potentials from the plantar muscles of the un-injected right hind foot. Individual responses were amplified and recorded using a TECA Synergy electromyograph system. For each animal, M-wave evoked responses elicited from distal (ankle) or proximal (sicatic notch) sites were captured as an average of 25 individual responses. Three averaged responses elicited from distal and proximal stimulation sites were recorded for each animal. Motor nerve conduction velocity (NCV) was calculated as the distance from notch-to-ankle divided by the average difference of the M-wave latency (notch–ankle). A heating pad was used to maintain the body temperature of the rat at 37°C.
Histology and Immunohistochemistry
Following nerve conduction studies performed at peak of disease, sciatic nerves (3-4 cm) were rapidly harvested, bisected, and immediately fixed in ice-cold 2% paraformaldehyde (PFA) – 2.5% glutaraldehyde phosphate-buffered solution for 24h at 4°C or snap frozen in Tissue Tek OCT and chilled isopentane and stored at −80°C. For histological analysis, PFA-fixed nerves were post-fixed in 1% osmium tetroxide, dehydrated, and embedded in Embed-812 (Electron Microscopy Sciences, Fort Washington, PA). Serial transverse sections (0.5 μm) were prepared with a Reichert Ultracut S microtome (W. Nuhsbaum Inc., McHenry, IL) and osmicated sections were stained with toluidine blue. Stained sections were viewed using a Leitz DMRB inverted phase contrast microscope. Representative images were captured with a Leica Wild MPS photographic system and scored, by a blinded investigator, using a four-point scoring system for pathological changes (demyelination, axonal damage, cellular infiltration, and edema) as previously described (Sarkey et al. 2007).
For immunohistochemical analysis of CD4+ and CD68+ infiltrates, 8 μm transverse sections of snap-frozen nerves were prepared with a cryostat and placed onto Superfrost Plus microscope slides. Sections were air dried, washed in PBS to remove OCT, and fixed in 3.7% paraformaldehyde (10 min, 23°C). Washed sections were blocked × 1h at 23°C in PBS containing 3% BSA, 1% normal goat serum, and 0.3% Triton X-100 (blocking buffer). Washed sections were incubated overnight at 4°C in blocking buffer supplemented with a 1:100 dilution of mouse anti-rat CD4 (clone W3/25, purified IgG) or mouse anti-rat CD68 (clone ED1, purified IgG) primary antibody (BioRad) and a 1:50 dilution of rabbit polyclonal CD31 primary antibody (ThermoFisher Scientific). Immunostained sections were washed and subsequently incubated × 2h in blocking buffer containing a 1:1000 dilution of AlexaFluor 488 conjugated goat anti-mouse IgG secondary antibody and AlexaFluor 568 conjugated goat anti-rabbit IgG secondary antibody (ThermoFisher Scientific), both of which have been highly cross-adsorbed against rat IgG. Sections were coverslipped with fluoroshield containing DAPI. Images were captured on a Nuhsbaum inverted confocal microscope.
Statistical Analysis
Data are expressed as the mean ± SEM of N observations unless noted otherwise. Statistical significance of parametric data was determined by Student's t-test. Significance between multiple experimental groups was determined by one- or two-way ANOVA with a Bonferroni multiple comparison post-hoc analysis. For statistical evaluation of clinical and neuropathological data, a Mann–Whitney non-parametric U-test analysis was performed. In each case, p < 0.05 was considered statistically significant.
RESULTS
In Vitro Characterization of Lovastatin-Encapsulating PLGA Nanoparticles
Nanoparticles with varying lactide:glycolide ratios (50:50 or 85:15; inherent viscosity 0.26-0.54 and 0.55–0.75, respectively; Durect Corp.) were prepared without (empty particles) or with lovastatin at a theoretical drug loading content of 6.25% (w/w) or 25.0% (w/w) and characterized by dynamic light scattering (particle size) and HPLC (drug loading efficiency). Independent of polymer composition or drug loading efficiency, PLGA nanoparticles exhibited a particle size of 260.0 ± 14.4 nm (Table 1). At 6.25% (w/w), encapsulation efficiencies ranged from 87.4 ± 4.4% to 104.3± 4.1, depending on the polymer composition (Table 1). The release of encapsulated lovastatin from PLGA nanoparticles into an aqueous environment was characterized in vitro by static dialysis. Empty or lovastatin-encapsulating PLGA nanoparticles (10 mg) were dialyzed against PBS at 37°C under gentle continuous mixing. At selected timed intervals, an aliquot of the dialysate was removed and analyzed by HPLC. The dialysate contained a single compound eluting on HPLC with a retention time of 5.6 min, identified by co-migration with authentic active (open-ring) β-hydroxy acid form of lovastatin (Fig. 1A, green and black traces). By comparison, dissolving lovastatin-encapsulating PLGA nanoparticles directly in DMSO yielded a single compound eluting on HPLC with a retention time of 8.5 min, identified by co-migration with authentic inactive prodrug (β-lactone) of lovastatin (Fig. 1A, red and blue traces). These findings suggest that encapsulated lovastatin remains as an inactive prodrug. Only after release, does the lactone ring of lovastatin become hydrolyzed thereby activating this compound. Cumulative release was quantified over a period of 30 days using an external standard curve prepared in PBS. In all cases, within 25.1 ± 4.9 days (n=9), release of encapsulated lovastatin was complete (Fig. 1B). At 6.25% (w/w) or 25% (w/w), lovastatin-encapsulating nanoparticles with a 85:15 lactide:glycolide ratio achieved 50% release by 10.7 ± 1.1 days or 9.7 ± 0.6 days, respectively (n=3). By comparison, nanoparticles prepared using a 50:50 lactide:glycolide ratio achieved 50% release by 17.3 ± 4.6 days (Fig. 1B).
Table 1. In vitro characterization of lovastatin-encapsulating PLGA nanoparticles.
