

Abstract
FKBP12 is a conserved member of the prolyl-isomerase enzyme family and serves as the intracellular receptor for FK506 that mediates immunosuppression in mammals and antimicrobial actions in fungi. To investigate the cellular functions of FKBP12 in Saccharomyces cerevisiae, we employed a high-throughput assay to identify mutations that are synthetically lethal with a mutation in the FPR1 gene, which encodes FKBP12. This screen identified a mutation in the HOM6 gene, which encodes homoserine dehydrogenase, the enzyme catalyzing the last step in conversion of aspartic acid into homoserine, the common precursor in threonine and methionine synthesis. Lethality of fpr1 hom6 double mutants was suppressed by null mutations in HOM3 or HOM2, encoding aspartokinase and aspartate β-semialdehyde dehydrogenase, respectively, supporting the hypothesis that fpr1 hom6 double mutants are inviable because of toxic accumulation of aspartate β-semialdehyde, the substrate of homoserine dehydrogenase. Our findings also indicate that mutation or inhibition of FKBP12 dysregulates the homoserine synthetic pathway by perturbing aspartokinase feedback inhibition by threonine. Because this pathway is conserved in fungi but not in mammals, our findings suggest a facile route to synergistic antifungal drug development via concomitant inhibition of FKBP12 and Hom6.
Prolyl isomerases are widely conserved, ubiquitous enzymes that catalyze cis-trans isomerization of peptidyl-prolyl bonds, a reaction that can be rate limiting for protein folding. Founding members of this group of enzymes are cyclophilin A, previously identified as the cyclosporine A receptor (25, 31, 75), and the structurally unrelated enzyme FK506 binding protein FKBP12 (35). A third family of prolyl isomerases, known as the parvulins, was discovered for bacteria (60, 61) and later was found to be conserved in many other organisms.
Cyclophilin A and FKBP12 mediate the immunosuppressive effects of cyclosporine A and FK506 in mammals by forming complexes with these drugs that bind to and inhibit the functions of calcineurin in T-cell activation (for a review, see reference 68). FKBP12 is also the receptor for the drug rapamycin, and the FKBP12-rapamycin complex inhibits the functions of the Tor proteins (36, 44). Both cyclophilin A and FKBP12 are conserved in budding yeast (where they are encoded by the CPR1 and FPR1 genes, respectively) and mediate calcineurin inhibition by cyclosporine A and FK506 (11, 26, 30, 37, 38, 52, 55, 78, 83) and Tor inhibition by rapamycin (36, 44). Saccharomyces cerevisiae expresses seven other cyclophilins (Cpr2 to Cpr8), three other FKBPs (Fpr2 to Fpr4), and a single parvulin (Ess1) (for a recent review, see reference 6).
With the exception of Ess1, all yeast prolyl isomerases are dispensable for growth (18, 32, 34). However, cyclophilin A does become essential in cells compromised for Ess1 function, suggesting a functional overlap between these two structurally unrelated prolyl isomerases (4). Thus far, the endogenous functions that have been defined for these proteins are relatively specific and devoted to restricted interaction partners. For example, cyclophilin A is required for glucose-stimulated transport of fructose-1,6-bisphosphatase into Vid (vacuole import and degradation) vesicles (12). In addition, cyclophilin A promotes proper subcellular localization of the essential zinc-finger protein Zpr1 (2). Cyclophilin A interacts with two different histone deacetylase complexes that regulate meiosis, the Sin3-Rpd3 and Set3 complexes, and recent studies have revealed a nuclear role for Cpr1 in controlling the expression of key meiosis-specific genes (4, 5, 58). Cpr3 is a mitochondrial cyclophilin that accelerates protein refolding after mitochondrial import (17, 19, 49, 66, 67). Cpr6 and Cpr7, like their human homolog cyclophilin 40, interact with and regulate the activity of the molecular chaperone Hsp90 (15, 20, 22, 47, 71, 76). Ess1, the first eukaryotic parvulin, was originally associated with pre-mRNA processing and termination (33, 34) and more recently with transcription and chromatin modification (4, 51, 84-86).
Yeast FKBP12 interacts with calcineurin in the absence of FK506, and genetic evidence implicates this interaction in negatively regulating calcineurin function, suggesting that this could be one of the cellular functions of this prolyl isomerase (14). A search for yeast proteins interacting with FKBP12 in the yeast two-hybrid system identified the enzyme aspartokinase (AK) as an FKBP12 binding partner (1). AK catalyzes the first reaction in the conversion of aspartic acid into the amino acid homoserine, a branch point in synthesis of threonine and methionine. Studies by Alarcon and Heitman (1) suggest that FKBP12 influences AK feedback inhibition by threonine, the main point of regulatory control in the aspartate pathway (3, 48, 57, 62).
More recently, a yeast synthetic lethal genetic screen with fpr1 mutations identified the HMO1 gene, which encodes a high-mobility-group (HMG) protein also conserved in humans (21, 45). Hmo1 is a nuclear protein that functions in stabilizing chromatin structure and plasmid maintenance (46) and as an RNA polymerase I factor (27). FKBP12 and Hmo1 interact physically, possibly to regulate Hmo1 self-association (21).
In this study, we extended the analysis of FKBP12 cellular functions by conducting a systematic search for yeast mutations that exhibit synthetic lethality with an fpr1 mutation. In this screen, we found that in addition to hmo1Δ, a mutation in the HOM6 gene encoding homoserine dehydrogenase also conferred lethality in an fpr1Δ mutant. Homoserine dehydrogenase catalyzes the last step in the synthesis of homoserine from aspartate. We present evidence that loss of FKBP12 function in a hom6 mutant leads to toxic accumulation of aspartate β-semialdehyde, the substrate of homoserine dehydrogenase, through deregulation of AK activity. Our results indicate that FKBP12 is a key component governing metabolic flux through the homoserine biosynthetic cascade.
MATERIALS AND METHODS
Oligonucleotides.
Oligonucleotides used in this work are listed in Table 1.
TABLE 1.
Oligonucleotides used in this study
| Oligonucleotide | 5′-3′ sequence |
|---|---|
| JOHE7593 | ACTCGAGTATAAGCAAAAAATCAATCAAAACAAGTAATAACATGGAGGCCCAGAATACCC |
| JOHE7594 | AAAAGCAGAAAGGCGGCTCAATTGATAGTACTTTGCTTCAGTATAGCGACCAGCATTCAC |
| JOHE11302 | AGAACGTAGATAGTATCATCAATCGAATAATAAAAAAAAACAGCTGAAGCTTCGTACGC |
| JOHE11303 | ATATAAATATACCTATGTTTTTATATGTCTGTTTACTGATGCATAGGCCACTAGTGGATCTG |
| JOHE8033 | CAGCTGAAGCTTCGTACGC |
| JOHE8034 | GCATAGGCCACTAGTGGATCTG |
| JOHE11689 | CGCGGATCCAGTGCAATTAAGAAACGTGTTACAAAGAAGGAAGTCTTCGGTTAACAATG |
| JOHE11690 | CGCGGATCCTCCAGAGTGAATCCGGGAAACATAATCGGGAAAATTCATTGTTAACCGAA |
Plasmids.
2μm plasmid pYJH23, expressing the wild-type FPR1 gene, and the control plasmid pSEY8 were described previously (37, 42). Centromere-based plasmid pMA-HOM6, expressing the wild-type HOM6 gene, was obtained by gap repair (65). With this aim, a synthetic, minimal hom6Δ allele was first obtained by PCR using 3′-complementary primer pairs JOHE11689 and JOHE11690, digested with BamHI and cloned into the BamHI site of the vector pRS316 (70). The resulting plasmid, pMA-hom6Δ, contains an insert consisting of two 40-bp-long segments, corresponding to sequences found 300 bp upstream and downstream of the HOM6 open reading frame, respectively, and flanking a unique HpaI restriction site. Plasmid pMA-HOM6 was rescued from yeast strain BY4741 transformed with plasmid pMA-hom6Δ, previously linearized by HpaI digestion. Two-hybrid expression vectors, pGBT9 and pGAD424, were as described previously (7). Plasmid pGBT9-Fpr1 expresses Fpr1 fused to the C terminus of the Gal4 DNA binding domain, as described previously (14). Plasmids pGBT9-AK and pGAD424-AK express wild-type AK fused to the C terminus of the Gal4 DNA binding domain or Gal4 activation domain, respectively, as described previously (1). Plasmid pMACR7 expresses the mutant HOM3-R7 allele (57), as described by I. Velasco et al. (unpublished data). Plasmid pGBT9-AK(E282D) expresses AKE282D fused to the C terminus of the Gal4 DNA-binding domain and was constructed by cloning in pGBT9-AK the SwaI-NdeI restriction fragment of pMACR7 containing the HOM3-R7 GAA846→GAT mutation determining the E282D amino acid substitution. Plasmid pGAD424-AK(E282D), expressing AKE282D fused to the C terminus of the Gal4 activation domain, was constructed by cloning the smaller BamHI-EcoRI fragment of pGBT9-AK(E282D) into plasmid pGAD424, previously digested with these enzymes. Centromere-based plasmids pFP101 and pFP102, expressing wild-type Fpr1 and the active-site mutant Fpr1F43Y, respectively, were as described previously (39). Centromere-based vector YCplac111 was described previously (28).
