

Abstract
The immune system is capable of distinguishing between danger- and non-danger signals, thus inducing either an appropriate immune response against pathogens and cancer or inducing self-tolerance to avoid autoimmunity and immunopathology. One of the mechanisms that have evolved to prevent destruction by the immune system, is to functionally silence effector T cells, termed T cell exhaustion, which is also exploited by viruses and cancers for immune escape In this review, we discuss some of the phenotypic markers associated with T cell exhaustion and we summarize current strategies to reinvigorate exhausted T cells by blocking these surface marker using monoclonal antibodies.
Keywords: Immunotherapy, PD-1, PD-L1, T cell exhaustion, Cancer
Background
Exhausted T cells can be distinguished from other T cell dysfunctions such as anergy and senescence based on their underlying molecular mechanisms [1]. Whereas anergy is introduced during priming due to the absence of costimulatory signals and senescence is growth arrest after extensive proliferation [2] exhausted T cells arise from cells, which initially gained effector function, but become gradually silenced due to continous T cell receptor (TCR) stimulation from persistent antigen [3].
T cell exhaustion has been initially observed in mice infected with the lymphocytic choriomeninigits virus (LCMV), where a chronically persistent virus strain rendered virus specific cytotoxic T cells non-functional. Using the same mouse model, reversibility of T cell exhaustion could be demonstrated [4, 5].
Exhausted T cells have also been observed in response to several other virus infections like simian immunodeficiency virus (SIV), human immunodeficiency virus (HIV), hepatitis B virus (HBV), hepatitis C virus (HCV) and human T lymphotropic virus 1 (HTLV1) [6–15]. However, mice with impeded T cell exhaustion develop severe spontaneous autoimmune diseases and succumb to fatal CD8 T cell-mediated immune pathologies during early systemic LCMV infection, showing that T cell exhaustion substantially contributes to peripheral tolerance and to moderate immune responses [16, 17]. In line with that, presence of exhausted T cells in patients with autoimmune diseases correlates with favorable prognosis [18]. T cell exhaustion has also been observed in tumor patients, where the exhaustion of tumor specific T cells is suggested to impede clearance of the tumor, thus contributing to tumor immune escape [19–23]. Characteristics of exhaustion are are continuous enhancement of T cell dysfunction due to persistent antigen exposure, an increased expression of multiple inhibitory receptors (IR), theprogressive loss of effector cytokine secretion (IL-2, Interferone gamma [IFNγ], Tumor necrosis factor alpha [TNFα]), analtered cell metabolism and a markedly different transcriptional profile [20, 21, 23–26]. The gradual dysfunction of exhausted T cells is accompanied by the expression of IRs, which wire inhibitory signals to the nucleus upon interaction with ligands on target cells (Fig. 1 and Table 1). However, recent reports reveal that T cells do not uniformly exhaust during chronic diseases or cancer, but that specific subsets with different memory-like or proliferative potentials emerge upon exposure to persisting anigen [27–29]. As blocking iR/ligand interactions (so called immune checkpoint inhibition) seems an appealing strategy to partially reverse T cell exhaustion and to possibly regain anti-cancer immunity, a set of most promising inhibitory receptors (although their expression is not exclusively restricted to exhausted T cells) and current approaches to impede their function in context of current cancer therapies are discussed in this review:
Fig. 1.
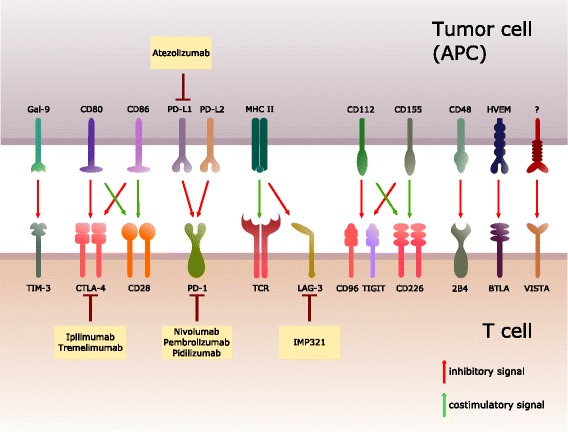
Inhibitory/costimulatory receptors and their corresponding ligands. Schematic overview of inhibitory/ costimulatory receptors expressed by T cells interacting with their counterpart on antigen-presenting cells (APCs) or tumor cells. Additionally, various blocking antibodies against inhibitory receptors or their ligands in clinical trials are depicted with the aim of reversing T cell exhaustion
Table 1.
Expression, ligands and signaling pathways of immune checkpoint molecules (based on [210] and [211])
| Immune checkpoint receptors (synonym) | Cellular expression | Ligand | Intracellular motif | Signaling pathways |
|---|---|---|---|---|
| CTLA-4 (CD152) | T cells | CD80, CD86 | YxxM | SHP2, LCK/ZAP70/PI3K PP2A/AKT |
| PD-1 (CD279) | T cells, B cells, DCs, NKT cells, Monocytes | PD-L1, PD-L2 | ITIM, ITSM | SHP1, PI3K/AKT SHP2, LCK/ZAP70/PI3K, RAS |
| TIGIT (VSIG9, VSTM3) | T cells, NK and NKT cells | CD155, CD112 | 2 × ITIM | NF-kB, PI3K and MAPK |
| LAG-3 (CD223) | T cells, B cells, DC, NK cells | MHCII | KIEELE | not determined |
| 2B4 (CD244) | T cells, NK cells, Monocytes, Basophiles | CD2, CD48 | ITSM | not determined |
| BTLA (CD272) | T cells, B cells, DC, Macrophages, Myeloid cells | HVEM, CD80 | ITIM, ITSM | SHP1, PI3K/AKT SHP2, LCK/ZAP70/PI3K |
| TIM3 (HAVCR2) | T cells, B cells, NK cells, NKT cells, DCs, Macrophages | Gal-9 | Y235, Y242 | PI3K BAT3/LCK |
| VISTA (PD1-H) | T cells, DCs, Macrophages, Monocytes, Neutrophils | not determined | not determined | not determined |
| CD96 (Tactile) | T cells, NK cells, Myeloid cells | CD155 | ITIM | not determined |
Inhibitory receptors associated with T cell exhaustion
Cytotoxic T-lymphocyte-associated Protein 4 (CTLA-4)
CTLA-4 counteracts the positive signal mediated by CD28 by competing for the same ligands (CD80/86) with higher affinity [30–32]. CTLA-4 transmits signals by intracellularily binding the phosphatases PP2A and SHP-2. In addition, CTLA-4 is able to entrap its ligands CD80/CD86 by trans-endocytosis followed by degradation [33, 34].
CTLA-4 is up-regulated upon activation on naïve T cells and constitutively expressed on regulatory T cells (Tregs), since CTLA-4 is a transcriptional target of Foxp3, a key transcriptional factor of this subset [35, 36]. The role of CTLA-4 in immune suppression and tolerance has been validated in autoimmune mouse models such as type I diabetes and multiple sclerosis, where CTLA-4 blockade results in increased severity of the inflammatory phenotype [37]. CTLA-4 knockout mice provide additional evidence for its role as negative regulator of the immune response, due to the enhanced lymphoproliferative disorder and multiorgan tissue destruction [38, 39]. Paradoxically, although CTLA-4 decreases effector functions of CD4+ and CD8+ T cells, it increases the suppressive capacity of Tregs. For example, specific CTLA-4 knockdown or blockade on Tregs results in T cell mediated autoimmune disease and contributes to antitumor immunity. Additionally, CTLA-4 expressing Tregs mediate the downregulation of CD80/CD86 on antigen presenting cells and thereby reduce activation of naïve T cells [40, 41]. In context of cancer, it is suggested that CTLA-4 expression on low-affinity tumor specific T cells attenuates their proliferation which could be possibly overcome by CTLA-4 blockade. In addition, CTLA-4 expression on tumor specific Tregs could contribute to tumor immune escape by increasing the suppressive anti-tumor immunity and by downregulating CD80/CD86 on antigen presenting cells [42].
Thus, CTLA-4 dampens T cell activation, decreases the efficacy of antigen presenting cells to activate T cells and augments Treg mediated immune suppression.
Programmed cell death 1 (PD-1)
Whereas CTLA-4 predominantly regulates initial T cell activation, the inhibitory receptor programmed cell death 1 (PD-1) is dampening effector T cell functions [43, 44]. Transient PD-1 cell surface expression is initiated upon T cell activation, but sustained expression is a characteristic marker of T cell exhaustion [45]. However, recent data show that PD-1 is not required for initiating T cell exhaustion and that absence of PD-1 even promotes accumulation of exhausted CD8+ T cells in mice [46]. The intracellular domain consists of an immunoreceptor tyrosine- based inhibitory motif (ITIM) and an immunoreceptor tyrosine- based switch motif (ITSM). PD-1 engagement with its ligand (PD-L1 or PD-L2) results in ITIM/ ITSM phosphorylation and subsequent recruitment of the phosphatases SHP1/ SHP2, which negatively regulate PI3K/ AKT and RAS signaling pathways [47–49]. In addition to CTLA-4 Tregs also express PD-1 on their cell surface [50]. During chronic infections such as LCMV, two subsets of exhausted T cells have been identified according to their transcriptional profile and expression of the inhibitory receptor PD-1 [51].
T cells with an increase in the transcription factor T-bet and an intermediate expression of PD-1 (T-bethigh PD-1int) retain residual secretion of IFNγ, TNFα and a limited proliferation rate. On the contrary, high levels of Eomesodermin (Eomes) and PD-1 (Eomeshigh PD-1high) exhibited higher Blimp1and granzyme B production, co-expression of additional inhibitory receptors (CD160, Lag-3, 2B4, Tim-3) and are associated with a severe state of exhaustion, despite of a greater cytotoxic activity compared to T-bethigh PD-1int T cells. Additionally, T-bethigh PD-1int give rise to Eomeshigh PD-1high in an antigen driven manner and therefore count as a progenitor subset [51]. However, opposing data show that during chronic infection, a small subset of CD8+ T cells which were T cel factor 1 (Tcf1)+, PD-1+ and Eomes+ sustained a memory-like T cell response [28].
The blockade of the PD-1/PD-L1 axes in chronic infected LCMV mice sufficiently induces an antiviral state, by which two subpopulations of CD8 cells were identified. Whereas Eomeshigh PD-1high T cells exhibit a poor response to PD-1 pathway blockade, T-bethigh PD-1int virus specific CD8 T cells efficiently reverse exhaustion and induce protective immunity in vivo suggesting that only a small fraction of exhausted T cells might overcome exhaustion by blocking PD-1 signaling [52].
T cell immunoreceptor with Ig and ITIM domains (TIGIT)
Genome wide search for genes specifically expressed on immune cells and consisting of an extracellular Ig domain, type I transmembrane region together with either ITIMs or immunoreceptor tyrosine-based activation motifs (ITAMs), have revealed the existence of an additional inhibitory receptor namely T cell immunoreceptor with Ig and ITIM domains (TIGIT) [53, 54]. It belongs to the type 1 transmembrane proteins with an cytoplasmatic tail containing an immunoglobulin tail tyrosine (ITT)- like phosphorylation motif and ITIM [55]. Its expression is widely distributed across various T cell subsets including follicular helper T cells (TFH), Tregs, activated/memory T cells, natural killer (NK) and natural killer T (NKT) cells [53, 54, 56]. TIGIT attachment to poliovirus receptors (PVR) CD155/ CD112 results in the Grb2 mediated- recruitment of the SHIP1 phosphatase and downstream inhibition of NF-kB, PI3K and MAPK pathways [57, 58]. PVRs are expressed on APCs, endothelial cells, epithelial cells, but also on a number of tumor cells, which are inducible by Ras activation, Toll-like receptor (TLR) engagement and genotoxic stress [59–64].
Similar to CTLA-4/CD28 interactions, TIGIT shares the same ligands as the costimulatory molecule CD226 and competes for ligation resulting in the inhibition of T cell activation [65]. Interestingly, TIGIT is also capable of directly preventing the homodimerization of CD226 [65] leading to impaired TIGIT/CD226 balance, which impedes CD8 and NK cell antitumor and antiviral T cell response [66, 67]. Additionally, experiments in CD226 deficient mice showed impaired T cell proliferation, reduced immunological synapse formation and antitumor cytotoxicity [68]. Whereas an agonistic TIGIT antibody decreases T cell activation via CD3/CD28 stimulation, TIGIT knockdown enhances T cell proliferation, effector cytokine production such as IFNγ, IL-2 while decreasing IL-10 levels [69]. Additionally, circulating TIGIT+ TFH cells produce higher levels of IL-21 and IL-4 and decreased IFNγ secretion compared to TIGIT− TFH cells promoting the differentiation and activation of B cells upon chronic stimulation [56]. Notably, the transcription factor FoxP3 regulates TIGIT expression and furthermore TIGIT+ Tregs exhibit higher suppressive functions compared to TIGIT− Tregs [70, 71]. Besides the expression of additional inhibitory receptors, TIGIT+ Tregs are promoting Th2 responses by attenuating the secretion of the pro-inflammatory cytokines IFNγ and IL-17 [71].
