

Abstract
The human gut contains trillions of commensal bacteria, and similar to pathogenic bacteria, the gut microbes and their products can be recognized by toll-like receptors (TLRs). It is well acknowledged that the interaction between gut microbiota and the local TLRs help to maintain the homeostasis of intestinal immunity. High-fat intake or obesity can weaken gut integrity leading to the penetration of gut microbiota or their bacterial products into the circulation, leading to the activation of TLRs on immune cells and subsequently low-grade systemic inflammation in host. Metabolic cells including hepatocytes and adipocytes also express TLRs. Although they are able to produce and secrete inflammatory molecules, the effectiveness remains low compared with the immune cells embedded in the liver and adipose tissue. The interaction of TLRs in these metabolic cells or organs with gut microbiota remains unclear, but a few studies have suggested that the functions of these TLRs are related to metabolism. Alteration of the gut microbiota is associated with body weight change and adiposity in human, and the interaction between the commensal gut microbiota and TLRs may possibly involve both metabolic and immunological regulation. In this review, we will summarize the current findings on the relationship between TLRs and gut microbiota with a focus on metabolic regulation and discuss how such interaction participates in host metabolism.
Keywords: Toll-like receptor, Gut microbiota, Metabolic regulation
Introduction
Trillions of commensal bacteria reside in our gastrointestinal tract, and the interactions between gut microbiota and the toll-like receptors (TLRs) on intestinal epithelial cells and immune cells help to maintain the homeostasis of our immune system [1]. TLRs are also expressed in hepatocytes and adipocytes. Although they are able to produce inflammatory molecules, the effectiveness remains low compared with the immune cells embedded in the liver and adipose tissue, rendering the function of TLRs in these cells elusive [2–4]. A few studies suggest that their function is metabolism-related [5]. Several global knockout models of TLRs or related pathways including TLR2, TLR5, interferon regulatory factor-3 (IRF3), and IRF5 which represent defects in immunity show an increase in body weight or fat mass regardless of other metabolic phenotypes [6–9]. The cell-specific knockout models also show the metabolic link, but the phenotypes are rather diverse. For example, hepatocyte-specific TLR4-knockout model shows an improvement in overall metabolic phenotypes upon high-fat diet challenge, but conversely, the same specific TLR5-knockout model displays an opposite phenotype including increased body weight, fatty liver, and fasting blood glucose [10, 11]. Inflammation mediated by TLR activation leads to downregulation of metabolism-related genes in the adipose tissue and liver [12]. Low-grade inflammation is often observed in obesity and metabolic diseases due to the increased gut permeability, and presumably, the penetration of molecules produced by gut microbiota can activate peripheral TLRs [13]. One of the functions of TLR pathway is to regulate intrinsic metabolism in immune cells in order to spare the energy for immune response [14]. Whether such energy relocation happens at an intercellular level or even cross-organ level remains unknown. The function of TLRs in innate and adaptive immunity and how TLRs modulate host immunity via the interaction with gut microbiota are reviewed elsewhere [15]. In this review, we mainly focus on the current findings of the relationship between TLRs and gut microbiota in terms of metabolic regulation and discuss how such interaction supports the hypothesis of intercellular energy relocation in the host, and its clinical implication in obesity and metabolic diseases.
Metabolic regulation by TLR pathway at cellular level
Glucose metabolism
It is well known that glycolysis plays a crucial role in macrophage polarization and dendritic cell activation. In the resting state, dendritic cells utilize lipid as their energy source through β-oxidation and oxidative phosphorylation [14, 16]. Engagement of TLR with its ligand activates the PI3K/Akt pathway and leads to a metabolic switch towards glycolysis to generate ATP [17]. Activation of the downstream kinases TBK1 and IKKε upon TLR binding induces phosphorylation of Akt, and activation of Akt triggers the enrichment of the rate-limiting enzyme for glycolysis, hexokinase-II, in the mitochondrial fraction and increases its activity [18]. During M1 macrophage polarization, a isoform switch of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (PFK2) from liver type (L-PFK2) to a more active ubiquitous type (u-PFK2) is observed, causing a higher glycolytic flux, and the switch is dependent on TLR (TLR2, 3, 4, and 9) pathway [19]. Conversely, during M2 polarization of macrophage in helminth infection, TLR2- and TLR4-dependent activation of MAPK cascade, and subsequently, CREB leads to IL-10 production and concurrent alteration of a series of metabolism-related genes including aconitase and ADP-dependent glucokinase [20]. Instead of aerobic glycolysis, the M2 macrophages rely on oxidative phosphorylation to utilize glucose as their energy source [20] (Fig. 1).
Fig. 1.