Empty- or lovastatin-encapsulating PLGA nanoparticles (NP) were prepared by oil-in-water emulsion. NP size was measured with dynamic light scattering and percent drug loading (DL) was quantified by HPLC. EE, percent encapsulation efficiency of drug loading. Data shown are the means ± SEM, n=3–4 sample preparations.
| PLGA ratio: | 85:15 | 85:15 | 85:15 | 50:50 |
|---|---|---|---|---|
| NP size (nm) | 252.9 ± 33.8 | 230.8 ± 5.9 | 299.6 ± 5.3 | 256.8 ± 9.8 |
| DL (Theoretical) | 6.25 | 25.0 | 0 | 6.25 |
| DL (Actual) | 6.5 ± 0.3 | 27.3± 4.2 | 0 | 5.5 ± 0.3 |
| EE | 104.3 ± 4.1 | 109.2 ± 4.2 | N/A | 87.4 ± 4.4 |
Figure 1. Higher lactide content facilitates release of lovastatin from encapsulated lactide:glycolide co-polymer.

(A) Representative composite HPLC chromatograms showing inactive lovastatin prodrug (red trace, retention time 8.5 min.) extracted from lovastatin-encapsulating nanoparticles or active lovastatin hydroxy acid recovered from dialysate (green trace, retention time 5.6 min.). For comparison, co-migration with authentic standards (blue trace, lovastatin prodrug; black trace, lovastatin hydroxy acid) are shown. (B) Time-dependent cumulative release of lovastatin hydroxy acid following static dialysis of empty- or lovastatin-encapsulating PLGA nanoparticles as a function of lactide:glycolide co-polymer composition. Release from 25% w/w drug loaded (DL) 85:15 co-polymer is shown for comparison. Data shown are the mean ± SEM.
To determine the functional activity of lovastatin released from PLGA nanoparticles, semi-confluent cultures of immortalized PNMECs were incubated × 24h in the presence of 40 μg nanoparticles and evaluated for changes in small GTPase mRNA expression. Previous studies have shown that treating cultured cells with statins limits the availability of isoprenoid metabolic intermediates leading to a marked compensatory increase in constitutive RhoA and inducible RhoB mRNA expression (Von Zee et al. 2009). Treating PNMECs cultures with activated lovastatin (10 μM, 24h) did not significantly alter constitutive levels of RhoA mRNA (Fig. 2A). By comparison, however, a 10–15 fold increase in inducible RhoB mRNA content was observed in PNMEC cultures treated with activated lovastatin (10 μM, 24h) when compared to vehicle-treated control (Fig. 2A). PNMECs cultured in the presence of 40 μg lovastatin-encapsulating PLGA nanoparticles exhibited a robust 20-fold increase in inducible RhoB mRNA content (Fig. 2B). Constitutive levels of RhoA mRNA, while increased in response to lovastatin-encapsulating nanoparticles, did not achieve statistical significance (Fig. 2B). PNMECs cultures treated with empty PLGA nanoparticles (40 μg) had no effect on either RhoA or RhoB mRNA expression (Fig. 2B). These findings suggest that encapsulating lovastatin within PLGA nanoparticles does not alter the ability of this prodrug to become functionally active under physiologic conditions.
Figure 2. Lovastatin-encapsulating PLGA nanoparticles functionally enhance Rho GTPase mRNA content in PNMEC cultures.

Confluent PNMEC cultures were incubated × 24h (A) in the absence (open bar, 0.01% ethanol) or presence (closed bar) of 10 μM activated lovastatin or with (B) 40 μg empty (open bar) or 40 μg lovastatin-encapsulating nanoparticles (closed bar). Relative changes in RhoA GTPase and RhoB GTPase mRNA content were quantified by RT-PCR. Data shown are the means ± SEM (n = 3) and are expressed as GAPDH-normalized fold-changes. *, p < 0.05, Student's t-test.
In Vivo Characterization of Lovastatin-Encapsulating PLGA Nanoparticles
To determine whether locally administered lovastatin-encapsulating PLGA nanoparticles can functionally alter expression of small GTPases mRNA content in vivo, healthy male Lewis rats were injected with 20 mg (250 μl, single bolus) of empty (left leg) and 20 mg (250 μl, single bolus) of 25.0% (w/w) lovastatin-encapsulating nanoparticles (right leg). Sciatic nerves (Fig. 3A) and adjacent muscle tissue (Fig. 3B) harvested from the right lovastatin-treated leg expressed a significant 5-fold increase in inducible RhoB mRNA content as compared to tissue harvested from the left control leg (Fig. 3 A, B) For each tissue, by comparison, lovastatin-encapsulating nanoparticles elicited a qualitatively similar increase in the constitutive expression of RhoA mRNA content compared to empty nanoparticles (Fig. 3A, B). These findings suggest that lovastatin-encapsulating nanoparticles act locally on adjacent tissue, rather than through systemic circulation. Consistent with this thesis, the levels of circulating lovastatin prodrug, or active β-hydroxy acid form, present in sera from rats receiving lovastatin-encapsulating nanoparticles was below the limit of HPLC detection. In contrast, circulating levels of active lovastatin present in serum samples prepared from blood of healthy male Lewis rats 1h after receiving oral (100 mg/kg) or intraperitoneal (10 mg/kg) lovastatin prodrug were 385.3 ± 53.7 ng/ml (n=6) and 937.5 ± 50.4 ng/ml (n=3), respectively (Table 2).
Figure 3. Lovastatin-encapsulating PLGA nanoparticles functionally enhance Rho GTPase mRNA expression in vivo without disrupting peripheral nerve function.

Healthy male Lewis rats received bilateral peri-neural injections of empty- (left leg, open bars) and lovastatin-encapsulating (25% w/w, right leg, closed bars) nanoparticles (20 mg each). One week later, relative changes in RhoA and RhoB mRNA content in (A) sciatic nerve and (B) muscle were quantified by RT-PCR. Data shown are the means ± SEM (n = 4–8) from two independent experiments, expressed as GAPDH-normalized fold-changes. *, p < 0.05, Student's t-test. (C, D) Prior to (open bar, day 0) or one week post peri-neural injection of lovastatin-encapsulating nanoparticles (closed bars, day 7), (C) ankle- or sciatic notch-evoked CMAP amplitudes were recorded from the right leg and (D) nerve conduction velocities calculated. Data shown are the means ± SEM (n = 8) from two independent experiments.
Table 2. Content of activated lovastatin in sera of rats receiving lovastatin-encapsulating PLGA nanoparticles.