Pre-clinical tumor studies showed that the specific co-inhibition of the TIGIT and PD-1 checkpoint axis causes a significant enhancement of anti-melanoma immune responses by increasing the effector function of cytotoxic T cells [72, 73]. Additionally, TIGIT positive tumor infiltrating CD8 T-cells could be detected in other solid-tumor entities such as small-cell lung carcinomas and colorectal carcinomas [65, 74]. Taken together, the combination of an anti-TIGIT and anti-PD-1 therapy could be a promising approach with associated stratified tumor entities in the future.
Lymphocyte-activated gene-3 (LAG-3)
The cell surface protein lymphocyte-activated gene-3 (LAG-3) shows structural homologies to CD4 and binds MHCII with a higher affinity compared to CD4 [75, 76]. LAG-3 was also shown to interact with LSECTin, a surface lectin of the DC-SIGN family which is expressed on dendritic cells and also on tumor tissue [77]. LAG-3 is expressed on various cells such as B-cells, NK-cells, plasmacytoid dentritic cells, activated CD4, Tregs and CD8 T cells [78–81]. In the case of T cells, LAG-3 is transiently expressed upon activation and becomes internalized and degraded in the lysosomal compartments [82]. On the cell surface, LAG-3 co-distributes with TCR-CD3, binds to MHCII and inhibits CD4-dependent downstream signaling via its cytoplasmatic KIEELE motif and interestingly, not by disrupting CD4- MHCII engagement [83, 84]. As a result, LAG-3 exhibits a negative impact on T cell activation and effector function in vivo and vitro. Upon LAG-3 blockade in vitro T cell proliferation and cytokine production (mainly Th1 cytokines) increases and LAG-3 deficient T cells generate a larger pool of memory cells due to a delayed cell cycle arrest [85, 86]. An additional subtype of Tregs has been described coexisting in parallel to the classical CD4+Foxp3+ Treg cells called type 1 regulatory T cells (Tr1), which are lacking the expression of the transcription factor Foxp3 [87]. Tr1 cells exhibit immunosuppressive functions such as IL-10 and TGF-β secretion, however, LAG-3 blockade results in decreased suppressive activity in vivo and vitro pointing out a role for LAG-3 in Treg induction and expansion [88]. Similar to other exhaustion markers, LAG-3 is up-regulated in cancer and chronic infections. During chronic LCMV infections in mouse models combinatorial blockade of PD-1 and LAG-3 initiates synergistic control of viral load and improves T cell response in vivo [89]. Also various human cancer entities as well as tumor mouse models exhibit co-expression of PD-1 and LAG-3 on tumor-infiltrating T cells (TILs) [90, 91]. Interestingly, single inhibition of either LAG-3 or PD-1 alone does not result in improved control of chronic infection or tumor growth, pointing out the complex interactions among inhibitory receptors, whereby dual blockade synergistically reverses the exhausted phenotype [89, 91].
2B4
The receptor 2B4 (CD244) belongs to the signaling lymphocyte activation molecule (SLAM) subfamily within the immunoglobulin superfamily (IgSV). All members of this family contain two or more immunoreceptor tyrosine-based switch motifs (ITSMs) in their cytoplasmatic tail including the receptors CD229, CS1, NTB-A and CD84 [92]. 2B4 is expressed by NK cells, γδ T cells basophils and monocytes, upon activation on CD8+ T cells and binds with high affinity to CD48 on lymphoid and myeloid cells [93–95]. An additional binding partner of CD48 is CD2, which is suggested to contribute to the formation of lipid rafts and provides costimulatory signals [96]. Similar to the situation of TIGIT, 2B4- CD48 interaction exhibits either direct intracellular signaling or disruption of CD2-CD48 engagement. Interestingly, 2B4 is not a simple inhibitory receptor, indeed it can also exert costimulatory functions, depending on various factors. For example, 2B4 expression level, usage of downstream adaptor proteins (SAP or EAT-2) and it depends also on which of the four ITSMs is posphorylated [97–99].
2B4 is associated with T cell exhaustion. Various studies revealed, that exhausted CD8+ T cells exhibit increased 2B4 expression during chronic human diseases such as LCMV, HBV, HCV, HIV and also melanoma [100–105]. Interestingly, the adaptor protein SAP contributes to a positive 2B4 signaling, which is higher expressed in effector T cells compared to exhausted T cells, whereas the exhausted ones display elevated 2B4 levels in chronic LCMV infection [100, 106]. This leads to the suggestion, that the SAP/2B4 ratio is decreased, contributing to the T cell dysfunction during chronic antigen exposure.
B and T lymphocyte attenuator (BTLA)
The cell surface protein B and T lymphocyte attenuator (BTLA) shares structural similarities with PD-1 and CTLA-4 and is expressed on T cells, B cells, macrophages and mature dentritic cells (DC) [107, 108]. Just like LAG-3, BTLA is transiently up-regulated upon TCR engagement and down-regulated on fully activated T cells, albeit retaining PD-1 and CTLA-4 expression [108]. Interestingly, only Th1 polarized cells maintain BTLA cell surface expression but not Th2 cells [107, 108]. The herpesvirus entry mediator (HVEM), which is expressed on various cell types (DCs, NK cells, T and B cells), binds to BTLA and also to the inhibitory receptor CD160 and the costimulatory receptor LIGHT [109, 110]. BTLA- HVEM engagement in T cells leads to tyrosine phosporylation on the conserved intracellular ITIM, inducing recruitment of the Src homology domain 2 (SH2)-containing protein tyrosine phosphatases SHP-1 and SHP-2 resulting in diminished CD3-induced secretion of IL-2 and T cell proliferation [108, 111].
Since BTLA is described as an inhibitory receptor, it is associated with peripheral tolerance. BTLA deficient mice develop autoimmune hepatitis- like disease with elevated levels of self antibodies, activated CD4+ T cells in the periphery, inflammatory cell infiltration of various organs and reduced survival [112]. Similar results have been achieved by the usage of BTLA-deficient T cells exhibiting increased susceptibility to experimental autoimmune encephalomyelitis EAE [108]. Interestingly, a single administration of agonistic BTLA antibodies at the time of autologous haematopoietic stem cell transplantation prevents the development of graft- versus- host disease by the inhibition of CD4+ Foxp3− effector T cell expansion [113]. Furthermore, agonistic BTLA antibodies prolong murine cardiac allograft survival by decreasing IL-2 and IFNγ production and shifting the differentiation towards the Treg phenotype [114]. Additionally to the function as receptor, BTLA can also behave as ligand. This have been proved by several studies, indicating that HVEM elicits pro- survival signal for effector and memory T cells expressing HVEM [115–117].
Overexpression in human cancer [118], especially in hematological tumors [119], is linked to impaired tumor specific T-cell activity [23, 120]. Focusing on malignant melanoma, the triple blockade of PD1, TIM3 and BTLA leads consecutively to an increased expansion, proliferation and cytokine production of tumor-associated antigen- specific CD8+ T-cells [121]. Comparably to malignant melanoma, a heterogeneous amount of PD-1, Tim-3, CTLA-4, LAG-3, and BTLA were expressed on intratumoral CD8+ T cells from 32 patients with NSCLC. Furthermore, these findings could be linked to progression of the disease [122]. Interestingly, this investigation could clearly demonstrate, that the expression of these immune checkpoint inhibitors was time-dependent showing an early PD-1 and late LAG-3/BTLA expression [122]. Another study with NSCLS could relate the expression of PD-L1, PD-L2, PD-1, TIM-3, B7-H3, BTLA and CTLA-4 to the carcinogenesis relevant epithelial-mesenchymal transition [123]. In another animal model, investigating thyroid carcinoma, a combination of vaccination with BTLA inhibition lead to tumor regression [124]. Furthermore, it was shown that BTLA plays a role in suppression of tumor-associated antigen-specific CD8+ T-cell kind allogeneic stem-cell transplantation [125].
T-cell immunoglobulin and mucin- containing protein 3 (TIM3)
The inhibitory receptor T-cell immunoglobulin and mucin- containing protein 3 (TIM-3) is regulated by the transcription factor T-bet and expressed on various T cell subsets including Th1, CD8+, Tregs but also on DCs, macrophages and monocytes [126, 127]. Although TIM-3 is thought to exhibit suppressive functions it does not contain an ITIM motif in its intracellular domain like PD-1 or TIGIT. It binds to the soluble molecule S-type lectin Galectin-9 (Gal-9), which is upregulated by IFNγ leading to the downstream recruitment of the Src family tyrosine kinase Fyn and the p85 phosphatidylinositol 3-kinase (PI3K) adaptor [128, 129]. As a result, Th1 mediated immunity is impaired by reducing IFNγ production, increased apoptosis in Th1 and cytotoxic CD8+ T cell in vitro [130, 131]. Other ligands for TIM3 are carcinoembryonic antigen cell adhesion molecule 1 (CEACAM1) [132], HMGB1 [133] and phosphatidylserine [134]. In preclinical studies, it could be shown that, blockade of TIM-3 signaling enhances the skewing from Th2 to Th1 subsets, thereby reducing allergen induced airway inflammation. Inhibition of Gal-9 amplifies symptoms of experimental autoimmune encephalomyelitis acute graft-versus host disease and type I diabetes in non-obese (NOD) mice [135–138]. The role of TIM-3 is currently being controversially discussed. Some studies display a negative impact on Th1 and Th17 polarization in vitro, while others suppose that Gal-9 triggers Treg differentiation or inhibits Th17 skewing in a TIM-3 independent manner [139–142]. Antagonistic TIM-3 antibodies increases the secretion of Th1 and Th17 effector cytokine production in vitro, elevated Th1 and Th17 differentiation in vivo and diminishes Treg conversion in vitro and in vivo [138, 143, 144]. TIM-3 expression on CD8+T cells is associated with high degree of dysfunction in various chronic infections, but also in lymphoma and melanoma patients [145–148]. As discussed in the last section, antagonizing TIM-3 signaling contributes to tumor regression and control of viral load, which can be potentiated by additional PD-1 blockade [146, 149–151].
V domain Ig suppressor of T cells activation (VISTA)
Cloning of a Treg specific transcript with homology to the Ig superfamily led to the discovery of the V domain Ig suppressor of T cells activation (VISTA) or also known as PD-1 homolog (PD-1H) [152, 153]. This type I transmembrane protein consists of 7 exons and shares 85,6% similarity between human and mouse [153]. Although it is suggested that VISTA shares homology with either PD-1 or PD-L1, it does not contain ITIMs or ITAMs [152, 154]. However, due to the fact that the cytoplasmatic tail contains two protein kinase C binding sites and proline residues, which potentially function as docking sites, VISTA may act as both receptor and ligand such as the inhibitory receptor BTLA [154]. Interestingly, the binding partner of VISTA is still unknown. VISTA expression is not limited to T cells. Indeed, is also expressed by DCs, macrophages, monocytes and neutrophils [152, 153, 155]. Besides CTLA-4, PD-1 and TIGIT, Tregs additionally express VISTA on their cell surface, which is suggested to contribute to Treg differentiation and to their suppressive function. Several studies offer solid evidence for VISTAs immunomodulatory role. Firstly, VISTA-fusion protein promotes Treg differentiation in vitro [155]. Secondly, blockade of VISTA impairs differentiation of tumor-specific Tregs, whereby decreasing Treg-mediated suppression and increases infiltration, proliferation and effector functions of tumor-specific T cells [156]. The role of VISTA as a negative regulator of T cell mediated immune response has been strengthened by the fact that VISTA deficient mice display elevated T cell activation, proliferation, secretion of inflammatory cytokines (IFNγ, TNFα, monocyte chemotactic protein-1 [MCP-1], IL-6), chemokines (interferone gamma induced protein-10 [IP-10], monocyte interferon gamma inducing factor [MIG], MCP-1) and multiorgan chronic inflammation. This inflammatory phenotype is synergistically enhanced by VISTA/PD-1 double knockout. In addition, VISTA single knockout mice exhibit resistance towards transplanted GL261 glioma [154, 157, 158]. Interestingly, compared to CTLA-4 knockout mice, VISTA knockout mice exhibit no signs for severe autoimmunity pointing out, that other inhibitory receptors compensate for loss of VISTA [157]. The role of VISTA in cancer immune evasion has been demonstrated in melanoma mouse models, where anti- VISTA antibody treatment resulted in enhanced effector function of tumor specific T cells and to decreased tumor growth [156].