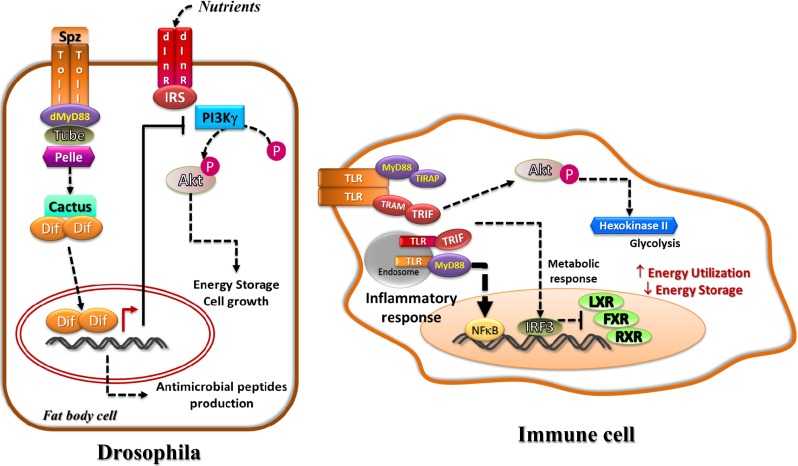
Intracellular energy relocation in fat body cell of Drosophila and immune cell of human. The fat body cell of Drosophila function as both metabolic and immune cell. Activation of toll and its adaptors, tube and dMyD88, results in the recruitment of Pelle kinase, subsequently inactivation of cactus (an orthologue of IκB), and the release of Dif (a transcription factor for antimicrobial genes). Simultaneously, insulin signaling is antagonized by the related proteins regulated by toll-induced Dif activation (left panel). Upon infection, activation of TLR in immune cell such as macrophage or dendritic cell stimulates inflammatory responses via the NFκB or IRF3 pathway, and IRF3 can also regulate metabolic response through interfering with the transcription activity of LXR, FXR, and RXR. In addition, phosphorylation of Akt mediated by the downstream kinases of TLR results in induction of glycolysis to generate ATP. Energy is being utilized to sustain the antimicrobial response (right panel)
Lipid metabolism
Activation of TLR3 and TLR4 in viral and bacterial infection suppresses the expression of liver X receptor (LXR)-dependent genes that regulate cholesterol efflux in macrophages [21]. The retained cholesterol acts as a reserve for the phagocytic process of macrophages [22], though in the case of atherosclerosis, it promotes foam cell formation [21, 23]. The activation of IRF3, a key downstream nuclear factor of TLR3 signaling, in response to viral infection on the one hand stimulates antiviral response through the production of interferons, and on the other hand suppresses metabolic response by downregulating retinoid X receptor-α (RXRα) [24]. RXRα can form heterodimers with other nuclear factors including peroxisome proliferator-activated receptor-γ (PPARγ), LXR, and farnesoid X receptor (FXR) which together construct a nuclear network regulating metabolism-related genes [25]. Such suppression is key to prevent viral assembly, as viruses can utilize host’s lipids to facilitate their own replication [26]. Injection of TLR3, TLR4, TLR5, TLR7, or TLR9 ligand in hepatitis B virus transgenic mice is shown to inhibit viral replication [27].
However, unlike acute response that inhibits storage and increases energy expenditure, chronic activation of TLR4 by subinfectious dose of LPS in macrophages facilitates fatty acid uptake and storage in the form of triglycerides with a parallel decrease in lipolysis and β-oxidation [28, 29]. The stimulated uptake and storage of triglycerides are also observed in adipose tissue macrophages during obesity, and the lipid accumulation is related to liposomal biogenesis [30]. The reason for a switch from glycolysis to lipid storage when low-grade inflammation sustains, and whether it is a physiological or pathophysiological phenotype is, however, unclear. It possibly serves as an adaptive mechanism to acquire an external source of energy to sustain inflammatory or antimicrobial responses, and prevent bacteria from utilizing pyruvate and acetyl-CoA for their growth, thereby limiting their growth.
Theory of energy relocation: from immunity to obesity
Fighting against infection requires high turnover of energy [18, 31]. It is hypothesized that organisms are able to free the energy from anabolism to immunological utilization upon infection. Such energy relocation has been reported in lower rank organisms; whereas in higher rank animals and human, the findings limit to the intrinsic behaviors within immune cells (Fig. 1) [32, 33]. The “toll” in TLR was originally derived from such protein found in Drosophila. The functions of TLR-related molecules in these lower organisms do not limit to immunity but also include embryonic development [34]. Toll locates at the fat body cells in Drosophila and facilitates biosynthetic and metabolic activities [34]. The fat body is analogous to the liver and adipose tissue in human [32]. However, the fat body cells not only store excess nutrient but also synthesize hemolymph proteins, circulating metabolites as well as antimicrobial peptides [32]. In Drosophila, activation of toll through genetic and fungal stimulation suppresses insulin signaling pathway resulting in decreased nutrient storage and growth, and sparing of energy for the induced immunity [33]. The fat body cell is a single compartment with multiple functions, and such intrinsic metabolic regulation by toll allows an internal shift of energy utilization from usual growth and storage to immunological activities.
