

Abstract
Helper T cell development and function must be tightly regulated to induce an appropriate immune response that eliminates specific pathogens yet prevents autoimmunity. Many approaches involving different model organisms have been utilized to understand the mechanisms controlling helper T cell development and function. However, studies using mouse models have proven to be highly informative due to the availability of genetic, cellular, and biochemical systems. One genetic approach in mice used by many labs involves retroviral transduction of primary helper T cells. This is a powerful approach due to its relative ease, making it accessible to almost any laboratory with basic skills in molecular biology and immunology. Therefore, multiple genes in wild type or mutant forms can readily be tested for function in helper T cells to understand their importance and mechanisms of action. We have optimized this approach and describe here the protocols for production of high titer retroviruses, isolation of primary murine helper T cells, and their transduction by retroviruses and differentiation toward the different helper subsets. Finally, the use of this approach is described in uncovering mechanisms utilized by microRNAs (miRNAs) to regulate pathways controlling helper T cell development and function.
Keywords: Immunology, Issue 117, Retroviral transduction, primary murine T cells, helper T cell isolation, helper T cell differentiation, flow cytometry, microRNAs
Introduction
The immune response must be highly regulated to eliminate infections but prevent attacks on self-tissue that lead to autoimmunity. Helper T cells play an essential role in regulating the immune response, and a great deal of effort has been undertaken to understand their development and function (illustrated in several recent reviews 1-3). However, many questions remain, and many approaches have been utilized to study the mechanisms controlling helper T cell development and function. These have ranged from the use of in vitro cell culture systems to whole animals. Cell culture systems, especially those using cell lines, offer the benefit of ease of use and the ability to generate large amount of material to do sophisticated biochemical analyses. However, they suffer from their limited ability to reproduce the actual conditions occurring in an immune response. In contrast, whole animal experiments offer the benefit of relevance, but they can suffer from difficulties in manipulation and the ability to perform precise controls in addition to their large costs and ethical implications. Nevertheless, the vast majority of helper T cells studies today still require the use of whole animal experiments involving primary T cells because of the inability of cell lines to duplicate the exact steps occurring in the whole animal. Therefore, it is essential to utilize cost effective approaches that are highly informative.
Genetics is one powerful tool to study helper T cell development and function, yet traditional methods involving gene knockouts or transgenes are time consuming and expensive so they are often out of reach of small labs. However, retroviral transduction offers a powerful, rapid and, cost effective genetic approach to study the mechanisms of specific gene products. Therefore, it is commonly used in papers studying helper T cell development and function.
We have optimized a procedure for retroviral transduction of helper T cells. It utilizes the pMIG (Murine stem cell virus-Internal ribosomal entry site-Green fluorescent protein) retroviral expression vector, in which the gene of interest can be cloned and thereby expressed from the retrovirus long terminal repeat (LTR) 4. In addition, downstream of the inserted gene of interest is an internal ribosome entry sequence (IRES) followed by the green fluorescent protein (GFP) gene so transduced cells can easily be followed by their expression of GFP. The vector was originally derived from the Murine Stem Cell Virus (MSCV) vectors, which contain mutations in repressor binding sites in the LTRs making them resistant to silencing and thus, giving high expression in many cell types including helper T cells 5,6. Production of high titer retrovirus requires a simple transient transfection protocol of human embryonic kidney (HEK) 293T cells with the MIG vector and a helper virus vector that expresses the retroviral GAG, Pol, and Env genes. For this the pCL-Eco helper virus vector 7 works well in producing high titer replication incompetent retroviruses.
Here these protocols for retroviral production and transduction of primary murine T cells are described in addition to some of our results using this approach to study miRNA regulation of gene expression controlling helper T cell differentiation. miRNAs are small RNAs of approximately 22 nucleotides in length that post-transcriptionally regulate gene expression by targeting homologous sequences in protein encoding messenger RNAs and suppressing translation and inducing message instability 8,9. miRNAs play critical roles in developmental gene regulation. They are essential in the earliest stages of development, as embryos that cannot produce miRNAs die at a very early stage 10. In addition miRNAs are important later on in the development of many tissues. They are thought to function by fine-tuning the expression of genes required for developmental programs 1. In helper T cells miRNAs play multiple roles and are required for regulatory T cell (Treg) development 11-14. We used retroviral transduction as a means to dissect the mechanisms of miRNA regulation of Treg differentiation 15. Through such studies important individual miRNAs were determined by retroviral-mediated overexpression. Subsequently, relevant genes regulated by these miRNAs were identified in order to understand the molecular pathways regulated by miRNAs in helper T cell differentiation.
Protocol
All mouse work performed in these protocols was undertaken according to the Animals Scientific Procedures Act, UK under the animal Project License 70/6965.
1. Retroviral Production
Prior to proceeding obtain all required approvals for producing genetically modified organisms and the use of retroviruses in mammalian cells.