Preclinical studies with inhibition of VISTA revealed a progression of autoimmune encephalomyelitis [152], whereby graft- versus-host-reaction could be inhibited by VISTA blockade [153]. In murine tumor models (such as fibrosarcoma [152] or melanoma [159]), VISTA blockade could significantly improve clinic-pathological aspects like tumor growth or overall survival rate. Additionally, this was paralleled by enhanced anti-tumor immunity with increased infiltration, proliferation, and effector function of T-cells [156]. Interestingly, the efficiency of the inhibition of VISTA is independent of missing VISTA expression on the tumor cells, and of the presence of high PD-L1 expression [156, 160].
CD96
CD96 (also known as Tactile (T cell activation, increased late expression)) is beside CD226 one of the ligands of CD155 [161]. The discovery of CD96 upregulation in T cells and NK cells within human tumors led to the the hypothesis that the inhibition of the CD155/CD96 could essentially influence the tumor elimination [162]. In particular, CD96−/− mice show increased NK-cell activity in response to immune challenge and significant resistance to cancer [163, 164]. In addition, further studies could highlight the role of CD96 in acute myeloid leukaemia (AML) as well as in congenital disease like C syndrome or opitz trigonocephaly [165, 166]. Furthermore CD96 plays a key role in chronic viral disease induced by Hepatitis B [167] or HIV-1 [168], where investigations could reveal that CD96 expression is pathogenetically linked to disease progression [168].
Clinical trials exploiting reinvigoration of T cells
Although checkpoint inhibition is relatively new, it has become a very attractive single therapy option or a combination partner with other standard care of treatment options. This chapter will summarize in a clear and concise manner recently published clinical trials dealing with checkpoint inhibition (for detailed information see Table 2). To do so, we will concentrate on efficacy and tolerability of the checkpoint inhibitors for CTLA-4, PD-1 and, PD-L1 (Fig. 1), due to the fact that there is too little or even no information about other immune checkpoints in clinical trials at the moment. To anticipate efficacy and possible immune related adverse effects (irAEs), it is important to consider which immune cells and T cell subsets are targeted by the respective therapeutic antibodies. As described in the previous chapters, expression of IRs are not solely restricted to exhausted CD8+ Tcells but may also be expressed on T helper, Treg or antigen presenting cells which could amplify or impede therapeutic effects. Hence, CTLA-4 and PD-1/PD-L1 specific antibodies differ in their mode of action. Whereas CTLA-4 antibodies lower the threshold for T cell activation (also of low affine tumor specific naive T cells), antibodies targeting the PD-1/PD-L axis aim at regulating effector T cell activity [42, 169]. In that sense, PD-1/PD-L antibodies do not merely target cytotoxic CD8+ T cell subsets but can impede tumor specific Tregs, thereby potentiating tumor specific cytolytic attacks [169]. Monoclonal antibodies that pharmaceutically inhibit CTLA-4 are ipilimumab and tremelimumab. Used as a single therapy, ipilimumab has mostly been investigated in the setting of malignant melanoma and non Hodgkin lymphomas (NHL). In 2015 Eggermont et al. stated in a phase III clinical trial when ipilimumab is given in an adjuvant manner in previously resected stage III melanoma, it significantly improved recurrence-free survival compared with placebo [170]. In combination with glycoprotein 100 (gp100) vaccination or with radiotherapy, ipilimumab improved overall survival or increased the duration of irradiated tumor response [171–173]. Moreover, in combination with the immunostimulator sargramostim, ipilimumab showed longer overall survival in the same setting [174]. Beashey et al. who treated patients suffering from aggressive NHL with ipilimumab after allogenic hematopoetic cell transplantation recorded antitumor responses as well [175]. Nevertheless, a phase II clinical trial in 2015revealed only little clinical activity for ipilimumab when given adjuvant after resection of advanced uveal melanoma [176].
Table 2.
Clinical trials for checkpoint inhibitors alone and compared to standard care of treatment
| Agent (inhibited checkpoint) | Setting | Phase | Treatment | Tumor response | OS (PFS) in MO | Toxicity (irAE grade ≥3) | Ref |
|---|---|---|---|---|---|---|---|
| Ipilimumab (CTLA-4) | Advanced uveal melanoma | II | Ipilimumap | SD 47% | 6.8 (2.8) | Colitis, diarrhea, elevated liver enzymes | [176] |
| After complete resection of advanced melanoma | III | Ipilimumab or placebo after complete resection | NM | (26.7 vs 17.1) | Diarrhea, colitis,rash, pruritus, hypo-physitis, elevated liver enzymes | [170] | |
| Advanced melanoma | II | Ipilimumap | CR 0% PR 10% SD 10% PD 65% |
8.7 (2.7) | Elevated liver enzymes | [205] | |
| Relapse of malignancy after allogeneic hematopoietic stemcell transplan-tation | I | Ipilimumab | ORR 6.9% CR 6.9% PR 3.4% |
24.7 | Arthritis, pneumonitis | [175] | |
| Relapsed and refractory B-cell NHL | I | Ipilimumap | NM | NM | Diarrhea, fatigue, | [206] | |
| Treme-limumap (CTLA-4) | Advanced melanoma | III | Tremeli-mumab vs. standard-of-care chemotherapy | NM | 12.6 vs 10.7 (at 6 MO 20.3%vs 18.1%) | Diarrhea, colitis, pruritus, rash |
[183] |
| Advanced melanoma | I | Anti-CD40 + Tremeli-mumab | NM | 26.1 (2.5) | Diarrhea, colitis, pruritus, rash | [212] | |
| Advanced gastric and esophageal adeno-carcinoma | II | Tremeli-mumap | PR 5.6% SD 22% |
4.8 (2.8) | Diarrhea, atrial fibrillation, increased liver enzymes | [177] | |
| Advanced (metastatic) colorectal carcinoma | II | Tremeli-mumap | PR 2.2% PD 95.6% | At 1a 4.8 vs 10.7% (at 6 MO 2.3 vs 2.1%) | Diarrhea, fatigue, colitis | [185] | |
| Advanced NSCLC | II | Tremeli-mumap vs. best supportive care | PR 4.8% SD 16.6% |
20.9% (34%) at 3 MO | Diarrhea, colitis | [213] | |
| HHC and chronic hepatitis C | II | Tremeli-mumap | SD 58.8% PR 17.6% |
8.2 (6.5) | Skin rash, diarrhea, syncope, diverticulitis, depression | [179] | |
| Advanced malignant mesothelioma | II | Tremeli-mumap | PR 3% SD 38% |
11.3 | Gastrointes-tinal events, dermatologi-cal events, fever | [214] | |
| Nivolumab (PD-1) | Advanced refractory squamous NSCLC | II | Nivolumab 3 mg/kg every 2 weeks until progression | PR 14.5% SD 26% PD 44% |
8.2 (1.9); 1a 40.1% | Fatigue, diarrhea, rash pruritus | [196] |
| Untreated melanoma (BRAF wild type vs mutated) | I | Nivolumab + Ipilimumab vs Ipilimumab + placebo | WT [BRAF+] ORR 61% vs 11% [3% vs 1%] CR 16% vs 0% [5% vs 0%] PR 28% vs 4% [7% vs 1%] SD 9% vs 13% [5% vs 7%] |
NM | Diarrhea rash. fatigue pruritus, elevated liver enzymes | [187] | |
| Untreated melanoma without BRAF mutation | III | Nivolumab vs Dacarbazine | ORR 40,0% vs 13,9% | 72.9% vs 42.1% at 1a (5.1 vs 2.2) | Fatigue, pruritus, nausea, diarrhea | [186] | |
| Advanced Squamous-Cell NSCLC | III | Nivolumab vs Docetaxel | ORR 20 vs 9% CR 1 vs 0% PR 26 vs 12% SD 39 vs 47% PD 56% vs 48% |
9.2 vs 6.0 (3.5 vs 2.8) | Fatigue, leukopenia | [191] | |
| Advanced non-Squamous-Cell NSCLC | III | Nivolumab vs Docetaxel | ORR 19% vs 12% CR 4 vs 1% PR 52% vs 35% SD 12;7% vs 21% PD 22.2% vs 14.6% |
12.2 vs 9.4 (2.3 vs 4.2) | Fatigue, nausea, diarrhea | [192] | |
| Relapsed or refractory Hodgkin 's lymphoma | I | Nivolumab | CR 17% PR 70% SD 13% |
NM | Leukopenia, stomatitis increased lipase levels, pancreatitis | [206] | |
| Pretreated advanced NSCLC (s and ns) | I | Nivolumab | ORR 17.1% (16.7% s vs 17.6% ns) | 9.9 | Rash, Colitis | [190] | |
| Untreated melanoma | III | Nivolumab vs Nivolumab + Ipilimumab vs Ipilimumab | ORR 14.6% vs 19.2% vs 6.3% CR 8.9% vs 11.5% vs 2.2% PR 34.8% vs 46.2% vs 16.8% SD 10.8% vs 13.1% vs 21.9% PD 37.7% vs 22.6% vs 48.9% |
11.5 vs 2.9 vs 6.9 | Diarrhea, fatigue, pruritus, rash | [188] | |
| Platinum resistant ovarian cancer | II | Ipilimumab | CR 10% PR 5% SD 30% PD 50% |
20 (3.5) | Lympho-cytopenia, anemia | [215] | |
| Advanced melanoma after anti CTLA-4 treatment | III | Nivolumab vs investigators choice of chemo | ORR 31.7% vs 10.6% CR 3.3% vs 0% PR 28.3% vs 10.6% SD 23.3% vs 34% PD 35% vs 31.9% |
(4.7 vs 4.2) | Anemia, fatigue, vomitting | [189] | |
| Advanced renal cell carcinoma | III | Nivolumab vs Everolimus | ORR 25% vs 5% CR 1% vs <1% | 25.0 vs 19.6 (4.6 vs 4.4) | Fatigue, diarrhea, rash | [216] | |
| Pembroli-zumab (PD-1) | Advanced NSCLC | I | Pembroli-zumab | ORR 19.4% | 12.0 (3.7) | Fatigue, rash, diarrhea | [217] |
| Advanced triple negative breast cancer | Ib | Pembroli-zumab | ORR 18.5% CR 3.7%; PR 14.8% SD 25.9% PD 48.1% | NM | Anemia, headache, | [218] | |
| Previously treated advanced non-small-cell lung cancer | II/III | Pembroli-zumab vs Docetaxel | NM | 10.4 vs 12.7 vs 8.5 (3.9 vs 4.0 vs 4.0) | Anemia, headache, | [193] | |
| Advanced melanoma | I | Pembroli-zumab | ORR 38.6% vs 28.6% | 23 (4) | Anemia, headache, | [194] | |
| Progressive metastatic carcinoma with or without mismatch repair-deficiency | II | Pembroli-zumab | ORR 40% vs 78% for mismatch repair-deficienct CRC and 0% vs 11% mismatch repair-proficient colorectal cancer | NM | Lympho-penia, anemia, diarrhea, bowel obstruction, elevated liver enzymes | [195] | |
| Advanced melanoma | III | Pembrolizumab vs Ipilimumab | ORR 89.4% vs 96.7% vs 87.9% | At 1a 74.1% vs 68.4% (at 6 MO 47.3%vs 46.4% vs 26.5%) | Lympho-penia, anemia, diarrhea, bowel obstruction, elevated liver enzymes | [219] | |
| Atezoli-zumab (PD-L1) | Previously treated metastatic uorthelial carcinoma | II | Atezoli-zumab | ORR 15% CR 5% PR 10% SD 19% PD 51% | NM | Fatigue, decreased appetite, dyspnoea, anemia, colitis | [202] |
| Previously treated NSCLC | II | Atezo-lizumab vs Docetaxel | NM | 12.6 vs 9.7 | Diarrhea, asthenia, neutropenia | [201] |
Abbreviations: CR complete response, HCC hepatocellular carcinoma, irAE immune related adverse effects, MO months, NM not mentioned, NSCLC non small cell lung cancer, ORR overall response rate, OS overall survival, PD progressive disease, PFS progression free survival, PR partial response, SD stable disease
Tremelimumab as well has been investigated not only in the setting of advanced malignant melanoma, but also in a number of other malignancies like advanced adenocarcinomas of the gastrointestinal tract, non small cell lung carcinoma (NSCLC) and hepatocellular carcinoma (HCC) as well as malignant mesothelioma [177–182]. Concerning malignant melanoma, in 2013 Ribas et al. were not able to demonstrate a statistically significant survival advantage for tremelimumab compared to standard-of-care chemotherapy in patients suffering from advanced melanoma [183]. But in combination with high dose interferon-α treatment of malignant melanomas showed significant therapeutic benefit [184]. The clinical phase II studies dealing with adenocarcinomas of the esophagus and the colon showed disappointing response rates, not supporting further investigations [177, 185]. In contrast, tremelimumab showed antitumor and antiviral effects in patients suffering from HCC on the basis of hepatitis C-virus infections [179].