Healthy male Lewis rats received a single peri-neural injection of lovastatin-encapsulating PLGA nanoparticles and one week later sera levels of active lovastatin were analyzed by HPLC and found to be below the level of detection. For comparison, levels of activated lovastatin in sera of rats that received lovastatin prodrug as a single intraperitoneal bolus or by oral gavage, as indicated, are shown. In all cases, levels of the inactive lovastatin prodrug were below the level of detection. Data shown are the means ± SEM, n=3–6.
| Dose and Route of Administration | Time | Lovastatin Hydroxy Acid (ng/ml) |
|---|---|---|
| 10 mg/kg, intraperitoneal | 1h | 937.5 ± 50.4 |
| 100 mg/kg, oral gavage | 1h | 385.3 ± 53.7 |
| 20 mg of 25% w/w lovastatin-encapsulating nanoparticles | 1 week | undetectable |
To determine whether peri-neural administration of lovastatin-encapsulating PLGA nanoparticles induces localized toxicity or injury to peripheral nerves, nerve function studies were performed on healthy male Lewis rats. Prior to (baseline, day 0) and 7 days following unilateral injection of 20 mg lovastatin-encapsulating nanoparticles (25% w/w), evoked compound muscle action potentials (CMAP) amplitudes were recorded and nerve conduction velocities (NCV) calculated. Evoked CMAP amplitudes (Fig. 3C) and NCV (Fig. 3D) determined from the injected right legs of rats receiving lovastatin-encapsulating nanoparticles were statistically indistinguishable from baseline (day 0) control responses. Evoked responses from the un-injected left legs did not differ from the injected right legs (data not shown). These findings suggest that peri-neural administration of lovastatin-encapsulating PLGA nanoparticles in healthy male Lewis rats produces minimal toxicity and trauma to adjacent nerve bundles.
Clinical Application of Lovastatin-Encapsulating PLGA Nanoparticles
Our group has previously shown that a short-term high-dose course of intraperitoneal administered lovastatin elicits a marked attenuation of EAN in adult male Lewis rats (Sarkey et al. 2007). Despite these advancements, the clinical application of systemically administered lovastatin for the management of inflammatory autoimmune diseases remains a challenge (Pihl-Jensen et al. 2015, Sorensen et al. 2011). A more viable option by which to achieve local concentrations of statins capable of mediating pleiotropic effects while minimizing systemic side effects may include the application of nanotechnology. To determine the therapeutic potential of localized administration of lovastatin-encapsulating PLGA nanoparticles in EAN, a bilateral peri-neural injection of empty nanoparticles or lovastatin-encapsulating nanoparticles (25% w/w, 20 mg each) was administered to adult male Lewis rats five days prior to disease induction. Rats receiving empty nanoparticles developed a monophasic course of EAN beginning on day 10.8 ± 0.4 and peaking on day 15.9 ± 0.3, post-immunization (Fig. 4A). In rats receiving lovastatin-encapsulating nanoparticles, a similar monophasic course of EAN developed beginning on day 10.8 ± 0.2 and peaking on day 15.5 ± 0.4, post-immunization. While lovastatin-encapsulating nanoparticles did not alter the course of EAN, the severity of EAN (peak score 3.7 ± 0.2 vs. 1.8 ± 0.1) was significantly attenuated (Fig. 4). Another independent measure of disease severity is weight loss. EAN rats that had received empty control nanoparticles exhibited weight loss that is characteristically observed with EAN development, reaching a peak weight loss of 10.6 ± 1.2% body weight on Day 17 (Fig. 4B). In contrast, EAN rats that received lovastatin-encapsulating nanoparticles exhibited significantly less weight loss (Fig. 4B). To confirm that lovastatin-encapsulating nanoparticles were most effective when administered locally, in a separate experiment empty control or lovastatin-encapsulating nanoparticles were injected subcutaneously in the thigh rather than peri-neurally. The course and severity of EAN (Fig. 4C) and degree of weight loss at peak of disease (Fig. 4D) between these two groups was significantly indistinguishable.
Figure 4. Lovastatin-encapsulating PLGA nanoparticles protect against clinical development of EAN.

Five days prior to induction of EAN, male Lewis rats received bilateral (A, B) peri-neural or (C, D) subcutaneous injections of empty-(open symbols & bars) or 25% w/w lovastatin-encapsulating (closed symbols & bars) nanoparticles (20 mg each). Clinical progression of (A, C) EAN and (B, D) body weight were assessed daily. Shown are the mean ± SEM (n=6 per group). *p<0.01, Mann-Whitney non-parametric U-test; #p<0.0001, Student's t-test.
The impact of lovastatin-encapsulating nanoparticles on preservation of peripheral nerve function was next assessed using evoked-response electrophysiology. The presence of nanoparticles, whether empty control or lovastatin encapsulated, had no effect on baseline evoked CMAP or calculated NCV (Fig. 3C, D and Fig. 5). At peak of disease, evoked CMAP amplitudes and calculated NCV in EAN rats that received empty control nanoparticles were significantly smaller and slower, respectively, compared to baseline (Fig. 5). In contrast, evoked CMAP amplitudes and calculated NCV in EAN rats that received lovastatin-encapsulating nanoparticles were statistically indistinguishable from baseline (Fig. 5). Toluidine blue stained transverse sections of sciatic nerves collected from EAN rats that received empty control nanoparticles showed neuropathological evidence of edema, demyelination, axonal damage, and cellular infiltrates (Fig. 6A). In contrast, stained sections of sciatic nerves collected from EAN rats that received lovastatin-encapsulating nanoparticles had qualitatively less severe neuropathological findings (Fig. 6B). Sections of sciatic nerves collected from EAN rats that received empty control nanoparticles stained heavily for the presence of perivascular CD4+ T cells (Fig. 6C) and CD68+ macrophages (Fig. 6E). In contrast, sections of sciatic nerves collected from lovastatin-encapsulating nanoparticles had qualitatively less CD4+ (Fig. 6D) and CD68+ (Fig. 6E) perivascular infiltrates. Collectively, these findings support a protective effect of locally administered lovastatin-encapsulating nanoparticles against EAN-associated deficits in peripheral nerve function.