As the complexity of biostructure increases along the evolution of higher rank organisms, the metabolic and immune cells/organs are separated in origin. The metabolic functions of TLR pathway have been observed in immune cells only, and such regulations remain to be immunity-related in higher animals and human (see previous section). Nonetheless, TLRs are expressed in non-immune cells including adipocytes, hepatocytes, and smooth muscle cells. Although the majority of studies reported that their functions are to produce inflammatory molecules, such kind of duplicate functions seems to be redundant owing to the embedded immune cells in these tissues. In fact, a few studies have shown the metabolic functions of TLR pathway in the metabolic organs and cells. For example, activation of TLR4 by lipopolysaccharides (LPS) suppresses the expression of phosphoenolpyruvate carboxykinase (PEPCK) in the liver and adipose tissue, and the authors suggest that such decrease would result in downregulated gluconeogenesis in the liver and lipogenesis in adipose tissue [12]. TLR4 pathway is also shown to inhibit lipogenesis in muscle during fasting, and TLR4-deficient mice display a significantly higher fat mass in fasted state compared with the wild type control [35]. Moreover, treatment with LPS in rat stimulates lipolysis in adipocytes in a TLR4-dependent manner [5] (Fig. 1). These findings are consistent with the outcome of intrinsic metabolic regulation by the TLR pathway that energy is released for immunological activities rather than being stored. However, a slight decrease in body weight and adiposity with improved inflammatory status is observed in TLR4-deficient mice fed a high saturated or monosaturated fat diet, which is against the molecular mechanism of TLR4-inhibited storage of energy [36]. It is important to note that inflammation induced by immune cells in obesity aggravates insulin resistance and metabolic defects. Such dilemma is possibly because the amelioration due to suppressed inflammatory response in immune cells outweighs the sequels of the absence of TLR-mediated catabolic events in metabolic cells during long-term overnutrition in the TLR4-knockout model. By contrast, another member, TLR5, is known to play a key role in regulating colonization of gut microbiota, and its knockout model shows a drastic increase in fat mass under both normal and high-fat diet compared with wild type [1, 7]. Unlike the TLR4-deficient model, TLR5-knockout mice show a higher serum IL-1β level (i.e., pro-inflammatory status) under high-fat diet compared with wild type, which indicates an absence of immunosuppressive effect. Therefore, the inhibition of catabolism observed in these TLR5-deficient mice dominates.
In other words, the systemic metabolic phenotypes observed in these TLR-knockout models would depend on the balance between the degree of altered inflammation and the direct metabolic functions of TLRs. This balance varies among different TLRs possibly because (1) the expressions of TLRs vary in the intestine, and their blockade would thus yield the penetration of different types and amount of gut microbial products [15]. (2) The types of immunological function can be affected by the subcellular location of TLRs. For example, endosomal TLRs such as TLR3, TLR7, and TLR9 and internalization of membrane TLR4 and TLR2 into endosome result in production of type I interferons which are usually considered as anti-inflammatory mediators [37, 38]. The production of anti-inflammatory mediators along with the distinct metabolic functions of TLRs may affect the overall metabolic phenotypes. (3) The TLR-mediated metabolic functions differ in degree in the liver, adipose tissue, or muscle due to the diversity in cell types and TLR expression in these organs, and ectopic accumulation of lipids or energy away from the normal storage sites possibly aggravates metabolic dysfunction. In order to make the concept of TLR agonism/antagonism pharmacologically applicable, it is important to comprehensively investigate the expressions of TLRs in different non-immune cells and organs. For example, understanding the roles of intestinal TLRs in blocking the penetration of bacterial products would allow us to predict the availability of TLR ligands to tissues in disease states. The evaluation of metabolic and immunological capacities of TLRs in different metabolic cells using cell-specific knockout models along with in vitro studies would give us insight on generation of specific TLR agonists or antagonists for different diseases [2, 39].
Interaction between gut microbiota and TLRs: possible intercellular energy relocation
The commensal microbes reside throughout our bodies including the skin, oral cavity, and gut, and the human gut contains 1014 bacteria which outnumber the total number of cells in all physiological compartments. Several TLRs have been found to affect the colonization of gut microbiota [1, 6]. For example, the first contact of gut microbiota with the intestinal lining triggers the activation of TLR5 in epithelial cells and dendritic cells, resulting in the recruitment of B cells and T cells, and subsequently the production of IgA to limit the overcolonization of gut microbiota [40]. TLR2 on T cells can sense the polysaccharide A on Bacteroides fragilis and control its colonization [41]. Instead of triggering major inflammation, TLRs in fact protect the host from hyperinflammation by limiting the access of bacterial products to cytosolic inflammasome, and by atypically inhibiting NFκB activation in intestinal epithelium [42, 43]. However, a study argues that the alteration of gut microbiota observed in TLR-deficient mice is due to the familial transmission attributed by housing environment or maternal transmission rather than the defects in immunity [44]. Nonetheless, regardless whether TLR has a role in alteration of gut microbiota, from the perspective of receptor-ligand dynamics, we are concerned about how much and what type of TLR ligands penetrate and what the final outcomes of TLR activation are. Certainly, we cannot rule out the possibility that change in diet or alteration of gut microbiota would also alter the expressions of TLRs, thus affecting the location of TLR-mediated catabolic events.
High-fat intake or obesity can weaken the gut integrity leading to penetration of gut microbes or their products into the circulation [45]. This low-grade endotoxemia triggered by the commensal microbiota is believed to be the cause of TLR activation. Body weight change and increased adiposity are associated with alteration of gut microbiota, and the interaction between gut microbiota and TLRs can be both immunological and metabolic. Although there is yet any direct evidence showing metabolic regulation by gut microbiota-induced TLR activation, several studies have demonstrated that the metabolic functions of TLR pathway can be triggered by low-dose LPS which is similar to the range found in obesity-induced metabolic endotoxemia [5, 12]. If the concept of intercellular energy relocation holds true, immune or inflammatory response would be stimulated during nutrient overload while anabolic event would simultaneously be suppressed, which creates an insulin resistance-like condition. Changes in gut microbiota and intestinal permeability occur rapidly after initiation and termination of high-fat diet [46]. Assuming that sensitivity of TLRs is different in immune cells and metabolic cells, it is possible that the TLRs in the metabolic cells are more sensitive to the gut microbiota-derived endotoxemia, thereby metabolic alteration would be engaged first. Such metabolic regulation facilitates intercellular or inter-organ relocation of energy to prepare for the future possible full-blown attack of microbes. It would be problematic in the situation of overnutrition when the energy is not stored in the proper locations such as adipose tissue (Fig. 2). As excessive intake of energy continues, other metabolic pathways are activated. Expansion of adipose tissue results in high turnover of adipocytes as well as lipid content, and for adaptation, both catabolic and anabolic rates remain high. Excess energy continuously overflows to other organs. Our body may be misled by the sustained metabolic endotoxemia that the infection has not been subsided, resulting in infiltration of immune cells to the organs, and the immune cells in turn store the energy to sustain the inflammatory responses [28–30] (Fig. 2). In fact, it is shown that inflammation does not contribute to metabolic dysfunction in short-term high-fat diet, and the observed early-onset insulin resistance is possibly caused by lipid overload in the liver and muscle instead [47].