Strains.
Yeast strains used in this work are listed in Table 2. With the exception of two-hybrid host strain PJ69-4A (41), all of the yeast strains used are derivatives of the isogenic S288C-derived strain BY4741, BY4742, or BY4743 (10). Strain MAY193 was obtained from strain BY4741 by disruption of the FPR1 gene with the nourseothricin resistance natMX4 module from plasmid pAG25 (29), PCR amplified with primers JOHE7593 and JOHE7594. Strain MAY308 was obtained from strain BY4742 by disruption of the HOM6 gene with the G418 resistance KanMX2 module from plasmid pFA6-KanMX2 (81), PCR amplified with primers JOHE11302 and JOHE11303. Strains MAY309, MAY310, and MAY313 were obtained as meiotic products of the corresponding hom2Δ/hom2Δ and hom3Δ/hom3Δ homozygous diploid strains (Saccharomyces Genome Deletion Project, distributed by Openbiosystems). Diploid strains MAYX118, MAYX119, and MAYX120 were obtained by crossing strain MAY193 with strains MAY308, MAY309, and MAY310, respectively. Strains MAYX119-4A and MAYX120-2D were obtained as meiotic products of strains MAYX119 and MAYX120, respectively. Strain MAY312 was obtained from strain MAY308 by substitution of the kanMX2 module in the hom6Δ::kanMX2 allele with the hygromycin B resistance hphMX4 module from plasmid pAG32 (29), which had been PCR amplified with primers JOHE8033 and JOHE8034. Strains MAYX122 and MAYX123 were obtained by crossing strain MAY312 with strains MAYX119-4A and MAYX120-2D, respectively. Strain MAYX123-2C was obtained as a meiotic product of strain MAYX123. Strain MAY315 was obtained from strain MAY313 by replacing the hom3Δ::kanMX4 allele of this strain with the HOM3-R7 allele carried in the HpaI-XbaI fragment of plasmid pMACR7. Strain MAYX125 was obtained by crossing strain MAYX123-2C with strain MAY315.
TABLE 2.
Yeast strains used in this study
| Strain | Genotype | Reference |
|---|---|---|
| PJ69-4A | MATatrp1-901 leu2-3,112 ura3-52 his3-200 gal4Δ gal80Δ LYS2::GAL1-HIS3 GAL2-ADE2 met2::GAL7-lacZ | 41 |
| BY4741 | MATahis3Δ1 leu2Δ0 met15Δ0 ura3Δ0 | 10 |
| BY4742 | MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 | 10 |
| MAY193 | MATahis3Δ1 leu2Δ0 met15Δ0 ura3Δ0 fpr1Δ::natMX4 | This study |
| MAY308 | MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 hom6Δ::kanMX2 | This study |
| MAY309 | MATα his3Δ1 leu2Δ0 met15Δ0 lys2Δ0 ura3Δ0 hom2Δ::kanMX4 | This study |
| MAY310 | MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 hom3Δ::kanMX4 | This study |
| MAYX118 | MATa/MATα his3Δ1/his3Δ1 leu2Δ0/leu2Δ0 met15Δ0/MET15 lys2Δ0/LYS2 ura3Δ0/ura3Δ0 FPR1/fpr1Δ::natMX4 HOM6/hom6Δ::kanMX2 | This study |
| MAYX119 | MATa/MATα his3Δ1/his3Δ1 leu2Δ0/leu2Δ0 met15Δ0/met15Δ0 lys2Δ0/LYS2 ura3Δ0/ura3Δ0 FPR1/fpr1Δ::natMX4 HOM2/hom2Δ::kanMX4 | This study |
| MAYX120 | MATa/MATα his3Δ1/his3Δ1 leu2Δ0/leu2Δ0 met15Δ0/MET15 lys2Δ0/LYS2 ura3Δ0/ura3Δ FPR1/fpr1Δ::natMX4 HOM3/hom3Δ::kanMX3 | This study |
| MAYX119-4A | MATahis3Δ1 leu2Δ0 ura3Δ0 fpr1Δ::natMX4 hom2Δ::kanMX4 | This study |
| MAYX120-2D | MATahis3Δ1 leu2Δ0 met15Δ0 lys2Δ0 ura3Δ0 fpr1Δ::natMX4 hom3Δ::kanMX4 | This study |
| MAY312 | MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 hom6Δ::hphMX4 | This study |
| MAYX122 | MATa/MATα his3Δ1/his3Δ1 leu2Δ0/leu2Δ0 LYS2/lys2Δ0 ura3Δ0/ura3Δ0 FPR1/fpr1Δ::natMX4 HOM2/hom2Δ::kanMX4 HOM6/hom6Δ::hphMX4 | This study |
| MAYX123 | MATa/MATα his3Δ1/his3Δ1 leu2Δ0/leu2Δ0 met15Δ0/MET15 lys2Δ0/lys2Δ0 ura3Δ0/ura3Δ0 FPR1/fpr1Δ::natMX4 HOM3/hom3Δ::kanMX4 HOM6/hom6Δ::hphMX4 | This study |
| MAYX123-2C | MATα his3Δ1 leu2Δ0 met15Δ0 lys2Δ0 ura3Δ0 hom3Δ::kanMX4 hom6Δ::hphMX4 | This study |
| MAY313 | MATahis3Δ1 leu2Δ0 met15Δ0 ura3Δ0 hom3Δ::kanMX4 | This study |
| MAY315 | MATahis3Δ1 leu2Δ0 met15Δ0 ura3Δ0 HOM3-R7 | This study |
| MAYX125 | MATa/MATα his3Δ1/his3Δ1 leu2Δ0/leu2Δ0 met15Δ0/met15Δ0 lys2Δ0/LYS2 ura3Δ0/ura3Δ0 HOM3-R7/hom3Δ::kanMX4 HOM6/hom6Δ::hphMX4 | This study |
Media.
Growth media for S. cerevisiae (synthetic minimal medium [YNB], synthetic complete medium [SC], and rich complex medium [YPD]) were described in reference 69. Sporulation medium was 1.5% potassium acetate (KAc) (pH 7.5), supplemented with uracil and the required amino acids.
Fpr1 affinity chromatography.
Affinity purification of the His6-Fpr1 protein and Fpr1 affinity chromatography were performed as described previously (14).
Two-hybrid interaction assays.
Yeast two-hybrid host strain PJ69-4A was cotransformed with plasmids expressing the Gal4 DNA binding-domain (BD) and Gal4 activation domain (AD) fusion proteins. Transformants were grown in liquid synthetic dextrose (SD) medium supplemented with adenine, uracil, methionine, and histidine or in the same medium with 1 g of l-threonine/liter or with 10 mg of FK506/liter, and induction of the lacZ reporter gene was measured as β-galactosidase activity as described previously (14).
Western blot analysis.
For Western blot analysis of expression of AK and Fpr1, yeast strains expressing these proteins were cultured in liquid YPD medium. Whole-cell protein extracts were prepared by glass bead disruption in lysis buffer A (20 mM HEPES [pH 7.4], 20 mM KCl, 0.5 mM EDTA, and a cocktail of protease inhibitors consisting of 0.5 mM phenylmethylsulfonyl fluoride, 1 μg of pepstatin ml−1, 1 mM benzamidine, and 0.001% aprotinin), using a FastPrep instrument (FP 120; Bio 101, Savant). Proteins were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, transferred to a polyvinylidene difluoride membrane (Immun-Blot; Bio-Rad), probed with rabbit polyclonal antiserum against Fpr1 (13) or with rabbit polyclonal antiserum against aspartokinase (59), kindly provided by S. Carl Falco. Reactions were detected with ECL (Amersham Biosciences).
AK purification and assays.
AK partial purification was as described previously (24). AK activity was measured with an enzymatic assay that couples ADP formation with NADH depletion, using the pyruvate kinase/lactate dehydrogenase system (63).
RESULTS
fpr1Δ hom6Δ double mutants are inviable.
To elucidate cellular functions of FKBP12 in yeast, we conducted a search for mutations in yeast genes that result in a lethal phenotype when combined with an fpr1Δ mutation. By using a high-throughput assay recently developed by Pan et al. (54), we identified a deletion of the HOM6 gene, which encodes homoserine dehydrogenase, as a candidate synthetic lethal mutation.