Figure 5. Lovastatin-encapsulating PLGA nanoparticles protect against EAN-induced peripheral nerve function deficits.

Five days prior to induction of EAN, male Lewis rats received bilateral peri-neural injections of empty- or 25% w/w lovastatin-encapsulating nanoparticles (20 mg each) as indicated. Prior to induction of EAN (open bars) or at peak of disease (closed bars) (A, B) ankle- or sciatic notch-evoked CMAP amplitudes were recorded and (C) nerve conduction velocities calculated. Data shown are the means ± SEM (n= 3-4). *, p < 0.01, two-way ANOVA with Bonferroni's post-hoc analysis.
Figure 6. Lovastatin-encapsulating PLGA nanoparticles protect against EAN-induced neuropathology.

Five days prior to induction of EAN, male Lewis rats received bilateral peri-neural injections of (A, C, E) empty- or (B, D, F) 25% w/w lovastatin-encapsulating nanoparticles (20 mg each). At peak of disease, sciatic nerves were harvested and processed for histological and immunehistochemical analyses. Shown are representative (A, B) toluidine blue stained histological or confocal (C, D) CD4+ (E, F) CD68+ immunostained transverse sections of sciatic nerves. Green, CD4+ or CD68+ immunostained infiltrates; red, PECAM immunostained endoneurial endothelial cells; blue, DAPI stained nuclei. Scale bar, 20 μm.
DISCUSSION
In this study, a well-established biodegradable nanotechnology-based drug delivery approach was implemented as a new and innovative strategy by which to locally administer therapeutic compounds exhibiting low bioavailability, high systemic toxicity, and/or poor water solubility for the management of systemic inflammatory peripheral nerve disorders. Here, we report that a one-time localized peri-neural application of lovastatin-encapsulating poly(lactic-co-glycolic) acid (PLGA) nanoparticles significantly attenuates the clinical development of systemic experimental autoimmune neuritis (EAN), a well-established animal model of AIDP/GBS.
Poly(lactic-co-glycolic) acid (PLGA) nanoparticles are ideal biodegradable candidates with which to efficiently encapsulate hydrophobic compounds for controlled and sustained release (Astete & Sabliov 2006, Kulkarni & Kompella 2014). Previous studies from our laboratory have shown that a short-term high-dose course of intraperitoneally-administered statins markedly restricts the transendothelial trafficking of autoreactive leukocytes into peripheral nerves of adult male Lewis rats and safely attenuates the development and progression of EAN. Despite these advancements, the clinical application of systemically-administered statins for the management of inflammatory disorders remains controversial due to disappointingly inconclusive phase trials.
To resolve these limitations, a previously described oil-in-water single emulsion technique was used to encapsulate variable amounts of lovastatin prodrug into PLGA nanoparticles having either a 50:50 or 85:15 lactide:glycolide ratio. In agreement with previous studies (Zaghloul et al. 2005), nanoparticles composed of 85:15 lactide:glycolide yielded slightly higher encapsulation efficiencies compared to those having a 50:50 ratio, possibly due to a higher hydrophobicity of the 85:15 co-polymer. While higher drug loading conditions have been reported to yield larger size nanoparticles (McAlvin et al. 2013), PLGA nanoparticles prepared using the oil-in-water single emulsion technique exhibited an average particle size of 260.0 ± 14.4 nm that was not influenced by polymer composition or drug loading efficiency, consistent with earlier reported findings (Astete & Sabliov 2006, Panyam et al. 2004).
Polymer composition was found to influence release of lovastatin from encapsulated PLGA nanoparticles. Previous studies have shown that PLGA nanoparticles prepared with a lower lactide content hydrolyze more quickly, thus releasing encapsulated compounds on a shorter time scale (Wischke & Schwendeman 2008). In our hands, however, at a drug load burden of 6.25%, lovastatin-encapsulating nanoparticles prepared with an 85:15 lactide:glycolide ratio released lovastatin into the dialysate at a near linear rate of ~5% per day. Nanoparticles prepared with a lower (50:50) content of lactide, by comparison, exhibited a bi-phasic release profile approximating ~2.5% per day over the first 10 days and a subsequent rate of release of nearly 5% per day. Both co-polymers preparations of lovastatin-encapsulating PLGA nanoparticles (50:50; 85:15) reached 85% cumulative release by days 20.3 ± 2.9 and 19.0 ± 1.8, respectively. Increasing the drug load to 25% measurably enhanced the rate of lovastatin release from 85:15 PLGA nanoparticles to ~6% per day.
When administered orally or by intraperitoneal injection, lovastatin prodrug is rapidly activated to the open ring hydroxy acid isoform by CYP3A4 cytochrome P450 (Romana et al. 2014). While cytochrome P450 is highly expressed in the liver, it is also expressed in other tissues including lung, small intestine, kidney, esophagus, and olfactory mucosa (Yang & Hwang 2006, McLaughlin et al. 2010, Ding & Kaminsky 2003). PLGA nanoparticles prepared using the oil-in-water single emulsion technique were found to encapsulate and retain lovastatin in its prodrug lactone confirmation. Upon release of the encapsulated prodrug, however, lovastatin is converted to the active hydroxy acid isoform, consistent with previous reports of non-enzymatic conversion of the lactone prodrug within an aqueous environment (Shen et al. 1996, Ye et al. 2000).
By limiting the availability of isoprenoid metabolic intermediates, we have previously shown that statins elicit a marked compensatory increase in constitutive RhoA and inducible RhoB mRNA expression (Stubbs & Von Zee 2012). PNMEC cultures responded robustly to lovastatin-encapsulating PLGA nanoparticles by significantly increasing Rho GTPase mRNA content in a manner similar to that seen following the addition of chemically-activated lovastatin hydroxy acid. Given their dimensions (~260 nm), it is unlikely that cultured cells physically respond to the PLGA nanoparticles themselves. We speculate that the observed effects elicited by the lovastatin-encapsulating nanoparticles are most likely due to release of the prodrug into the culture media with subsequent non-enzymatic, or metabolic, conversion to its hydroxy acid active isoform.