The PD-1 inhibiting agents, Nivolumab and Pembrolizumab, were also used in clinical trials to treat malignant melanoma. In a phase III clinical trial, performed by Robert et al., nivolumab showed significant improvements in overall survival and progression free survival compared with dacarbazine. This trial setting focused on untreated melanoma without BRAF mutation [186]. Additionally, Postow et al. and others demonstrated that the combination of nivolumab and ipilimumab had significant advantages over single nivolumab therapy or placebo alone concerning progression-free survival [187, 188]. Even as a second line therapy nivolumab seems to improve outcome in malignant melanoma. In this phase III trial, ipilumumab pretreated advanced melanoma patients were either treated with nivolumab or investigators choice of chemotherapy. In this setting nivolumab demonstrated higher objective response rates than the alternative available chemotherapy [189]. In the setting of squamous or non squamous NSCLC, nivolumab seems to improve survival rates in previously heavily treated patients [190]. It even showed a better performance compared to docetaxel [191, 192]. Similar to that, pembrolizumab prolonged overall survival compared to docetaxel in NSCLC in a phase II/III clinical trial [193]. Obviously, patients with malignant melanoma were treated with pembrolizumab in a clinical trial as well. Ribas et al. were able to show that pembrolizumab prolonged progression-free survival and overall survival compared to ipilimumab. In another phase I clinical trial pembrolizumab improved objective response and survival rates [194]. In addition, Le et al. showed another very interesting feature of pembrolizumab. They performed a phase II clinical trial in which they were able to investigate that mismatch-repair deficiency predicted clinical effect of pembrolizumab in patients suffering from colorectal carcinoma [195], implying that response rates and clinical benefit from anti-PD1 therapies is correlating with high non-synonymous mutation load, which associates with the presence of tumor associated neoantigens [195, 196]. It was suggested that there is a general correlation of mutation load within tumor DNA and efficacy of immune checkpoint inhibition, irrespective of targeting PD-1 or its ligand, likely by an increased expression of tumor associated neoantigens [195–197]. While tumors with deficiencies in DNA mismatch-repair were found to have a better response toPD-1 blockade [195], it will certainly be clinically relevant to assess other surrogate markers which predict response to immune checkpoint blockade. These markers could likely be mutations in other DNA repair genes but also expression levels of DNA-mutating enzymes, such as family members of the AID/APOBEC deaminases, which could lead to increased mutation load in tumor DNA [198]. In addition, a similar correlation of treatment response and mutation load has been shown for melanoma patients treated with CTLA-4 [194, 195].
Pidilizumab, another PD-1 inhibitor, was used in a combination therapy in two different phase II clinical studies. Relapsed follicular lymphoma patients treated with pidilizumab in combination with rituximab exhibited an overall response rate of 66% and a complete response rate of 52% [199]. In the setting of diffuse large B cell lymphoma, patients treated with pidilizumab after hematopoietic stem cell transplantation showed an overall response rate of 51% and complete response in 34%, although 37% of patients showed a progressive disease in the same clinical trial [200].
Unlike PD-1 targeting antibodies, the PD-L1 specific antibody atezolizumab is not primarily used in the setting of melanoma. In previously treated NSCLC patients, atezolizumab improved survival compared with docetaxel in correlation with PD-L1 expression in the tumor and in tumor infiltrating immune cells [201]. Similar effects on survival were seen in another study dealing with previously metastatic urothelial carcinoma [202]. In combination with cobimetinib, a selective mitogen activated protein kinase (MAP2K1) inhibitor, atezolizumab ameliorated response rates even in mismatch repair proficient metastatic colorectal cancer [203].
Regarding the immune related adverse events of checkpoint inhibitors, all mentioned antibodies show similar immune related adverse events (irAEs, see Tables 2 and 3). Adverse events of grade 3 or higher affected most of the gastrointestinal tract, the skin, the liver function and the hematopoietic system (for more details see Tables 2 and 3). Diarrhea or colitis was observed in almost all clinical trials. However, the majority of adverse events were acceptable and mostly easy to manage [204–206]. Compared to standard chemotherapy, some investigators stated a much better tolerability for checkpoint inhibitors [189, 192, 201]. Moreover, a combination of checkpoint inhibition with ipilimumab and radiotherapy did not show an increase in adverse events [172]. Furthermore, clinical trials investigating combination therapies with standard of care therapies like exemestane in breast cancer, bicalutamide in prostate cancer, rituximab in follicular lymphoma or gemcitabine in pancreatic cancer, showed usually a satisfactory adverse events profile [199, 207–209]).
Table 3.
Clinical trials for checkpoint inhibitors in combination with standard care of treatment
| Agent (inhibited check-point) | Setting | Phase | Treatment | Tumor response | OS (PFS) in months | Toxicity irAE grade ≥3 | Ref. |
|---|---|---|---|---|---|---|---|
| Ipilimumab (CTLA-4) | Advanced melanoma | III | Ipilimumab or Ipilimumab + glycoprotein 100 or glycoprotein 100 only | NM | 10 vs 10.1 vs 6.4 (2.76 vs 2.86 vs 2.76) | Diarrhea, nausea, constipation, vomiting, abdominal pain | [171] |
| Advanced melanoma | Retrospective | Ipilimumab or maintenance + median 30 Gy | NM | 9 vs 39 | NM | [172] | |
| Advanced melanoma | Retrospective | Ipilimumab vs Ipilimumab + radiotherapy | NM | 10.2 vs 19.6 | Rash, colitis, GI, fatigue | [173] | |
| Advanced melanoma | I | Ipilimumab plus radiotherapy | NM | 10.7 (3.8) | Anemia, diarrhea, colitis | [220] | |
| Metastatic melanoma | II | Ipilimumab + sargramostim vs Ipilimumab alone | NM | 17.5 vs 12.7 (3.1 vs 3.1) | Diarrhea, rash, colitis, elevated liver enzymes | [174] | |
| Metastatic NSCLC | I | Ipilimumab + Paclitaxel vs Ipilimumab + Carboplatin | NM | NM | Adrenal insuffiency, enterocolitis | [221] | |
| Advanced, bone metastasis, castration-resistant prostate cancer | III | Ipilimumab or placebo after 8 GY | NM | 11.2 vs 10.2 (4.0 vs 3.1; at 6 MO 30.7% vs 18.1%) | Diarrhea, colitis | [222] | |
| Tremel-imumap (CTLA-4) | Prostate cancer (PSA-recurrent) | I | Tremeli-mumab + Bicalutamide | NM | NM | Colitis | [208] |
| Advanced breast cancer | I | Tremeli-mumab + Exemestane | SD 42% | NM | Diarrhea, rash | [207] | |
| Metastatic pancreatic cancer | I | Tremeli-mumab + Gemcitabine | PR 10.5% | 7.4 | Asthenia, nausea, diarrhea | [223] | |
| Advanced melanoma (or solid tumors) | I | Tremeli-mumab + PF-3512676 (CPG 7909) = Toll like receptor 9 inhibitor | NM | 19 | Diarrhea, hypophy-sitis, colitis, nausea, vomiting, pruritus, rash, neutropenia, rectal Bleeding | [224] | |
| Advanced melanoma | II | Trimilimumab + high dose INFalpha (HDI) | ORR 24% CR 11% PR 14% SD 38% | 21 (6.4) | Diarrhea, colitis, elevated liver enzymes, rash, fatigue, anxiety/depression | [184] | |
| Metastatic renal cell carcinoma | I | Tremeli-mumab + sunitinib | PR 42.8%; SD 9.5% | 2.8–18.2MO | Fatigue, mucositis, dypnea | [225] | |
| Nivolumab (PD-1) | Resected advanced melanoma | II | Adjuvant Nivolumab + multi-peptide vaccine (gp100, MART-1 & NY-ESO-1 with Montanide ISA 51 VG) | NM | At 1a 87% At 2a 82% |
Colitis, enteritis, rash, hypokalemia | [226] |
| Pidilizumab (PD-1) | Relapsed follicular lymphoma | II | Pidilizumab + Rituximab | ORR 66% CR 52% PR 14% | NM | No grade 3 or higher irAE | [199] |
| DLBCL | II | Pidilizumab after autologous hematopoietic stem- cell transplan-tation | ORR 51% CR 34% PR 17% SD 37% PD 11% | At 16 MO 0.85% (at 16 MO 0,72%) | Thrombo-cytopenia, anemia, pyrexia, renal failure, | [200] | |
| Atezoli-zumab (PD-L1) | Microsatellite stable metastatic colorectal cancer | Ib | Combination of cobimetinib and ateolizumab | ORR 17% and 20% in KRAS-mutant tumors | At 6 MO 72% | NM | [203] |
Abbreviations: CR complete response, irAE immune related adverse effects, MO months, NM not mentioned, NSCLC non small cell lung cancer, ORR overall response rate, OS overall survival, PD progressive disease, PFS progression free survival, PR partial response, SD stable disease
Conclusions
The results of numerous clinical trials using immune checkpoint inhibitors are very encouraging. Blocking antibodies for CTLA-4, PD-1 or PD-L1 seem to have a strong therapeutic potential when given alone or in combination with standard care of treatment in many different tumor entities. Additionally, checkpoint inhibitors adverse events profiles do not seem to be much worse than profiles of standard chemotherapies, but due to the fact that recently published clinical trials were in phase I or II, these encouraging data needs to be verified in more phase III clinical trials with longer follow up and larger numbers of patients. In addition, future challenges will be to elucidate proper pretreatments or combination therapies to increase clinical benefit of checkpoint inhibition also in cancer with initial low non-synonymous mutation load or low neoantigen expression.
Acknowledgements
Not applicable.
Funding
RG receives support from the Austrian science fund FWF, grant P24619 and grant P28201.
KC and RG receive support from the LIMCR-SCRI, the province and the city of Salzburg.
Availability of data and materials
Not applicable.
Authors' contributions
CK, EK, DN and RG wrote the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Abbreviations
- AKT
proteinkinase B
- BTLA
B and T lymphocyte attenuator
- CR
complete response
- CTLA-4
cytotoxic T-lymphocyte-associated protein 4
- EAE
experimental autoimmune encephalomyelitis
- Eomes
eomesodermin
- Gal-9
galectin-9
- HBV
hepatitis B virus
- HCC
hepatocellular carcinoma
- HCC
hepatocellular carcinoma
- HCV
hepatitis C virus
- HIV
human immunodeficiency virus
- HTLV1
human T lymphotropic virus 1
- HVEM
herpesvirus entry mediator
- IgSV
immunoglobulin superfamily
- IR
inhibitory receptor
- irAE
immune related adverse effects
- ITAM
immunoreceptor tyrosine-based activation motif
- ITIM
immunoreceptor tyrosine- based inhibitory motif
- ITSM
immunoreceptor tyrosine- based switch motif
- ITT
immunoglobulin tail tyrosine
- LAG-3
lymphocyte-activated gene-3
- LCMV
lymphocytic choriomeninigits virus
- MO
months
- NHL
non Hodgkin lymphoma
- NK
natural killer cell
- NKT
natural killer T cell
- NM
not mentioned
- NOD
non-obese diabetic
- NSCLC
non small cell lung cancer
- NSCLC
non-small cell lung cancer
- ORR
overall response rate
- OS
overall survival
- PD
progressive disease
- PD-1
programmed cell death 1
- PD-1H
PD-1 homolog
- PD-L1
programmed cell death-ligand 1
- PD-L2
programmed cell death-ligand 1
- PFS
progression free survival
- PI3K
phosphatidylinositide 3-kinases
- PR
partial response
- PVR
poliovirus receptors
- SD
stable disease
- SIV
simian immunodeficiency virus
- SLAM
signaling lymphocyte activation molecule
- T-bet
T-box transcription factor TBX21
- TCR
T cell receptor
- TFH
follicular helper T cells
- TIGIT
T cell immunoreceptor with Ig and ITIM domains
- TILs
tumor-infiltrating T cell
- TIM-3
T-cell immunoglobulin and mucin- containing protein 3
- TLR
toll-like receptor
- Tr1
type 1 regulatory T cells
- Treg
regulatory T cells
- Tregs
regulatory T cells
- VISTA
V domain Ig suppressor of T cells activation
Contributor Information
Kemal Catakovic, Email: k.catakovic@salk.at.
Eckhard Klieser, Email: e.klieser@salk.at.
Daniel Neureiter, Email: d.neureiter@salk.at.
Roland Geisberger, Phone: +43 (0) 57255 25847, Email: r.geisberger@salk.at.