To validate these results, the synthetic lethal interaction between the fpr1Δ and hom6Δ mutations was tested by classic tetrad analysis. To this end, fpr1Δ and hom6Δ single mutants were first constructed by replacing the entire FPR1 and HOM6 open reading frames with nourseothricin and G418 resistance modules, respectively. The resulting fpr1Δ::nat and hom6Δ::kan strains were crossed to obtain an FPR1/fpr1Δ::nat HOM6/hom6Δ::kan doubly heterozygous mutant diploid strain. As shown in Fig. 1A, this diploid strain sporulated to produce haploid meiotic progeny that were resistant to nourseothricin (Natr) or to G418 (G418r) but not to both drugs. This finding indicates that the fpr1Δ hom6Δ double mutant is inviable and supports the results obtained in the high-throughput screen. Microscopic observation of meiotic products with an inferred fpr1Δ hom6Δ genotype (deduced from the genotype of their tetrad siblings) revealed that these spores germinate and undergo a limited number of cell divisions prior to growth cessation (data not shown). Neither the fpr1Δ mutation nor the hom6Δ mutation exhibited synthetic lethality, with the met15Δ0 or lys2Δ0 mutation also segregating in this cross (data not shown).
FIG. 1.

fpr1Δ and hom6Δ mutations are synthetically lethal. (A) Tetrad analysis of an FPR1/fpr1Δ::nat HOM6/hom6Δ::kan diploid. Spores from strain MAYX118 were dissected on solid YPD medium, and each tetrad was arrayed in a column. Plates were incubated at 30°C for 3 days, photographed, and replica plated to YPD plates containing 200 μg of G418/ml, 70 μg of nourseothricin (Nat)/ml, or both. Replica plates were incubated for 2 days and photographed. (B) Tetrad analysis of strain MAYX118 transformed with URA3 plasmids expressing FPR1 (pYJH23) or HOM6 (pMA-HOM6) or with a URA3 control vector (pSEY8). Transformants were grown in medium selective for the plasmids (SC-uracil) and transferred to sporulation medium, and spores were dissected and analyzed as for panel A. SC-uracil and 5-fluoro-orotic acid plates were included in this assay.
Viability of fpr1Δ hom6Δ double mutants was rescued by ectopic expression of plasmid-borne copies of FPR1 or HOM6, indicating that lethality of the double mutant is attributable to deficiencies in FPR1- and HOM6-encoded functions. The FPR1/fpr1Δ::nat HOM6/hom6Δ::kan diploid strain was transformed with URA3-selectable plasmids expressing either the wild-type FPR1 gene or the HOM6 gene, and the resulting strains produced Ura+ Natr G418r segregants (Fig. 1B). All Ura+ Natr G418r meiotic segregants were sensitive to counterselection of the URA3 plasmid-borne marker with 5-fluoro-orotic acid (9), indicating that the FPR1- or HOM6-expressing plasmids are required for viability. In control experiments, sporulation of the FPR1/fpr1Δ::nat HOM6/hom6Δ::kan diploid strain transformed with a URA3 control vector failed to produce any viable fpr1 hom6 (Ura+ Natr G418r) meiotic products (Fig. 1B).
Expression of an FKBP12 mutant protein with reduced prolyl-isomerase activity restores viability of fpr1Δ hom6Δ double mutants.
We next addressed whether FKBP12 enzymatic activity is required for function. The FPR1/fpr1Δ::nat HOM6/hom6Δ::kan diploid strain was transformed with a centromere-based LEU2 plasmid expressing the Fpr1F43Y mutant, altered in an amino acid residue conserved in mammalian FKBP12 and important for prolyl-isomerase activity (77), and analyzed by tetrad dissection. As shown in Fig. 2, expression of Fpr1F43Y rescued viability of fpr1Δ hom6Δ double mutants, indicating that full FKBP12 prolyl-isomerase activity is not required for viability of these mutants. We note that the growth rate of fpr1Δ hom6Δ colonies expressing Fpr1F43Y was lower than that of those expressing wild-type Fpr1 from the same LEU2 vector in a control experiment (Fig. 2), and this result could be attributable to reduced expression of the Fpr1F43Y mutant, as previously observed (39).
FIG. 2.

FKBP12 active-site mutant restores viability of fpr1Δ hom6Δ double mutants. Diploid strain MAYX118 was transformed with plasmids expressing wild-type FPR1 (pFP101) or the FPR1-F43Y mutant (pFP102), or with a control vector (YCplac111), and spores were dissected and analyzed as described above.
fpr1Δ is not synthetically lethal with other hom mutations.
The yeast HOM6 gene encodes homoserine dehydrogenase, which catalyzes the last step in conversion of aspartic acid to homoserine, the common precursor in synthesis of threonine and methionine. The first two steps in this pathway are catalyzed by AK and aspartate β-semialdehyde dehydrogenase, which are encoded by the HOM3 and HOM2 genes, respectively (Fig. 3A). hom3, hom2, and hom6 mutants are all auxotrophic for threonine and methionine and must therefore import these amino acids from the culture medium to survive. Because the fpr1Δ mutation was not synthetically lethal with a met15 mutation causing methionine auxotrophy, one possible model to explain the synthetic lethal phenotype of fpr1Δ hom6Δ double mutants is that FKBP12 might be required for efficient threonine uptake in yeast. One prediction of this model is that fpr1Δ hom3Δ and fpr1Δ hom2Δ double mutants would exhibit a lethal phenotype, similar to that observed for fpr1Δ hom6Δ double mutants. To test this, we constructed G418-resistant hom3Δ::kan and hom2Δ::kan single mutants, mated these with the fpr1Δ::nat strain described above, and isolated FPR1/fpr1Δ::nat HOM3/hom3Δ::kan and FPR1/fpr1Δ::nat HOM2/hom2Δ::kan diploid strains. As shown in Fig. 3B, sporulation of these strains produced viable Natr G418r spores that exhibited no growth defect, indicating that the fpr1Δ hom3Δ and fpr1Δ hom2Δ double mutants are viable and therefore capable of efficient threonine uptake. Thus, the synthetic lethal interaction observed between hom6 and fpr1 is gene specific and is not observed with other hom mutations.
FIG. 3.

fpr1Δ is not synthetically lethal with the hom3Δ or hom2Δ mutation. (A) Aspartate pathway in yeast. A diagram of the synthesis of threonine and methionine is shown. Only the genes, enzymes, and metabolic intermediates relevant to this study are represented. The dotted line symbolizes feedback inhibition of AK activity by threonine. AsD, aspartate β-semialdehyde dehydrogenase; HD, homoserine dehydrogenase; Asp-P, β-aspartyl phosphate; ASA, aspartate β-semialdehyde. (B) fpr1Δ hom3Δ and fpr1Δ hom2Δ double mutants are viable. Spores from FPR1/fpr1Δ::natMX4 HOM3/hom3Δ::kanMX4 (MAYX120) and FPR1/fpr1Δ::natMX4 HOM2/hom2Δ::kanMX4 (MAYX119) diploid strains were analyzed as described in Fig. 1.
Deletion of HOM3 or HOM2 suppresses lethality of fpr1Δ hom6Δ double mutants.
An alternative model to explain the lethal phenotype of fpr1Δ hom6Δ double mutants is that these strains accumulate toxic levels of the substrate of homoserine dehydrogenase (Hom6), aspartate β-semialdehyde (ASA). In this model, introduction of a mutation earlier in the pathway will block ASA formation and restore viability of fpr1Δ hom6Δ mutant strains. We therefore tested whether hom3Δ and hom2Δ mutations suppress lethality of the fpr1Δ hom6Δ double mutant. fpr1 hom3 and fpr1 hom2 double-mutant strains were crossed with a hom6Δ::hph mutant, and FPR1/fpr1 HOM3/hom3 HOM6/hom6 and FPR1/fpr1 HOM2/hom2 HOM6/hom6 diploid strains heterozygous at three loci were isolated. Sporulation of these diploids produced no viable fpr1 hom6 double mutants (Natr G418s Hygr), confirming synthetic lethality of fpr1 and hom6Δ mutations in these crosses (Fig. 4). In contrast, viable fpr1Δ hom3Δ hom6Δ and fpr1Δ hom2Δ hom6Δ triple-mutant strains (Natr G418r Hygr) were readily isolated, and the growth of these triple mutants was indistinguishable from that of the wild type. These results support a model in which ASA accumulation is toxic and results in the lethal phenotype observed in fpr1Δ hom6Δ mutants.
FIG. 4.

Interrupting the aspartate pathway suppresses lethality of fpr1Δ hom6Δ double mutants. Spores from FPR1/fpr1Δ::natMX4 HOM3/hom3Δ::kanMX4 HOM6/hom6Δ::hphMX4 (MAYX123) and FPR1/fpr1Δ::natMX4 HOM2/hom2Δ::kanMX4 HOM6/hom6Δ::hphMX4 (MAYX122) diploid strains were dissected as described above, and the resulting colonies were analyzed by replica plating them to YPD medium containing G418, nourseothricin, 100 μg of hygromycin B (Hyg)/ml, or all three drugs combined at the same concentrations.
