

Abstract
There is growing appreciation that cellular metabolic and bioenergetic pathways do not play merely passive roles in activated leukocytes. Rather, metabolism plays important roles in controlling cellular activation, differentiation, survival and effector function. Much of this work has been performed in T cells; however, there is still very little information regarding mast cell metabolic reprogramming and its effect on cellular function. Mast cells perform important barrier functions and help control type 2 immune responses. Here we show here that murine bone marrow derived mast cells rapidly alter their metabolism in response to stimulation through the Fc receptor for IgE (FcεRI). We also demonstrate that specific metabolic pathways appear to be differentially required for control of mast cell function. Manipulation of metabolic pathways may represent a novel point for manipulation of mast cell activation.
Introduction
Mast cells are important leukocytes at the interface of innate and adaptive (particularly type 2) immunity (1). These cells play critical roles in the expulsion of parasitic worms, by virtue of their sensitivity to antigenic crosslinking of IgE pre-bound to FcεRI-bearing mast cells. Mast cells are also involved in the pathology caused by dysregulated type 2 immunity associated with many allergic or atopic diseases. In response to FcεRI crosslinking by Ag and IgE, mast cells are rapidly activated. This is a multi-phasic response, including early release of granules that contain pre-packaged histamine and TNF-α, and later phases that include de novo formation of lipid mediators and induction of transcription that leads to production of multiple cytokines, chemokines and other effector molecules (2, 3).
While metabolism and nutrient sensing represent key pathways that govern cellular homeostasis, it is now clear that, especially in leukocytes, these pathways interest heavily with cellular fate and function (4). This has been most extensively studied in T cells, which undergo dynamic and complex metabolic reprogramming in response to activation, cytokine stimulation, and other changes in their microenvironment (5). Metabolic manipulation can lead to substantial alterations not only in acute lymphocyte effector function, but also the differentiation of helper subsets and memory responses (6). Similar themes have been explored in natural killer, B, and dendritic cells, which also have complex differentiation and effector program changes. By contrast, there is still limited information on how metabolism is modulated in post-mitotic, terminally differentiated effector cell types, like antigen-stimulated primary murine mast cells (7–11).
Here we show mast cells, too, utilize metabolic reprogramming to influence their effector function. Activation of mast cells through the FcεR results in rapid induction of glycolysis reminiscent of immediate reprogramming induced by lymphocyte activation. Modulation of downstream metabolic pathways results in inhibition of certain, but not all, effector functions.
Materials and Methods
Bone marrow derived mast cell culture
Bone marrow derived mast cells (BMMCs) were generated by culturing bone marrow from C57BL/6 or Nur77GFP mice for 4–6 weeks in IL-3 supplemented media, after which cells were at least 90% positive for the growth factor receptor c-kit and the multi-chain activating receptor FcεRI.
Seahorse metabolic flux assays
For direct in-Seahorse measurement of the immediate effects of Ag stimulation (Fig. 1), BMMCs (100,000/well) were pre-sensitized for three hours with 1 μg/ml DNP-specific IgE (clone SPE-7, Sigma). After 30 min of basal measurements, indicated amounts of high (DNP32-HSA) or low (DNP5-BSA) valency antigen were injected using the Seahorse metabolic flux analyzer and metabolic readings were taken as indicated. For Seahorse metabolic “stress tests” (Fig. 2), BMMCs were IgE-sensitized (1 μg/ml) overnight and stimulated with indicated amounts of high (DNP32-HSA) or low (DNP5-BSA) valency antigens for 2.5 hours, prior to Seahorse metabolic flux analysis, as described (7). Basal measurements were taken for 30 minutes prior to sequential injections of 1 μM oligomycin, which inhibits mitochondrial ATP production and stimulates glycolysis, 0.5 μM FCCP, which uncouples the respiratory chain and stimulates maximal oxygen consumption, and 0.5 μM rotenone/antimycin A, which inhibits complex I and complex III, respectively, to inhibit mitochondrial oxygen consumption altogether. Seahorse media included 2 mM glucose and 2 mM glutamine in minimal, unbuffered, DMEM.
Figure 1.

IgE/Ag-activated mast cells undergo a rapid increase in glycolysis. (A–B) WT BMMCs were sensitized with IgE for three hours and activated in-Seahorse with the indicated concentrations of DNP32 and DNP5 for two hours. Data in panel A show the change in glycolysis (ECAR), while panel B shows mitochondrial respiration (OCR). (C–D) WT and Tim-3 KO BMMCs were sensitized with IgE for three hours and stimulated with indicated antigens directly in-Seahorse for two hours. Arrow indicated when antigen was injected. Data are representative of two (E, F) and three independent (A–D) experiments.
