

Abstract
Fluorescence-activated cell sorting (FACS) is a technique to purify specific cell populations based on phenotypes detected by flow cytometry. This method enables researchers to better understand the characteristics of a single cell population without the influence of other cells. Compared to other methods of cell enrichment, such as magnetic-activated cell sorting (MCS), FACS is more flexible and accurate for cell separation due to the ability of phenotype detection by flow cytometry. In addition, FACS is usually capable of separating multiple cell populations simultaneously, which improves the efficiency and diversity of experiments. Although FACS has some limitations, it has been broadly used to purify cells for functional studies in both in vitro and in vivo settings. Here we report a protocol using fluorescence-activated cell sorting to isolate a very rare population of immune cells, plasmacytoid dendritic cells (pDC), with high purity from the bone marrow of lupus-prone mice for in vitro functional studies of pDC.
Keywords: Immunology, Issue 117, FACS, mouse, bone marrow, plasmacytoid dendritic cells (pDC), IFNα, lupus
Introduction
Efficient separation of a cell population of choice from other cells enables studies of the population that may not be possible otherwise. Fluorescence-activated cell sorting (FACS) is a method to enrich an interesting cell population with high purity. 1,2 Different cell types usually express unique molecules, or a unique combination of several molecules, on the plasma membrane that can distinguish one cell population from another. Upon binding of these cell surface molecules by specific fluorescence-conjugated antibodies, a detecting machine called flow cytometer/sorter is able to excite and detect the light signals of different fluorescent dyes that represent different molecule markers on the cells at the single cell level. The combined information consisting of either the presence of a light signal (representing positive expression of the corresponding surface molecule) or the absence of a light signal (representing negative expression of a molecule) defines the phenotype of the cell. After passing through the detector, cells with the same phenotype of interest are diverted towards a designated collecting tube based on electrical charge.
FACS is broadly applied in various studies as long as the population to be enriched is labeled with fluorescence.3-7 It has been used to separate immunoglobulin (Ig)A-coated bacteria from non-IgA coated bacteria in the gut microbiota 8 and sort genetically engineered cell populations expressing fluorescent proteins. 9 Importantly, it has the capacity to separate more than one population simultaneously, which not only saves time and reagents but also allows for more sophisticated study designs. 10 However, FACS also has its limitations. If a population of interest is very rare (less than 1%), the sorting efficiency may be reduced, causing significant cell loss. In addition, some antibody binding may activate intracellular signal transduction that induces functional changes of the sorted cell population. 11 Therefore, the phenotype used for sorting should be selected carefully.
Other methods exist besides FACS that are also based on cell surface markers for the enrichment of specific cell populations, such as magnetic-activated cell sorting (MCS). 12 Similar to FACS, magnetic beads-conjugated antibodies can target specific cell surface molecules. Upon antibody-antigen interaction, magnetic beads-coated cells can be separated from non-coated cells after passing through a magnetic field. However, only a limited number of molecules can be targeted in MCS, as magnetic beads are, unlike various fluorescent colors in FACS, undistinguishable. It is thus difficult for MCS to define a cell phenotype with a complicated combination of surface markers. 13,14 In addition, MCS is also able to cause unintended activation of target cells.
In our studies of a mouse model of systemic lupus erythematosus (SLE), 15 we intended to purify plasmacytoid dendritic cells (pDC) to investigate their functional changes with disease progression. We first used MCS to enrich pDC from the bone marrow by targeting PDCA-1, a molecule highly and uniquely expressed on murine pDC at steady state. 16 However, the cell purity was unexpectedly low, likely due to the upregulation of PDCA-1 on other cell populations in an inflammatory environment such as SLE.16 Ultimately, we have used FACS with a combination of four surface markers (CD11c, CD11b, B220 and PDCA-1) to separate high-purity pDC as CD11c+CD11b-B220+PDCA-1+ population. Murine pDC has another specific surface marker Siglec-H. We decided not to use Siglec-H, as antibody binding of this molecule represses the function of pDC to produce IFNα. 11
Protocol
NOTE: MRL/Mp-Faslpr (MRL/lpr) lupus-prone mice were bred and maintained in a specific pathogen-free facility following the requirements of Institutional Animal Care and Use Committee (IACUC) at Virginia Tech (Animal Welfare Assurance Number: A3208-01). This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. All animal experiments were conducted under IACUC protocol #12-062.