Deletion of HOM6 is deleterious to strains expressing an AK mutant resistant to feedback inhibition.
In yeast, flux through the homoserine biosynthetic pathway is governed through feedback inhibition of AK by threonine (Fig. 3A). Mutations that render AK resistant to feedback inhibition lead to threonine overproduction (3, 48, 57, 62, 63, 72).
Interestingly, AK was identified as a binding partner of FKBP12 during a search for yeast proteins interacting with FKBP12 in the yeast two-hybrid system (1). In these studies, yeast cells with a deletion of the FPR1 gene or exposed to the FKBP12-specific inhibitor FK506 exhibited resistance to the toxic threonine analog hydroxynorvaline. Mutant cells expressing a feedback-resistant AK are also resistant to hydroxynorvaline (62), suggesting that FKBP12 regulates AK inhibition by threonine (1). If loss of FKBP12 function results in deregulation of AK activity, deletion of FPR1 in a hom6Δ mutant could lead to ASA accumulation, in accordance with our results. In addition, this model predicts that a HOM3 mutant allele encoding a feedback-resistant AK would also exhibit synthetic lethality with a hom6Δ mutation. To test this model, we constructed a yeast strain expressing a feedback-resistant AK by integrating the HOM3-R7 mutant allele, which was originally isolated from a threonine-overproducing yeast strain (57; Velasco et al., unpublished). The HOM3-R7 mutant strain was then crossed with a hom3 hom6 double mutant to obtain a HOM3-R7/hom3::Kan HOM6/hom6::hyg diploid strain. Tetrad analysis of this diploid revealed that most HOM3-R7 hom6Δ recombinants were inviable, and those few that did survive grew poorly (Fig. 5). Taken together, these results indicate that dysregulation of AK activity becomes toxic to cells defective in homoserine dehydrogenase activity and support the hypothesis that accumulation of ASA impairs growth.
FIG. 5.
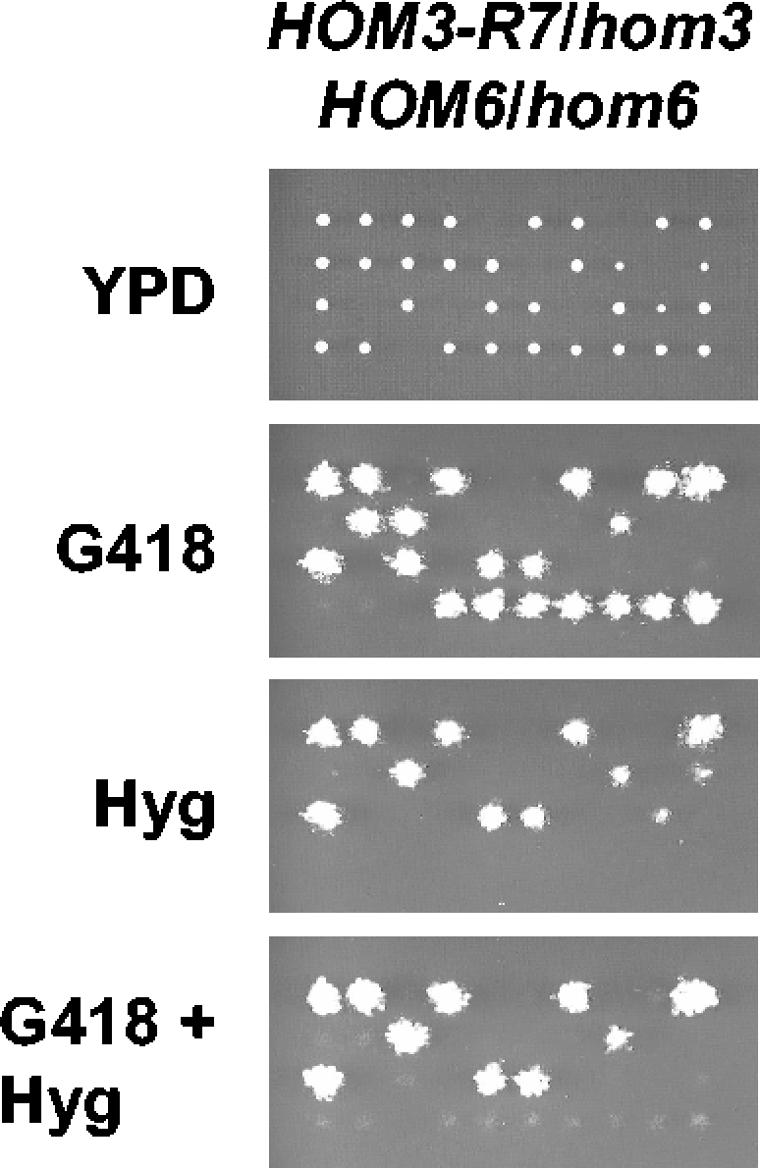
Deletion of HOM6 is deleterious to strains expressing a mutant AK resistant to feedback inhibition. Spores from the HOM3-R7/hom3Δ::kanMX4 HOM6/hom6Δ::hphMX4 diploid strain (MAYX125) were analyzed as described above.
FKBP12-AK interaction is induced by threonine, and reduced by a mutation in HOM3 that renders AK resistant to feedback inhibition.
FKBP12 and AK have been shown to physically interact both in vivo and in vitro (1). To study whether this interaction influences AK feedback inhibition by threonine, we studied the effect of threonine on FKBP12 binding to wild-type and feedback-resistant mutant AK. We first studied FKBP12-AK interaction in vitro by affinity chromatography by assaying binding of wild-type and mutant AK to recombinant His6-Fpr1 protein that had been produced in bacteria and coupled to an agarose matrix. As shown in Fig. 6A, both wild-type and feedback-resistant AK interacted with FKBP12 in the absence of exogenous threonine. Addition of threonine to the binding reactions increased FKBP12 interaction with wild-type AK but not with feedback-resistant AK, suggesting that threonine enhances FKBP12 binding to wild-type AK but less so with a feedback-resistant AK mutant.
FIG. 6.

Aspartokinase interacts with itself and FKBP12. (A) FKBP12-AK and AK-AK interaction by in vitro affinity chromatography. Crude protein extracts obtained from a wild-type strain (BY4741) or from a strain expressing the HOM3-R7 mutant allele (MAY315) were incubated with His6-tagged yeast FKBP12 coupled to agarose beads in the presence or absence of 30 mM l-threonine. Bound proteins were eluted and analyzed by Western blotting with antibodies against yeast AK. Binding reactions with agarose beads alone were included as controls. (B) Fpr1-AK and AK-AK interactions in vivo. Transformants from yeast two-hybrid host strain PJ69-4A coexpressing Gal4 DNA-binding domain (BD) fusion proteins from plasmid pGBT9-Fpr1 (BD-Fpr1), pGBT9-AK (BD-AK), or pGBT9-AK(E282D) (BD-AKE282D) and Gal4 activation domain (AD) fusion proteins from plasmids pGAD424-AK (AD-AK) or pGAD424-AK(E282D) (AD-AKE282D) were grown in SD medium supplemented with uracil, adenine, methionine, and histidine (YNB) or in the same medium plus 1 g of l-threonine/liter (YNB+Thr) or 10 μg of FK506/ml (YNB+FK506), and induction of the lacZ reporter gene was quantified by assaying β-galactosidase activity. PJ69-4A cotransformed with vectors pGBT9 (BD) and pGAD424 (AD) was assayed as a control.
We further analyzed FKBP12-AK interactions in vivo with the yeast two-hybrid system. The two-hybrid host strain PJ69-4A, coexpressing Gal4 DNA binding domain-Fpr1 (Gal4BD-Fpr1) and Gal4 activation domain-AK (Gal4AD-AK) fusion proteins, was grown in a synthetic medium with or without threonine. Interactions between the fusion proteins were detected and quantified by measuring expression of the lacZ reporter gene. Similar experiments were conducted with cells coexpressing Gal4BD-Fpr1 and a Gal4AD-AKE282D feedback-resistant fusion protein. Results of these experiments are shown in Fig. 6B. In the absence of exogenous threonine, the interaction detected between FKBP12 and wild-type AK was considerably greater than that detected between FKBP12 and AKE282D, indicating that the mutant AK binds FKBP12 with less affinity than wild-type AK in vivo in the presence of threonine at normal intracellular concentrations (Fig. 6B). Addition of threonine to the culture medium increased both FKBP12-AK and FKBP12-AKE282D binding, lending support to the hypothesis that threonine enhances these interactions. FKBP12-AK binding was nearly abolished by the presence of FK506 (Fig. 6B), in accord with previous observations (1).
In addition, we detected a low but significant level of self-interaction between both wild-type and mutant AKs in the two-hybrid assay (Fig. 6B). These results suggest that yeast AK might form homodimers, or homo-oligomers, as has been reported for this protein in other organisms (8, 23, 56, 80). AK-AK and AKE282D-AKE282D interactions were also detected in the presence of FK506, indicating that neither FKBP12 binding nor prolyl-isomerase activity is required for AK self-interaction.