When evaluated in vivo, we found that unilateral peri-neural administration of lovastatin-encapsulating PLGA nanoparticles to naïve healthy rats produced a qualitatively similar increase in ipsilateral sciatic nerve and muscle tissue Rho GTPase mRNA content. By comparison, peri-neural co-administration of empty nanoparticles within the contralateral limb had no effect on adjacent sciatic nerve or muscle tissue Rho GTPase mRNA expression, supporting a localized, rather than systemic, effect of the lovastatin-encapsulating nanoparticles. The content of lovastatin prodrug, or activated isomer, in the serum of rats receiving peri-neural lovastatin-encapsulating nanoparticles was below the level of detection by HPLC. Peri-neural administered lovastatin-encapsulating nanoparticles remained localized as a globular deposit adjacent to the nerve fiber bundle, presumably optimizing their functional efficacy while minimizing previously encountered systemic complications. Of particular clinical relevance, in vivo peri-neural administration of lovastatin-encapsulating nanoparticles did not significantly alter peripheral nerve evoked CMAP amplitudes or conduction velocities in healthy animals.
Previous studies have shown that a short-term high-dose regimen of intraperitoneally administered lovastatin significantly attenuates the progression of EAN in adult male Lewis rats (Sarkey et al. 2007), possibly by limiting CCL2-mediated transendothelial migration of autoreactive leukocytes into peripheral nerve (Langert et al. 2013b). Given that lovastatin-encapsulating nanoparticles administered adjacent to healthy peripheral nerve locally disrupted Rho GTPase signaling without adversely affecting peripheral nerve function, we evaluated the ability of these nanoparticles to protect against the development and progression of EAN. Rats that received bilateral peri-neural administration of lovastatin-encapsulating nanoparticles developed a significantly less severe course of EAN compared to rats receiving empty nanoparticles. Peripheral nerve function studies supported a protective effect of lovastatin-encapsulating nanoparticles against the development of EAN.
The mechanism by which these nanoparticles exert their protective effect is suggested by a reduced presence of CD68+ macrophage and CD4+ T cell infiltrates in sciatic nerves compared to nerves harvested from empty nanoparticle treated EAN rats. It is well established that pro-inflammatory CD8+ and CD4+ T cell subsets play a penultimate role in the development of both EAN and AIDP/GBS (Calik et al. 2015, Zhang et al. 2013). Similarly, depending on disease stage, macrophage subsets (M1, M2) may initiate induction of, or facilitate recovery from, EAN (Shin et al. 2013, Kiefer et al. 2001). It remains to be determined whether lovastatin-encapsulating nanoparticles elicit protection against EAN by selectively altering the distribution of infiltrating macrophage (M1, M2) and/or CD8+ and CD4+ T cell (Th1, Th2, Th17, Tregs) subsets during the induction or effector phase of this disease.
In this study, we report the preparation and characterization of PLGA nanoparticles encapsulating lovastatin. We demonstrate in vitro and in vivo that lovastatin-encapsulating nanoparticles afford meaningful protection against the clinical development of EAN, possibly by a mechanism that involves disruption of autoreactive leukocyte trafficking at the blood-nerve barrier. Limitations of this study include (i) prophylactic proof-of-concept and (ii) localized administration of nanoparticles, which do not address therapeutic relevance. Systemic administration of functionalized nanoparticles engineered to recognize and adhere to the activated endoneurial endothelium offers a promising approach by which to address therapeutic relevance for the management of advanced, more extensively involved, inflammatory neuropathies. This study provides the first proof-of-concept approach for the application of a nanoparticle-based local drug delivery platform for the management of inflammatory demyelinating diseases, including AIDP/GBS.
ACKNOWLEDGEMENTS
This work was supported, in part, by grants from the Department of Veterans Affairs (I21RX001553, E.B.S.; IK1RX001159, K.A.L.) and NIH (IR21NS085420, E.B.S.).
Abbreviations used
- AIDP
Acute inflammatory demyelinating polyneuropathy
- CCL2
chemokine (C-C motif) ligand 2
- CMAP
compound muscle action potential
- EAN
experimental autoimmune neuritis
- NCV
nerve conduction velocity
- PVA
polyvinyl alcohol
- GBS
Guillain-Barré Syndrome
- PNMECs
peripheral nerve microvascular endoneurial endothelial cells
Footnotes
ARRIVE guidelines have been followed:
Yes
=> if No, skip complete sentence
=> if Yes, insert “All experiments were conducted in compliance with the ARRIVE guidelines.”
Conflicts of interest: none
=> if `none', insert “The authors have no conflict of interest to declare.”
=> otherwise insert info unless it is already included
We declare that there are no financial or other relationships associated with this study that may result in a conflict of interest.