1. Cell Culture Medium and Sorting Buffer
Prepare complete cell culture medium (C10) using RPMI 1640 supplemented with 10% fetal bovine serum, 1 mM sodium pyruvate, 1% 100 MEM non-essential amino acids, 10 mM HEPES, 55 µM 2-mercaptoethanol, 2 mM L-glutamine and 100 U/ml penicillin-streptomycin.
Prepare sorting buffer (HBSS-full) using 1x Hank's Balanced Salt Solution (HBSS) supplemented with 10 mM HEPES, 2.5 mg/ml bovine serum albumin, 0.05 mM MgCl2 and 0.2 U/ml DNase I.
2. Mouse Dissection
Prepare two 6-cm diameter dishes containing 4 ml C10. Place the dishes on ice.
Euthanize the mouse by CO2 inhalation.
- Harvest the spleen and keep it in a dish with C10 on ice.
- Pin the mouse down with the belly facing up on the dissection plate. Sterilize the body by spraying 70% ethanol.
- Cut the skin and separate the skin from the muscle wall underneath along the ventral midline from the pubic symphysis to the neck.
- Cut the skin along the hind limb from the first incision to the ankle.
- Pin the skin back on the sides and cut the muscle wall along the sides from the first incision place to diaphragm.
- Pin the muscle wall back on the right side of the head.
- Locate the spleen on the right side of the mouse underneath the small intestine and beside the stomach. Pick up one end of the spleen and separate it from the stomach.
- Cut both hind limbs out, including femur and tibia, and remove all muscles as much as possible.
- Cut the muscles along the hind limb from the pelvic/hip joint to the ankle with a sharp scissor, trying to avoid blood vessels.
- Separate the hind limb out by cutting at the pelvic/hip joint and ankle/foot joint without the exposure of bone marrow contents.
- Clean remaining muscles on the bones by scratching with a razor blade repeatedly and cutting the muscles at the joint points.
Keep the bones in a 6 cm dish containing 4 ml C10 on ice. Proceed to dissecting the next mouse.
3. Splenocyte Isolation
Homogenize the spleen gently with a syringe plunger against a 70-µm cell strainer on top of the dish containing 4 ml C10 until no red pieces are visible.
Add additional 6 ml C10 into the dish through the strainer and then transfer the total 10 ml cell suspension to a 15 ml conical tube.
Centrifuge the conical tube at 800 x g for 5 min, 4 °C.
After centrifugation, aspirate the supernatant and resuspend the cell pellet in 2 ml 1 x red blood cell (RBC) lysis buffer. Incubate at RT for 5 min.
After incubation, centrifuge the conical tube at 800 x g for 5 min, 4 °C.
After centrifugation, aspirate the supernatant and resuspend the cell pellet in 1 ml C10. Discard any big clumps (dead cells) and keep the tube on ice for later. NOTE: If there are many small clumps, use a 70-µm cell strainer to filter the cell suspension once.
Count the cell number with trypan blue using an automated cell counter.
4. Bone Marrow Cell Isolation
Transfer the bones with 4 ml C10 into a mortar and add 6 ml C10 to make a volume of 10 ml C10 in total.
Crack the bones gently in the mortar by using a pestle.
Stir gently with the pestle to release the bone marrow into C10.
Transfer the 10 ml C10 containing bone marrow into a 50 ml conical tube on ice through a 70-µm cell strainer. Add 10 ml fresh C10 into the mortar still containing the bones. NOTE: Pipette the solution containing bone marrow up and down several times before passing through the strainer if red clumps are visible.
Repeat steps 4.2 to 4.4 twice. After the last wash, the bones should appear white.
Centrifuge the 50 ml conical tube at 800 x g for 5 min, 4 °C. Meanwhile, prepare 5 ml ready-to-use density gradient medium in a 15 ml conical centrifuge tube at RT.
After centrifugation, aspirate the supernatant and resuspend the cell pellet in 10 ml C10.
Slowly layer the 10 ml cell suspension on top of the 5 ml density gradient medium, maintaining clear interface between the two phases. Then centrifuge the 15 ml conical tube at 1,363 g for 30 min at RT (20 °C) with acceleration set as 9 and deceleration set as 0 (or Brake OFF).