The hom6Δ mutation confers sensitivity to FK506.
Because FK506 prevented FKBP12-AK interaction in the two hybrid assay, we tested whether FK506 mimics the lethal effect of an fpr1Δ mutation in a homoserine dehydrogenase-deficient mutant. As shown in Fig. 7A and B, FK506 indeed inhibited growth of the hom6Δ mutant. By microscopic examination, hom6Δ mutant cells exposed to FK506 exhibited abnormal morphologies, indicative of a cytokinesis defect (Fig. 7C). Drugs that target both FKBP12 and Hom6 might therefore be expected to exhibit a synergistic lethal effect (see Discussion).
FIG. 7.

hom6Δ mutation confers sensitivity to FK506. (A) Approximately 107 cells from hom6Δ mutant strain MAY308 were plated on solid YPD medium and exposed to 250 μg of FK506 diffusing from a paper disk. A control paper disk containing only the FK506 solvent (90% EtOH plus 10% Tween 20) was included. The plate was incubated at 30°C for 2 days and photographed. As a control, the same assay was conducted with isogenic wild-type strain BY4742. (B) Liquid YPD cultures of hom6 (MAY308) and wild-type (BY4742) strains were exposed to 10 μg of FK506/ml or left untreated as controls, and growth was measured based on optical density at 600 nm (OD600). (C) hom6Δ cells from the FK506-exposed culture described for panel B were photographed using differential interference contrast microscopy after 22 h of incubation with the drug.
Aspartokinase from an fpr1Δ mutant is inhibited by threonine in vitro.
The results obtained thus far indicate a role for FKBP12 in the regulation of AK activity, as proposed originally by Alarcon and Heitman (1). To test this hypothesis, we assayed AK activity (in the presence and absence of threonine) from wild-type and fpr1, hom6, or HOM3-R7 feedback-resistant mutant cells. AK expression and AK partial purification was analyzed by Western blotting, revealing no significant differences between the strains (Fig. 8A). As shown in Fig. 8B, all strains exhibited similar levels of AK activity in the absence of threonine. In the presence of 1 mM threonine, the AK activities from wild-type and fpr1Δ strains were inhibited to comparable levels, indicating that FKBP12 is not required for AK inhibition in vitro by threonine. Threonine also inhibited AK activity of the hom6Δ mutant, suggesting that homoserine dehydrogenase does not play a role in AK feedback inhibition. AK activity in the wild-type, fpr1Δ, or hom6Δ strain was completely feedback inhibited by the addition of 5 mM threonine, whereas the feedback-resistant AK exhibited no inhibition (Fig. 8B). Addition of recombinant His6-Fpr1 to these assays did not have any detectable effect on feedback inhibition of AK from wild-type or the fpr1Δ mutant (data not shown).
FIG. 8.

AK inhibition by threonine. (A) Western blot analysis of AK and FKBP12 in protein extracts and partially purified AK preparations obtained from the wild type (BY4741) or from an fpr1Δ (MAY193), hom6Δ (MAY308), or HOM3-R7 (MAY315) mutant strain or from a hom3Δ mutant strain (MAY310) as a control. (B) AK activity in partially purified extracts obtained from the strains described above was assayed in the presence or absence of threonine (1 or 5 mM) as described in Materials and Methods and in Results.
One possible role for FKBP12 in AK inhibition could be to bind and stabilize threonine-AK complexes, modulating the dynamics of AK response to changes in intracellular threonine concentrations. In this model, FKBP12 might function to maintain AK in an inhibited conformation, preventing it from returning to the active state. To investigate this, we assayed AK from the wild-type and fpr1Δ strains following preincubation of the enzyme preparations with threonine in the presence or absence of bacterially expressed His6-Fpr1. Threonine-treated AK preparations were then mixed with the AK assay reagents, reducing the threonine concentration, and the AK activity was then compared to that of similar AK reactions containing the same final threonine concentrations but in which AK was not preexposed to threonine. These experiments revealed no increase in threonine inhibition in AK preincubated with threonine, indicating that AK inhibition by threonine, even in the presence of FKBP12, is fully reversible in vitro. In these assays, there was no significant difference in threonine inhibition of AK from the wild type compared to results with the fpr1Δ mutant in either the presence or the absence of His6-Fpr1 (data not shown).
DISCUSSION
Here we investigated the cellular functions of yeast FKBP12 by a systematic search for mutations that are lethal when combined with an fpr1 mutation. In addition to revealing the fpr1-hom6 synthetically lethal interaction which is the subject of this report, this assay also confirmed the fpr1-hmo1 synthetically lethal interaction previously reported by Dolinski and Heitman (21).
Our results are consistent with toxic accumulation of ASA, or an ASA derivative, as being the cause of lethality in fpr1Δ hom6Δ double-mutant strains. The hypothesis that ASA accumulation inhibits growth of fpr1Δ hom6Δ double mutants is supported by the fact that hom3Δ and hom2Δ mutations, which both abolish ASA formation, rescue the viability of fpr1Δ hom6Δ double mutants. In this model, FKBP12 regulates metabolic flux to prevent ASA accumulation. Deletion of HOM6 alone does not lead to lethal levels of ASA, likely because exogenous threonine imported from the medium feedback inhibits AK activity, reduces Asp-P, and limits ASA formation (Fig. 3A). hom6Δ mutants expressing a feedback-resistant AK enzyme are severely growth impaired or inviable, even though these strains express FKBP12, again supporting the model that increasing flux though the pathway is lethal when the pathway is blocked by mutation at the Hom6 step.
In solution, ASA is an unstable compound that polymerizes (16, 50, 64, 79). We tested the hypothesis that ASA might cyclize to produce a compound analogous to l-azetidine-2-carboxylic acid, a toxic proline analogue. Recent studies have shown that yeast strains in the Σ1278b background are naturally resistant to l-azetidine-2-carboxylic acid and that this resistance is due to the expression of MPR1 or MPR2, which are homologous genes encoding acetyltransferases that detoxify this proline analogue (43, 74). MPR1 and MPR2 are not present in strains in the S288C background studied here. A plasmid expressing MPR1 did not rescue viability of fpr1Δ hom6Δ double mutants, suggesting that ASA conversion into l-azetidine-2-carboxylic acid is not the basis for ASA toxicity. The mechanism of ASA toxicity is currently unknown and might be addressed through different genetic approaches, including isolation of additional mutations suppressing lethality of fpr1Δ hom6Δ double mutants or screening for high-copy suppressors of these double-mutant strains. We have observed morphological changes in FK506-exposed hom6Δ mutant cells that appear to be the result of cytokinesis defects.
Interestingly, our studies reveal a potential new target for antifungal drug therapy. We show that the widely used immunosuppressant FK506 inhibits growth of a hom6Δ mutant, probably by blocking FKBP12-AK interaction, and thereby mimicking the effects of an fpr1Δ mutation. In theory, combination of FK506 (or preferably, a nonimmunosuppressive derivative of FK506) with a homoserine dehydrogenase-specific inhibitor could reproduce the lethal effect of an fpr1Δ hom6Δ double mutation. Such a combination therapy would target a biosynthetic pathway that is conserved in fungi but not in mammals. The natural compound 5-hydroxy-4-oxonorvaline (HON), a toxic amino acid produced by Streptomyces akiyoshiensis, is a very efficient inhibitor of homoserine dehydrogenase (40, 82, 87, 88). HON inhibits growth by blocking synthesis of homoserine, and it has shown activity against different fungal organisms (87). In addition, HON has established activity in treating systemic Candida infections in mice (87). Preliminary assays revealed no inhibitory effect of HON on fpr1 mutants in YPD medium, indicating that HON uptake is reduced in this medium. Further studies will be necessary to identify other homoserine dehydrogenase inhibitors with potential use in combination with FK506 in new antifungal therapies. Indeed, combination therapies with FK506 and azole antifungals have proven highly effective against Aspergillus fumigatus and Candida species (53, 73).
We have observed that threonine enhances FKBP12-AK interactions, indicating that this amino acid induces structural changes in AK that result in increased affinity for FKBP12. These results, together with the genetic data described above and the observations reported by Alarcon and Heitman (1), suggest a role for FKBP12 in AK feedback regulation. In support of this model, FKBP12 shows reduced affinity for a mutant AK carrying the E282D substitution, affecting a highly conserved amino acid residue required for feedback regulation by threonine (Velasco et al., unpublished). The E282D substitution could decrease AK affinity for threonine, rather than for FKBP12, because an increasing threonine concentration has a clear positive effect on FKBP12-AKE282D binding in vivo.