REFERENCES
- Archelos JJ, Maurer M, Jung S, Toyka KV, Hartung HP. Suppression of experimental allergic neuritis by an antibody to the intracellular adhesion molecule ICAM-1. Brain. 1993;116(Pt 5):1043–1058. doi: 10.1093/brain/116.5.1043. [DOI] [PubMed] [Google Scholar]
- Astete CE, Sabliov CM. Synthesis and characterization of PLGA nanoparticles. Journal of biomaterials science. Polymer edition. 2006;17:247–289. doi: 10.1163/156856206775997322. [DOI] [PubMed] [Google Scholar]
- Bao L, Lindgren JU, Zhu Y, Ljunggren HG, Zhu J. Exogenous soluble tumor necrosis factor receptor type I ameliorates murine experimental autoimmune neuritis. Neurobiol Dis. 2003;12:73–81. doi: 10.1016/s0969-9961(02)00007-4. [DOI] [PubMed] [Google Scholar]
- Blum S, Reddel S, Spies J, McCombe P. Clinical features of patients with Guillain-Barre syndrome at seven hospitals on the East Coast of Australia. J Peripher Nerv Syst. 2013;18:316–320. doi: 10.1111/jns5.12045. [DOI] [PubMed] [Google Scholar]
- Cai A, Zhou Y, Li L. Rho-GTPase and Atherosclerosis: Pleiotropic Effects of Statins. Journal of the American Heart Association. 2015;4 doi: 10.1161/JAHA.115.002113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calik MW, Shankarappa SA, Langert KA, Stubbs EB., Jr. Forced Exercise Preconditioning Attenuates Experimental Autoimmune Neuritis by Altering Th1 Lymphocyte Composition and Egress. ASN neuro. 2015;7 doi: 10.1177/1759091415595726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chataway J, Schuerer N, Alsanousi A, et al. Effect of high-dose simvastatin on brain atrophy and disability in secondary progressive multiple sclerosis (MS-STAT): a randomised, placebo-controlled, phase 2 trial. Lancet. 2014;383:2213–2221. doi: 10.1016/S0140-6736(13)62242-4. [DOI] [PubMed] [Google Scholar]
- Csurhes PA, Sullivan AA, Green K, Pender MP, McCombe PA. T cell reactivity to P0, P2, PMP-22, and myelin basic protein in patients with Guillain-Barre syndrome and chronic inflammatory demyelinating polyradiculoneuropathy. Journal of neurology, neurosurgery, and psychiatry. 2005;76:1431–1439. doi: 10.1136/jnnp.2004.052282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding X, Kaminsky LS. Human extrahepatic cytochromes P450: function in xenobiotic metabolism and tissue-selective chemical toxicity in the respiratory and gastrointestinal tracts. Annual review of pharmacology and toxicology. 2003;43:149–173. doi: 10.1146/annurev.pharmtox.43.100901.140251. [DOI] [PubMed] [Google Scholar]
- Flachenecker P. Epidemiology of neuroimmunological diseases. J Neurol. 2006;253:v2–v8. doi: 10.1007/s00415-006-5001-3. [DOI] [PubMed] [Google Scholar]
- Frenzen PD. Economic cost of Guillain-Barre syndrome in the United States. Neurology. 2008;71:21–27. doi: 10.1212/01.wnl.0000316393.54258.d1. [DOI] [PubMed] [Google Scholar]
- Garrett IR, Gutierrez GE, Rossini G, Nyman J, McCluskey B, Flores A, Mundy GR. Locally delivered lovastatin nanoparticles enhance fracture healing in rats. J Orthop Res. 2007;25:1351–1357. doi: 10.1002/jor.20391. [DOI] [PubMed] [Google Scholar]
- Gold R, Hartung HP, Toyka KV. Animal models for autoimmune demyelinating disorders of the nervous system. Mol Med Today. 2000;6:88–91. doi: 10.1016/s1357-4310(99)01639-1. [DOI] [PubMed] [Google Scholar]
- Goldstein LB. Statins for stroke prevention. Current atherosclerosis reports. 2007;9:305–311. doi: 10.1007/s11883-007-0037-0. [DOI] [PubMed] [Google Scholar]
- Greenwood J, Walters CE, Pryce G, Kanuga N, Beraud E, Baker D, Adamson P. Lovastatin inhibits brain endothelial cell Rho-mediated lymphocyte migration and attenuates experimental autoimmune encephalomyelitis. Faseb J. 2003;17:905–907. doi: 10.1096/fj.02-1014fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn AF. Experimental allergic neuritis (EAN) as a model for the immune-mediated demyelinating neuropathies. Rev Neurol (Paris) 1996;152:328–332. [PubMed] [Google Scholar]
- Han F, Luo B, Shi R, Han C, Zhang Z, Xiong J, Jiang M, Zhang Z. Curcumin ameliorates rat experimental autoimmune neuritis. Journal of neuroscience research. 2014;92:743–750. doi: 10.1002/jnr.23357. [DOI] [PubMed] [Google Scholar]
- Hartung HP, Kieseier BC, Kiefer R. Progress in Guillain-Barre syndrome. Curr Opin Neurol. 2001;14:597–604. doi: 10.1097/00019052-200110000-00008. [DOI] [PubMed] [Google Scholar]
- Hartung HP, Schafer B, Heininger K, Stoll G, Toyka KV. The role of macrophages and eicosanoids in the pathogenesis of experimental allergic neuritis. Serial clinical, electrophysiological, biochemical and morphological observations. Brain. 1988;111(Pt 5):1039–1059. doi: 10.1093/brain/111.5.1039. [DOI] [PubMed] [Google Scholar]
- Ho MH, Chiang CP, Liu YF, Kuo MY, Lin SK, Lai JY, Lee BS. Highly efficient release of lovastatin from poly(lactic-co-glycolic acid) nanoparticles enhances bone repair in rats. J Orthop Res. 2011;29:1504–1510. doi: 10.1002/jor.21421. [DOI] [PubMed] [Google Scholar]
- Howard MD, Lu X, Jay M, Dziubla TD. Optimization of the lyophilization process for long-term stability of solid-lipid nanoparticles. Drug development and industrial pharmacy. 2012;38:1270–1279. doi: 10.3109/03639045.2011.645835. [DOI] [PubMed] [Google Scholar]
- Ifergan I, Wosik K, Cayrol R, et al. Statins reduce human blood-brain barrier permeability and restrict leukocyte migration: relevance to multiple sclerosis. Ann Neurol. 2006;60:45–55. doi: 10.1002/ana.20875. [DOI] [PubMed] [Google Scholar]