After centrifugation, remove the top 8 ml solution carefully and collect the 2 ml buffy coat at the interface (a layer of mononuclear cells) into a new 15 ml conical tube.
Add 12 ml ice cold C10 to the 15 ml conical tube containing bone marrow mononuclear cells, cap and invert several times to mix well, then centrifuge the tube at 800 x g for 10 min, 4 °C.
5. Cell Surface Staining for FACS
- Sample staining
- Prepare anti-mouse CD16/32 antibody solution (1 to 100 dilution in HBSS-full, 5 µg/ml final concentration).
- After centrifugation in step 4.10, aspirate the supernatant and resuspend the cell pellet in 100 µl anti-mouse CD16/32 antibody solution to block Fc receptor on the mononuclear cells. Incubate on ice for 10 min.
- Prepare fluorescence-conjugated antibody mixture (anti-mouse CD11c-PE 1:40 dilution, anti-mouse CD11b-APC-CY7 1:80 dilution, anti-mouse PDCA-1-FITC 1:40 dilution and anti-mouse B220-V500 1:40 dilution in HBSS-full as recommended by the manufacturer at 100 µl final volume per sample). NOTE: It is important to titrate antibodies before use.
- After 10 min incubation in step 5.1.2, add 4 ml ice cold HBSS-full and centrifuge at 800 x g for 5 min, 4 °C to remove unbound anti-mouse CD16/32.
- After centrifugation, aspirate the supernatant and resuspend the cell pellet in 100 µl fluorescence-conjugated antibody mixture. Incubate on ice for 15 min in the dark.
- After 15 min incubation, add 4 ml ice cold HBSS-full and centrifuge at 800 x g for 5 min, 4 °C to remove unbound fluorescence-conjugated antibodies.
- Prepare FACS loading solution (500 ng/ml DAPI in HBSS-full).
- After centrifugation in step 5.1.6, aspirate the supernatant and resuspend the cell pellet in 500 µl FACS loading solution. Transfer the cell suspension into a 12 x 75 mm round bottom tube (FACS tube) and keep the tube on ice in the dark until sorting within 1 hr.
- Staining for compensation
- Label 6 FACS tubes as PE, APC-CY7, FITC, V500, DAPI or unstained. Aliquot splenocytes at 1 x 106 cells/tube into the FACS tubes, and wash with 4 ml HBSS-full at 800 x g for 5 min, 4 °C. NOTE: Use DNA-binding fluorescence dye, DAPI, to distinguish live cells from dead cells. DAPI can pass through the plasma membrane of dead cells (but not of live cells) and bind chromosome DNA.
- Aspirate the supernatant and resuspend the cell pellet in 100 µl HBSS-full.
- In each corresponding FACS tube, add one of the following antibodies: anti-mouse CD19-PE 1:40 dilution, anti-mouse CD11b-APC-CY7 1:80 dilution, anti-mouse PDCA-1-FITC 1:40 dilution or anti-mouse B220-V500 1:40 dilution as recommended by the manufacturer. Leave the 5th (DAPI) and 6th (unstained) FACS tubes as they are. Mix well by shaking and incubate all the FACS tubes on ice in the dark for 15 min.
- After incubation, add 4 ml ice cold HBSS-full into each tube and centrifuge the FACS tubes at 800 x g for 5 min, 4 °C.
- After centrifugation, aspirate the supernatant and resuspend the cell pellet in 500 µl ice cold HBSS-full except for the pellet in the tube labeled DAPI, which is resuspended in 500 µl FACS loading solution. Keeping all 6 tubes on ice in the dark until sorting.
6. Sorting on the Cell Sorter
NOTE: Operation of sorting procedure on the cytometer and software is standardized with a detailed instruction provided by the company. Briefly, we use 100 micron nozzle at 20 psi, set target cell concentration between 5 - 10 million per ml, and adjust efficiency to 70% or higher (i.e., conflicts kept under 30%).
Prepare collection FACS tubes with 500 µl FBS/tube containing 100 U/ml penicillin-streptomycin.
- Adjust compensation parameters of the cell sorter using single-stain compensation tubes from step 5.2.
- Record 10,000 cells in unstained tube to set negative gate for each fluorescent intensity.