Thus, one possibility is that AK feedback regulation involves threonine-induced interaction with FKBP12. However, our results indicate that AK inhibition by threonine in vitro does not require FKBP12, because AK activities from the wild type and an fpr1Δ mutant exhibit similar threonine sensitivities. These results indicate that FKBP12 is not required for providing AK with a conformational state competent for subsequent threonine inhibition and suggest that FKBP12-AK interaction is not necessary for AK feedback regulation under these in vitro conditions. Western blot analysis of the partially purified extracts from the wild-type strain used in the AK assays revealed the presence of FKBP12, although the fractionation procedure used for AK enrichment might result in reduced FKBP12/AK concentration ratios in the partially purified AK preparations. However, addition of bacterially expressed, His6-tagged yeast FKBP12 to the AK reactions did not increase threonine sensitivity of AK obtained from the wild-type or fpr1Δ strain (data not shown).
What then is the role of FKBP12 in AK feedback inhibition? FKBP12 might interact with threonine-inhibited AK complexes to stabilize them in an inactive state. In our studies, AK preincubation with threonine in the presence of endogenous Fpr1 or bacterially expressed His6-Fpr1 did not result in a detectable increase in AK inhibition. In this regard, we cannot rule out the possibility that the His6-Fpr1 fusion protein used in these studies fails to promote AK inhibition, despite the fact that His6-Fpr1 interacts with AK physically in vitro. Alternatively, FKBP12-AK interaction might be reduced in the chemical environment where AK activity is routinely assayed in vitro or feedback inhibition might require additional proteins that are lost during AK partial purification. Future studies will address the molecular mechanism by which FKBP12 exerts a regulatory influence over AK activity in vivo.
Acknowledgments
We thank Hiroshi Takagi, S. Carl Falco, and Robert L. White for generously supplying us with MPR1-expressing plasmids, antibodies against aspartokinase, and HON, respectively. We thank John McCusker, Toshiaki Harashima, Alex Idnurm, and Pablo Marina for critical reading of the manuscript. We also thank Cristl Arndt for excellent technical assistance.
This work was supported by R01 grants AI50438 from the NIAID to J.H. and HG002432 to J.D.B. X.P. is supported in part by a Leukemia & Lymphoma Society Fellowship. Joseph Heitman is an investigator of the Howard Hughes Medical Institute and a Burroughs Wellcome Scholar in Molecular Pathogenic Mycology.
REFERENCES
- 1.Alarcon, C., and J. Heitman. 1997. FKBP12 physically and functionally interacts with aspartokinase in Saccharomyces cerevisiae. Mol. Cell. Biol. 17:5968-5975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ansari, H., G. Greco, and J. Luban. 2002. Cyclophilin A peptidyl-prolyl isomerase activity promotes Zpr1 nuclear export. Mol. Cell. Biol. 22:6993-7003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arevalo-Rodriguez, M., I. L. Calderon, and S. Holmberg. 1999. Mutations that cause threonine sensitivity identify catalytic and regulatory regions of the aspartate kinase of Saccharomyces cerevisiae. Yeast 15:1331-1345. [DOI] [PubMed] [Google Scholar]
- 4.Arevalo-Rodriguez, M., M. E. Cardenas, X. Wu, S. D. Hanes, and J. Heitman. 2000. Cyclophilin A and Ess1 interact with and regulate silencing by the Sin3-Rpd3 histone deacetylase. EMBO J. 19:3739-3749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arevalo-Rodriguez, M., and J. Heitman. Cyclophilin A is nuclear localized and controls meiosis in yeast. (Submitted for publication.)
- 6.Arevalo-Rodriguez, M., X. Wu, S. D. Hanes, and J. Heitman. 2004. Prolyl isomerases in yeast. Front. Biosci. 9:2420-2446. [DOI] [PubMed] [Google Scholar]
- 7.Bartel P.L., C.-T. Chien, R. Sternglanz, and S. Fields. 1993. Using the two-hybrid system to detect protein-protein interactions, p. 153-179. In H. D. A. (ed.), Cellular interactions in development: a practical approach. Oxford University Press, Oxford, United Kingdom.
- 8.Bearer, C. F., and K. E. Neet. 1978. Threonine inhibition of the aspartokinase-homoserine dehydrogenase I of Escherichia coli. Threonine binding studies. Biochemistry 17:3512-3516. [DOI] [PubMed] [Google Scholar]
- 9.Boeke, J. D., F. LaCroute, and G. R. Fink. 1984. A positive selection for mutants lacking orotidine-5′-phosphate decarboxylase activity in yeast: 5-fluoro-orotic acid resistance. Mol. Gen. Genet. 197:345-346. [DOI] [PubMed] [Google Scholar]
- 10.Brachmann, C. B., A. Davies, G. J. Cost, E. Caputo, J. Li, P. Hieter, and J. D. Boeke. 1998. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast 14:115-132. [DOI] [PubMed] [Google Scholar]
- 11.Breuder, T., C. S. Hemenway, N. R. Movva, M. E. Cardenas, and J. Heitman. 1994. Calcineurin is essential in cyclosporin A- and FK506-sensitive yeast strains. Proc. Natl. Acad. Sci. USA 91:5372-5376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brown, C. R., D.-Y. Cui, G. G.-C. Hung, and H.-L. Chiang. 2001. Cyclophilin A mediates Vid22p function in the import of fructose-1,6-bisphosphatase into Vid vesicles. J. Biol. Chem. 276:48017-48026. [DOI] [PubMed] [Google Scholar]
- 13.Cardenas, M., and J. Heitman. 1995. FKBP12-rapamycin target TOR2 is a vacuolar protein with an associated phosphatidylinositol-4 kinase activity. EMBO J. 14:5892-5907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cardenas, M. E., C. Hemenway, R. S. Muir, R. Ye, D. Fiorentino, and J. Heitman. 1994. Immunophilins interact with calcineurin in the absence of exogenous immunosuppressive ligands. EMBO J. 13:5944-5957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chang, H., and S. Lindquist. 1994. Conservation of Hsp90 macromolecular complexes in Saccharomyces cerevisiae. J. Biol. Chem. 269:24983-24988. [PubMed] [Google Scholar]
- 16.Coulter, C. V., J. A. Gerrard, J. A. E. Kraunsoe, D. J. Moore, and A. J. Pratt. 1996. (S)-Aspartate semi-aldehyde: Synthetic and structural studies. Tetrahedron 52:7127-7136. [Google Scholar]
- 17.Davis, E. S., A. Becker, J. Heitman, M. N. Hall, and M. B. Brennan. 1992. A yeast cyclophilin gene essential for lactate metabolism at high temperature. Proc. Natl. Acad. Sci. USA 89:11169-11173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dolinski, K., S. Muir, M. Cardenas, and J. Heitman. 1997. All cyclophilins and FK506 binding proteins are, individually and collectively, dispensable for viability in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 94:13093-13098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dolinski, K., C. Scholz, R. S. Muir, S. Rospert, F. X. Schmid, M. E. Cardenas, and J. Heitman. 1997. Functions of FKBP12 and mitochondrial cyclophilin active site residues in vitro and in vivo in Saccharomyces cerevisiae. Mol. Biol. Cell 8:2267-2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dolinski, K. J., M. E. Cardenas, and J. Heitman. 1998. CNS1 encodes an essential p60/Sti1 homolog in Saccharomyces cerevisiae that suppresses cyclophilin 40 mutations and interacts with Hsp90. Mol. Cell. Biol. 18:7344-7352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dolinski, K. J., and J. Heitman. 1999. Hmo1p, a high mobility group 1/2 homolog, genetically and physically interacts with the yeast FKBP12 prolyl isomerase. Genetics 151:935-944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Duina, A. A., H.-C. J. Chang, J. A. Marsh, S. Lindquist, and R. F. Gaber. 1996. A cyclophilin function in Hsp90-dependent signal transduction. Science 274:1713-1715. [DOI] [PubMed] [Google Scholar]
- 23.Falcoz-Kelly, F., J. Janin, J. C. Saari, M. Veron, P. Truffa-Bachi, and G. N. Cohen. 1972. Revised structure of aspartokinase I-homoserine dehydrogenase I of Escherichia coli K12. Evidence for four identical subunits. Eur. J. Biochem. 28:507-519. [DOI] [PubMed] [Google Scholar]