Fig. 2.
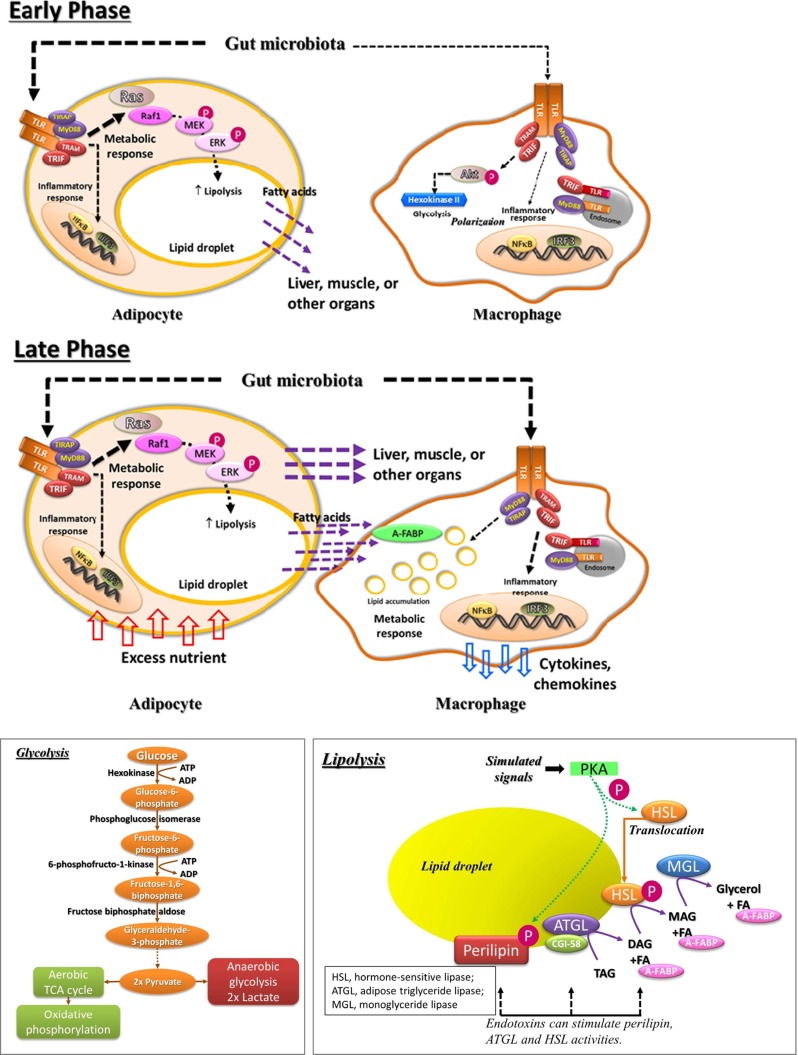
Hypothesis of intercellular energy relocation triggered by gut microbiota. Dietary alteration can result in change of gut microbiota and weakening of gut integrity, subsequently penetration of gut microbes and their products. The TLRs on metabolic cell such as adipocyte can sense the low amount of gut bacteria-derived TLR ligands, which leads to catabolic events including lipolysis. Energy or nutrient is shunted to other locations. Activation of the TLR pathway turns on glycolysis in immune cell (e.g., macrophage) to initiate polarization (upper panel). When the change in diet persists, e.g., overeating in obesity, other metabolic mechanisms are activated. Expansion of adipose tissue results in high turnover of lipids in adipocytes. Excess energy continuously overflows to other organs. More immune cells infiltrate into those organs, and energy or lipids are now taken up by the adjacent macrophages for their own utilization to sustain inflammation, which aggravates metabolic dysfunction (middle panel). The glycolysis and lipolysis pathways are briefly depicted in the boxes at the bottom (bottom panel)
Clinical implication of interfering the crosstalk between gut microbiota and TLRs
The concept of energy relocation between immunity and metabolism is based on a nutrient- or energy-scarce environment [33]. Nonetheless, the current issue faced by humankind is the long-term medical problems due to overnutrition. Obesity is often associated with metabolic endotoxemia which stimulates local and systemic inflammation and eventually aggravates metabolic dysfunction and cardiovascular risks. As a result, anti-inflammation seems to be a therapeutic option for obesity-associated diseases. However, the clinical benefit is yet conclusive, and in the case of diabetes, use of anti-inflammatory drug even increases the risk of cardiovascular event. [48] In addition, in spite of the consistent beneficial effects on metabolic functions shown in TLR-deficient animal models, no conclusive association between TLR polymorphism and metabolic diseases can be drawn from existing human data [49, 50]. A study reported that among 1894 patients without acute myocardial infarction but requiring coronary angiography, the prevalence of diabetes was 7 % lower in those with the TLR4 polymorphism (Asp299Gly) variant allele compared with the wild type allele [51]. In contrast, another study selecting a subpopulation of 722 subjects in the Cooperative Health Research in the Augsburg Region (KORA) Survey 2000 found no association with type 2 diabetes, impaired glucose tolerance, or other components of metabolic syndrome in the heterozygous and homozygous TLR4 variant alleles [50]. On the other hand, a nonsense TLR5 polymorphism prevents weight gain but imposes risk for diabetes [52]. Two studies on the association of TLR2 polymorphism (rs3804100, 1350 T/C) with type 1 diabetes showed conflicting results [53, 54]. These discrepancies may be partially related to the geographic, socioeconomic, or epigenetic influence on gut microbiota of the individuals.