- Record 10,000 cells in other single-stain compensation tubes to set positive gate for each fluorescent intensity. NOTE: The software automatically calculates the compensation parameters.
Use one sample from step 5.1 to set the gates of sorted cell populations by recording 3,000 cells. Using fluorescent intensity as parameters of X and Y-axis graph, gate target cell population manually as a group of dots apparently separated from other dots. NOTE: This gating set is flexible and arbitrary based on the requirements of researchers. If researchers expect higher purity based on one or more markers, move the gate higher based on the corresponding fluorescent intensity.
Load a collection tube and start to sort the target cell population.
Keep the collection tube on ice after sorting until all sample tubes are done.
Add ice cold C10 to the collection tube up to 4 ml, cap and invert to mix.
Centrifuge the collection tube at 800 x g for 5 min, 4 °C, and then aspirate the supernatant, leaving around 200 µl with cell pellet untouched.
Add 1 ml ice cold C10 to resuspend the cell pellet and transfer all cell suspension into a 1.5 ml centrifuge tube.
Centrifuge the 1.5 ml tube at 800 x g for 5 min, 4 °C and aspirate the supernatant, leaving 100 µl with the cell pellet untouched.
Store the sorted cell populations on ice until use.
Representative Results
We aimed to enrich bone marrow pDC with high purity, and without the influence of other cell types, from MRL/lpr lupus-prone mice of both young and old age to study the functional changes of pDC regarding their ability to produce IFNα. The first purification strategy used was MCS, which, as Figure 1 shows, led to only 7.75% purity after the enrichment. Compared to MCS, FACS enriched pDC with purity as high as 96.4%. To ensure high purity, a step-by-step gating strategy was performed. As shown in Figure 2, mononuclear cells were gated and separated from debris by forward scatter (FSC) and side scatter (SSC) parameters. Using FSC-width (FSC-W) vs. FSC-height (FSC-H) and SSC-W vs. SSC-H, single cells were gated to avoid the false positive signals produced by aggregates. Live single cells were then gated as the DAPI- population (DAPI+ cells are dead or apoptotic). From live single cells, CD11c+CD11b- population was gated, from which B220+PDCA-1+ population was further gated as pDC. The sorted pDC were not only of high purity but also functionally normal. They had the ability to produce IFNα in vitro upon CpG stimulation as shown in Figure 3 (Re-print with permission from our previous publication17.
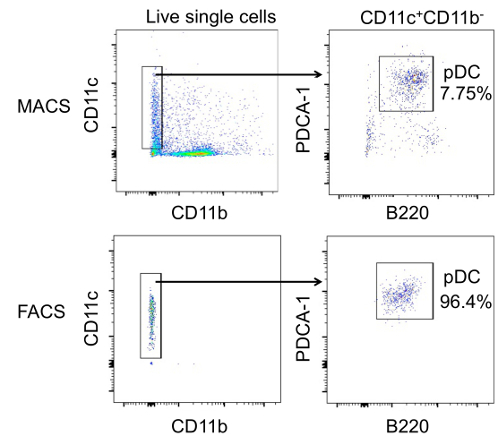 Figure 1. The Purity of pDC Sorted through either MCS and FACS Method. Representative flow cytometry figures showing the percentage of pDC (CD11c+CD11b-B220+PDCA-1+) in sorted cells by MCS (top) and FACS (bottom), respectively. The CD11c+CD11b- population was gated from total live cells (left), and then pDC as B220+PDCA-1+ cells were gated within the CD11c+CD11b- population (right). Please click here to view a larger version of this figure.
Figure 1. The Purity of pDC Sorted through either MCS and FACS Method. Representative flow cytometry figures showing the percentage of pDC (CD11c+CD11b-B220+PDCA-1+) in sorted cells by MCS (top) and FACS (bottom), respectively. The CD11c+CD11b- population was gated from total live cells (left), and then pDC as B220+PDCA-1+ cells were gated within the CD11c+CD11b- population (right). Please click here to view a larger version of this figure.