Figure 2.
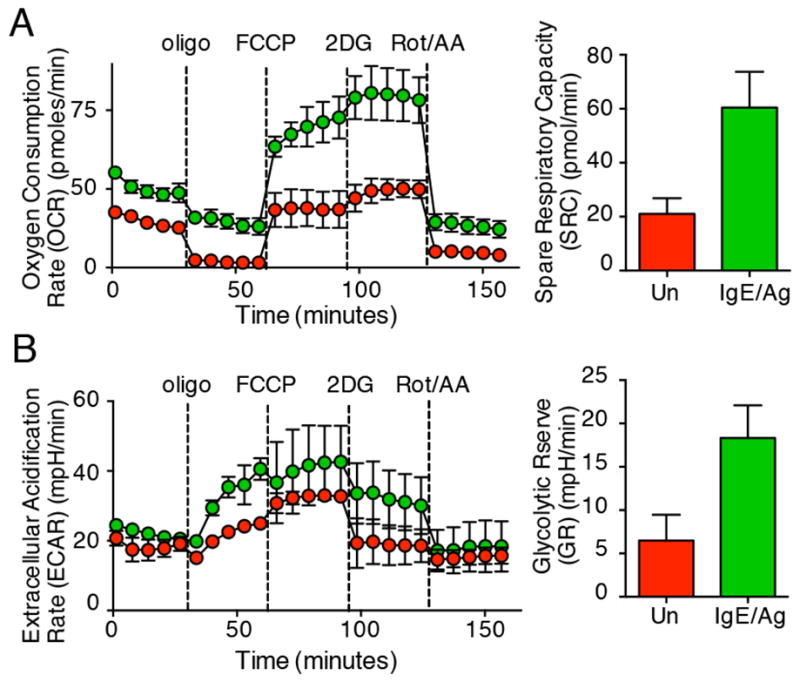
Pre-stimulated mast cells increase both glycolytic potential and mitochondrial respiration. WT BMMCs were sensitized with 1μg/ml IgE overnight and stimulated with DNP32 (high) and DNP5 (low) valency for 2.5 hours prior to stress test by Seahorse flux analyzer. (A) Spare respiratory capacity (SRC) was calculated as the difference between FCCP-uncoupled and basal OCR, while in (B) glycolytic reserve (GR) was calculated as the difference between oligo-stimulated and basal ECAR. Results are representative of three independent experiments.
Mast cell functional assays
Mast cell degranulation, cytokine production and Nur77GFP induction were carried out as previously described (12). Briefly, cells were pre-loaded with anti-DNP IgE (1 μg/ml) overnight. The next day, cells were washed and pre-incubated for 30 minutes with the indicated inhibitors, before stimulating the cells with 50 ng/ml DNP32-HSA. For degranulation, a flow cytometry based assay was used (12). This short-term assay relies not only on PS exposure as vesicles are fused with the plasma membrane (read-out by Annexin V staining), but also on a concomitant decrease in Lysotracker staining, which also occurs when exocytic granules fuse with the plasma membrane. Cells were monitored for cell death, using an exclusion dye (Ghost Dye, Tonbo Biosciences), and only live cells were analyzed for degranulation.
Results and Discussion
We set out to measure the relative state of the major bioenergetic pathways in mast cells, before and after activation of the cells by IgE and antigen (Ag). We first wanted to determine whether antigen crosslinking triggers immediate changes in metabolic responses in mast cells. As shown by others (13), and in our recent paper (12), mast cell function is linked to FcεRI signal strength in a complex fashion, through the Src family kinase Lyn. Stimulation with a high-valency antigen leads to initially strong signaling, followed by rapid down-regulation, due to recruitment of inhibitory phosphatases to the FcεRI signalosome. By contrast, low valency antigen only engages the positive effects of Lyn, leading to initially weaker signaling that is not subject to the same negative feedback, resulting in more sustained signaling. Thus, when we directly stimulated IgE-sensitized bone marrow derived mast cells (BMMCs) “in-Seahorse” with high- vs. low-valency antigen, we observed that the former (DNP32) led to an immediate, robust increase in glycolysis, which peaked within ten minutes and persisted for over two hours. However, stimulation with lower valency antigen (DNP5) did not result in a significant change in glycolysis (Fig. 1A). Antigen concentration was equally important in engaging this glycolytic switch (Fig. 1A). Antigen cross-linking did not immediately alter the state of oxidative phosphorylation in mast cells, as OCR was relatively unchanged despite a small decrease upon antigen injection (Fig. 1B). As shown in our recent report (12), signal strength through FcεRI can also be modulated by the receptor Tim-3, so we asked whether the Tim-3 expression could also modulate the acute changes in metabolic flux that we observed in mast cells. Thus, high antigen valency stimulated an immediate increase in glycolysis in WT BMMCs, which did not occur with low valency antigen and was also reduced in Tim3-deficient BMMCs (Fig. 1C), while again OCR was unaffected (Fig. 1D). Interestingly, the sensitivity of increased glycolysis, as read-out by ECAR, was also reflected in a detectable, although delayed, increase in ECAR when mast cells were treated with IgE alone, in the absence of Ag (Supplemental Fig. 1A). However, no increase in either ECAR or OCR was noted in the complete absence of Ag and IgE (Supplemental Fig. 1A–B).