References
- 1.Crespo J, et al. T cell anergy, exhaustion, senescence, and stemness in the tumor microenvironment. Curr Opin Immunol. 2013;25(2):214–21. doi: 10.1016/j.coi.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961;25:585–621. doi: 10.1016/0014-4827(61)90192-6. [DOI] [PubMed] [Google Scholar]
- 3.Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. 2015;15(8):486–99. doi: 10.1038/nri3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Angelosanto JM, et al. Progressive loss of memory T cell potential and commitment to exhaustion during chronic viral infection. J Virol. 2012;86(15):8161–70. doi: 10.1128/JVI.00889-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brooks DG, McGavern DB, Oldstone MB. Reprogramming of antiviral T cells prevents inactivation and restores T cell activity during persistent viral infection. J Clin Invest. 2006;116(6):1675–85. doi: 10.1172/JCI26856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Day CL, et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature. 2006;443(7109):350–4. doi: 10.1038/nature05115. [DOI] [PubMed] [Google Scholar]
- 7.Dyavar Shetty R, et al. PD-1 blockade during chronic SIV infection reduces hyperimmune activation and microbial translocation in rhesus macaques. J Clin Invest. 2012;122(5):1712–6. doi: 10.1172/JCI60612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Petrovas C, et al. SIV-specific CD8+ T cells express high levels of PD1 and cytokines but have impaired proliferative capacity in acute and chronic SIVmac251 infection. Blood. 2007;110(3):928–36. doi: 10.1182/blood-2007-01-069112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yamamoto T, et al. Surface expression patterns of negative regulatory molecules identify determinants of virus-specific CD8+ T-cell exhaustion in HIV infection. Blood. 2011;117(18):4805–15. doi: 10.1182/blood-2010-11-317297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gruener NH, et al. Sustained dysfunction of antiviral CD8+ T lymphocytes after infection with hepatitis C virus. J Virol. 2001;75(12):5550–8. doi: 10.1128/JVI.75.12.5550-5558.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Radziewicz H, et al. Liver-infiltrating lymphocytes in chronic human hepatitis C virus infection display an exhausted phenotype with high levels of PD-1 and low levels of CD127 expression. J Virol. 2007;81(6):2545–53. doi: 10.1128/JVI.02021-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reignat S, et al. Escaping high viral load exhaustion: CD8 cells with altered tetramer binding in chronic hepatitis B virus infection. J Exp Med. 2002;195(9):1089–101. doi: 10.1084/jem.20011723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Urbani S, et al. Virus-specific CD8+ lymphocytes share the same effector-memory phenotype but exhibit functional differences in acute hepatitis B and C. J Virol. 2002;76(24):12423–34. doi: 10.1128/JVI.76.24.12423-12434.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abdelbary NH, et al. Reduced Tim-3 expression on human T-lymphotropic virus type I (HTLV-I) Tax-specific cytotoxic T lymphocytes in HTLV-I infection. J Infect Dis. 2011;203(7):948–59. doi: 10.1093/infdis/jiq153. [DOI] [PubMed] [Google Scholar]
- 15.Ezinne CC, et al. HTLV-1 specific CD8+ T cell function augmented by blockade of 2B4/CD48 interaction in HTLV-1 infection. PLoS One. 2014;9(2) doi: 10.1371/journal.pone.0087631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frebel H, et al. Programmed death 1 protects from fatal circulatory failure during systemic virus infection of mice. J Exp Med. 2012;209(13):2485–99. doi: 10.1084/jem.20121015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nishimura H, et al. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity. 1999;11(2):141–51. doi: 10.1016/S1074-7613(00)80089-8. [DOI] [PubMed] [Google Scholar]
- 18.McKinney EF, et al. T-cell exhaustion, co-stimulation and clinical outcome in autoimmunity and infection. Nature. 2015;523(7562):612–6. doi: 10.1038/nature14468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dong H, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8(8):793–800. doi: 10.1038/nm730. [DOI] [PubMed] [Google Scholar]
- 20.Fourcade J, et al. PD-1 is a regulator of NY-ESO-1-specific CD8+ T cell expansion in melanoma patients. J Immunol. 2009;182(9):5240–9. doi: 10.4049/jimmunol.0803245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gassner FJ, et al. Chemotherapy-induced augmentation of T cells expressing inhibitory receptors is reversed by treatment with lenalidomide in chronic lymphocytic leukemia. Haematologica. 2014;99(5):67–9. doi: 10.3324/haematol.2013.098459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee PP, et al. Characterization of circulating T cells specific for tumor-associated antigens in melanoma patients. Nat Med. 1999;5(6):677–85. doi: 10.1038/9525. [DOI] [PubMed] [Google Scholar]
- 23.Baitsch L, et al. Exhaustion of tumor-specific CD8(+) T cells in metastases from melanoma patients. J Clin Invest. 2011;121(6):2350–60. doi: 10.1172/JCI46102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gros A, et al. PD-1 identifies the patient-specific CD8(+) tumor-reactive repertoire infiltrating human tumors. J Clin Invest. 2014;124(5):2246–59. doi: 10.1172/JCI73639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Radoja S, et al. CD8(+) tumor-infiltrating T cells are deficient in perforin-mediated cytolytic activity due to defective microtubule-organizing center mobilization and lytic granule exocytosis. J Immunol. 2001;167(9):5042–51. doi: 10.4049/jimmunol.167.9.5042. [DOI] [PubMed] [Google Scholar]
- 26.Zenz T. Exhausting T cells in CLL. Blood. 2013;121(9):1485–6. doi: 10.1182/blood-2013-01-475939. [DOI] [PubMed] [Google Scholar]
- 27.Im SJ, et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature. 2016;537(7620):417–421. doi: 10.1038/nature19330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Utzschneider DT, et al. T Cell Factor 1-Expressing Memory-like CD8(+) T Cells Sustain the Immune Response to Chronic Viral Infections. Immunity. 2016;45(2):415–27. doi: 10.1016/j.immuni.2016.07.021. [DOI] [PubMed] [Google Scholar]
- 29.He R, et al. Follicular CXCR5-expressing CD8+ T cells curtail chronic viral infection. Nature. 2016;537(7620):412–428. doi: 10.1038/nature19317. [DOI] [PubMed] [Google Scholar]
- 30.Freeman GJ, et al. Cloning of B7-2: a CTLA-4 counter-receptor that costimulates human T cell proliferation. Science. 1993;262(5135):909–11. doi: 10.1126/science.7694363. [DOI] [PubMed] [Google Scholar]
- 31.Hathcock KS, et al. Identification of an alternative CTLA-4 ligand costimulatory for T cell activation. Science. 1993;262(5135):905–7. doi: 10.1126/science.7694361. [DOI] [PubMed] [Google Scholar]
- 32.Azuma M, et al. B70 antigen is a second ligand for CTLA-4 and CD28. Nature. 1993;366(6450):76–9. doi: 10.1038/366076a0. [DOI] [PubMed] [Google Scholar]
- 33.Rudd CE, Taylor A, Schneider H. CD28 and CTLA-4 coreceptor expression and signal transduction. Immunol Rev. 2009;229(1):12–26. doi: 10.1111/j.1600-065X.2009.00770.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Qureshi OS, et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science. 2011;332(6029):600–3. doi: 10.1126/science.1202947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alegre ML, et al. Regulation of surface and intracellular expression of CTLA4 on mouse T cells. J Immunol. 1996;157(11):4762–70. [PubMed] [Google Scholar]
- 36.Takahashi T, et al. Immunologic self-tolerance maintained by CD25(+)CD4(+) regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J Exp Med. 2000;192(2):303–10. doi: 10.1084/jem.192.2.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bour-Jordan H, et al. Intrinsic and extrinsic control of peripheral T-cell tolerance by costimulatory molecules of the CD28/ B7 family. Immunol Rev. 2011;241(1):180–205. doi: 10.1111/j.1600-065X.2011.01011.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tivol EA, et al. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. 1995;3(5):541–7. doi: 10.1016/1074-7613(95)90125-6. [DOI] [PubMed] [Google Scholar]
- 39.Waterhouse P, et al. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science. 1995;270(5238):985–8. doi: 10.1126/science.270.5238.985. [DOI] [PubMed] [Google Scholar]
- 40.Wing K, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science. 2008;322(5899):271–5. doi: 10.1126/science.1160062. [DOI] [PubMed] [Google Scholar]
- 41.Peggs KS, et al. Blockade of CTLA-4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti-CTLA-4 antibodies. J Exp Med. 2009;206(8):1717–25. doi: 10.1084/jem.20082492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Intlekofer AM, Thompson CB. At the bench: preclinical rationale for CTLA-4 and PD-1 blockade as cancer immunotherapy. J Leukoc Biol. 2013;94(1):25–39. doi: 10.1189/jlb.1212621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ishida Y, et al. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992;11(11):3887–95. doi: 10.1002/j.1460-2075.1992.tb05481.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Freeman GJ, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192(7):1027–34. doi: 10.1084/jem.192.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Barber DL, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439(7077):682–7. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- 46.Odorizzi PM, et al. Genetic absence of PD-1 promotes accumulation of terminally differentiated exhausted CD8+ T cells. J Exp Med. 2015;212(7):1125–37. doi: 10.1084/jem.20142237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yokosuka T, et al. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J Exp Med. 2012;209(6):1201–17. doi: 10.1084/jem.20112741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Parry RV, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol. 2005;25(21):9543–53. doi: 10.1128/MCB.25.21.9543-9553.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Patsoukis N, et al. Selective effects of PD-1 on Akt and Ras pathways regulate molecular components of the cell cycle and inhibit T cell proliferation. Sci Signal. 2012;5(230):ra46. doi: 10.1126/scisignal.2002796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Park HJ, et al. PD-1 upregulated on regulatory T cells during chronic virus infection enhances the suppression of CD8+ T cell immune response via the interaction with PD-L1 expressed on CD8+ T cells. J Immunol. 2015;194(12):5801–11. doi: 10.4049/jimmunol.1401936. [DOI] [PubMed] [Google Scholar]
- 51.Paley MA, et al. Progenitor and terminal subsets of CD8+ T cells cooperate to contain chronic viral infection. Science. 2012;338(6111):1220–5. doi: 10.1126/science.1229620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Blackburn SD, et al. Selective expansion of a subset of exhausted CD8 T cells by alphaPD-L1 blockade. Proc Natl Acad Sci U S A. 2008;105(39):15016–21. doi: 10.1073/pnas.0801497105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yu X, et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat Immunol. 2009;10(1):48–57. doi: 10.1038/ni.1674. [DOI] [PubMed] [Google Scholar]
- 54.Stanietsky N, et al. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc Natl Acad Sci U S A. 2009;106(42):17858–63. doi: 10.1073/pnas.0903474106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Le Mercier I, Lines JL, Noelle RJ. Beyond CTLA-4 and PD-1, the Generation Z of Negative Checkpoint Regulators. Front Immunol. 2015;6:418. doi: 10.3389/fimmu.2015.00418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Godefroy E, et al. TIGIT-positive circulating follicular helper T cells display robust B-cell help functions: potential role in sickle cell alloimmunization. Haematologica. 2015;100(11):1415–25. doi: 10.3324/haematol.2015.132738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li M, et al. T-cell immunoglobulin and ITIM domain (TIGIT) receptor/poliovirus receptor (PVR) ligand engagement suppresses interferon-gamma production of natural killer cells via beta-arrestin 2-mediated negative signaling. J Biol Chem. 2014;289(25):17647–57. doi: 10.1074/jbc.M114.572420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu S, et al. Recruitment of Grb2 and SHIP1 by the ITT-like motif of TIGIT suppresses granule polarization and cytotoxicity of NK cells. Cell Death Differ. 2013;20(3):456–64. doi: 10.1038/cdd.2012.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Levin SD, et al. Vstm3 is a member of the CD28 family and an important modulator of T-cell function. Eur J Immunol. 2011;41(4):902–15. doi: 10.1002/eji.201041136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Carlsten M, et al. DNAX accessory molecule-1 mediated recognition of freshly isolated ovarian carcinoma by resting natural killer cells. Cancer Res. 2007;67(3):1317–25. doi: 10.1158/0008-5472.CAN-06-2264. [DOI] [PubMed] [Google Scholar]