- Growth of HEK 293T Cells
- Grow HEK 293T cells on 10 cm tissue culture dishes in HEK 293T medium (Dulbecco's Modified Eagle's Medium (DMEM) with 10% fetal bovine serum (FBS), 100 units/ml Penicillin 0.1 mg/ml Streptomycin, and 2 mM L-Glutamine). For general cell passage, split cells 1:10 when they reach confluence by removing medium and pipetting the cells off the plate with fresh HEK 293T medium. NOTE: Cells should reach confluence in 2-3 days. 293T HEK cells attach loosely to the plate so trypsinization is not required.
- 24 hr prior to transfection, split a confluent plate of cells 1:10 to a 10 cm plate (one plate per transfection) so that the next day the cells are ~50% confluent.
- Transfection by Calcium Phosphate Precipitation
- Approximately 1 hr prior to transfection, remove medium from cells and carefully add 9 ml of fresh HEK 293T medium to prevent cells detaching from the plate.
- Prepare 2x Hepes Buffered Saline (2x HBS) and a fresh 2.5 M CaCL2 solution as described in Table 1. Thaw DNA stocks. For the most efficient transfections use plasmid prep quality DNA.
- In a 6 ml round bottom or 15 ml conical tube add 5 µg of retroviral DNA (pMIG)4, 5 µg of Helper virus DNA (pCL-Eco)7, and H2O to 420 µl. Add 80 µl of 2.5 M CaCl2 bringing the final volume to 500 µl. Note: This along with the next step (addition of 2x HBS) can be done on a regular bench as long as general sterile technique is used.
- Slowly add 500 µl of 2x HBS drop by drop to the above DNA mixture while gently vortexing so that the solution is continuously mixed and the CaPO4 precipitates in even-sized crystals.
- Transfer to a tissue culture hood and slowly add the CaPO4 DNA mixture (1 ml) dropwise to the 9 ml of medium covering the cells while constantly and gently swirling the plate. Once again be careful not to detach cells from the plate. Place the cells back into the incubator. NOTE: At this point the CaPO4 precipitate will be readily visible under a microscope as small crystals about the size of bacteria. These are easily seen after 30-60 min when the crystals have had a chance to settle to the bottom of the plate.
- Collection of Virus in Culture Supernatants.
- The following day (anywhere from 12-24 hr after transfection) remove the medium and feed the cells with 10 ml of fresh HEK 293T medium once again being careful not to dislodge the cells from the plate. NOTE: This dilutes out lymphocyte inhibitory substances that the HEK 293T cells secrete into the medium during the transfection.
- In the evening of the second day (~24 hr post transfection) feed cells with 3.5 ml of fresh HEK 293T medium. Be careful that the plate is placed exactly level in the incubator so that one side is not left dry.
- Collect medium ~12 hr later (store at 4 °C) and feed with 3.5 ml of fresh HEK 293T medium. Repeat collection 2 more times at roughly 12 hr intervals so that about 10 ml of medium is collected. This is the virus stock.
- Spin out any residual HEK 293T cells and cellular debris by centrifuging at 600 x g for 5 min. Alternatively, filter using a 0.45 µm, low protein binding, syringe filter. Store virus at 4 °C, and for best titers, use within a day or two, as the titer will slowly diminish at 4 °C. Do not freeze, as this significantly decreases the titer.
2. Isolation of Primary Naïve CD4+ T Cells
- Isolation of Leukocyte Cells
- Euthanize mice by cervical dislocation, CO2 asphyxiation, or other humane protocol appropriate to the animal handling rules of the institution.
- Dissect freshly euthanized mice and remove spleen and desired lymph nodes 15. Place the organs in wells of a 6 well tissue culture plate containing R10 medium (RPMI medium with 10% heat inactivated FBS, 100 units/ml Penicillin, 0.1 mg/ml Streptomycin, 2 mM L-Glutamine, and 0.1% β-mercaptoethanol). Use a cell culture hood for dissection of mice and all subsequent manipulations to minimize chance of contamination in cultures. NOTE: Keep the R10 medium cold and perform all the downstream purification steps at 4 °C.
- Place a 70 µm cell culture strainer into a 50 ml conical tube containing approximately 5 ml of R10 medium. Use one strainer for the spleen and lymph nodes of up to three mice.
- Add spleen and lymph nodes to the cell strainer and macerate using the end of a 5 ml syringe plunger. Rinse with 1-2 ml of R10 medium.
- Transfer cells to a 15 ml conical tube and bring the volume up to 14 ml with R10. Centrifuge at 600 x g for 5 min at 4 °C. Discard supernatant by pouring it off with one clean movement to prevent disturbing the cell pellet.
- Add 2 ml of cold (4 °C) red cell lysis buffer (Table 1) to each tube. Gently mix for 3.5 min, keeping the tube on ice. NOTE: The timing of red cell lysis is critical to prevent death of lymphocytes. Therefore, the exact timing of each batch of Red cell lysis buffer will need to be optimized.
- Add ≈12-13 ml of R10 medium. Immediately centrifuge and discard supernatant as in step 2.1.5. Perform the washing steps 2x more with R10 medium to remove any residual red cell lysis buffer from the cells and avoid death of lymphocytes.
- Count cells in a hemocytometer diluting 1:20 in trypan blue to determine the yield of viable cells. Expect approximately 8-10 x 107 cells per mouse.