Next, we measured both glycolysis and oxidative phosphorylation of un-manipulated mast cells, or cells that were pre-stimulated for 2.5 hours with IgE/Ag. Thus, we subjected BMMCs from normal mice to metabolic “stress tests,” using the Seahorse metabolic flux analyzer. As shown in Figure 2, mast cells that were pre-stimulated with IgE/Ag (green symbols) increased both their ability to carry out oxidative phosphorylation (as determined by oxygen consumption rate (OCR); Fig. 2A) and spare respiratory capacity (SRC)) as well as their capacity for glycolysis, determined by the extracellular acidification rate (ECAR) and glycolytic reserve (GR) (Fig. 2B). Thus, pre-activation of mast cells results in a broader increase in the metabolic potential (as determined by SRC and GR) of these cells, compared with what we observed upon acute stimulation in Figure 1. Overall, the results in Figures 1–2 indicate that a rapid glycolytic switch in mast cells is closely associated with intensity of antigen receptor signaling and co-stimulation, e.g. through Tim-3. Furthermore, results in Figure 1 suggest that the change in mitochondrial respiration shown in Figure 2A (occurring after several hours of pre-stimulation) requires more complex, e.g. transcriptional, reprogramming, since it did not occur with acute “in-Seahorse” stimulation.
We next determined whether downstream metabolic pathways are important for mast cell effector function in response to stimulation with IgE/Ag. We took advantage of well-characterized pharmacological inhibitors of key metabolic enzymes. First we employed dichloroacetate (DCA), which inhibits pyruvate dehydrogenase kinase (PDHK), which itself phosphorylates and inhibits pyruvate dehydrogenase (14). Thus, DCA treatment increases the conversion of pyruvate into acetyl CoA, at the expense of lactate production, while preserving oxidative phosphorylation. As shown in Figure 3A, DCA inhibited mast cell degranulation at non-toxic doses of this compound (Fig. 3A–B). Production of IL-6 by mast cells in response to IgE/Ag stimulation was also inhibited by DCA in a similar dose range (Fig. 3D). However, these effects did not appear to be due to general inhibition of FcεRI signaling, since activation of a transgenic Nur77GFP reporter, a transcriptional readout for calcium-dependent signaling in mast cells and lymphocytes (12, 15), was not affected by doses of DCA that significantly inhibited both cytokine production and degranulation (Fig. 3C). Another compound that results in a relative shift from glycolysis to oxidative phosphorylation is 2-deoxyglucose (2-DG), which inhibits hexokinase, a key early enzyme in the glycolytic pathway. Although 2-DG was not as potent as DCA, it nevertheless led to a modest, but statistically significant, decrease in both degranulation and cytokine production upon activation of mast cells with IgE/Ag (Supplemental Fig. 2A–B).
Figure 3.

Mast cells require glycolysis for Ag-induced degranulation and cytokine production. (A–B) WT BMMCs were loaded with Lysotracker Deep Red and sensitized with IgE prior to stimulation with Ag alone, Ag together with DMSO or the indicated concentrations of DCA. Degranulation was assessed by flow cytometry analysis of loss of positive Annexin V staining and decreased Lysotracker staining. A representative degranulation assay is shown in panel A, and the combined results of three experiments are shown in panel B. (C) BMMCs generated from Nur77GFP Tg mice were sensitized with IgE and stimulated with antigen, plus varying concentrations of DCA, or with vehicle control. Nur77GFP expression was measured as an indication of antigen-induced FcεRI activation. Line colors correspond to the bars in panel D. (D) IL-6 release was analyzed by ELISA of culture supernatant harvested after six hours of stimulation. Results are representative of three independent experiments. *p<0.05; **p<0.005.