Likewise, even though it is well accepted that alteration of gut microbiota can contribute to obesity and metabolic dysfunction, results from different laboratories appear to be inconsistent. The evolutionary purpose of gut microbiota is related to enhanced energy harvesting in host [55], and depletion of gut microbiota using antibiotics can promote browning of white adipose tissue and thus prevent obesity [56]. However, antibiotics have been wildly used in agriculture to stimulate weight gain of livestock in recent decades [57]. Use of antibiotics in early stage of life is associated with childhood obesity. Also, even within the same genus of Lactobacillus, the strain Lactobacillus plantarum promotes weight loss whereas Lactobacillus ingluiviei and Lactobacillus acidophilus induce weight gain [58, 59]. It is no doubt that the idea of manipulating gut microbiota to regulate body weight and metabolism requires further detailed investigation due to the complex relationship between the host and gut microbiota.
Gut microbiota pattern can be shaped by diet and substantial differences are observed in carnivores, omnivores, and herbivores [60]. Low-grade systemic inflammation induced by high-fat diet may be an evolutional protective mechanism against food-borne pathogens particularly derived from ingesting animal fat where cross infection is highly possible. In modern medicine, antagonists of TLR for metabolic and cardiovascular diseases have been explored because of the beneficial effects yielded by immunosuppression. However, if gut microbiota-derived molecules in these chronic diseases can activate TLRs, catabolism in the host would be predicted. Inhibition of the TLR pathway in such scenario would promote the energy storage; however, considering the variation in types of penetrated bacterial products and expression of TLRs in different organs and cell types, it might result in undesirable anabolic events in certain location, which would exacerbate the metabolic dysfunction. In addition, there are concerns of suppressing host TLR activity because it increases the vulnerability to infection which is also a contemporary medical issue. A thorough investigation on the functions of the TLR pathway and the interaction between TLRs and gut microbiota will allow us to better evaluate on the clinical application of agonism/antagonism of TLRs in chronic diseases.
Reference
- 1.Chassaing B, Ley RE, Gewirtz AT. Intestinal epithelial cell toll-like receptor 5 regulates the intestinal microbiota to prevent low-grade inflammation and metabolic syndrome in mice. Gastroenterology. 2014;147(1363–77) doi: 10.1053/j.gastro.2014.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schwabe RF, Seki E, Brenner DA. Toll-like receptor signaling in the liver. Gastroenterology. 2006;130:1886–1900. doi: 10.1053/j.gastro.2006.01.038. [DOI] [PubMed] [Google Scholar]
- 3.Kopp A, Buechler C, Neumeier M, Weigert J, Aslanidis C, Scholmerich J, Schaffler A. Innate immunity and adipocyte function: ligand-specific activation of multiple toll-like receptors modulates cytokine, adipokine, and chemokine secretion in adipocytes. Obesity (Silver Spring) 2009;17:648–656. doi: 10.1038/oby.2008.607. [DOI] [PubMed] [Google Scholar]
- 4.Caesar R, Tremaroli V, Kovatcheva-Datchary P, Cani PD, Backhed F. Crosstalk between gut microbiota and dietary lipids aggravates WAT inflammation through TLR signaling. Cell Metab. 2015;22:658–668. doi: 10.1016/j.cmet.2015.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zu L, He J, Jiang H, Xu C, Pu S, Xu G. Bacterial endotoxin stimulates adipose lipolysis via toll-like receptor 4 and extracellular signal-regulated kinase pathway. J Biol Chem. 2009;284:5915–5926. doi: 10.1074/jbc.M807852200. [DOI] [PubMed] [Google Scholar]
- 6.Caricilli AM, Picardi PK, de Abreu LL, Ueno M, Prada PO, Ropelle ER, Hirabara SM, Castoldi A, Vieira P, Camara NO, et al. Gut microbiota is a key modulator of insulin resistance in TLR 2 knockout mice. PLoS Biol. 2011;9 doi: 10.1371/journal.pbio.1001212. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 7.Vijay-Kumar M, Aitken JD, Carvalho FA, Cullender TC, Mwangi S, Srinivasan S, Sitaraman SV, Knight R, Ley RE, Gewirtz AT. Metabolic syndrome and altered gut microbiota in mice lacking toll-like receptor 5. Science. 2010;328:228–231. doi: 10.1126/science.1179721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang XA, Zhang R, She ZG, Zhang XF, Jiang DS, Wang T, Gao L, Deng W, Zhang SM, Zhu LH, et al. Interferon regulatory factor 3 constrains IKKbeta/NF-kappaB signaling to alleviate hepatic steatosis and insulin resistance. Hepatology. 2014;59:870–885. doi: 10.1002/hep.26751. [DOI] [PubMed] [Google Scholar]
