

Abstract
A method to rapidly generate gene replacement constructs by fusion PCR is described for Aspergillus nidulans. The utility of the approach is demonstrated by green fluorescent protein (GFP) tagging of A. nidulans ndc80 to visualize centromeres through the cell cycle. The methodology makes possible large-scale GFP tagging, promoter swapping, and deletion analysis of A. nidulans.
With the completion of 13X genome sequence coverage of Aspergillus nidulans, completing large-scale gene tagging with targeted gene replacements has become a possibility (Aspergillus Sequencing Project, Center for Genome Research, http://www.broad.mit.edu). However, the generation of targeting constructs is a limiting factor because of the necessity for >1 kb of homologous DNA for efficient gene targeting by homologous recombination. Here, we describe a fusion PCR-based method (7) to efficiently generate green fluorescent protein (GFP) tagging constructs which contain ∼2 kb of flanking targeting sequences. After the transformation into A. nidulans, high-frequency (averaging >40% efficiency) homologous recombination generates C-terminally GFP-tagged genes. The tagged version replaces the wild-type allele and is expressed from its own promoter, minimizing the potential for artifacts caused by over- or underexpression. The method is described in detail, as are the approaches used to confirm site-specific gene replacements. Using this methodology, we have tagged A. nidulans ndc80 (An-ndc80) with a C-terminal GFP and examined the localization of this centromeric protein through the cell cycle.
The gene replacement approach necessitates in-frame integration of GFP just before the stop codon of the target gene. We therefore confirmed the stop codon predicted by automated gene annotation by using rapid amplification of cDNA ends PCR before generating targeting constructs.
Our initial experiments failed to generate strains that were successfully tagged after gene replacement. We considered the possibility that a hinge region between the target gene and GFP may improve the functionality of GFP chimeras, as seen previously (8). We therefore generated a cassette containing a hinge region encoding five Gly-plus-Ala repeats (GA-5) in frame at the N terminus of GFP (4), followed by the Aspergillus fumigatus pyrG (Af-pyrG) gene (3) as a selectable nutritional marker (Fig. 1A). Using A. fumigatus pyrG helps minimize the rate of integration at the native pyrG locus. Utilizing this GA-5-GFP construct, we have successfully tagged numerous genes.
FIG. 1.

Construction of gene replacement constructs using fusion PCR. (A) Shown is the gene replacement cassette in which GA-5 is fused to GFP to act as a hinge between the target protein and GFP. The Af-pyrG gene (A. fum. pyrG) functions as a selectable nutritional marker. (B) The first PCR amplifies the GFP-pyrG cassette while incorporating 5′ and 3′ extensions complementary to 30 bp before and after the stop codon of the target gene with primers GFP1 and GFP2. (C) Two regions flanking the target gene stop codon are amplified with gene-specific primers 1 plus 2 and 3 plus 4 as indicated. (D) Fusion PCR is completed with the three amplified fragments and gene-specific primers 1 and 4.
An outline of the fusion PCR approach is shown in Fig. 1B through D. Three fragments are first amplified. For the first fragment, the GA-5-GFP-Af-pyrG cassette is amplified with primers GFP1 and GFP2. Primer GFP1 has a 5′ extension of 30 bp identical to the last 10 codons of the target gene upstream of the stop codon (Fig. 1B). Primer GFP2 primes at the 3′ end of the pyrG portion of the cassette and has a 5′ 30-bp extension corresponding to the reverse complement of the sequence immediately following the stop codon of the target gene (Fig. 1B).
For the second fragment, an upstream targeting region (Fig. 1C) of ∼2 kb is amplified with gene-specific primer 1 (GSP1) and GSP2. The sequence of GSP2 is complementary to the 5′ extension of GFP1.
For the third fragment, a downstream targeting region (Fig. 1C) of ∼2 kb is amplified with primers GSP3 and GSP4. The sequence of GSP3 is complementary to the 5′ extension of GFP2.