- 61.Masson D, et al. Overexpression of the CD155 gene in human colorectal carcinoma. Gut. 2001;49(2):236–40. doi: 10.1136/gut.49.2.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hirota T, et al. Transcriptional activation of the mouse Necl-5/Tage4/PVR/CD155 gene by fibroblast growth factor or oncogenic Ras through the Raf-MEK-ERK-AP-1 pathway. Oncogene. 2005;24(13):2229–35. doi: 10.1038/sj.onc.1208409. [DOI] [PubMed] [Google Scholar]
- 63.Kamran N, et al. Toll-like receptor ligands induce expression of the costimulatory molecule CD155 on antigen-presenting cells. PLoS One. 2013;8(1) doi: 10.1371/journal.pone.0054406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Soriani A, et al. ATM-ATR-dependent up-regulation of DNAM-1 and NKG2D ligands on multiple myeloma cells by therapeutic agents results in enhanced NK-cell susceptibility and is associated with a senescent phenotype. Blood. 2009;113(15):3503–11. doi: 10.1182/blood-2008-08-173914. [DOI] [PubMed] [Google Scholar]
- 65.Johnston RJ, et al. The immunoreceptor TIGIT regulates antitumor and antiviral CD8(+) T cell effector function. Cancer Cell. 2014;26(6):923–37. doi: 10.1016/j.ccell.2014.10.018. [DOI] [PubMed] [Google Scholar]
- 66.Tahara-Hanaoka S, et al. Tumor rejection by the poliovirus receptor family ligands of the DNAM-1 (CD226) receptor. Blood. 2006;107(4):1491–6. doi: 10.1182/blood-2005-04-1684. [DOI] [PubMed] [Google Scholar]
- 67.Welch MJ, et al. CD8 T cell defect of TNF-alpha and IL-2 in DNAM-1 deficient mice delays clearance in vivo of a persistent virus infection. Virology. 2012;429(2):163–70. doi: 10.1016/j.virol.2012.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ramsbottom KM, et al. Cutting edge: DNAX accessory molecule 1-deficient CD8+ T cells display immunological synapse defects that impair antitumor immunity. J Immunol. 2014;192(2):553–7. doi: 10.4049/jimmunol.1302197. [DOI] [PubMed] [Google Scholar]
- 69.Zhang T, et al. Increased expression of TIGIT on CD4+ T cells ameliorates immune-mediated bone marrow failure of aplastic anemia. J Cell Biochem. 2014;115(11):1918–27. doi: 10.1002/jcb.24862. [DOI] [PubMed] [Google Scholar]
- 70.Zhang Y, et al. Genome-wide DNA methylation analysis identifies hypomethylated genes regulated by FOXP3 in human regulatory T cells. Blood. 2013;122(16):2823–36. doi: 10.1182/blood-2013-02-481788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Joller N, et al. Treg cells expressing the coinhibitory molecule TIGIT selectively inhibit proinflammatory Th1 and Th17 cell responses. Immunity. 2014;40(4):569–81. doi: 10.1016/j.immuni.2014.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mahnke K, Enk AH. TIGIT-CD155 Interactions in Melanoma: A Novel Co-Inhibitory Pathway with Potential for Clinical Intervention. J Invest Dermatol. 2016;136(1):9–11. doi: 10.1016/j.jid.2015.10.048. [DOI] [PubMed] [Google Scholar]
- 73.Inozume T, et al. Melanoma Cells Control Antimelanoma CTL Responses via Interaction between TIGIT and CD155 in the Effector Phase. J Invest Dermatol. 2016;136(1):255–63. doi: 10.1038/JID.2015.404. [DOI] [PubMed] [Google Scholar]
- 74.Kurtulus S, et al. TIGIT predominantly regulates the immune response via regulatory T cells. J Clin Invest. 2015;125(11):4053–62. doi: 10.1172/JCI81187. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 75.Huard B, et al. CD4/major histocompatibility complex class II interaction analyzed with CD4- and lymphocyte activation gene-3 (LAG-3)-Ig fusion proteins. Eur J Immunol. 1995;25(9):2718–21. doi: 10.1002/eji.1830250949. [DOI] [PubMed] [Google Scholar]
- 76.Triebel F, et al. LAG-3, a novel lymphocyte activation gene closely related to CD4. J Exp Med. 1990;171(5):1393–405. doi: 10.1084/jem.171.5.1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Xu F, et al. LSECtin expressed on melanoma cells promotes tumor progression by inhibiting antitumor T-cell responses. Cancer Res. 2014;74(13):3418–28. doi: 10.1158/0008-5472.CAN-13-2690. [DOI] [PubMed] [Google Scholar]
- 78.Baixeras E, et al. Characterization of the lymphocyte activation gene 3-encoded protein. A new ligand for human leukocyte antigen class II antigens. J Exp Med. 1992;176(2):327–37. doi: 10.1084/jem.176.2.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Huang CT, et al. Role of LAG-3 in regulatory T cells. Immunity. 2004;21(4):503–13. doi: 10.1016/j.immuni.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 80.Kisielow M, et al. Expression of lymphocyte activation gene 3 (LAG-3) on B cells is induced by T cells. Eur J Immunol. 2005;35(7):2081–8. doi: 10.1002/eji.200526090. [DOI] [PubMed] [Google Scholar]
- 81.Workman CJ, et al. LAG-3 regulates plasmacytoid dendritic cell homeostasis. J Immunol. 2009;182(4):1885–91. doi: 10.4049/jimmunol.0800185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bae J, et al. Trafficking of LAG-3 to the surface on activated T cells via its cytoplasmic domain and protein kinase C signaling. J Immunol. 2014;193(6):3101–12. doi: 10.4049/jimmunol.1401025. [DOI] [PubMed] [Google Scholar]
- 83.Hannier S, Triebel F. The MHC class II ligand lymphocyte activation gene-3 is co-distributed with CD8 and CD3-TCR molecules after their engagement by mAb or peptide-MHC class I complexes. Int Immunol. 1999;11(11):1745–52. doi: 10.1093/intimm/11.11.1745. [DOI] [PubMed] [Google Scholar]
- 84.Workman CJ, Dugger KJ, Vignali DA. Cutting edge: molecular analysis of the negative regulatory function of lymphocyte activation gene-3. J Immunol. 2002;169(10):5392–5. doi: 10.4049/jimmunol.169.10.5392. [DOI] [PubMed] [Google Scholar]
- 85.Macon-Lemaitre L, Triebel F. The negative regulatory function of the lymphocyte-activation gene-3 co-receptor (CD223) on human T cells. Immunology. 2005;115(2):170–8. doi: 10.1111/j.1365-2567.2005.02145.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Workman CJ, et al. Lymphocyte activation gene-3 (CD223) regulates the size of the expanding T cell population following antigen activation in vivo. J Immunol. 2004;172(9):5450–5. doi: 10.4049/jimmunol.172.9.5450. [DOI] [PubMed] [Google Scholar]
- 87.Groux H, et al. A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature. 1997;389(6652):737–42. doi: 10.1038/39614. [DOI] [PubMed] [Google Scholar]
- 88.Durham NM, et al. Lymphocyte Activation Gene 3 (LAG-3) modulates the ability of CD4 T-cells to be suppressed in vivo. PLoS One. 2014;9(11) doi: 10.1371/journal.pone.0109080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Blackburn SD, et al. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol. 2009;10(1):29–37. doi: 10.1038/ni.1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Matsuzaki J, et al. Tumor-infiltrating NY-ESO-1-specific CD8+ T cells are negatively regulated by LAG-3 and PD-1 in human ovarian cancer. Proc Natl Acad Sci U S A. 2010;107(17):7875–80. doi: 10.1073/pnas.1003345107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Woo SR, et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012;72(4):917–27. doi: 10.1158/0008-5472.CAN-11-1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.McNerney ME, Lee KM, Kumar V. 2B4 (CD244) is a non-MHC binding receptor with multiple functions on natural killer cells and CD8+ T cells. Mol Immunol. 2005;42(4):489–94. doi: 10.1016/j.molimm.2004.07.032. [DOI] [PubMed] [Google Scholar]
- 93.Brown MH, et al. 2B4, the natural killer and T cell immunoglobulin superfamily surface protein, is a ligand for CD48. J Exp Med. 1998;188(11):2083–90. doi: 10.1084/jem.188.11.2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Garni-Wagner BA, et al. A novel function-associated molecule related to non-MHC-restricted cytotoxicity mediated by activated natural killer cells and T cells. J Immunol. 1993;151(1):60–70. [PubMed] [Google Scholar]
- 95.Nakajima H, et al. Activating interactions in human NK cell recognition: the role of 2B4-CD48. Eur J Immunol. 1999;29(5):1676–83. doi: 10.1002/(SICI)1521-4141(199905)29:05<1676::AID-IMMU1676>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 96.Muhammad A, et al. Sequential cooperation of CD2 and CD48 in the buildup of the early TCR signalosome. J Immunol. 2009;182(12):7672–80. doi: 10.4049/jimmunol.0800691. [DOI] [PubMed] [Google Scholar]
- 97.Chlewicki LK, et al. Molecular basis of the dual functions of 2B4 (CD244) J Immunol. 2008;180(12):8159–67. doi: 10.4049/jimmunol.180.12.8159. [DOI] [PubMed] [Google Scholar]
- 98.Eissmann P, et al. Molecular basis for positive and negative signaling by the natural killer cell receptor 2B4 (CD244) Blood. 2005;105(12):4722–9. doi: 10.1182/blood-2004-09-3796. [DOI] [PubMed] [Google Scholar]
- 99.Bloch-Queyrat C, et al. Regulation of natural cytotoxicity by the adaptor SAP and the Src-related kinase Fyn. J Exp Med. 2005;202(1):181–92. doi: 10.1084/jem.20050449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wherry EJ, et al. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity. 2007;27(4):670–84. doi: 10.1016/j.immuni.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 101.Raziorrouh B, et al. The immunoregulatory role of CD244 in chronic hepatitis B infection and its inhibitory potential on virus-specific CD8+ T-cell function. Hepatology. 2010;52(6):1934–47. doi: 10.1002/hep.23936. [DOI] [PubMed] [Google Scholar]
- 102.Bengsch B, et al. Coexpression of PD-1, 2B4, CD160 and KLRG1 on exhausted HCV-specific CD8+ T cells is linked to antigen recognition and T cell differentiation. PLoS Pathog. 2010;6(6) doi: 10.1371/journal.ppat.1000947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Aldy KN, et al. 2B4+ CD8+ T cells play an inhibitory role against constrained HIV epitopes. Biochem Biophys Res Commun. 2011;405(3):503–7. doi: 10.1016/j.bbrc.2011.01.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Casado JG, et al. CD8 T cells expressing NK associated receptors are increased in melanoma patients and display an effector phenotype. Cancer Immunol Immunother. 2005;54(12):1162–71. doi: 10.1007/s00262-005-0682-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Enose-Akahata Y, et al. High expression of CD244 and SAP regulated CD8 T cell responses of patients with HTLV-I associated neurologic disease. PLoS Pathog. 2009;5(12) doi: 10.1371/journal.ppat.1000682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.West EE, et al. Tight regulation of memory CD8(+) T cells limits their effectiveness during sustained high viral load. Immunity. 2011;35(2):285–98. doi: 10.1016/j.immuni.2011.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Han P, et al. An inhibitory Ig superfamily protein expressed by lymphocytes and APCs is also an early marker of thymocyte positive selection. J Immunol. 2004;172(10):5931–9. doi: 10.4049/jimmunol.172.10.5931. [DOI] [PubMed] [Google Scholar]
- 108.Watanabe N, et al. BTLA is a lymphocyte inhibitory receptor with similarities to CTLA-4 and PD-1. Nat Immunol. 2003;4(7):670–9. doi: 10.1038/ni944. [DOI] [PubMed] [Google Scholar]
- 109.Sedy JR, et al. B and T lymphocyte attenuator regulates T cell activation through interaction with herpesvirus entry mediator. Nat Immunol. 2005;6(1):90–8. doi: 10.1038/ni1144. [DOI] [PubMed] [Google Scholar]
- 110.Cai G, et al. CD160 inhibits activation of human CD4+ T cells through interaction with herpesvirus entry mediator. Nat Immunol. 2008;9(2):176–85. doi: 10.1038/ni1554. [DOI] [PubMed] [Google Scholar]
- 111.Gonzalez LC, et al. A coreceptor interaction between the CD28 and TNF receptor family members B and T lymphocyte attenuator and herpesvirus entry mediator. Proc Natl Acad Sci U S A. 2005;102(4):1116–21. doi: 10.1073/pnas.0409071102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Oya Y, et al. Development of autoimmune hepatitis-like disease and production of autoantibodies to nuclear antigens in mice lacking B and T lymphocyte attenuator. Arthritis Rheum. 2008;58(8):2498–510. doi: 10.1002/art.23674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Albring JC, et al. Targeting of B and T lymphocyte associated (BTLA) prevents graft-versus-host disease without global immunosuppression. J Exp Med. 2010;207(12):2551–9. doi: 10.1084/jem.20102017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Uchiyama M, et al. An agonistic anti-BTLA mAb (3C10) induced generation of IL-10-dependent regulatory CD4+ T cells and prolongation of murine cardiac allograft. Transplantation. 2014;97(3):301–9. doi: 10.1097/01.TP.0000438204.96723.8b. [DOI] [PubMed] [Google Scholar]