- 9.Dalmas E, Toubal A, Alzaid F, Blazek K, Eames HL, Lebozec K, Pini M, Hainault I, Montastier E, Denis RG, et al. Irf5 deficiency in macrophages promotes beneficial adipose tissue expansion and insulin sensitivity during obesity. Nat Med. 2015;21:610–618. doi: 10.1038/nm.3829. [DOI] [PubMed] [Google Scholar]
- 10.Jia L, Vianna CR, Fukuda M, Berglund ED, Liu C, Tao C, Sun K, Liu T, Harper MJ, Lee CE, et al. Hepatocyte toll-like receptor 4 regulates obesity-induced inflammation and insulin resistance. Nat Commun. 2014;5:3878. doi: 10.1038/ncomms4878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Etienne-Mesmin L, Vijay-Kumar M, Gewirtz AT, Chassaing BH. Toll-like receptor 5 promotes bacterial clearance and protects mice against high-fat diet-induced liver disease. CMGH in press. 2016 doi: 10.1016/j.jcmgh.2016.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feingold KR, Moser A, Shigenaga JK, Grunfeld C. Inflammation inhibits the expression of phosphoenolpyruvate carboxykinase in liver and adipose tissue. Innate Immun. 2012;18:231–240. doi: 10.1177/1753425911398678. [DOI] [PubMed] [Google Scholar]
- 13.Cullender TC, Chassaing B, Janzon A, Kumar K, Muller CE, Werner JJ, Angenent LT, Bell ME, Hay AG, Peterson DA, et al. Innate and adaptive immunity interact to quench microbiome flagellar motility in the gut. Cell Host Microbe. 2013;14:571–581. doi: 10.1016/j.chom.2013.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kelly B, O'Neill LA. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015;25:771–784. doi: 10.1038/cr.2015.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carvalho FA, Aitken JD, Vijay-Kumar M, Gewirtz AT. Toll-like receptor-gut microbiota interactions: perturb at your own risk! Annu Rev Physiol. 2012;74:177–198. doi: 10.1146/annurev-physiol-020911-153330. [DOI] [PubMed] [Google Scholar]
- 16.Dong H, Bullock TN. Metabolic influences that regulate dendritic cell function in tumors. Front Immunol. 2014;5:24. doi: 10.3389/fimmu.2014.00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krawczyk CM, Holowka T, Sun J, Blagih J, Amiel E, DeBerardinis RJ, Cross JR, Jung E, Thompson CB, Jones RG, Pearce EJ. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood. 2010;115:4742–4749. doi: 10.1182/blood-2009-10-249540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Everts B, Amiel E, Huang SC, Smith AM, Chang CH, Lam WY, Redmann V, Freitas TC, Blagih J, van der Windt GJ, et al. TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKvarepsilon supports the anabolic demands of dendritic cell activation. Nat Immunol. 2014;15:323–332. doi: 10.1038/ni.2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rodriguez-Prados JC, Traves PG, Cuenca J, Rico D, Aragones J, Martin-Sanz P, Cascante M, Bosca L. Substrate fate in activated macrophages: a comparison between innate, classic, and alternative activation. J Immunol. 2010;185:605–614. doi: 10.4049/jimmunol.0901698. [DOI] [PubMed] [Google Scholar]
- 20.Sanin DE, Prendergast CT, Mountford AP. IL-10 production in macrophages is regulated by a TLR-driven CREB-mediated mechanism that is linked to genes involved in cell metabolism. J Immunol. 2015;195:1218–1232. doi: 10.4049/jimmunol.1500146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Castrillo A, Joseph SB, Vaidya SA, Haberland M, Fogelman AM, Cheng G, Tontonoz P. Crosstalk between LXR and toll-like receptor signaling mediates bacterial and viral antagonism of cholesterol metabolism. Mol Cell. 2003;12:805–816. doi: 10.1016/S1097-2765(03)00384-8. [DOI] [PubMed] [Google Scholar]
- 22.de Chastellier C, Thilo L. Cholesterol depletion in Mycobacterium avium-infected macrophages overcomes the block in phagosome maturation and leads to the reversible sequestration of viable mycobacteria in phagolysosome-derived autophagic vacuoles. Cell Microbiol. 2006;8:242–256. doi: 10.1111/j.1462-5822.2005.00617.x. [DOI] [PubMed] [Google Scholar]
- 23.Yvan-Charvet L, Welch C, Pagler TA, Ranalletta M, Lamkanfi M, Han S, Ishibashi M, Li R, Wang N, Tall AR. Increased inflammatory gene expression in ABC transporter-deficient macrophages: free cholesterol accumulation, increased signaling via toll-like receptors, and neutrophil infiltration of atherosclerotic lesions. Circulation. 2008;118:1837–1847. doi: 10.1161/CIRCULATIONAHA.108.793869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chow EK, Castrillo A, Shahangian A, Pei L, O'Connell RM, Modlin RL, Tontonoz P, Cheng G. A role for IRF3-dependent RXRalpha repression in hepatotoxicity associated with viral infections. J Exp Med. 2006;203:2589–2602. doi: 10.1084/jem.20060929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vaz B, de Lera AR. Advances in drug design with RXR modulators. Expert Opin Drug Discov. 2012;7:1003–1016. doi: 10.1517/17460441.2012.722992. [DOI] [PubMed] [Google Scholar]