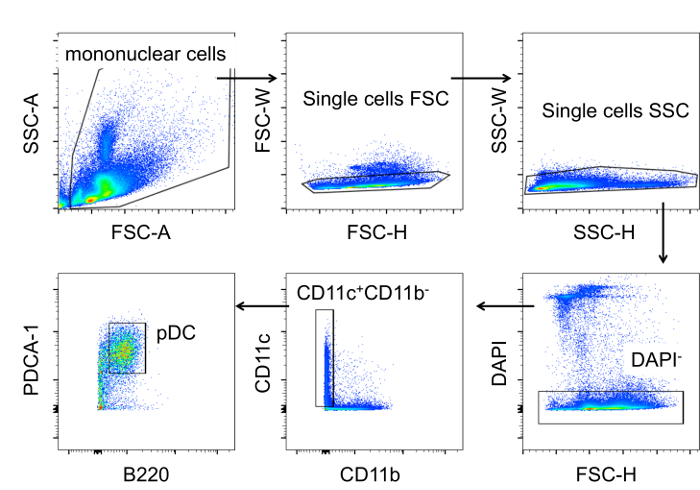 Figure 2. Gating Strategy for pDC Enrichment by FACS. A step-by-step gating strategy in the following order: mononuclear cells to single cells FSC, to single cells SSC, to DAPI- live cells, to CD11c+CD11b-, and to B220+PDCA-1+ as live pDC. pDC in total single cells is 2.95 ± 1.42 %; sorted pDC number is 0.88 ± 0.28 x 105 per mouse with a recovery rate around 50%. (n= 12 MRL/lpr mice, data are shown as mean ± SD in 95% CI of the mean). Please click here to view a larger version of this figure.
Figure 2. Gating Strategy for pDC Enrichment by FACS. A step-by-step gating strategy in the following order: mononuclear cells to single cells FSC, to single cells SSC, to DAPI- live cells, to CD11c+CD11b-, and to B220+PDCA-1+ as live pDC. pDC in total single cells is 2.95 ± 1.42 %; sorted pDC number is 0.88 ± 0.28 x 105 per mouse with a recovery rate around 50%. (n= 12 MRL/lpr mice, data are shown as mean ± SD in 95% CI of the mean). Please click here to view a larger version of this figure.
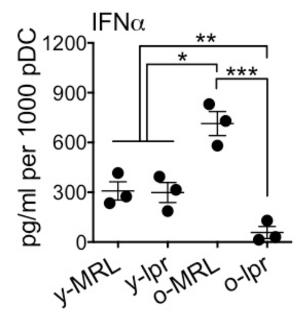 Figure 3. IFNα Production by FACS-sorted pDC. Sorted bone marrow pDC were treated with class A CpG (ODN1585; 5 μM) for 6 hr in vitro. In the figure, "y" stands for younger, 6-week-old mice; "o" stands for older, 16-week-old mice. MRL stands for MRL mice as healthy control mice; lpr stands for MRL/lpr mice as lupus-prone mice. The concentration of IFNα in the culture supernatant was measured with ELISA. *P< 0.05, **P< 0.01, ***P< 0.001, one-way ANOVA. Data are shown as mean ± standard error of the mean (n= 3 mice per group). This figure is reproduced from our publication in Journal of Immunology with permission. 17
Please click here to view a larger version of this figure.
Figure 3. IFNα Production by FACS-sorted pDC. Sorted bone marrow pDC were treated with class A CpG (ODN1585; 5 μM) for 6 hr in vitro. In the figure, "y" stands for younger, 6-week-old mice; "o" stands for older, 16-week-old mice. MRL stands for MRL mice as healthy control mice; lpr stands for MRL/lpr mice as lupus-prone mice. The concentration of IFNα in the culture supernatant was measured with ELISA. *P< 0.05, **P< 0.01, ***P< 0.001, one-way ANOVA. Data are shown as mean ± standard error of the mean (n= 3 mice per group). This figure is reproduced from our publication in Journal of Immunology with permission. 17
Please click here to view a larger version of this figure.