Each amplification was completed with a Boehringer Mannheim Expand Long Template PCR kit by using 50-μl PCR mixtures and the following conditions: 10 ng of the GA-5-GFP-Af-pyrG plasmid or 100 ng of genomic DNA, a 0.3 μM concentration of each primer, a 500 μM concentration of the deoxynucleoside triphosphates, 1× buffer 3, and 0.75 μl of Expand Long Template enzyme mix. The PCR cycling conditions were 94°C for 2 min and then 10 cycles of 94°C for 10 s, 60°C for 1 min, and 68°C for 4 min, followed by 15 cycles of 94°C for 10 s, 60°C for 30 s, and 68°C for 4 min. The 4-min extension time at 68°C in these 15 cycles was increased by 20 s with each cycle up to 7 min at 68°C, which was followed by a 4°C soak. The annealing temperature and extension times may vary with specific primers and fragment sizes being amplified (see the manufacturer protocol for details). Two examples of the amplification of these three fragments is shown in Fig. 2A.
FIG. 2.
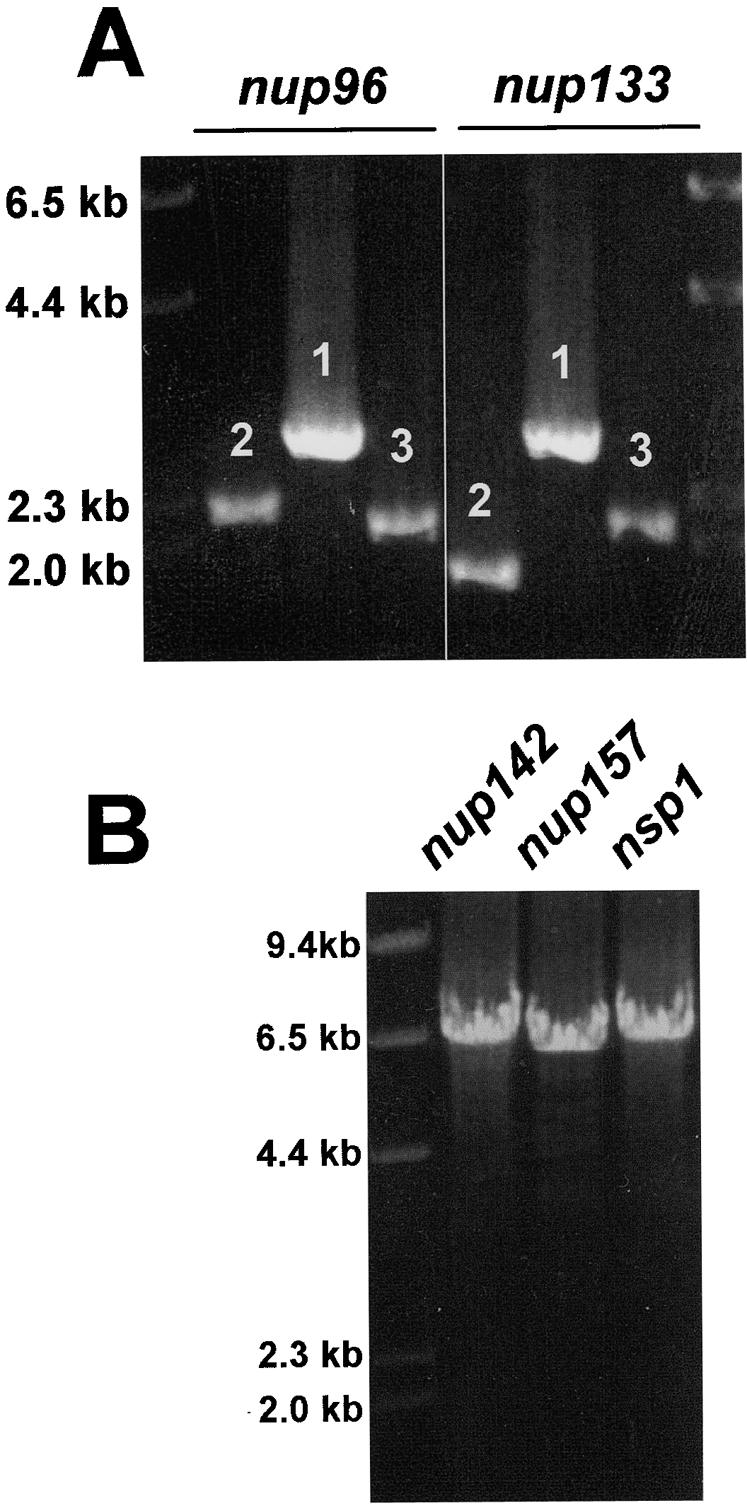
Generation of PCR fragments and a gene replacement construct. (A) Two examples of the three PCR fragments amplified for fusion PCR are shown. Fragments 1, 2, and 3 correspond to the fragments depicted in Fig. 1B and C. (B) Three examples of the final constructs generated by fusion PCR for GFP gene replacements.