- Isolation of CD4+ T Cells Using Magnetic Beads Kit to Remove CD8+, CD11b+, CD16/32+, CD45R+, MHC class II+ and Ter-119+ Cells.
- Aliquot 8-10 x 107 cells from step 2.1.8 per 15 ml conical tube and centrifuge at 600 x g for 5 min at 4 °C. Pour off the supernatant with one clean movement to prevent disturbing the cell pellet. Resuspend the cells in each tube in 100 µl of heat inactivated FBS then add 100 µl of antibody mix from the kit. Incubate for 30 min at 4 °C with gentle mixing on a roller.
- While the above cells are incubating, prepare the beads.
- Resuspend beads in the vial by vortexing for >30 sec then transfer 1 ml of beads per 8-10 x 107 prepared cells to a 15 ml conical tube. Add 2 ml of R10 medium per ml of beads and gently mix.
- Place the tube in the magnet for 1 min then discard the supernatant. Remove tube from the magnet and resuspend beads in 4 ml of R10 medium. This amount of beads is sufficient for two rounds of incubation.
- Wash the cells at the end of the antibody incubation (Step 2.2.1) by adding 10 ml of R10 medium per tube. Gently mix well with swirling of the tube and centrifuge cells at 600 x g for 5 min at 4 °C. Pour off the supernatant with one clean movement to prevent disturbing the cell pellet. Resuspend cells in 1 ml of R10 medium.
- Add 2 ml of the washed beads from step 2.2.2.2 (½ the amount prepared for each tube) to each tube of antibody treated cells and incubate for 30 min at 4 °C with gentle mixing on a roller.
- Resuspend the cell-bead mixture by gently pipetting 5 times with a pipet containing a narrow tip opening. Avoid foaming. Place the tube in the beads magnet for 2 min then transfer the supernatant containing the negatively selected cells to a new tube. Keep these cells, as they are the CD4+ population.
- Repeat negative selection one more time. Spin cells from step 2.2.5 at 600 x g for 5 min at 4 °C. Pour off the supernatant as in step 2.2.3 and resuspend in 1 ml of R10 medium. Follow steps 2.2.4 and 2.2.5 again. After the final magnetic bead selection, count the cells to determine recovery (typical yields are 15-20 x 106 cells per 108 leukocytes).
- Isolation of Naïve CD4+ T Cells (CD25- and CD62Lhigh) Using Cell Separation Columns
- Centrifuge CD4+ T cells at 600 x g for 5 min at 4° C and resuspend in 150-200 µl of R10 medium per 108 cells. Add 2 µl of stock biotinylated CD25 monoclonal antibody (clone 7D4). Incubate for 30 min at 4° C with gentle mixing on a roller.
- Wash cells by adding 10 ml of cell separation column buffer (Table 1). Centrifuge at 600 x g for 5 min at 4 °C. Remove as much supernatant as possible and estimate the volume of buffer/cells remaining. Resuspend to a final volume of approximately 90 µl per 107 cells.
- Add 20 µl of Streptavidin beads per 107 cells and mix with gentle flicks. Incubate for 15 min at 4 °C.
- While cells are incubating, prepare the medium cell separation (MS) columns for the manual approach. Each MS column retains 107 cells, and since CD25+ cells typically represent about 10% of total CD4+ cells, use 1 column per 108 CD4+ T cells. Add 500 µl of cell separation column buffer to the column and let flow through. Repeat 2 more times.
- At end of cell/bead incubation, wash cells by adding 10 ml of cell separation column buffer and centrifuging at 600 x g for 5 min at 4 °C. Resuspend cells in 500 µl of cell separation column buffer per 108 cells, but still use 500 µl if there are fewer cells.
- Apply 500 µl of cell/bead suspension to the column and collect the flow through. Pass this over the column 1 more time and collect the flow through once again. Rinse the column 3 times with 500 µl of cell separation column buffer, adding each flow through from these rinses to the collected cells. These are the CD25- cells. Count cells to determine yield.
- If Tregs (CD25+ cells) are desired, isolate as follows.
- Remove the column from the separator and hold it above an open universal tube taking care to maintain sterility. Quickly pipette 500 µl of cell separation buffer onto the column and firmly flush out the positive fraction, using the plunger supplied with the column.
- Repeat the flushing procedure 2 more times. Pass the positive fraction over a fresh MS cell separation column and discard this flow-through. Firmly flush out the positive cells three times, as before and centrifuge cells at 600 x g for 5 min at 4° C. Resuspend cells in 1 ml of R10 medium and count to determine the yield.
- To obtain CD25- CD62Lhigh T cells, centrifuge the CD25- T cells isolated above in step 2.3.6 at 600 x g for 5 min at 4° and resuspend in 150-200 µl of R10 medium per 107 cells. Add 5 µl of stock biotinylated CD62L monoclonal antibody (clone MEL-14) and incubate for 30 min at 4° C with gentle mixing on a roller as before.
- Perform wash, Streptavidin bead binding, and cell separation column preparation as described for Treg isolation protocol (2.3.7). This time use the large cell separation (LS) column, which has a capacity of 108 cells.