- 115.Deppong C, et al. B and T lymphocyte attenuator regulates T cell survival in the lung. J Immunol. 2008;181(5):2973–9. doi: 10.4049/jimmunol.181.5.2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Steinberg MW, et al. A crucial role for HVEM and BTLA in preventing intestinal inflammation. J Exp Med. 2008;205(6):1463–76. doi: 10.1084/jem.20071160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Flynn R, et al. CD8 T cell memory to a viral pathogen requires trans cosignaling between HVEM and BTLA. PLoS One. 2013;8(10) doi: 10.1371/journal.pone.0077991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Pasero C, et al. The HVEM network: new directions in targeting novel costimulatory/co-inhibitory molecules for cancer therapy. Curr Opin Pharmacol. 2012;12(4):478–85. doi: 10.1016/j.coph.2012.03.001. [DOI] [PubMed] [Google Scholar]
- 119.M'Hidi H, et al. High expression of the inhibitory receptor BTLA in T-follicular helper cells and in B-cell small lymphocytic lymphoma/chronic lymphocytic leukemia. Am J Clin Pathol. 2009;132(4):589–96. doi: 10.1309/AJCPPHKGYYGGL39C. [DOI] [PubMed] [Google Scholar]
- 120.Derre L, et al. BTLA mediates inhibition of human tumor-specific CD8+ T cells that can be partially reversed by vaccination. J Clin Invest. 2010;120(1):157–67. doi: 10.1172/JCI40070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Fourcade J, et al. CD8(+) T cells specific for tumor antigens can be rendered dysfunctional by the tumor microenvironment through upregulation of the inhibitory receptors BTLA and PD-1. Cancer Res. 2012;72(4):887–96. doi: 10.1158/0008-5472.CAN-11-2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Thommen DS, et al. Progression of Lung Cancer Is Associated with Increased Dysfunction of T Cells Defined by Coexpression of Multiple Inhibitory Receptors. Cancer Immunol Res. 2015;3(12):1344–55. doi: 10.1158/2326-6066.CIR-15-0097. [DOI] [PubMed] [Google Scholar]
- 123.Lou Y, et al. Epithelial-Mesenchymal Transition Is Associated with a Distinct Tumor Microenvironment Including Elevation of Inflammatory Signals and Multiple Immune Checkpoints in Lung Adenocarcinoma. Clin Cancer Res. 2016;22(14):3630–42. doi: 10.1158/1078-0432.CCR-15-1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lasaro MO, et al. Active immunotherapy combined with blockade of a coinhibitory pathway achieves regression of large tumor masses in cancer-prone mice. Mol Ther. 2011;19(9):1727–36. doi: 10.1038/mt.2011.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hobo W, et al. B and T lymphocyte attenuator mediates inhibition of tumor-reactive CD8+ T cells in patients after allogeneic stem cell transplantation. J Immunol. 2012;189(1):39–49. doi: 10.4049/jimmunol.1102807. [DOI] [PubMed] [Google Scholar]
- 126.Anderson AC, et al. T-bet, a Th1 transcription factor regulates the expression of Tim-3. Eur J Immunol. 2010;40(3):859–66. doi: 10.1002/eji.200939842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Monney L, et al. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature. 2002;415(6871):536–41. doi: 10.1038/415536a. [DOI] [PubMed] [Google Scholar]
- 128.Asakura H, et al. Selective eosinophil adhesion to fibroblast via IFN-gamma-induced galectin-9. J Immunol. 2002;169(10):5912–8. doi: 10.4049/jimmunol.169.10.5912. [DOI] [PubMed] [Google Scholar]
- 129.Lee J, et al. Phosphotyrosine-dependent coupling of Tim-3 to T-cell receptor signaling pathways. Mol Cell Biol. 2011;31(19):3963–74. doi: 10.1128/MCB.05297-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zhu C, et al. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat Immunol. 2005;6(12):1245–52. doi: 10.1038/ni1271. [DOI] [PubMed] [Google Scholar]
- 131.Sehrawat S, et al. Galectin-9/TIM-3 interaction regulates virus-specific primary and memory CD8 T cell response. PLoS Pathog. 2010;6(5) doi: 10.1371/journal.ppat.1000882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Huang YH, et al. CEACAM1 regulates TIM-3-mediated tolerance and exhaustion. Nature. 2015;517(7534):386–90. doi: 10.1038/nature13848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Chiba S, et al. Tumor-infiltrating DCs suppress nucleic acid-mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat Immunol. 2012;13(9):832–42. doi: 10.1038/ni.2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Freeman GJ, et al. TIM genes: a family of cell surface phosphatidylserine receptors that regulate innate and adaptive immunity. Immunol Rev. 2010;235(1):172–89. doi: 10.1111/j.0105-2896.2010.00903.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kearley J, McMillan SJ, Lloyd CM. Th2-driven, allergen-induced airway inflammation is reduced after treatment with anti-Tim-3 antibody in vivo. J Exp Med. 2007;204(6):1289–94. doi: 10.1084/jem.20062093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Lee SY, Goverman JM. The influence of T cell Ig mucin-3 signaling on central nervous system autoimmune disease is determined by the effector function of the pathogenic T cells. J Immunol. 2013;190(10):4991–9. doi: 10.4049/jimmunol.1300083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Veenstra RG, et al. Contrasting acute graft-versus-host disease effects of Tim-3/galectin-9 pathway blockade dependent upon the presence of donor regulatory T cells. Blood. 2012;120(3):682–90. doi: 10.1182/blood-2011-10-387977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Sanchez-Fueyo A, et al. Tim-3 inhibits T helper type 1-mediated auto- and alloimmune responses and promotes immunological tolerance. Nat Immunol. 2003;4(11):1093–101. doi: 10.1038/ni987. [DOI] [PubMed] [Google Scholar]
- 139.He W, et al. Galectin-9 significantly prolongs the survival of fully mismatched cardiac allografts in mice. Transplantation. 2009;88(6):782–90. doi: 10.1097/TP.0b013e3181b47f25. [DOI] [PubMed] [Google Scholar]
- 140.Seki M, et al. Galectin-9 suppresses the generation of Th17, promotes the induction of regulatory T cells, and regulates experimental autoimmune arthritis. Clin Immunol. 2008;127(1):78–88. doi: 10.1016/j.clim.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 141.Chou FC, et al. Overexpression of galectin-9 in islets prolongs grafts survival via downregulation of Th1 responses. Cell Transplant. 2013;22(11):2135–45. doi: 10.3727/096368912X657891. [DOI] [PubMed] [Google Scholar]
- 142.Oomizu S, et al. Galectin-9 suppresses Th17 cell development in an IL-2-dependent but Tim-3-independent manner. Clin Immunol. 2012;143(1):51–8. doi: 10.1016/j.clim.2012.01.004. [DOI] [PubMed] [Google Scholar]
- 143.Boenisch O, et al. TIM-3: a novel regulatory molecule of alloimmune activation. J Immunol. 2010;185(10):5806–19. doi: 10.4049/jimmunol.0903435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Hastings WD, et al. TIM-3 is expressed on activated human CD4+ T cells and regulates Th1 and Th17 cytokines. Eur J Immunol. 2009;39(9):2492–501. doi: 10.1002/eji.200939274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Golden-Mason L, et al. Negative immune regulator Tim-3 is overexpressed on T cells in hepatitis C virus infection and its blockade rescues dysfunctional CD4+ and CD8+ T cells. J Virol. 2009;83(18):9122–30. doi: 10.1128/JVI.00639-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Jin HT, et al. Cooperation of Tim-3 and PD-1 in CD8 T-cell exhaustion during chronic viral infection. Proc Natl Acad Sci U S A. 2010;107(33):14733–8. doi: 10.1073/pnas.1009731107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Yang ZZ, et al. IL-12 upregulates TIM-3 expression and induces T cell exhaustion in patients with follicular B cell non-Hodgkin lymphoma. J Clin Invest. 2012;122(4):1271–82. doi: 10.1172/JCI59806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Fourcade J, et al. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J Exp Med. 2010;207(10):2175–86. doi: 10.1084/jem.20100637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Sakuishi K, et al. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med. 2010;207(10):2187–94. doi: 10.1084/jem.20100643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Zhou Q, et al. Coexpression of Tim-3 and PD-1 identifies a CD8+ T-cell exhaustion phenotype in mice with disseminated acute myelogenous leukemia. Blood. 2011;117(17):4501–10. doi: 10.1182/blood-2010-10-310425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Takamura S, et al. Premature terminal exhaustion of Friend virus-specific effector CD8+ T cells by rapid induction of multiple inhibitory receptors. J Immunol. 2010;184(9):4696–707. doi: 10.4049/jimmunol.0903478. [DOI] [PubMed] [Google Scholar]
- 152.Wang L, et al. VISTA, a novel mouse Ig superfamily ligand that negatively regulates T cell responses. J Exp Med. 2011;208(3):577–92. doi: 10.1084/jem.20100619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Flies DB, et al. Cutting edge: A monoclonal antibody specific for the programmed death-1 homolog prevents graft-versus-host disease in mouse models. J Immunol. 2011;187(4):1537–41. doi: 10.4049/jimmunol.1100660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Flies DB, et al. Coinhibitory receptor PD-1H preferentially suppresses CD4(+) T cell-mediated immunity. J Clin Invest. 2014;124(5):1966–75. doi: 10.1172/JCI74589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Lines JL, et al. VISTA is an immune checkpoint molecule for human T cells. Cancer Res. 2014;74(7):1924–32. doi: 10.1158/0008-5472.CAN-13-1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Le Mercier I, et al. VISTA Regulates the Development of Protective Antitumor Immunity. Cancer Res. 2014;74(7):1933–44. doi: 10.1158/0008-5472.CAN-13-1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Wang L, et al. Disruption of the immune-checkpoint VISTA gene imparts a proinflammatory phenotype with predisposition to the development of autoimmunity. Proc Natl Acad Sci U S A. 2014;111(41):14846–51. doi: 10.1073/pnas.1407447111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Liu J, et al. Immune-checkpoint proteins VISTA and PD-1 nonredundantly regulate murine T-cell responses. Proc Natl Acad Sci U S A. 2015;112(21):6682–7. doi: 10.1073/pnas.1420370112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Sorensen MR, et al. Adenoviral vaccination combined with CD40 stimulation and CTLA-4 blockage can lead to complete tumor regression in a murine melanoma model. Vaccine. 2010;28(41):6757–64. doi: 10.1016/j.vaccine.2010.07.066. [DOI] [PubMed] [Google Scholar]
- 160.Lines JL, et al. VISTA is a novel broad-spectrum negative checkpoint regulator for cancer immunotherapy. Cancer Immunol Res. 2014;2(6):510–7. doi: 10.1158/2326-6066.CIR-14-0072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Bottino C, et al. Identification of PVR (CD155) and Nectin-2 (CD112) as cell surface ligands for the human DNAM-1 (CD226) activating molecule. J Exp Med. 2003;198(4):557–67. doi: 10.1084/jem.20030788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Fuchs A, et al. Cutting edge: CD96 (tactile) promotes NK cell-target cell adhesion by interacting with the poliovirus receptor (CD155) J Immunol. 2004;172(7):3994–8. doi: 10.4049/jimmunol.172.7.3994. [DOI] [PubMed] [Google Scholar]
- 163.Chan CJ, et al. The receptors CD96 and CD226 oppose each other in the regulation of natural killer cell functions. Nat Immunol. 2014;15(5):431–8. doi: 10.1038/ni.2850. [DOI] [PubMed] [Google Scholar]
- 164.Blake SJ, et al. Molecular Pathways: Targeting CD96 and TIGIT for Cancer Immunotherapy. Clin Cancer Res. 2016;22(21):5183–5188. doi: 10.1158/1078-0432.CCR-16-0933. [DOI] [PubMed] [Google Scholar]
- 165.Hosen N, et al. CD96 is a leukemic stem cell-specific marker in human acute myeloid leukemia. Proc Natl Acad Sci U S A. 2007;104(26):11008–13. doi: 10.1073/pnas.0704271104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Kaname T, et al. Mutations in CD96, a member of the immunoglobulin superfamily, cause a form of the C (Opitz trigonocephaly) syndrome. Am J Hum Genet. 2007;81(4):835–41. doi: 10.1086/522014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Gong J, et al. Establishment of an enzyme-linked immunosorbent assay system for determining soluble CD96 and its application in the measurement of sCD96 in patients with viral hepatitis B and hepatic cirrhosis. Clin Exp Immunol. 2009;155(2):207–15. doi: 10.1111/j.1365-2249.2008.03829.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Eriksson EM, et al. Differential expression of CD96 surface molecule represents CD8(+) T cells with dissimilar effector function during HIV-1 infection. PLoS One. 2012;7(12) doi: 10.1371/journal.pone.0051696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.He J, et al. Development of PD-1/PD-L1 Pathway in Tumor Immune Microenvironment and Treatment for Non-Small Cell Lung Cancer. Sci Rep. 2015;5:13110. doi: 10.1038/srep13110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Eggermont AM, et al. Adjuvant ipilimumab versus placebo after complete resection of high-risk stage III melanoma (EORTC 18071): a randomised, double-blind, phase 3 trial. Lancet Oncol. 2015;16(5):522–30. doi: 10.1016/S1470-2045(15)70122-1. [DOI] [PubMed] [Google Scholar]