- Kiefer R, Kieseier BC, Stoll G, Hartung HP. The role of macrophages in immune-mediated damage to the peripheral nervous system. Prog Neurobiol. 2001;64:109–127. doi: 10.1016/s0301-0082(00)00060-5. [DOI] [PubMed] [Google Scholar]
- Kim YS, Ahn Y, Hong MH, et al. Rosuvastatin suppresses the inflammatory responses through inhibition of c-Jun N-terminal kinase and Nuclear Factor-kappaB in endothelial cells. Journal of cardiovascular pharmacology. 2007;49:376–383. doi: 10.1097/FJC.0b013e31804a5e34. [DOI] [PubMed] [Google Scholar]
- Kulkarni SS, Kompella UB. Potential drug delivery approaches for XFS-associated and XFS-associated glaucoma. Journal of glaucoma. 2014;23:S77–79. doi: 10.1097/IJG.0000000000000109. [DOI] [PubMed] [Google Scholar]
- Langert KA, Pervan CL, Stubbs EB., Jr. Novel role of Cdc42 and RalA GTPases in TNF-alpha mediated secretion of CCL2. Small GTPases. 2014;5 doi: 10.4161/sgtp.29260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langert KA, Von Zee CL, Stubbs EB. Tumour necrosis factor alpha enhances CCL2 and ICAM-1 expression in peripheral nerve microvascular endoneurial endothelial cells. ASN Neuro. 2013a;5 doi: 10.1042/AN20120048. art:e00104.doi:00110.01042/AN20120048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langert KA, Von Zee CL, Stubbs EB., Jr. Cdc42 GTPases facilitate TNF-alpha-mediated secretion of CCL2 from peripheral nerve microvascular endoneurial endothelial cells. J Peripher Nerv Syst. 2013b;18:199–208. doi: 10.1111/jns5.12032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XL, Dou YC, Liu Y, Shi CW, Cao LL, Zhang XQ, Zhu J, Duan RS. Atorvastatin ameliorates experimental autoimmune neuritis by decreased Th1/Th17 cytokines and up-regulated T regulatory cells. Cellular immunology. 2011;271:455–461. doi: 10.1016/j.cellimm.2011.08.015. [DOI] [PubMed] [Google Scholar]
- Lin R, Liu J, Peng N, Gan W, Wang W, Han C, Ding C. Lovastatin reduces apoptosis and downregulates the CD40 expression induced by TNF-alpha in cerebral vascular endothelial cells. Current neurovascular research. 2006;3:41–47. doi: 10.2174/156720206775541796. [DOI] [PubMed] [Google Scholar]
- Lindenbaum Y, Kissel JT, Mendell JR. Treatment approaches for Guillain-Barre syndrome and chronic inflammatory demyelinating polyradiculoneuropathy. Neurologic clinics. 2001;19:187–204. doi: 10.1016/s0733-8619(05)70012-8. [DOI] [PubMed] [Google Scholar]
- Maher BM, Dhonnchu TN, Burke JP, Soo A, Wood AE, Watson RW. Statins alter neutrophil migration by modulating cellular Rho activity--a potential mechanism for statins-mediated pleotropic effects? Journal of leukocyte biology. 2009;85:186–193. doi: 10.1189/jlb.0608382. [DOI] [PubMed] [Google Scholar]
- Mao XJ, Zhang XM, Zhang HL, Quezada HC, Mix E, Yang X, Winblad B, Adem A, Zhu J. TNF-alpha receptor 1 deficiency reduces antigen-presenting capacity of Schwann cells and ameliorates experimental autoimmune neuritis in mice. Neuroscience letters. 2010;470:19–23. doi: 10.1016/j.neulet.2009.12.045. [DOI] [PubMed] [Google Scholar]
- McAlvin JB, Reznor G, Shankarappa SA, Stefanescu CF, Kohane DS. Local toxicity from local anesthetic polymeric microparticles. Anesthesia and analgesia. 2013;116:794–803. doi: 10.1213/ANE.0b013e31828174a7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCall RL, Sirianni RW. PLGA nanoparticles formed by single- or double-emulsion with vitamin E-TPGS. J Vis Exp. 2013:51015. doi: 10.3791/51015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCombe PA, Csurhes PA. T cells from patients with Guillain-Barre syndrome produce interferon-gamma in response to stimulation with the ganglioside GM1. Journal of clinical neuroscience : official journal of the Neurosurgical Society of Australasia. 2010;17:537–538. doi: 10.1016/j.jocn.2009.07.096. [DOI] [PubMed] [Google Scholar]
- McGrogan A, Madle GC, Seaman HE, de Vries CS. The epidemiology of Guillain-Barre syndrome worldwide. A systematic literature review. Neuroepidemiology. 2009;32:150–163. doi: 10.1159/000184748. [DOI] [PubMed] [Google Scholar]
- McLaughlin LA, Ronseaux S, Finn RD, Henderson CJ, Roland Wolf C. Deletion of microsomal cytochrome b5 profoundly affects hepatic and extrahepatic drug metabolism. Molecular pharmacology. 2010;78:269–278. doi: 10.1124/mol.110.064246. [DOI] [PubMed] [Google Scholar]
- Panyam J, Williams D, Dash A, Leslie-Pelecky D, Labhasetwar V. Solid-state solubility influences encapsulation and release of hydrophobic drugs from PLGA/PLA nanoparticles. Journal of pharmaceutical sciences. 2004;93:1804–1814. doi: 10.1002/jps.20094. [DOI] [PubMed] [Google Scholar]
- Pihl-Jensen G, Tsakiri A, Frederiksen JL. Statin treatment in multiple sclerosis: a systematic review and meta-analysis. CNS drugs. 2015;29:277–291. doi: 10.1007/s40263-015-0239-x. [DOI] [PubMed] [Google Scholar]
- Romana B, Batger M, Prestidge CA, Colombo G, Sonvico F. Expanding the therapeutic potential of statins by means of nanotechnology enabled drug delivery systems. Current topics in medicinal chemistry. 2014;14:1182–1193. doi: 10.2174/1568026614666140329232252. [DOI] [PubMed] [Google Scholar]
- Sarkey JP, Richards MP, Stubbs EB., Jr. Lovastatin attenuates nerve injury in an animal model of Guillain-Barre syndrome. J Neurochem. 2007;100:1265–1277. doi: 10.1111/j.1471-4159.2006.04309.x. [DOI] [PubMed] [Google Scholar]