- Since CD62Lhigh T cells typically represent about 60-70% of CD25- cells, use 1 LS cell separation column per 108 cells. Add cell/bead mixture to LS cell separation column as described in step 2.3.6 – this time discarding the final flow through and column rinses. Collect the CD62Lhigh T cells as described in step 2.3.7 for the CD25+ cell collection.
- Centrifuge cells at 600 x g for 5 min at 4° C. Resuspend cells in 1 ml of R10 and count to determine yield. Dilute to 0.75-1 X106 cells per ml in R10 medium.
- Analyze Purity of Cells by Fluorescent-activated Cell Sorting (FACS).
- FACS staining strategy
- For cells pre and post all stages of isolation, stain with CD4-FITC, CD8a-PerCP-Cy5.5 (helper and cytotoxic T cells), and MHCII-PE (MHC class II expressing cells).
- For cells pre and post CD25 isolation, stain with CD4-FITC and CD25-PE (Tregs versus other helper T cells).
- For cells pre and post CD62L isolation stain with CD4-FITC, CD62L-PE and CD44-APC (naïve versus memory and effector helper T cells).
- For each analysis place 105 cells in a well of a 96 well U-bottom plate. Centrifuge at 600 x g for 5 min at 4 °C. Pour off the medium and wash the cells once with 200 µl DPBS. Centrifuge as above and pour off DPBS.
- Add 50 µl of DPBS per well and 0.5 µl of each antibody (as indicated in 2.4.1) and incubate at RT for 30 min in the dark to avoid bleaching of fluorescent label.
- Wash cells 2x with DPBS as in 2.4.2 and immediately analyze on a flow cytometer using protocols and guidelines specific to the instrument at hand. NOTE: After the final isolation step of a good preparation, 90-95% of cells will be CD4+ CD25- CD62Lhigh T cells.
3. Retroviral Transduction of Activated CD4+ T Cells and their Differentiation into Specific T Helper Subsets
- Retroviral Transduction NOTE: Retroviral integration and expression of genes requires cell division. Therefore, T cells must be activated O/N.
- While isolating the naïve T cells, coat a 24 well tissue culture plate with anti-CD3 and anti-CD28 antibodies diluted in DPBS. Add 250 µl per well. Use anti anti-CD3 at 1µg/ml. Use anti-CD28 at 2 µg/ml if cells will ultimately be differentiated under Th0, Th1, Th2 and iTregs conditions. Use anti-CD28 at 10 µg/ml for Th9 and Th17 conditions. Incubate ~2 hr at 37 °C in a tissue culture incubator.
- Just prior to culturing the cells, remove the antibody solution and wash the plate with ~250 µl of DPBS per well to remove unbound antibody. Be careful not to let the plate dry out so process a maximum of 6 wells at a time. Remove DPBS wash and add 1 ml of naïve CD4+ T cells in R10 medium at 0.75-1 x 106 cells per ml. Activate cells O/N (14-16 hr).
- The following day, centrifuge cells in the plate at 900 x g for 5 min at 30° C to attach cells to the bottom of the wells.
- Collect and save the medium being careful not to displace the cells, and replace with 1 ml virus culture supernatant prepared from the virus production protocol. Do not let the cells dry out so process a maximum of 4 wells at a time.
- To each well add 1 µl of 8 mg/ml polybrene and 10 µl of 1M HEPES pH 7.5 to aid virus uptake and prevent significant alkalization in the ambient CO2 during the following centrifugation. Centrifuge cells in plate at 900 x g for 90 min at 30° C.
- Differentiation of Cells into Specific Subsets
- Carefully remove the virus culture supernatant and replace with 1 ml of the medium collected above in step 3.1.4, as it contains IL-2 and other T cell growth factors. Add reagents indicated in Table 2 and culture cells for 3-4 days to differentiate cells into specific subsets.
- Analysis of Cells
- For intracellular staining of cytokines, treat cells with 1 µg/ml each of Phorbol 12-myristate 13-acetate (PMA) and ionomycin for 4 hr. During the last 2 hr of stimulation, add 1 µg/ml of brefeldin A.
- Resuspend cells from the bottom of each well by pipetting up and down several times. Transfer approximately 105 cells to a 96 well U bottom plate (usually 100 µl from the 1 ml of culture of Th cells) for fixation and staining.
- For GFP staining, fix cells with 100 µl of 2% paraformaldehyde at RT for exactly 5 min, as a longer incubation time will bleach the GFP and interfere with Foxp3 staining. Wash cells by adding 100 µl of DPBS to each well and centrifuge at 600 x g at 22 °C for 5 min. Pour off the supernatant. Caution: Paraformaldehyde is toxic. Handle it in a fume hood with skin and eye protection.
- Fix the cells again using 100 µl 1x Fixation buffer made from the Foxp3 staining kit (1 volume of Fixation buffer to 3 volumes of diluent). Incubate the cells for 45 min at RT or alternatively incubate at 4 °C for 16 hr.
- After fixation, permeabilize cells by adding 100 µl of permeabilization buffer from the same kit. Incubate at RT for 30-45 min then centrifuge at 600 x g at 22 °C for 5 min.