The three amplified fragments are cleaned with QIAGEN or Promega PCR cleaning kits and quantitated after serial dilution and gel electrophoresis by comparison to known concentrations of marker DNA fragments.
The fusion PCR contains all three amplified fragments, along with primers GSP1 and GSP4 (Fig. 1D). The Boehringer Mannheim Expand Long Template PCR kit is again employed in 50-μl PCR mixtures containing 25 to 100 ng of each of the three PCR fragments as amplified above, a 0.3 μM concentration of GSP1, a 0.3 μM concentration of GSP4, a 500 μM concentration of the deoxynucleoside triphosphates, 1× buffer 3, and 0.75 μl of Expand Long Template enzyme mix. The PCR cycling conditions are 94°C for 2 min, and then 10 cycles of 94°C for 10 s, 61°C for 30 s, and 68°C for 8 min, followed by 15 cycles of 94°C for 10 s, 61°C for 30 s, and 68°C for 8 min. The 8-min extension time at 68°C in these 15 cycles is increased by 20 s with each cycle, up to 7 min at 68°C, which is followed by a 4°C soak. Three examples of the amplification of the final gene replacement construct are shown in Fig. 2B.
The final construct is gel purified by using a QIAquick gel extraction kit before transformation into A. nidulans by standard techniques (9). Crossover with each of the two ∼2-kb targeting fragments and their homologous domains within the genome generates a clean gene replacement (Fig. 3A).
FIG. 3.

Analysis of a strain with a putative gene replacement by diagnostic PCR. (A) The targeting construct consists of 2 kb of sequence before the stop codon of the target gene (red) and the GFP-pyrG cassette (green) and 2 kb of sequence after the stop codon of the target gene (blue). After homologous recombination between the targeting domains, a gene replacement is generated with the target gene tagged at its C terminus by GFP. (B) One primer anchored outside the targeting sequence (SSP-1) and another within GFP (SSP-2) were used in PCRs containing wild-type DNA or DNA from seven strains transformed with a gene replacement construct. No amplified band is expected for the wild type, and a 3.4-kb band is expected for strains with replacement genes. The arrow indicates a band amplified from a transformant (lane 4) in which the linear replacement construct circularized before integration as explained in the text. (C) Two primers that anchored just outside the upstream (SSP-3) and downstream (SSP-4)targeting regions as shown were used to amplify DNA from nine transformants and a wild-type control. A 4.4-kb band is expected for the wild-type DNA PCR, which is replaced by a 7.0-kb band in strains with gene replacements. Larger amplified bands (lanes 1 and 4) or no bands (lane 3) indicate circularization of the targeting construct before integration, as explained in the text.
PCR approaches have been used to confirm correct gene replacements. One primer was anchored in the genome, just outside the flanking targeting sequence used in the targeting construct (site-specific primer 1 [SSP-1]), and the other was anchored within the GFP sequence (SSP-2). In the example shown (Fig. 3B), a 3.4-kb fragment should be generated if a double crossover and gene replacement occurred. A fragment of this size was observed in six of the seven transformants tested (Fig. 3B), but one strain generated a larger fragment of 7.4 kb with the same primers (Fig. 3B, lane 4).