- 171.Hodi FS, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–23. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Barker CA, et al. Concurrent radiotherapy and ipilimumab immunotherapy for patients with melanoma. Cancer Immunol Res. 2013;1(2):92–8. doi: 10.1158/2326-6066.CIR-13-0082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Qin R, et al. Safety and Efficacy of Radiation Therapy in Advanced Melanoma Patients Treated With Ipilimumab. Int J Radiat Oncol Biol Phys. 2016;96(1):72–7. doi: 10.1016/j.ijrobp.2016.04.017. [DOI] [PubMed] [Google Scholar]
- 174.Hodi FS, et al. Ipilimumab plus sargramostim vs ipilimumab alone for treatment of metastatic melanoma: a randomized clinical trial. JAMA. 2014;312(17):1744–53. doi: 10.1001/jama.2014.13943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Bashey A, et al. CTLA4 blockade with ipilimumab to treat relapse of malignancy after allogeneic hematopoietic cell transplantation. Blood. 2009;113(7):1581–8. doi: 10.1182/blood-2008-07-168468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Zimmer L, et al. Phase II DeCOG-study of ipilimumab in pretreated and treatment-naive patients with metastatic uveal melanoma. PLoS One. 2015;10(3) doi: 10.1371/journal.pone.0118564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Ralph C, et al. Modulation of lymphocyte regulation for cancer therapy: a phase II trial of tremelimumab in advanced gastric and esophageal adenocarcinoma. Clin Cancer Res. 2010;16(5):1662–72. doi: 10.1158/1078-0432.CCR-09-2870. [DOI] [PubMed] [Google Scholar]
- 178.Antonia S, et al. Safety and antitumour activity of durvalumab plus tremelimumab in non-small cell lung cancer: a multicentre, phase 1b study. Lancet Oncol. 2016;17(3):299–308. doi: 10.1016/S1470-2045(15)00544-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Sangro B, et al. A clinical trial of CTLA-4 blockade with tremelimumab in patients with hepatocellular carcinoma and chronic hepatitis C. J Hepatol. 2013;59(1):81–8. doi: 10.1016/j.jhep.2013.02.022. [DOI] [PubMed] [Google Scholar]
- 180.Duffy AG, et al. Tremelimumab in Combination with Ablation in Patients with Advanced Hepatocellular Carcinoma. J Hepatol. 2016. doi: 10.1016/j.jhep.2016.10.029. [DOI] [PMC free article] [PubMed]
- 181.Calabro L, et al. Tremelimumab for patients with chemotherapy-resistant advanced malignant mesothelioma: an open-label, single-arm, phase 2 trial. Lancet Oncol. 2013;14(11):1104–11. doi: 10.1016/S1470-2045(13)70381-4. [DOI] [PubMed] [Google Scholar]
- 182.Guazzelli A, et al. Tremelimumab for the treatment of malignant mesothelioma. Expert Opin Biol Ther. 2015;15(12):1819–29. doi: 10.1517/14712598.2015.1116515. [DOI] [PubMed] [Google Scholar]
- 183.Ribas A, et al. Phase III randomized clinical trial comparing tremelimumab with standard-of-care chemotherapy in patients with advanced melanoma. J Clin Oncol. 2013;31(5):616–22. doi: 10.1200/JCO.2012.44.6112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Tarhini AA, et al. Safety and efficacy of combination immunotherapy with interferon alfa-2b and tremelimumab in patients with stage IV melanoma. J Clin Oncol. 2012;30(3):322–8. doi: 10.1200/JCO.2011.37.5394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Chung KY, et al. Phase II study of the anti-cytotoxic T-lymphocyte-associated antigen 4 monoclonal antibody, tremelimumab, in patients with refractory metastatic colorectal cancer. J Clin Oncol. 2010;28(21):3485–90. doi: 10.1200/JCO.2010.28.3994. [DOI] [PubMed] [Google Scholar]
- 186.Robert C, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372(4):320–30. doi: 10.1056/NEJMoa1412082. [DOI] [PubMed] [Google Scholar]
- 187.Postow MA, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med. 2015;372(21):2006–17. doi: 10.1056/NEJMoa1414428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Larkin J, et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med. 2015;373(1):23–34. doi: 10.1056/NEJMoa1504030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Weber JS, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015;16(4):375–84. doi: 10.1016/S1470-2045(15)70076-8. [DOI] [PubMed] [Google Scholar]
- 190.Gettinger SN, et al. Overall Survival and Long-Term Safety of Nivolumab (Anti-Programmed Death 1 Antibody, BMS-936558, ONO-4538) in Patients With Previously Treated Advanced Non-Small-Cell Lung Cancer. J Clin Oncol. 2015;33(18):2004–12. doi: 10.1200/JCO.2014.58.3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Brahmer J, et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N Engl J Med. 2015;373(2):123–35. doi: 10.1056/NEJMoa1504627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Borghaei H, et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N Engl J Med. 2015;373(17):1627–39. doi: 10.1056/NEJMoa1507643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Herbst RS, et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet. 2016;387(10027):1540–50. doi: 10.1016/S0140-6736(15)01281-7. [DOI] [PubMed] [Google Scholar]
- 194.Ribas A, et al. Association of Pembrolizumab With Tumor Response and Survival Among Patients With Advanced Melanoma. JAMA. 2016;315(15):1600–9. doi: 10.1001/jama.2016.4059. [DOI] [PubMed] [Google Scholar]
- 195.Le DT, et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med. 2015;372(26):2509–20. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Rizvi NA, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348(6230):124–8. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Roszik J, et al. Novel algorithmic approach predicts tumor mutation load and correlates with immunotherapy clinical outcomes using a defined gene mutation set. BMC Med. 2016;14(1):168. doi: 10.1186/s12916-016-0705-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Rebhandl S, et al. AID/APOBEC deaminases and cancer. Oncoscience. 2015;2(4):320–33. doi: 10.18632/oncoscience.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Westin JR, et al. Safety and activity of PD1 blockade by pidilizumab in combination with rituximab in patients with relapsed follicular lymphoma: a single group, open-label, phase 2 trial. Lancet Oncol. 2014;15(1):69–77. doi: 10.1016/S1470-2045(13)70551-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Armand P, et al. Disabling immune tolerance by programmed death-1 blockade with pidilizumab after autologous hematopoietic stem-cell transplantation for diffuse large B-cell lymphoma: results of an international phase II trial. J Clin Oncol. 2013;31(33):4199–206. doi: 10.1200/JCO.2012.48.3685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Fehrenbacher L, et al. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): a multicentre, open-label, phase 2 randomised controlled trial. Lancet. 2016;387(10030):1837–46. doi: 10.1016/S0140-6736(16)00587-0. [DOI] [PubMed] [Google Scholar]
- 202.Rosenberg JE, et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet. 2016;387(10031):1909–20. doi: 10.1016/S0140-6736(16)00561-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Bendell JC. Cobimetinib Plus Atezolizumab Active in Microsatellite Stable mCRC - See more at: http://global.onclive.com/conference-coverage/2016-world-gi/cobimetinib-plus-atezolizumab-active-in-microsatellite-stable-mcrc?p=2#sthash.djKjryZZ.dpuf. 2016 World Congress on GI Cancer 2016.
- 204.Rizvi NA, et al. Activity and safety of nivolumab, an anti-PD-1 immune checkpoint inhibitor, for patients with advanced, refractory squamous non-small-cell lung cancer (CheckMate 063): a phase 2, single-arm trial. Lancet Oncol. 2015;16(3):257–65. doi: 10.1016/S1470-2045(15)70054-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Yamazaki N, et al. Phase II study of ipilimumab monotherapy in Japanese patients with advanced melanoma. Cancer Chemother Pharmacol. 2015;76(5):997–1004. doi: 10.1007/s00280-015-2873-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Ansell SM, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin's lymphoma. N Engl J Med. 2015;372(4):311–9. doi: 10.1056/NEJMoa1411087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Vonderheide RH, et al. Tremelimumab in combination with exemestane in patients with advanced breast cancer and treatment-associated modulation of inducible costimulator expression on patient T cells. Clin Cancer Res. 2010;16(13):3485–94. doi: 10.1158/1078-0432.CCR-10-0505. [DOI] [PubMed] [Google Scholar]
- 208.McNeel DG, et al. Phase I trial of tremelimumab in combination with short-term androgen deprivation in patients with PSA-recurrent prostate cancer. Cancer Immunol Immunother. 2012;61(7):1137–47. doi: 10.1007/s00262-011-1193-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Wang-Gillam A, et al. A phase I study of IMP321 and gemcitabine as the front-line therapy in patients with advanced pancreatic adenocarcinoma. Invest New Drugs. 2013;31(3):707–13. doi: 10.1007/s10637-012-9866-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol. 2013;13(4):227–42. doi: 10.1038/nri3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Thaventhiran T, Sethu S, Yeang HXA, Al-Huseini L, Hamdam J, Sathish JG. T Cell Co-inhibitory Receptors: Functions and Signalling Mechanisms. J Clin Cell Immunol. 2012;S12:004. doi:10.4172/2155-9899.S12-004.
- 212.Bajor DLM, e.a.,.CT137 – Combination of Agonistic CD40 Monoclonal Antibody CP-870,893 aNM Anti-CTLA-4 Antibody Tremelimumab in Patients with Metastatic Melanoma. Proceedings, Part 2: Clinical Trials aNM Late-Breaking Abstracts. Clinical Trials Plenary Session: Combinations of Therapeutic Agents. AACR. Vol. Part 2. I. Philadelphia, PA: AACR.org; 2015.
- 213.Zatloukal P, Heo DS, Park K, Kang J, Butts C, Bradford D, Graziano S, Huang B, Healey D. Randomized phase II clinical trial comparing tremelimumab (CP-675,206) with best supportive care (BSC) following first-line platinum-based therapy in patients (pts) with advanced non-small cell lung cancer (NSCLC). J Clin Oncol. 2009;27(15_suppl):8071.
- 214.Calabro L, et al. Efficacy and safety of an intensified schedule of tremelimumab for chemotherapy-resistant malignant mesothelioma: an open-label, single-arm, phase 2 study. Lancet Respir Med. 2015;3(4):301–9. doi: 10.1016/S2213-2600(15)00092-2. [DOI] [PubMed] [Google Scholar]
- 215.Hamanishi J, et al. Safety and Antitumor Activity of Anti-PD-1 Antibody, Nivolumab, in Patients With Platinum-Resistant Ovarian Cancer. J Clin Oncol. 2015;33(34):4015–22. doi: 10.1200/JCO.2015.62.3397. [DOI] [PubMed] [Google Scholar]
- 216.Motzer RJ, et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N Engl J Med. 2015;373(19):1803–13. doi: 10.1056/NEJMoa1510665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Garon EB, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015;372(21):2018–28. doi: 10.1056/NEJMoa1501824. [DOI] [PubMed] [Google Scholar]
- 218.Nanda R, et al. Pembrolizumab in Patients With Advanced Triple-Negative Breast Cancer: Phase Ib KEYNOTE-012 Study. J Clin Oncol. 2016;34(21):2460–7. doi: 10.1200/JCO.2015.64.8931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Robert C, et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N Engl J Med. 2015;372(26):2521–32. doi: 10.1056/NEJMoa1503093. [DOI] [PubMed] [Google Scholar]
- 220.Twyman-Saint Victor C, et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature. 2015;520(7547):373–7. doi: 10.1038/nature14292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Horinouchi H, et al. Phase I study of ipilimumab in phased combination with paclitaxel and carboplatin in Japanese patients with non-small-cell lung cancer. Invest New Drugs. 2015;33(4):881–9. doi: 10.1007/s10637-015-0243-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Kwon ED, et al. Ipilimumab versus placebo after radiotherapy in patients with metastatic castration-resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184-043): a multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol. 2014;15(7):700–12. doi: 10.1016/S1470-2045(14)70189-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Aglietta M, et al. A phase I dose escalation trial of tremelimumab (CP-675,206) in combination with gemcitabine in chemotherapy-naive patients with metastatic pancreatic cancer. Ann Oncol. 2014;25(9):1750–5. doi: 10.1093/annonc/mdu205. [DOI] [PubMed] [Google Scholar]
- 224.Millward M, et al. Phase I study of tremelimumab (CP-675 206) plus PF-3512676 (CPG 7909) in patients with melanoma or advanced solid tumours. Br J Cancer. 2013;108(10):1998–2004. doi: 10.1038/bjc.2013.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Rini BI, et al. Phase 1 dose-escalation trial of tremelimumab plus sunitinib in patients with metastatic renal cell carcinoma. Cancer. 2011;117(4):758–67. doi: 10.1002/cncr.25639. [DOI] [PubMed] [Google Scholar]
- 226.Gibney GT, et al. Safety, correlative markers, and clinical results of adjuvant nivolumab in combination with vaccine in resected high-risk metastatic melanoma. Clin Cancer Res. 2015;21(4):712–20. doi: 10.1158/1078-0432.CCR-14-2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.