- 24.Farfán, M. J., E. Martín-Rendón, and I. L. Calderón. 1996. Effect of gene amplification on threonine production by yeast. Biotechnol. Bioeng. 49:667-674. [DOI] [PubMed] [Google Scholar]
- 25.Fischer, G., B. Wittmann-Liebold, K. Lang, T. Kiefhaber, and F. X. Schmid. 1989. Cyclophilin and peptidyl-prolyl cis-trans isomerase are probably identical proteins. Nature 337:476-478. [DOI] [PubMed] [Google Scholar]
- 26.Foor, F., S. A. Parent, N. Morin, A. M. Dahl, N. Ramadan, G. Chrebet, K. A. Bostian, and J. B. Nielsen. 1992. Calcineurin mediates inhibition by FK506 and cyclosporin of recovery from alpha-factor arrest in yeast. Nature 360:682-684. [DOI] [PubMed] [Google Scholar]
- 27.Gadal, O., S. Labarre, C. Boschiero, and P. Thuriaux. 2002. Hmo1, an HMG-box protein, belongs to the yeast ribosomal DNA transcription system. EMBO J. 21:5498-5507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gietz, R. D., and A. Sugino. 1988. New yeast-Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair restriction sites. Gene 74:527-534. [DOI] [PubMed] [Google Scholar]
- 29.Goldstein, A. L., and J. H. McCusker. 1999. Three new dominant drug resistance cassettes for gene disruption in Saccharomyces cerevisiae. Yeast 15:1541-1553. [DOI] [PubMed] [Google Scholar]
- 30.Haendler, B., R. Keller, P. C. Hiestand, H. P. Kocher, G. Wegmann, and N. R. Movva. 1989. Yeast cyclophilin: isolation and characterization of the protein, cDNA and gene. Gene 83:39-46. [DOI] [PubMed] [Google Scholar]
- 31.Handschumacher, R. E., M. W. Harding, J. Rice, R. J. Drugge, and D. W. Speicher. 1984. Cyclophilin: a specific cytosolic binding protein for cyclosporin A. Science 226:544-547. [DOI] [PubMed] [Google Scholar]
- 32.Hanes, S. D., P. R. Shank, and K. A. Bostian. 1989. Sequence and mutational analysis of ESS1, a gene essential for growth in Saccharomyces cerevisiae. Yeast 5:55-72. [DOI] [PubMed] [Google Scholar]
- 33.Hani, J., B. Schelbert, A. Bernhardt, H. Domdey, G. Fischer, K. Wiebauer, and J. U. Rahfeld. 1999. Mutations in a peptidyl prolyl-cis/trans-isomerase gene lead to a defect in 3′-end formation of a pre-mRNA in Saccharomyces cerevisiae. J. Biol. Chem. 274:108-116. [DOI] [PubMed] [Google Scholar]
- 34.Hani, J., G. Stumpf, and H. Domdey. 1995. PTF1 encodes an essential protein in Saccharomyces cerevisiae, which shows strong homology with a new putative family of PPIases. FEBS Lett. 365:198-202. [DOI] [PubMed] [Google Scholar]
- 35.Harding, M. W., A. Galat, D. E. Uehling, and S. L. Schreiber. 1989. A receptor for the immunosuppressant FK506 is a cis-trans peptidyl-prolyl isomerase. Nature 341:758-760. [DOI] [PubMed] [Google Scholar]
- 36.Heitman, J., N. R. Movva, and M. N. Hall. 1991. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science 253:905-909. [DOI] [PubMed] [Google Scholar]
- 37.Heitman, J., N. R. Movva, P. C. Hiestand, and M. N. Hall. 1991. FK 506-binding protein proline rotamase is a target for the immunosuppressive agent FK 506 in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 88:1948-1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hemenway, C. S., K. Dolinski, M. E. Cardenas, M. A. Hiller, E. W. Jones, and J. Heitman. 1995. vph6 mutants of Saccharomyces cerevisiae require calcineurin for growth and are defective in vacuolar H+-ATPase assembly. Genetics 141:833-844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hemenway, C. S., and J. Heitman. 1996. Immunosuppressant target protein FKBP12 is required for P-glycoprotein function in yeast. J. Biol. Chem. 271:18527-18534. [DOI] [PubMed] [Google Scholar]
- 40.Jacques, S. L., I. A. Mirza, L. Ejim, K. Koteva, D. W. Hughes, K. Green, R. Kinach, J. F. Honek, H. K. Lai, A. M. Berghuis, and G. D. Wright. 2003. Enzyme-assisted suicide: molecular basis for the antifungal activity of 5-hydroxy-4-oxonorvaline by potent inhibition of homoserine dehydrogenase. Chem. Biol. 10:989-995. [DOI] [PubMed] [Google Scholar]
- 41.James, P., J. Halladay, and E. A. Craig. 1996. Genomic libraries and a host strain designed for highly efficient two-hybrid selection in yeast. Genetics 144:1425-1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jungmann, J., H. Reins, J. Lee, A. Romeo, R. Hassett, D. Kosman, and S. Jentsch. 1993. MAC1, a nuclear regulatory protein related to Cu-dependent transcription factors is involved in Cu/Fe utilization and stress resistance in yeast. EMBO J. 12:5051-5056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kimura, Y., S. Nakamori, and H. Takagi. 2002. Polymorphism of the MPR1 gene required for toxic proline analogue resistance in the Saccharomyces cerevisiae complex species. Yeast 19:1437-1445. [DOI] [PubMed] [Google Scholar]
- 44.Koltin, Y., L. Faucette, D. J. Bergsma, M. A. Levy, R. Cafferkey, P. L. Koser, R. K. Johnson, and G. P. Livi. 1991. Rapamycin sensitivity in Saccharomyces cerevisiae is mediated by a peptidyl-prolyl cis-trans isomerase related to human FK506-binding protein. Mol. Cell. Biol. 11:1718-1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Landsman, D., and M. Bustin. 1993. A signature for the HMG-1 box DNA-binding proteins. Bioessays 15:539-546. [DOI] [PubMed] [Google Scholar]
- 46.Lu, J., R. Kobayashi, and S. J. Brill. 1996. Characterization of a high mobility group 1/2 homolog in yeast. J. Biol. Chem. 271:33678-33685. [DOI] [PubMed] [Google Scholar]
- 47.Marsh, J. A., H. M. Kalton, and R. F. Gaber. 1998. Cns1 is an essential protein associated with the Hsp90 chaperone complex in Saccharomyces cerevisiae that can restore cyclophilin 40-dependent functions in cpr7Δ cells. Mol. Cell. Biol. 18:7353-7359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Martin-Rendon, E., M. J. Farfan, C. Ramos, and I. L. Calderon. 1993. Isolation of a mutant allele that deregulates the threonine biosynthesis in Saccharomyces cerevisiae. Curr. Genet. 24:465-471. [DOI] [PubMed] [Google Scholar]
- 49.Matouschek, A., S. Rospert, K. Schmid, B. Glick, and G. Schatz. 1995. Cyclophilin catalyzes protein folding in yeast mitochondria. Proc. Natl. Acad. Sci. USA 92:6319-6323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Meffre, P. 1999. Syntheses of optically active 2-amino-4-oxobutyric acid and N,O-protected derivatives. Amino Acids 16:251-272. [DOI] [PubMed] [Google Scholar]
- 51.Myers, J. K., D. P. Morris, A. L. Greenleaf, and T. G. Oas. 2001. Phosphorylation of RNA polymerase II CTD fragments results in tight binding to the WW domain from the yeast prolyl isomerase Ess1. Biochemistry 40:8479-8486. [DOI] [PubMed] [Google Scholar]
- 52.Nakamura, T., Y. Liu, D. Hirata, H. Namba, S. Harada, T. Hirokawa, and T. Miyakawa. 1993. Protein phosphatase type 2B (calcineurin)-mediated, FK506-sensitive regulation of intracellular ions in yeast is an important determinant for adaptation to high salt stress conditions. EMBO J. 12:4063-4071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Onyewu, C., J. R. Blankenship, M. Del Poeta, and J. Heitman. 2003. Ergosterol biosynthesis inhibitors become fungicidal when combined with calcineurin inhibitors against Candida albicans, Candida glabrata, and Candida krusei. Antimicrob. Agents Chemother. 47:956-964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pan, X., D. S. Yuan, D. Xiang, X. Wang, S. Sookhai-Mahadeo, J. S. Bader, P. Hieter, F. Spencer, and J. D. Boeke. A robust toolkit for functional profiling of the yeast genome. Mol. Cell, in press. [DOI] [PubMed]
- 55.Parent, S. A., J. B. Nielsen, N. Morin, G. Chrebet, N. Ramadan, A. M. Dahl, M. J. Hsu, K. A. Bostian, and F. Foor. 1993. Calcineurin-dependent growth of an FK506- and CsA-hypersensitive mutant of Saccharomyces cerevisiae. J. Gen. Microbiol. 139:2973-2984. [DOI] [PubMed] [Google Scholar]