Previous studies of A. nidulans have shown that linear fragments can sometimes circularize prior to site-specific recombination (1). If our constructs circularized, subsequent recombination could occur either via the upstream targeting sequence or the downstream targeting sequence. Integration of the circularized construct via a single recombination event within the coding region of the target gene (the upstream targeting sequence) would generate a GFP-tagged version of the gene and the expected PCR fragment with primers SSP-1 and SSP-2. However, even though the target gene is tagged with GFP, this is not a clean gene replacement, as the two targeting domains are duplicated after the GFP-tagged target gene.
If, after circularization, integration occurs via the downstream targeting sequence, then instead of the expected 3.4-kb fragment being amplified by SSP-1 and SSP-2, a 7.4-kb fragment would be amplified, as was seen for one of the seven strains (Fig. 3B, lane 4). In this strain, a duplication of the targeting sequences is also generated, but the endogenous gene is not tagged with GFP.
Because of the potential for circularization of the targeting constructs before integration, diagnostic PCR with one primer anchored outside the targeting sequence and another within GFP will not necessarily confirm a gene replacement. We therefore analyzed potential GFP-tagged strains using primers anchored outside the targeting sequences (primers SSP-3 and SSP-4). An example is shown in Fig. 3C. In the wild type, a 4.4-kb fragment is amplified with primers SSP-3 and SSP-4 (Fig. 3C), whereas strains with a replaced gene will generate a 7.0-kb fragment. When circularization of the construct occurs prior to integration, an 11.4-kb fragment will be amplified. In the example shown, of the nine transformants picked at random, six have gene replacements (Fig. 3C, lanes 2, 5, 6, 7, 8, and 9) and two have integrations of the circularized construct (lanes 1 and 4). The PCR for the strain loaded in lane 3 failed to amplify a band. This could be due to the integration of more than one copy of the construct after circularization, making the fragment to be amplified too large to be generated under the PCR conditions employed. Using primers anchored just outside the targeting domains, therefore, allows confirmation of specific gene replacement.
To confirm that the GFP-tagged protein of the correct size is being synthesized, protein extracts were analyzed by Western blotting using anti-GFP antibodies. Western blotting confirmed expression of fusion proteins of the expected sizes in all cases (seven) so far examined after PCR analysis indicated that a clean gene replacement had occurred.
The fusion PCR methodology as described above was used to complete a GFP gene replacement of the NDC80 homologue in A. nidulans. In Saccharomyces cerevisiae and Schizosaccharomyces pombe, Ndc80p locates to the centromere. In both yeasts, all centromeres are located adjacent to the spindle pole body during interphase (12), and Ndc80-GFP locates to a single dot within nuclei during interphase. Similarly, during interphase, a single dot per nucleus was seen in A. nidulans strains containing An-ndc80-GFP (Fig. 4A through D). During mitosis, S. pombe Ndc80-GFP (Sp-Ndc80-GFP) is located at the kinetochore and can be seen as up to six small dots representing the three duplicated chromosomes of this species (12). During A. nidulans mitosis, An-Ndc80-GFP was also seen as distinct dots dispersed throughout nuclei which likely represent the 16 kinetochores of the eight duplicated chromosomes of this species (Fig. 4E through H). As anaphase progresses in S. pombe, the Sp-Ndc80-GFP dots coalesce into two dots at either end of the cell. Exactly the same pattern of An-Ndc80-GFP was observed during A. nidulans telophase, with the two dots being located at the ends of long telophase spindles (Fig. 4I through L).
FIG. 4.

Location of the centromeric protein An-Ndc80 through the cell cycle. Interphase (A through D), medial mitotic (E through H), and telophase (I through L) cells are shown. The location of An-Ndc80-GFP (A, E, and I), microtubules (B, F, and J), DNA (C, G, and K), and a merge of all three (D, H, and L) during normal mitosis are shown. Notice the dramatic change in distribution of An-Ndc80 during the progression from interphase through mitosis.
The distribution seen for An-Ndc80-GFP through the cell cycle is identical to that observed previously for Sp-Ndc80-GFP, indicating that An-Ndc80 resides at the centromere throughout the cell cycle of A. nidulans.