To assess the functional role of mitochondrial respiration in mast cell activation we used rotenone, an inhibitor of the mitochondrial electron transport chain (16). Thus, titrating in doses of rotenone that were not toxic to mast cells (not shown), we observed that rotenone significantly inhibited antigen-stimulated cytokine (IL-6) production by bone marrow mast cells (Fig. 4A). In addition, IgE/Ag-induced degranulation of mast cells was also inhibited in a similar dose range (Fig. 4B). However, despite inhibition of both these functions, induction of the Nur77GFP transcriptional reporter was not inhibited by rotenone at theses doses (data not shown). Combined inhibition of both metabolic pathways further reduced the ability of mast cell to degranulate (Fig 4D) and to produce cytokines (Fig. 4C). These results suggest that mast cells require both glycolysis and oxidative phosphorylation for their immediate and late-phase responses. However, despite a role for general mitochondrial respiration, fatty acid oxidation appears to not be required for mast cell activation, since etomoxir, which inhibits this process (17), did not affect IgE/Ag-induced Nur77GFP induction (Fig. 5B), nor production of IL-6 (Fig. 5A) or degranulation (Fig. 5C).
Figure 4.

Mast cells require oxidative phosphorylation for Ag-induced degranulation and cytokine production. (A) WT BMMCs were stimulated as shown, together with DMSO or the indicated concentrations of Rotenone. IL-6 release was analyzed by ELISA of culture supernatant harvested after six hours of stimulation. (B) WT BMMCs were loaded with Lysotracker Deep Red and sensitized with IgE prior to stimulation with Ag alone, Ag. Degranulation was assessed by flow cytometry analysis of loss of positive Annexin V staining and decreased Lysotracker staining. (C–D) IL-6 secretion and degranulation were measured as above, in the presence of DCA or Rotenone alone, or the two together. The combined results of three experiments are shown. *p<0.05; **p<0.005.
Figure 5.
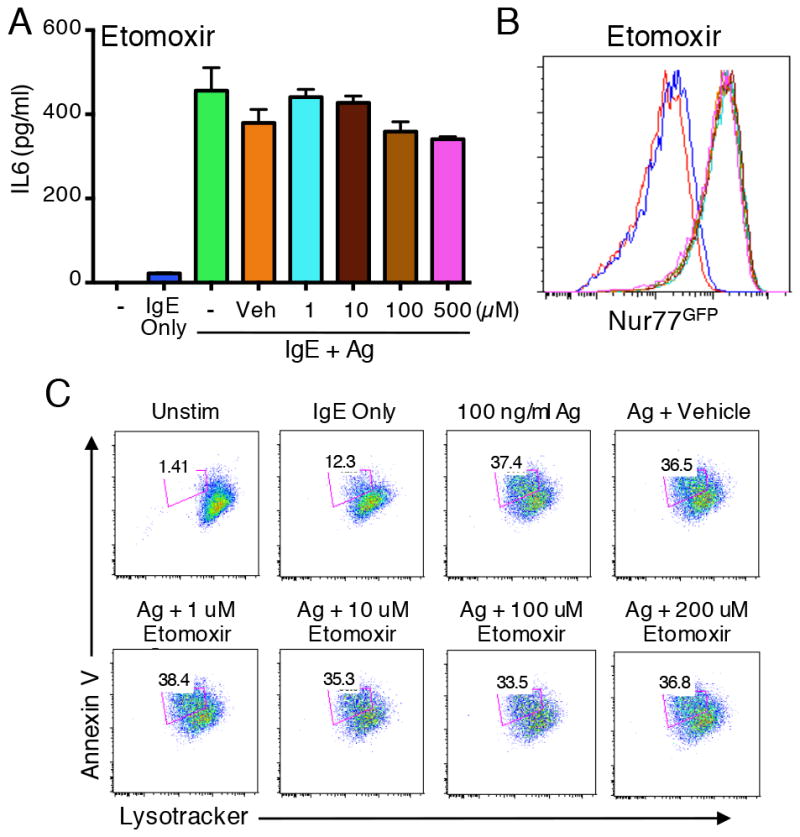
Fatty acid oxidation is dispensable for mast cell activation. (A) WT BMMCs were stimulated as shown, together with DMSO or the indicated concentrations of etomoxir. IL-6 release was analyzed by ELISA of culture supernatants harvested after six hours of stimulation. (B) BMMCs generated from Nur77GFP Tg mice were sensitized with IgE and stimulated with antigen, along with varying concentrations of etomoxir, or with vehicle control. Nur77GFP expression was measured as a readout of antigen-induced FcεRI activation. Line colors correspond to the bars in panel A. (C) WT BMMCs were loaded with Lysotracker Deep Red and sensitized with IgE prior to stimulation as indicated. Degranulation was assessed by flow cytometry analysis of loss of positive Annexin V staining and decreased Lysotracker staining. *p<0.05; **p<0.005.