- 26.Li Q, Pene V, Krishnamurthy S, Cha H, Liang TJ. Hepatitis C virus infection activates an innate pathway involving IKK-alpha in lipogenesis and viral assembly. Nat Med. 2013;19:722–729. doi: 10.1038/nm.3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Isogawa M, Robek MD, Furuichi Y, Chisari FV. Toll-like receptor signaling inhibits hepatitis B virus replication in vivo. J Virol. 2005;79:7269–7272. doi: 10.1128/JVI.79.11.7269-7272.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang YL, Morales-Rosado J, Ray J, Myers TG, Kho T, Lu M, Munford RS. Toll-like receptor agonists promote prolonged triglyceride storage in macrophages. J Biol Chem. 2014;289:3001–3012. doi: 10.1074/jbc.M113.524587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kazemi MR, McDonald CM, Shigenaga JK, Grunfeld C, Feingold KR. Adipocyte fatty acid-binding protein expression and lipid accumulation are increased during activation of murine macrophages by toll-like receptor agonists. Arterioscler Thromb Vasc Biol. 2005;25:1220–1224. doi: 10.1161/01.ATV.0000159163.52632.1b. [DOI] [PubMed] [Google Scholar]
- 30.Xu X, Grijalva A, Skowronski A, van Eijk M, Serlie MJ, Ferrante AW Jr (2013) Obesity activates a program of lysosomal-dependent lipid metabolism in adipose tissue macrophages independently of classic activation. Cell Metab 18:816–830 [DOI] [PMC free article] [PubMed]
- 31.Buttgereit F, Burmester GR, Brand MD. Bioenergetics of immune functions: fundamental and therapeutic aspects. Immunol Today. 2000;21:192–199. doi: 10.1016/S0167-5699(00)01593-0. [DOI] [PubMed] [Google Scholar]
- 32.Arrese EL, Soulages JL. Insect fat body: energy, metabolism, and regulation. Annu Rev Entomol. 2010;55:207–225. doi: 10.1146/annurev-ento-112408-085356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.DiAngelo JR, Bland ML, Bambina S, Cherry S, Birnbaum MJ. The immune response attenuates growth and nutrient storage in Drosophila by reducing insulin signaling. Proc Natl Acad Sci U S A. 2009;106:20853–20858. doi: 10.1073/pnas.0906749106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Leulier F, Lemaitre B. Toll-like receptors—taking an evolutionary approach. Nat Rev Genet. 2008;9:165–178. doi: 10.1038/nrg2303. [DOI] [PubMed] [Google Scholar]
- 35.Pang S, Tang H, Zhuo S, Zang YQ, Le Y. Regulation of fasting fuel metabolism by toll-like receptor 4. Diabetes. 2010;59:3041–3048. doi: 10.2337/db10-0418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Orr JS, Puglisi MJ, Ellacott KL, Lumeng CN, Wasserman DH, Hasty AH. Toll-like receptor 4 deficiency promotes the alternative activation of adipose tissue macrophages. Diabetes. 2012;61:2718–2727. doi: 10.2337/db11-1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kagan JC, Su T, Horng T, Chow A, Akira S, Medzhitov R. TRAM couples endocytosis of toll-like receptor 4 to the induction of interferon-beta. Nat Immunol. 2008;9:361–368. doi: 10.1038/ni1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stack J, Doyle SL, Connolly DJ, Reinert LS, O'Keeffe KM, McLoughlin RM, Paludan SR, Bowie AG. TRAM is required for TLR2 endosomal signaling to type I IFN induction. J Immunol. 2014;193:6090–6102. doi: 10.4049/jimmunol.1401605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schaffler A, Scholmerich J, Salzberger B. Adipose tissue as an immunological organ: toll-like receptors, C1q/TNFs and CTRPs. Trends Immunol. 2007;28:393–399. doi: 10.1016/j.it.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 40.Uematsu S, Fujimoto K, Jang MH, Yang BG, Jung YJ, Nishiyama M, Sato S, Tsujimura T, Yamamoto M, Yokota Y, Kiyono H, Miyasaka M, Ishii KJ, Akira S. Regulation of humoral and cellular gut immunity by lamina propria dendritic cells expressing toll-like receptor 5. Nat Immunol. 2008;9:769–776. doi: 10.1038/ni.1622. [DOI] [PubMed] [Google Scholar]
- 41.Round JL, Lee SM, Li J, Tran G, Jabri B, Chatila TA, Mazmanian SK. The toll-like receptor 2 pathway establishes colonization by a commensal of the human microbiota. Science. 2011;332:974–977. doi: 10.1126/science.1206095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carvalho FA, Aitken JD, Gewirtz AT, Vijay-Kumar M. TLR5 activation induces secretory interleukin-1 receptor antagonist (sIL-1Ra) and reduces inflammasome-associated tissue damage. Mucosal Immunol. 2011;4:102–111. doi: 10.1038/mi.2010.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell. 2004;118:229–241. doi: 10.1016/j.cell.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 44.Ubeda C, Lipuma L, Gobourne A, Viale A, Leiner I, Equinda M, Khanin R, Pamer EG. Familial transmission rather than defective innate immunity shapes the distinct intestinal microbiota of TLR-deficient mice. J Exp Med. 2012;209:1445–1456. doi: 10.1084/jem.20120504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, Burcelin R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes. 2008;57:1470–1481. doi: 10.2337/db07-1403. [DOI] [PubMed] [Google Scholar]