- Sejvar JJ, Baughman AL, Wise M, Morgan OW. Population incidence of Guillain-Barre syndrome: a systematic review and meta-analysis. Neuroepidemiology. 2011;36:123–133. doi: 10.1159/000324710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen P, Shiao M, Chung H, Lee K, Chao Y, Hunt V. Liquid Chromatographic Determination of 3-Hydroxy-3-methylglutaryl Coenzyme A Reductase Inhibitors. Journal of the Chinese Chemical Society. 1996;43:451–457. [Google Scholar]
- Shin T, Ahn M, Matsumoto Y, Moon C. Mechanism of experimental autoimmune neuritis in Lewis rats: the dual role of macrophages. Histology and histopathology. 2013;28:679–684. doi: 10.14670/HH-28.679. [DOI] [PubMed] [Google Scholar]
- Sirtori CR. The pharmacology of statins. Pharmacological research. 2014;88:3–11. doi: 10.1016/j.phrs.2014.03.002. [DOI] [PubMed] [Google Scholar]
- Sorensen PS, Lycke J, Eralinna JP, et al. Simvastatin as add-on therapy to interferon beta-1a for relapsing-remitting multiple sclerosis (SIMCOMBIN study): a placebo-controlled randomised phase 4 trial. The Lancet. Neurology. 2011;10:691–701. doi: 10.1016/S1474-4422(11)70144-2. [DOI] [PubMed] [Google Scholar]
- Stach K, Nguyen XD, Lang S, Elmas E, Weiss C, Borggrefe M, Fischer J, Kalsch T. Simvastatin and atorvastatin attenuate VCAM-1 and uPAR expression on human endothelial cells and platelet surface expression of CD40 ligand. Cardiology journal. 2012;19:20–28. doi: 10.5603/cj.2012.0005. [DOI] [PubMed] [Google Scholar]
- Stanislaus R, Gilg AG, Singh AK, Singh I. Immunomodulation of experimental autoimmune encephalomyelitis in the Lewis rats by Lovastatin. Neurosci Lett. 2002;333:167–170. doi: 10.1016/s0304-3940(02)00943-6. [DOI] [PubMed] [Google Scholar]
- Stanislaus R, Singh AK, Singh I. Lovastatin treatment decreases mononuclear cell infiltration into the CNS of Lewis rats with experimental allergic encephalomyelitis. J Neurosci Res. 2001;66:155–162. doi: 10.1002/jnr.1207. [DOI] [PubMed] [Google Scholar]
- Stubbs EB, Jr., Von Zee CL. Prenylation of Rho G-proteins: a novel mechanism regulating gene expression and protein stability in human trabecular meshwork cells. Molecular neurobiology. 2012;46:28–40. doi: 10.1007/s12035-012-8249-x. [DOI] [PubMed] [Google Scholar]
- Takeuchi S, Kawashima S, Rikitake Y, Ueyama T, Inoue N, Hirata K, Yokoyama M. Cerivastatin suppresses lipopolysaccharide-induced ICAM-1 expression through inhibition of Rho GTPase in BAEC. Biochemical and biophysical research communications. 2000;269:97–102. doi: 10.1006/bbrc.2000.2238. [DOI] [PubMed] [Google Scholar]
- Von Zee CL, Richards MP, Bu P, Perlman JI, Stubbs EB., Jr. Increased RhoA and RhoB protein accumulation in cultured human trabecular meshwork cells by lovastatin. Investigative ophthalmology & visual science. 2009;50:2816–2823. doi: 10.1167/iovs.08-2466. [DOI] [PubMed] [Google Scholar]
- Willison HJ. The immunobiology of Guillain-Barre syndromes. J Peripher Nerv Syst. 2005;10:94–112. doi: 10.1111/j.1085-9489.2005.0010202.x. [DOI] [PubMed] [Google Scholar]
- Wischke C, Schwendeman SP. Principles of encapsulating hydrophobic drugs in PLA/PLGA microparticles. International journal of pharmaceutics. 2008;364:298–327. doi: 10.1016/j.ijpharm.2008.04.042. [DOI] [PubMed] [Google Scholar]
- Wu K, Tian S, Zhou H, Wu Y. Statins protect human endothelial cells from TNF-induced inflammation via ERK5 activation. Biochemical pharmacology. 2013;85:1753–1760. doi: 10.1016/j.bcp.2013.04.009. [DOI] [PubMed] [Google Scholar]
- Xiao J, Zhai H, Yao Y, Wang C, Jiang W, Zhang C, Simard AR, Zhang R, Hao J. Chrysin attenuates experimental autoimmune neuritis by suppressing immuno-inflammatory responses. Neuroscience. 2014;262:156–164. doi: 10.1016/j.neuroscience.2014.01.004. [DOI] [PubMed] [Google Scholar]
- Yang DJ, Hwang LS. Study on the conversion of three natural statins from lactone forms to their corresponding hydroxy acid forms and their determination in Pu-Erh tea. Journal of chromatography. A. 2006;1119:277–284. doi: 10.1016/j.chroma.2005.12.031. [DOI] [PubMed] [Google Scholar]
- Ye LY, Firby PS, Moore MJ. Determination of lovastatin in human plasma using reverse-phase high-performance liquid chromatography with UV detection. Therapeutic drug monitoring. 2000;22:737–741. doi: 10.1097/00007691-200012000-00014. [DOI] [PubMed] [Google Scholar]
- Yuan F, Yosef N, Lakshmana Reddy C, Huang A, Chiang SC, Tithi HR, Ubogu EE. CCR2 gene deletion and pharmacologic blockade ameliorate a severe murine experimental autoimmune neuritis model of Guillain-Barre syndrome. PloS one. 2014;9:e90463. doi: 10.1371/journal.pone.0090463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaghloul AA, Mustafa F, Siddiqu A, Khan M. Biodegradable microparticulates of beta-estradiol: preparation and in vitro characterization. Drug development and industrial pharmacy. 2005;31:803–811. doi: 10.1080/03639040500217624. [DOI] [PubMed] [Google Scholar]
- Zapolska-Downar D, Siennicka A, Kaczmarczyk M, Kolodziej B, Naruszewicz M. Simvastatin modulates TNFalpha-induced adhesion molecules expression in human endothelial cells. Life sciences. 2004;75:1287–1302. doi: 10.1016/j.lfs.2004.03.005. [DOI] [PubMed] [Google Scholar]
- Zhang HL, Zheng XY, Zhu J. Th1/Th2/Th17/Treg cytokines in Guillain-Barre syndrome and experimental autoimmune neuritis. Cytokine & growth factor reviews. 2013;24:443–453. doi: 10.1016/j.cytogfr.2013.05.005. [DOI] [PubMed] [Google Scholar]
- Zhang ZY, Zhang Z, Zug C, Nuesslein-Hildesheim B, Leppert D, Schluesener HJ. AUY954, a selective S1P(1) modulator, prevents experimental autoimmune neuritis. Journal of neuroimmunology. 2009;216:59–65. doi: 10.1016/j.jneuroim.2009.09.010. [DOI] [PubMed] [Google Scholar]