- For antibody staining, remove fixation/permeabilization buffer and add 50 µl of fresh permeabilization buffer. Add the required antibody diluted in permeabilization buffer at 0.5 µl per 50 µl buffer per sample. Incubate for 30-45 min at RT in dark to avoid bleaching of fluorescent label.
- Add 150 µl permeabilization buffer and centrifuge at 600 x g at 22 °C for 5 min. Discard the permeabilization buffer and add 200 µl DPBS. Analyze on a flow cytometer using protocols and guidelines specific to the instrument at hand. NOTE: Be sure to include proper controls for cytokine staining such as staining with isotype control antibodies, analyzing non-activated or Th0 activated cells, etc.
Representative Results
The success of this experimental system requires highly pure populations of T cells and high titer retrovirus preparations. Representative results are shown here as examples of successful experiments. Figure 1 shows the typical purity of pre- and post-selected populations at each stage of the naïve helper T cell isolation protocol. Figure 2 and 3 illustrate the analysis of retrovirus production through GFP expression in the transfected HEK 293T cells (Figure 2) and transduced T cells (Figure 3). Transfection efficiencies of the HEK 293T cells can vary significantly with different retroviral constructs, but this often doesn't correlate with the level of retrovirus production observed with the number of GFP+ T cells. Also, the number of GFP+ T cells can vary depending on the polarization conditions. Furthermore, the mean expression level of GFP and the inserted gene can vary depending on the number of virus copies integrated, the effect of the integration site on transcription, and post-transcriptional regulatory mechanisms affecting the viral transcript.
Finally, Figure 4 shows some typical results we have observed with helper T cell differentiation when the miRNAs miR-15b/16 are overexpressed. These results show some of the variability that can occur within an individual experiment so true effects must be substantiated by statistical analysis of multiple repeat experiments using different preparations of helper T cells. In these experiments Th2 responses can be difficult to observe in the C57BL/6 line used here because they are prone to Th1 responses. Likewise, IL-9 staining can be difficult to detect above background. Therefore, it is imperative to do isotype controls and set up proper compensation to ensure correct gating of cytokine expression. In our results we have found that miR-15b/16 enhances iTreg induction by inhibiting the mTOR signaling pathway through suppressing the expression of the components Rictor and mTOR 15. miR-15b/16 can sometimes influence Th0, Th1, and Th17 differentiation in individual experiments, but there is no significant effect when examined in multiple repeat experiments. In contrast miR-15b/16 overexpression does significantly suppress Th9 differentiation (see reference 18).
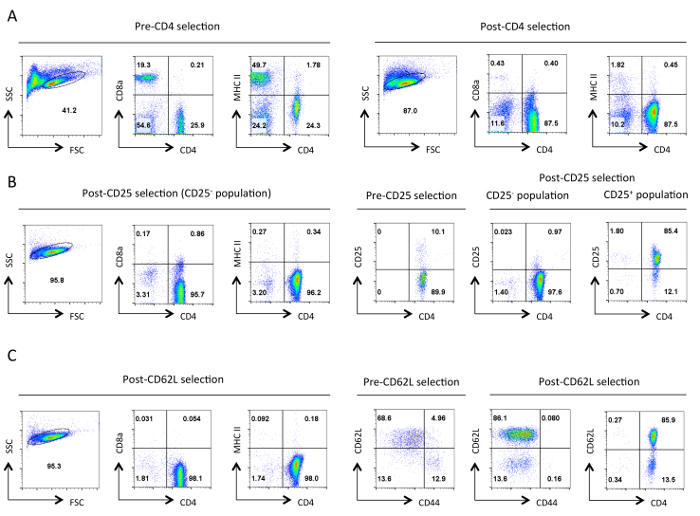 Figure 1. Typical purity of helper T cells at each stage of isolation. Representative flow cytometry results of the indicated antigens are shown from the gate of live cells designated in the Forward Scatter (FSC) and Side Scatter (SSC) plots. (A) Pre- and post-CD4 negative selection. Expression profiles of CD4, CD8a, and MHCII are shown. These illustrate the enrichment of helper T cells and the loss of cytotoxic T cells and MHC class II expressing cells. A good purification should result in ~90% CD4+ T cells at this stage. (B) CD25 selection. On the left are the expression profiles of CD4, CD8a, and MHCII, and on the right are the CD4 and CD25 expression profiles pre and post selection. At this point >95% of CD25 negatively selected cells should be CD4+ CD25-. (C) CD62L selection. CD4, CD8a, and MHCII expression profiles are shown on the left. On the right the expression profiles for CD62L and CD44 are shown for pre- and post-CD62L selected cells along with CD4 and CD62L expression profile of post selected cells. After CD62L selection virtually all memory cells (CD44+) are removed leaving a highly enriched population of naïve helper T cells that contains 10-15% effector cells (CD62Llow). For all FACS profiles, equivalent settings and scales for a specific parameter were maintained throughout. Numbers represent the percentage of cells within a gated population. The slight decrease in size of the cells after the initial selection is presumably due to mechanical stress during the protocol. Please click here to view a larger version of this figure.