Discussion
The protocol described in this manuscript is for high purity enrichment of live pDC that retain the ability to produce IFNα. The applications of this protocol include, but are not limited to, purification of pDC and/or any other mononuclear cells from the bone marrow of MRL/lpr and any other mouse strains for studies of cellular and molecular functions. Several critical steps in this protocol are to ensure high viability and purity of the sorted pDC. The first key step is the release of bone marrow from bones. To minimize damaging bone marrow cells, it is important to gently crack the bones and stir the pestle instead of smashing the bones. The second key step is the enrichment of mononuclear cells with density gradient medium. It is critical to establish a clear interface between the lower phase (density gradient medium) and the upper phase (cell suspension) while loading the cell suspension. The centrifugation condition is also critical for the formation of the buffy coat layer that contains mainly mononuclear cells. After centrifugation, bone debris, red blood cells and high-density neutrophils are excluded from the buffy coat layer and stay at the bottom of the tube. The third key step is incubating the cells with fluorescence-conjugated antibodies at a low temperature (on ice or 4 °C). After the staining, it is also important to wash the cells extensively to ensure the specific antibody binding with minimal false positives or background signals easily detectable by the cell sorter. Finally, the single-stain compensation tubes are important and necessary for the adjustment of compensation parameters to ensure only the correct signals are measured and analyzed on the cell sorter. Besides using isolated splenocytes for the compensation, commercially available compensation beads can be an alternative if cells are limited. In this particular study, as we needed to enrich as many pDC as possible for further functional studies, to avoid bone marrow cell loss, we used splenocytes instead of bone marrow cells for the compensation. Also, it needs to be noted that surface marker like CD11c are expressed on rare populations, so they are not the best choice for compensation. Instead, an alternative marker expressed at a relatively high level on a major population can be used for compensation purpose as long as the antibody is conjugated with the same fluorescence dye. For example, in this study, CD19-PE is used to replace CD11c-PE in the compensation staining of splenocytes. Besides single color controls, fluorescence minus one (FMO) controls are important for beginners, especially to sort a population with markers not apparently separated from other populations. However, in this study, single color controls are adequate as pDC are a distinct population clearly defined and separated from other cells. In addition, we could set the gate slightly smaller than the actual population to improve purity.
In our studies, we only sorted a single population by FACS. In fact, this sorting method can be used broadly not only to enrich several different cell populations simultaneously, but also to isolate any interesting particle targets within a certain size range decided by the cell sorter, as long as they can be labeled with fluorescence. MCS is another cell enrichment method similar to FACS by principle that both are based on antibody detection of cell surface markers. Even though MCS is also widely used, it may not be efficient in various situations. For example, the phenotype of a cell population may change with the microenvironment in vivo or culture condition in vitro. MCS is prone to errors when the cell phenotype changes, as it usually detects only one surface molecule. On the contrary, FACS is based on the combination of several adjustable markers that reflect a more specific and accurate phenotype of a cell population in dynamic conditions. In addition, the gating strategy of FACS gives it a more sophisticated sorting ability, such as sorting Ly6C+ monocytes into Ly6Chigh and Ly6Cmedium subsets, which cannot be done with MCS.
However, FACS has its limitations as well. As it separates cell populations according to the analysis of fluorescent signals, if the cells or particles of interest have high autofluorescence, it will be difficult to sort the targets. In addition, a designated professional staff is usually required to operate the cell sorter properly. Moreover, the binding of fluorescence-conjugated antibodies to their specific ligands on cells may change the function of sorted cells, affecting the outcomes of subsequent experiments. For example, Siglec-H as a pDC specific surface molecule can be used to sort pDC, but the binding of Siglec-H by its specific antibody has been demonstrated to suppress the ability of pDC to produce IFNα. Therefore, selection of markers for cell sorting needs to be cautious and based on the purpose of the study.
Furthermore, the high-throughput cell sorter is limited to sorting, with high efficiency, only target cells of certain percentage within the total cell population. In this study, we first used density gradient centrifugation to increase the percentage of pDC through removing red blood cells, high dense granulocytes and debris, which was a better choice than simply adding the red blood cell lysis buffer. If the population is so rare that is less than 1%, the sorting efficiency will drop with significant cell loss. Hence, extremely rare population can be enriched first by other methods, such as negative selection with MCS to remove some of the other cells, and then sorted by FACS to achieve high purity with minimal cell loss. In summary, FACS is an efficient and accurate method to enrich functional pDC from the mouse bone marrow with high purity.
Disclosures
The authors declare that there is no conflict of interest regarding the publication of this paper.
Acknowledgments
We thank Flow Cytometry Laboratory at Virginia-Maryland College of Veterinary Medicine for the use of flow cytometry core facility. This work was supported by XML's startup funds. XL is a Stamps Fellow in the Biomedical and Veterinary Sciences graduate program.