In summary, we have demonstrated here that the activation of mast cells by IgE and antigen is associated with a rapid increase in glycolysis, but not necessarily in mitochondrial respiration, effects reminiscent of what has been reported for T cells (18, 19). Nonetheless, while not connected with immediate mast cell activation, oxidative phosphorylation is important for mast cell effector function, since its inhibition drastically decreases degranulation and cytokine production. Intriguingly, our data suggest that these metabolic changes are actively induced by mast cells to carry out specific tasks. Our findings are intriguing in light of the observations that mast cell mitochondria translocate to the site of exocytosis during degranulation (20), and that mitochondrial STAT3 is responsible for induction of mitochondrial oxidative phosphorylation, ATP synthesis and subsequent degranulation (21).
Notably, the mast cell metabolic reprogramming that we observed, both glycolytic and oxidative, appeared to be dispensable for upregulation of Nur77, a direct readout of activation downstream of FcεRI. This suggests that mast cells utilize these metabolic pathways to modulate and amplify the cellular immune response, but they are not obligate for activation. In T cells, translation of IFN-gamma, for instance, is directly influenced by the glycolytic machinery (7). Since mast cells are terminally differentiated, they may utilize these metabolic pathways to further shape their cellular responses, to accommodate changes in the environment. Importantly, barrier surfaces are metabolically distinct, having decreased oxygen tension as well as lower nutrient levels, especially at sites of inflammation. Thus, metabolic intervention may represent a strategy to modulate mast cell effector function while preserving their differentiation status and cellularity at barrier sites.
Supplementary Material
Acknowledgments
B.P., L.A., G.M.D. and L.P.K. designed and analyzed experiments. B.P., L.A. and A.V.M. performed experiments. B.P. and L.P.K. wrote the manuscript, and G.M.D. edited the manuscript. The authors declare that they have no conflicts of interest to disclose.
Footnotes
This work was supported by PHS grant R56AI067544 (to L.P.K.) and by the Sidney Kimmel Foundation for Cancer Research (SKF-015-039, to G.M.D.).
References
- 1.Voehringer D. Protective and pathological roles of mast cells and basophils. Nature reviews Immunology. 2013;13:362–375. doi: 10.1038/nri3427. [DOI] [PubMed] [Google Scholar]
- 2.Gilfillan AM, Rivera J. The tyrosine kinase network regulating mast cell activation. Immunological reviews. 2009;228:149–169. doi: 10.1111/j.1600-065X.2008.00742.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Metcalfe DD, Peavy RD, Gilfillan AM. Mechanisms of mast cell signaling in anaphylaxis. The Journal of allergy and clinical immunology. 2009;124:639–646. doi: 10.1016/j.jaci.2009.08.035. quiz 647–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pearce EL, Poffenberger MC, Chang CH, Jones RG. Fueling immunity: insights into metabolism and lymphocyte function. Science. 2013;342:1242454. doi: 10.1126/science.1242454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Delgoffe GM, Powell JD. Feeding an army: The metabolism of T cells in activation, anergy, and exhaustion. Molecular immunology. 2015;68:492–496. doi: 10.1016/j.molimm.2015.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pollizzi KN, Powell JD. Integrating canonical and metabolic signalling programmes in the regulation of T cell responses. Nature reviews Immunology. 2014;14:435–446. doi: 10.1038/nri3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chang CH, Curtis JD, Maggi LB, Jr, Faubert B, Villarino AV, O’Sullivan D, Huang SC, van der Windt GJ, Blagih J, Qiu J, Weber JD, Pearce EJ, Jones RG, Pearce EL. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell. 2013;153:1239–1251. doi: 10.1016/j.cell.2013.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kitahata Y, Nunomura S, Terui T, Ra C. Prolonged culture of mast cells with high-glucose medium enhances the Fc epsilon RI-mediated degranulation response and leukotriene C4 production. International archives of allergy and immunology. 2010;152(Suppl 1):22–31. doi: 10.1159/000312122. [DOI] [PubMed] [Google Scholar]