Figure 1. Typical purity of helper T cells at each stage of isolation. Representative flow cytometry results of the indicated antigens are shown from the gate of live cells designated in the Forward Scatter (FSC) and Side Scatter (SSC) plots. (A) Pre- and post-CD4 negative selection. Expression profiles of CD4, CD8a, and MHCII are shown. These illustrate the enrichment of helper T cells and the loss of cytotoxic T cells and MHC class II expressing cells. A good purification should result in ~90% CD4+ T cells at this stage. (B) CD25 selection. On the left are the expression profiles of CD4, CD8a, and MHCII, and on the right are the CD4 and CD25 expression profiles pre and post selection. At this point >95% of CD25 negatively selected cells should be CD4+ CD25-. (C) CD62L selection. CD4, CD8a, and MHCII expression profiles are shown on the left. On the right the expression profiles for CD62L and CD44 are shown for pre- and post-CD62L selected cells along with CD4 and CD62L expression profile of post selected cells. After CD62L selection virtually all memory cells (CD44+) are removed leaving a highly enriched population of naïve helper T cells that contains 10-15% effector cells (CD62Llow). For all FACS profiles, equivalent settings and scales for a specific parameter were maintained throughout. Numbers represent the percentage of cells within a gated population. The slight decrease in size of the cells after the initial selection is presumably due to mechanical stress during the protocol. Please click here to view a larger version of this figure.
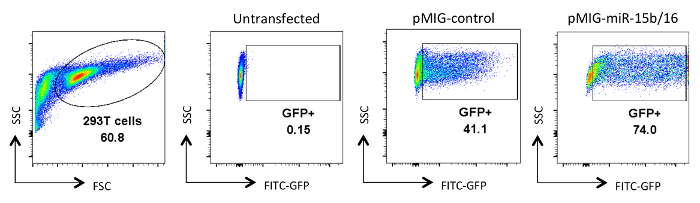 Figure 2. Analysis of retrovirus-transfected HEK 293T cells. GFP expression is shown in HEK 293T cells that were either untransfected or transfected and analyzed after collection of viral culture supernatants. GFP analysis was done on live cells from the gate on the FSC and SSC plot in the first panel. Numbers represent the percentage of GFP+ cells within the gated region. Typical transfection efficiencies range between 30-90%. Please click here to view a larger version of this figure.
Figure 2. Analysis of retrovirus-transfected HEK 293T cells. GFP expression is shown in HEK 293T cells that were either untransfected or transfected and analyzed after collection of viral culture supernatants. GFP analysis was done on live cells from the gate on the FSC and SSC plot in the first panel. Numbers represent the percentage of GFP+ cells within the gated region. Typical transfection efficiencies range between 30-90%. Please click here to view a larger version of this figure.
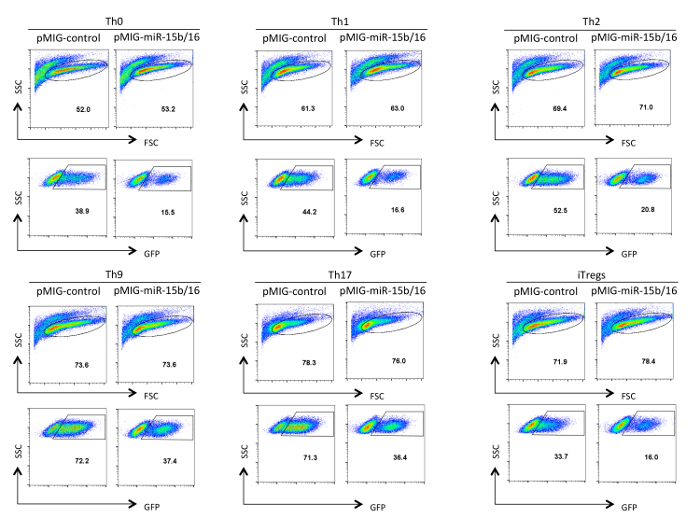 Figure 3. Analysis of retrovirus transduced helper T cells. GFP expression is shown in retroviral-transduced helper T cells after differentiation in Th0, Th1, Th2, Th9, Th17, and Treg polarization conditions for three days. Analysis was gated on live and activated cells indicated in the FSC/SSC panel. Transduction efficiencies can vary between 10-75% depending on the construct and the polarization conditions. Likewise, the mean fluorescent intensity of GFP expression can vary. Please click here to view a larger version of this figure.
Figure 3. Analysis of retrovirus transduced helper T cells. GFP expression is shown in retroviral-transduced helper T cells after differentiation in Th0, Th1, Th2, Th9, Th17, and Treg polarization conditions for three days. Analysis was gated on live and activated cells indicated in the FSC/SSC panel. Transduction efficiencies can vary between 10-75% depending on the construct and the polarization conditions. Likewise, the mean fluorescent intensity of GFP expression can vary. Please click here to view a larger version of this figure.