References
- Bonner WA, Hulett HR, Sweet RG, Herzenberg LA. Fluorescence activated cell sorting. Rev Sci Instrum. 1972;43(3):404–409. doi: 10.1063/1.1685647. [DOI] [PubMed] [Google Scholar]
- Herzenberg LA, et al. The history and future of the fluorescence activated cell sorter and flow cytometry: a view from Stanford. Clin Chem. 2002;48(10):1819–1827. [PubMed] [Google Scholar]
- Brown M, Wittwer C. Flow cytometry: principles and clinical applications in hematology. Clin Chem. 2000;46(8 Pt 2):1221–1229. [PubMed] [Google Scholar]
- Laerum OD, Farsund T. Clinical application of flow cytometry: a review. Cytometry. 1981;2(1):1–13. doi: 10.1002/cyto.990020102. [DOI] [PubMed] [Google Scholar]
- Alvarez-Barrientos A, Arroyo J, Canton R, Nombela C, Sanchez-Perez M. Applications of flow cytometry to clinical microbiology. Clin Microbiol Rev. 2000;13(2):167–195. doi: 10.1128/cmr.13.2.167-195.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattanovich D, Borth N. Applications of cell sorting in biotechnology. Microb Cell Fact. 2006;5(12) doi: 10.1186/1475-2859-5-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldahlawi AM, Elshal MF, Damiaiti LA, Damanhori LH, Bahlas SM. Analysis of CD95 and CCR7 expression on circulating CD4(+) lymphocytes revealed disparate immunoregulatory potentials in systemic lupus erythematosus. Saudi J Biol Sci. 2016;23(1):101–107. doi: 10.1016/j.sjbs.2015.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullender TC, et al. Innate and adaptive immunity interact to quench microbiome flagellar motility in the gut. Cell Host Microbe. 2013;14(5):571–581. doi: 10.1016/j.chom.2013.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez AG, et al. High-throughput fluorescence-based isolation of live C. elegans larvae. Nat Protoc. 2012;7(8):1502–1510. doi: 10.1038/nprot.2012.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai M, et al. DC-SIGN expression on podocytes and its role in inflammatory immune response of lupus nephritis. Clin Exp Immunol. 2016;183(3):317–325. doi: 10.1111/cei.12723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasius A, et al. A cell-surface molecule selectively expressed on murine natural interferon-producing cells that blocks secretion of interferon-alpha. Blood. 2004;103(11):4201–4206. doi: 10.1182/blood-2003-09-3108. [DOI] [PubMed] [Google Scholar]
- Welzel G, Seitz D, Schuster S. Magnetic-activated cell sorting (MCS) can be used as a large-scale method for establishing zebrafish neuronal cell cultures. Sci Rep. 2015;5:7959. doi: 10.1038/srep07959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valli H, et al. Fluorescence- and magnetic-activated cell sorting strategies to isolate and enrich human spermatogonial stem cells. Fertil Steril. 2014;102(2):566–580. doi: 10.1016/j.fertnstert.2014.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashiro S, et al. Phenotypic and functional change of cytokine-activated neutrophils: inflammatory neutrophils are heterogeneous and enhance adaptive immune responses. J Leukoc Biol. 2001;69(5):698–704. [PubMed] [Google Scholar]
- Apostolidis SA, Lieberman LA, Kis-Toth K, Crispin JC, Tsokos GC. The dysregulation of cytokine networks in systemic lupus erythematosus. J Interferon Cytokine Res. 2011;31(10):769–779. doi: 10.1089/jir.2011.0029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asselin-Paturel C, Brizard G, Pin JJ, Briere F, Trinchieri G. Mouse strain differences in plasmacytoid dendritic cell frequency and function revealed by a novel monoclonal antibody. J Immunol. 2003;171(12):6466–6477. doi: 10.4049/jimmunol.171.12.6466. [DOI] [PubMed] [Google Scholar]
- Liao R, et al. Tacrolimus Protects Podocytes from Injury in Lupus Nephritis Partly by Stabilizing the Cytoskeleton and Inhibiting Podocyte Apoptosis. PLoS One. 2015;10(7):e0132724. doi: 10.1371/journal.pone.0132724. [DOI] [PMC free article] [PubMed] [Google Scholar]