- 56.Paris, S., P. M. Wessel, and R. Dumas. 2002. Overproduction, purification, and characterization of recombinant bifunctional threonine-sensitive aspartate kinase-homoserine dehydrogenase from Arabidopsis thaliana. Protein Expr. Purif. 24:105-110. [DOI] [PubMed] [Google Scholar]
- 57.Pedersen, J. O., M. A. Rodriguez, M. Praetorius-Ibba, T. Nilsson-Tillgren, I. L. Calderon, and S. Holmberg. 1997. Locus-specific suppression of ilv1 in Saccharomyces cerevisiae by deregulation of CHA1 transcription. Mol. Gen. Genet. 255:561-569. [DOI] [PubMed] [Google Scholar]
- 58.Pijnappel, W. W., D. Schaft, A. Roguev, A. Shevchenko, H. Tekotte, M. Wilm, G. Rigaut, B. Seraphin, R. Aasland, and A. F. Stewart. 2001. The S. cerevisiae SET3 complex includes two histone deacetylases, Hos2 and Hst1, and is a meiotic-specific repressor of the sporulation gene program. Genes Dev. 15:2991-3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rafalski, J. A., and S. C. Falco. 1988. Structure of the yeast HOM3 gene which encodes aspartokinase. J. Biol. Chem. 263:2146-2151. [PubMed] [Google Scholar]
- 60.Rahfeld, J. U., K. P. Rucknagel, B. Schelbert, B. Ludwig, J. Hacker, K. Mann, and G. Fischer. 1994. Confirmation of the existence of a third family among peptidyl-prolyl cis/trans isomerases. Amino acid sequence and recombinant production of parvulin. FEBS Lett. 352:180-184. [DOI] [PubMed] [Google Scholar]
- 61.Rahfeld, J. U., A. Schierhorn, K. Mann, and G. Fischer. 1994. A novel peptidyl-prolyl cis/trans isomerase from Escherichia coli. FEBS Lett. 343:65-69. [DOI] [PubMed] [Google Scholar]
- 62.Ramos, C., and I. L. Calderon. 1992. Overproduction of threonine by Saccharomyces cerevisiae mutants resistant to hydroxynorvaline. Appl. Environ. Microbiol. 58:1677-1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ramos, C., M. A. Delgado, and I. L. Calderon. 1991. Inhibition by different amino acids of the aspartate kinase and the homoserine kinase of the yeast Saccharomyces cerevisiae. FEBS Lett. 278:123-126. [DOI] [PubMed] [Google Scholar]
- 64.Roberts, S. J., J. C. Morris, R. C. J. Dobson, and J. A. Gerrard. 2003. The preparation of (S)-aspartate semi-aldehyde appropriate for use in biochemical studies. Bioorg. Med. Chem. Lett. 13:265-267. [DOI] [PubMed] [Google Scholar]
- 65.Rothstein, R. 1991. Targeting, disruption, replacement, and allele rescue: integrative DNA transformation in yeast. Methods Enzymol. 194:281-301. [DOI] [PubMed] [Google Scholar]
- 66.Scholz, C., P. Maier, K. Dolinski, J. Heitman, and F. X. Schmid. 1999. R73A and H144Q mutants of the yeast mitochondrial cyclophilin Cpr3 exhibit a low prolyl isomerase activity in both peptide and protein-folding assays. FEBS Lett. 443:367-369. [DOI] [PubMed] [Google Scholar]
- 67.Scholz, C., T. Schindler, K. Dolinski, J. Heitman, and F. X. Schmid. 1997. Cyclophilin active site mutants have native prolyl isomerase activity with a protein substrate. FEBS Lett. 414:69-73. [DOI] [PubMed] [Google Scholar]
- 68.Schreiber, S. L., and G. R. Crabtree. 1992. The mechanism of action of cyclosporin A and FK506. Immunol. Today 13:136-142. [DOI] [PubMed] [Google Scholar]
- 69.Sherman, F. 2002. Getting started with yeast. Methods Enzymol. 350:3-41. [DOI] [PubMed] [Google Scholar]
- 70.Sikorski, R. S., and P. Hieter. 1989. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122:19-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Siligardi, G., B. Panaretou, P. Meyer, S. Singh, D. N. Woolfson, P. W. Piper, L. H. Pearl, and C. Prodromou. 2002. Regulation of Hsp90 ATPase activity by the co-chaperone Cdc37p/p50cdc37. J. Biol. Chem. 277:20151-20159. [DOI] [PubMed] [Google Scholar]
- 72.Stadtman, E. R., G. N. Cohen, G. LeBras, and H. de Robichon-Szulmajster. 1961. Feed-back inhibition and repression of aspartokinase activity in Escherichia coli and Saccharomyces cerevisiae. J. Biol. Chem. 236:2033-2038. [Google Scholar]
- 73.Steinbach, W. J., W. A. Schell, J. R. Blankenship, C. Onyewu, J. Heitman, and J. R. Perfect. 2004. In vitro interactions between antifungals and immunosuppressants against Aspergillus fumigatus. Antimicrob. Agents Chemother. 48:1664-1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Takagi, H., M. Shichiri, M. Takemura, M. Mohri, and S. Nakamori. 2000. Saccharomyces cerevisiae Sigma 1278b has novel genes of the N-acetyltransferase gene superfamily required for l-proline analogue resistance. J. Bacteriol. 182:4249-4256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Takahashi, N., T. Hayano, and M. Suzuki. 1989. Peptidyl-prolyl cis-trans isomerase is the cyclosporin A-binding protein cyclophilin. Nature 337:473-475. [DOI] [PubMed] [Google Scholar]
- 76.Tesic, M., J. A. Marsh, S. B. Cullinan, and R. F. Gaber. 2003. Functional interactions between Hsp90 and the co-chaperones Cns1 and Cpr7 in Saccharomyces cerevisiae. J. Biol. Chem. 278:32692-32701. [DOI] [PubMed] [Google Scholar]
- 77.Timerman, A. P., G. Wiederrecht, A. Marcy, and S. Fleischer. 1995. Characterization of an exchange reaction between soluble FKBP-12 and the FKBP-ryanodine receptor complex. J. Biol. Chem. 270:2451-2459. [DOI] [PubMed] [Google Scholar]
- 78.Tropschug, M., I. B. Barthelmess, and W. Neupert. 1989. Sensitivity to cyclosporin A is mediated by cyclophilin in Neurospora crassa and Saccharomyces cerevisiae. Nature 342:953-955. [DOI] [PubMed] [Google Scholar]
- 79.Tudor, D. W., T. Lewis, and D. J. Robins. 1993. Synthesis of the trifluoroacetate salt of aspartic acid β-semialdehyde, an intermediate in the biosynthesis of L-lysine, L-threonine, and L-methionine. Synthesis 11:1061-1062. [Google Scholar]
- 80.Vickers, L. P., G. K. Ackers, and J. W. Ogilvie. 1978. Aspartokinase I-homoserine dehydrogenase I of Escherichia coli K12. Concentration-dependent dissociation to dimers in the presence of L-threonine. J. Biol. Chem. 253:2155-2160. [PubMed] [Google Scholar]
- 81.Wach, A., A. Brachat, R. Pohlmann, and P. Philippsen. 1994. New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae. Yeast 10:1793-1808. [DOI] [PubMed] [Google Scholar]
- 82.White, R. L., K. C. Smith, and A. C. DeMarco. 1994. Biosynthesis of 5-hydroxy-4-oxo-L-norvaline in Streptomyces akiyoshiensis. Can. J. Chem. 72:1645-1655. [Google Scholar]
- 83.Wiederrecht, G., L. Brizuela, K. Elliston, N. Sigal, and J. Siekierka. 1991. FKB1 encodes a nonessential FK 506-binding protein in Saccharomyces cerevisiae and contains regions suggesting homology to the cyclophilins. Proc. Natl. Acad. Sci. USA 88:1029-1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wu, X., A. Chang, M. Sudol, and S. D. Hanes. 2001. Genetic interactions between the ESS1 prolyl-isomerase and the RSP5 ubiquitin ligase reveal opposing effects on RNA polymerase II function. Curr. Genet. 40:234-242. [DOI] [PubMed] [Google Scholar]
- 85.Wu, X., A. Rossettini, and S. D. Hanes. 2003. The ESS1 prolyl isomerase and its suppressor BYE1 interact with RNA Pol II to inhibit transcription elongation in Saccharomyces cerevisiae. Genetics 165:1687-1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wu, X., C. B. Wilcox, G. Devasahayam, R. L. Hackett, M. Arevalo-Rodriguez, M. E. Cardenas, J. Heitman, and S. D. Hanes. 2000. The Ess1 prolyl isomerase is linked to chromatin remodeling complexes and the general transcription machinery. EMBO J. 19:3727-3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yamaguchi, H., K. Uchida, T. Hiratani, T. Nagate, N. Watanabe, and S. Omura. 1988. RI-331, a new antifungal antibiotic. Ann. N. Y. Acad. Sci. 544:188-190. [DOI] [PubMed] [Google Scholar]
- 88.Yamaki, H., M. Yamaguchi, T. Tsuruo, and H. Yamaguchi. 1992. Mechanism of action of an antifungal antibiotic, RI-331, (S) 2-amino-4-oxo-5-hydroxypentanoic acid; kinetics of inactivation of homoserine dehydrogenase from Saccharomyces cerevisiae. J. Antibiot. (Tokyo) 45:750-755. [DOI] [PubMed] [Google Scholar]