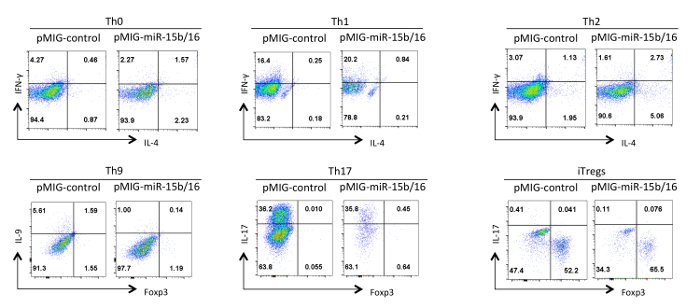 Figure 4. Effect of miR-15b/16 overexpression on helper T cell differentiation in different polarization conditions. Representative cytokine profiles are shown on the GFP+ population of cells from Figure 3. Please click here to view a larger version of this figure.
Figure 4. Effect of miR-15b/16 overexpression on helper T cell differentiation in different polarization conditions. Representative cytokine profiles are shown on the GFP+ population of cells from Figure 3. Please click here to view a larger version of this figure.
 Table 1:
Buffers used in these protocols.
Please click here to download this table as an Excel spreadsheet.
Table 1:
Buffers used in these protocols.
Please click here to download this table as an Excel spreadsheet.
| Helper T cell polarization conditions | ||
| Th0 | anti-IL-4 | 5 µg/ml |
| anti-IFN-γ | 5 µg/ml | |
| Th1 | recombinant-IL-12 | 20 ng/ml |
| anti-IL-4 | 5 µg/ml | |
| Th2 | recombinant IL-4 | 40 ng/ml |
| anti-IFN-γ | 5 µg/ml | |
| Th9 | recombinant TGF-β | 2.5 ng/ml |
| recombinant IL-4 | 40 ng/ml | |
| anti-IFN-γ | 10 µg/ml | |
| Th17 | recombinant TGF-β | 2.5 ng/ml |
| recombinant IL-6 | 50 ng/ml | |
| anti-IFN-γ | 5 µg/ml | |
| anti-IL-4 | 5 µg/ml | |
| anti-IL-2 | 5 µg/ml | |
| Tregs | recombinant TGF-β | 2.5 ng/ml |
| recombinant IL-2 | 5 ng/ml |
Table 2: Helper T cell subset polarization conditions.
Discussion
Retroviral mediated overexpression of genes is a powerful way to analyze function in helper T cells, as their development and function is often determined by the expression level of key regulators. However, cautious interpretation of the results is required because expression levels significantly above those of the endogenous gene can introduce many artifacts. Therefore, this technique should be combined with others to verify the relevance of function. For example, overexpression should be complemented by reduced expression using siRNAs or gene knockouts if available. With miRNAs, we complemented overexpression experiments with those of blocking by using viruses that overexpressed artificial miRNA targeting sites that acted as competitive inhibitors for a miRNA 15. Retroviral transduced cells can also be utilized in biochemical assays involving RNA and protein analysis. However, a major limitation of these experiments is efficiency of transduction resulting in a mixed population of transduced and untransduced cells. Therefore, these assays will most likely require sorting of the GFP+ population. Finally, in vitro differentiation assays should be combined with in vivo experiments, and one way this can be achieved is by adoptively transferring the transduced T cells into mice and following their differentiation and their effect on the immune response.
One of the key limitations to this system is the size of the RNA genome that can be packaged into the retroviral capsid. In our experience, the maximum insert size for MIG retroviral system that gives good virus production is 3-3.5 kb. Therefore, larger genes cannot be analyzed with this system, as they give poor virus titers. However, most genes are smaller than this size so this system is useful for a wide variety of gene studies.
With retroviral transduction, several alternatives within these protocols have been used. Many researchers have utilized packaging cell lines that stably express the retroviral genes (for example reference 16). However, we have obtained the highest titers using standard HEK 293T cells with co-transfection of the pCL-Eco helper virus vector. Isolation of naïve helper T cells can also be achieved through cell sorting rather than the magnetic bead and cell separation column protocol, but this requires access to a cell sorter, and the costs for sort time are typically higher than the bead reagents. Finally, there are variations on activation conditions used to differentiate helper T cells into the different subsets. For example, TCR stimulation of cells for too long before exposure to Treg inducing conditions can inhibit their induction 16. This can be a problem because retroviral expression requires cell division induced by stimulation of cells. Nevertheless, we have found efficient Treg induction using this protocol with O/N activation prior to retroviral transduction.
Within these protocols, successful application requires several factors. High titer retrovirus preparations need efficient transfection of the HEK 293T cells so high quality DNA and precisely prepared 2x HBS are important. In addition, the cell density of the HEK 293T cells needs to be roughly 50% at point of transfection because good expression of the transfected DNA requires that the cells are actively growing, and this will be inhibited if the cells are too sparse or dense. Cells at the optimal density during transfection should reach confluence at some point during the virus collection steps, but they will continue producing high titer virus stocks all the way through to the last collection. Efficient differentiation of the helper T cells requires good cell quality so ensure that isolated cells are to the purity illustrated in Figure 1. Likewise, the quality of the cells is dependent on the mice from which they were isolated. For these studies, we have used 6-8 week old C57BL/6 mice. Older mice can have less naïve cells, and other strains may differ in their differentiation. For example, BALB/c mice are more prone to Th2 responses than C57BL/6 mice 17 so as stated above, C57BL/6 T cells can be difficult to induce a Th2 response. In addition, any of the differentiation conditions may vary slightly from lab to lab, and the effect of gene overexpression may only become apparent in sub-optimal conditions so cytokine concentrations in the various polarization conditions may need to be titrated. Finally, effects of the overexpressed gene or the polarization conditions on cell proliferation can influence the transduction efficiency so measuring effects of the gene of interest may require optimizing the timing and concentration of polarizing reagents. Optimizing all these factors should lead to informative results with this system.