We have successfully completed 15 GFP gene replacements, with >40% of the transformants tested having gene replacements. In addition to generating cassettes for GFP tagging, we have generated monomeric red fluorescent protein tagging (2), tandem affinity purification tagging (10), and S tagging (5, 6) cassettes. All will be made available at the Fungal Genetics Stock Center (http://www.fgsc.net/). It should also be possible to generate promoter replacement cassettes which could be used to generate conditional mutations based upon the alcA promoter (11), which can be regulated. Additionally, we have utilized the approach as originally described (7) to generate gene deletion constructs. With the fusion PCR approach and methods outlined, large-scale gene replacement projects are now more feasible with A. nidulans.
ADDENDUM IN PROOF
Yu et al. (Fungal Genet. Biol., in press) have recently reported deletion of 26 genes from A. nidulans using similar PCR approaches to generate gene replacement cassettes.
Acknowledgments
We thank Reinhard Fischer for providing plasmids.
This work was supported by the National Institutes of Health grant GM 42564.
REFERENCES
- 1.Bussink, H. J., and S. A. Osmani. 1999. A mitogen-activated protein kinase (MPKA) is involved in polarized growth in the filamentous fungus, Aspergillus nidulans. FEMS Microbiol. Lett. 173:117-125. [DOI] [PubMed] [Google Scholar]
- 2.Campbell, R. E., O. Tour, A. E. Palmer, P. A. Steinbach, G. S. Baird, D. A. Zacharias, and R. Y. Tsien. 2002. A monomeric red fluorescent protein. Proc. Natl. Acad. Sci. USA 99:7877-7882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chaveroche, M. K., J. M. Ghigo, and C. d'Enfert. 2000. A rapid method for efficient gene replacement in the filamentous fungus Aspergillus nidulans. Nucleic Acids Res. 28:E97. [Online.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fernandez-Abalos, J. M., H. Fox, C. Pitt, B. Wells, and J. H. Doonan. 1998. Plant-adapted green fluorescent protein is a versatile vital reporter for gene expression, protein localization and mitosis in the filamentous fungus, Aspergillus nidulans. Mol. Microbiol. 27:121-130. [DOI] [PubMed] [Google Scholar]
- 5.Kim, J. S., and R. T. Raines. 1993. Ribonuclease S-peptide as a carrier in fusion proteins. Protein Sci. 2:348-356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim, J. S., and R. T. Raines. 1994. Peptide tags for a dual affinity fusion system. Anal. Biochem. 219:165-166. [DOI] [PubMed] [Google Scholar]
- 7.Kuwayama, H., S. Obara, T. Morio, M. Katoh, H. Urushihara, and Y. Tanaka. 2002. PCR-mediated generation of a gene disruption construct without the use of DNA ligase and plasmid vectors. Nucleic Acids Res. 30:E2. [Online.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Labib, K., J. F. Diffley, and S. E. Kearsey. 1999. G1-phase and B-type cyclins exclude the DNA-replication factor Mcm4 from the nucleus. Nat. Cell Biol. 1:415-422. [DOI] [PubMed] [Google Scholar]
- 9.Osmani, S. A., G. S. May, and N. R. Morris. 1987. Regulation of the mRNA levels of nimA, a gene required for the G2-M transition in Aspergillus nidulans. J. Cell Biol. 104:1495-1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rigaut, G., A. Shevchenko, B. Rutz, M. Wilm, M. Mann, and B. Seraphin. 1999. A generic protein purification method for protein complex characterization and proteome exploration. Nat. Biotechnol. 17:1030-1032. [DOI] [PubMed] [Google Scholar]
- 11.Waring, R. B., G. S. May, and N. R. Morris. 1989. Characterization of an inducible expression system in Aspergillus nidulans using alcA and tubulin-coding genes. Gene 79:119-130. [DOI] [PubMed] [Google Scholar]
- 12.Wigge, P. A., and J. V. Kilmartin. 2001. The Ndc80p complex from Saccharomyces cerevisiae contains conserved centromere components and has a function in chromosome segregation. J. Cell Biol. 152:349-360. [DOI] [PMC free article] [PubMed] [Google Scholar]