- 9.Lowman MA, Benyon RC, Church MK. Characterization of neuropeptide-induced histamine release from human dispersed skin mast cells. British journal of pharmacology. 1988;95:121–130. doi: 10.1111/j.1476-5381.1988.tb16555.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ryu H, Walker JK, Kim S, Koo N, Barak LS, Noguchi T, Kang BY, Kim KM. Regulation of M2-type pyruvate kinase mediated by the high-affinity IgE receptors is required for mast cell degranulation. British journal of pharmacology. 2008;154:1035–1046. doi: 10.1038/bjp.2008.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sumbayev VV, Yasinska I, Oniku AE, Streatfield CL, Gibbs BF. Involvement of hypoxia-inducible factor-1 in the inflammatory responses of human LAD2 mast cells and basophils. PloS one. 2012;7:e34259. doi: 10.1371/journal.pone.0034259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Phong BL, Avery L, Sumpter TL, Gorman JV, Watkins SC, Colgan JD, Kane LP. Tim-3 enhances FcεRI-proximal signaling to modulate mast cell activation. The Journal of experimental medicine. 2015:212. doi: 10.1084/jem.20150388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xiao W, Nishimoto H, Hong H, Kitaura J, Nunomura S, Maeda-Yamamoto M, Kawakami Y, Lowell CA, Ra C, Kawakami T. Positive and negative regulation of mast cell activation by Lyn via the FcepsilonRI. Journal of immunology. 2005;175:6885–6892. doi: 10.4049/jimmunol.175.10.6885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gerriets VA, Kishton RJ, Nichols AG, Macintyre AN, Inoue M, Ilkayeva O, Winter PS, Liu X, Priyadharshini B, Slawinska ME, Haeberli L, Huck C, Turka LA, Wood KC, Hale LP, Smith PA, Schneider MA, MacIver NJ, Locasale JW, Newgard CB, Shinohara ML, Rathmell JC. Metabolic programming and PDHK1 control CD4+ T cell subsets and inflammation. The Journal of clinical investigation. 2015;125:194–207. doi: 10.1172/JCI76012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zikherman J, Parameswaran R, Weiss A. Endogenous antigen tunes the responsiveness of naive B cells but not T cells. Nature. 2012;489:160–164. doi: 10.1038/nature11311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Onishi T. Mechanism of electron transport and energy conservation in the site I region of the respiratory chain. Biochimica et biophysica acta. 1973;301:105–128. [PubMed] [Google Scholar]
- 17.Ratheiser K, Schneeweiss B, Waldhausl W, Fasching P, Korn A, Nowotny P, Rohac M, Wolf HP. Inhibition by etomoxir of carnitine palmitoyltransferase I reduces hepatic glucose production and plasma lipids in non-insulin-dependent diabetes mellitus. Metabolism. 1991;40:1185–1190. doi: 10.1016/0026-0495(91)90214-h. [DOI] [PubMed] [Google Scholar]
- 18.MacIver NJ, Michalek RD, Rathmell JC. Metabolic regulation of T lymphocytes. Annual review of immunology. 2013;31:259–283. doi: 10.1146/annurev-immunol-032712-095956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pearce EL, Pearce EJ. Metabolic pathways in immune cell activation and quiescence. Immunity. 2013;38:633–643. doi: 10.1016/j.immuni.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang B, Alysandratos KD, Angelidou A, Asadi S, Sismanopoulos N, Delivanis DA, Weng Z, Miniati A, Vasiadi M, Katsarou-Katsari A, Miao B, Leeman SE, Kalogeromitros D, Theoharides TC. Human mast cell degranulation and preformed TNF secretion require mitochondrial translocation to exocytosis sites: relevance to atopic dermatitis. The Journal of allergy and clinical immunology. 2011;127:1522–1531 e1528. doi: 10.1016/j.jaci.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Erlich TH, Yagil Z, Kay G, Peretz A, Migalovich-Sheikhet H, Tshori S, Nechushtan H, Levi-Schaffer F, Saada A, Razin E. Mitochondrial STAT3 plays a major role in IgE-antigen-mediated mast cell exocytosis. The Journal of allergy and clinical immunology. 2014;134:460–469. doi: 10.1016/j.jaci.2013.12.1075. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.