Disclosures
The authors have no conflicting interest in the publication of this work.
Acknowledgments
This work was supported by a Biotechnology and Biological Sciences Research Council (BBSRC) grant (BB/H018573/1) and a BD Biosciences grant.
References
- Baumjohann D, Ansel KM. MicroRNA-mediated regulation of T helper cell differentiation and plasticity. Nat Rev Immunol. 2013;13(9):666–678. doi: 10.1038/nri3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson CB, Rowell E, Sekimata M. Epigenetic control of T-helper-cell differentiation. Nat Rev Immunol. 2009;9(2):91–105. doi: 10.1038/nri2487. [DOI] [PubMed] [Google Scholar]
- Pearce EL, Poffenberger MC, Chang CH, Jones RG. Fueling immunity: insights into metabolism and lymphocyte function. Science. 2013;342(6155):1242454. doi: 10.1126/science.1242454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry SR, Biniszkiewicz D, Van Parijs L, Baltimore D, Jaenisch R. Retroviral Expression in Embryonic Stem Cells and Hematopoietic Stem Cells. Mol and Cell Biol. 2000;20(20):7419–7426. doi: 10.1128/mcb.20.20.7419-7426.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller AD, Rosman GJ. Improved retroviral vectors for gene transfer and expression. BioTechniques. 1989;7(9):980–990. [PMC free article] [PubMed] [Google Scholar]
- Grez M, Akgün E, Hilberg F, Ostertag W. Embryonic stem cell virus, a recombinant murine retrovirus with expression in embryonic stem cells. Proc Nat Acad Sci USA. 1990;87(23):9202–9206. doi: 10.1073/pnas.87.23.9202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naviaux RK, Costanzi E, Haas M, Verma IM. The pCL vector system: rapid production of helper-free, high-titer, recombinant retroviruses. J Virol. 1996;70(8):5701–5705. doi: 10.1128/jvi.70.8.5701-5705.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipowicz W, Bhattacharyya SN, Sonenberg N. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat Rev Genetics. 2008;9(2):102–114. doi: 10.1038/nrg2290. [DOI] [PubMed] [Google Scholar]
- He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genetics. 2004;5(7):522–531. doi: 10.1038/nrg1379. [DOI] [PubMed] [Google Scholar]
- Bernstein E, Kim SY, et al. Dicer is essential for mouse development. Nat Genetics. 2003;35(3):215–217. doi: 10.1038/ng1253. [DOI] [PubMed] [Google Scholar]
- Cobb BS, Hertweck A, et al. A role for Dicer in immune regulation. J Exp Med. 2006;203(11):2519–2527. doi: 10.1084/jem.20061692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong MMW, Rasmussen JP, Rudensky AY, Rundensky AY, Littman DR. The RNAseIII enzyme Drosha is critical in T cells for preventing lethal inflammatory disease. J Exp Med. 2008;205(9):2005–2017. doi: 10.1084/jem.20081219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liston A, Lu LF, O'Carroll D, Tarakhovsky A, Rudensky AY. Dicer-dependent microRNA pathway safeguards regulatory T cell function. J Exp Med. 2008;205(9):1993–2004. doi: 10.1084/jem.20081062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Jeker LT, et al. Selective miRNA disruption in T reg cells leads to uncontrolled autoimmunity. J Exp Med. 2008;205(9):1983–1991. doi: 10.1084/jem.20080707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh Y, Garden OA, Lang F, Cobb BS. MicroRNA-15b/16 Enhances the Induction of Regulatory T Cells by Regulating the Expression of Rictor and mTOR. J Immunol. 2015;195(12):5667–5677. doi: 10.4049/jimmunol.1401875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer S, Bruno L, et al. T cell receptor signaling controls Foxp3 expression via PI3K, Akt, and mTOR. Proc Nat Acad Sci USA. 2008;105(22):7797–7802. doi: 10.1073/pnas.0800928105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagi J, Arimura Y, Takatori H, Nakajima H, Iwamoto I, Uchiyama T. Genetic background influences Th cell differentiation by controlling the capacity for IL-2-induced IL-4 production by naive CD4+ T cells. Int Immunol. 2006;18(12):1681–1690. doi: 10.1093/intimm/dxl102. [DOI] [PubMed] [Google Scholar]
- Singh Y, Garden OA, Lang F, Cobb BS. MicroRNAs regulate T-cell production of interleukin-9 and identify hypoxia-inducible factor-2a as an important regulator of T helper 9 and regulatory T-cell differentiation. Immunology. 2016;149:74–86. doi: 10.1111/imm.12631. [DOI] [PMC free article] [PubMed] [Google Scholar]