- 46.David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, Ling AV, Devlin AS, Varma Y, Fischbach MA, et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2014;505:559–563. doi: 10.1038/nature12820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee YS, Li P, Huh JY, Hwang IJ, Lu M, Kim JI, Ham M, Talukdar S, Chen A, Lu WJ, Bandyopadhyay GK, et al. Inflammation is necessary for long-term but not short-term high-fat diet-induced insulin resistance. Diabetes. 2011;60:2474–2483. doi: 10.2337/db11-0194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Coxib and traditional NSAID Trialists’ (CNT) Collaboration. Bhala N, Emberson J, Merhi A, Abramson S, Arber N, Baron JA, Bombardier C, Cannon C, Farkouh ME, et al (2013) Vascular and upper gastrointestinal effects of non-steroidal anti-inflammatory drugs: meta-analyses of individual participant data from randomised trials. Lancet 382:769–779 [DOI] [PMC free article] [PubMed]
- 49.Dasu MR, Devaraj S, Park S, Jialal I. Increased toll-like receptor (TLR) activation and TLR ligands in recently diagnosed type 2 diabetic subjects. Diabetes Care. 2010;33:861–868. doi: 10.2337/dc09-1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Illig T, Bongardt F, Schopfer A, Holle R, Muller S, Rathmann W, Koenig W, Meisinger C, HE W, Kolb H, Group KS The endotoxin receptor TLR4 polymorphism is not associated with diabetes or components of the metabolic syndrome. Diabetes. 2003;52:2861–2864. doi: 10.2337/diabetes.52.11.2861. [DOI] [PubMed] [Google Scholar]
- 51.Kolek MJ, Carlquist JF, Muhlestein JB, Whiting BM, Horne BD, Bair TL, Anderson JL. Toll-like receptor 4 gene Asp299Gly polymorphism is associated with reductions in vascular inflammation, angiographic coronary artery disease, and clinical diabetes. Am Heart J. 2004;148:1034–1040. doi: 10.1016/j.ahj.2004.05.049. [DOI] [PubMed] [Google Scholar]
- 52.Al-Daghri NM, Clerici M, Al-Attas O, Forni D, Alokail MS, Alkharfy KM, Sabico S, Mohammed AK, Cagliani R, Sironi M. A nonsense polymorphism (R392X) in TLR5 protects from obesity but predisposes to diabetes. J Immunol. 2013;190:3716–3720. doi: 10.4049/jimmunol.1202936. [DOI] [PubMed] [Google Scholar]
- 53.Bjornvold M, Munthe-Kaas MC, Egeland T, Joner G, Dahl-Jorgensen K, Njolstad PR, Akselsen HE, Gervin K, Carlsen KC, Carlsen KH, Undlien DE. A TLR2 polymorphism is associated with type 1 diabetes and allergic asthma. Genes Immun. 2009;10:181–187. doi: 10.1038/gene.2008.100. [DOI] [PubMed] [Google Scholar]
- 54.Santin I, Bilbao JR, de Nanclares GP, Calvo B, Castano L. No association of TLR2 and TLR4 polymorphisms with type I diabetes mellitus in the Basque population. Ann N Y Acad Sci. 2006;1079:268–272. doi: 10.1196/annals.1375.040. [DOI] [PubMed] [Google Scholar]
- 55.Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–1031. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- 56.Suarez-Zamorano N, Fabbiano S, Chevalier C, Stojanovic O, Colin DJ, Stevanovic A, Veyrat-Durebex C, Tarallo V, Rigo D, Germain S, et al. Microbiota depletion promotes browning of white adipose tissue and reduces obesity. Nat Med. 2015;21:1497–1501. doi: 10.1038/nm.3994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dibner JJ, Richards JD. Antibiotic growth promoters in agriculture: history and mode of action. Poult Sci. 2005;84:634–643. doi: 10.1093/ps/84.4.634. [DOI] [PubMed] [Google Scholar]
- 58.Lee K, Paek K, Lee HY, Park JH, Lee Y. Antiobesity effect of trans-10,cis-12-conjugated linoleic acid-producing Lactobacillus plantarum PL62 on diet-induced obese mice. J Appl Microbiol. 2007;103:1140–1146. doi: 10.1111/j.1365-2672.2007.03336.x. [DOI] [PubMed] [Google Scholar]
- 59.Angelakis E, Merhej V, Raoult D. Related actions of probiotics and antibiotics on gut microbiota and weight modification. Lancet Infect Dis. 2013;13:889–899. doi: 10.1016/S1473-3099(13)70179-8. [DOI] [PubMed] [Google Scholar]
- 60.Ley RE, Hamady M, Lozupone C, Turnbaugh PJ, Ramey RR, Bircher JS, Schlegel ML, Tucker TA, Schrenzel MD, Knight R, Gordon JI. Evolution of mammals and their gut microbes. Science. 2008;320:1647–1651. doi: 10.1126/science.1155725. [DOI] [PMC free article] [PubMed] [Google Scholar]