

Abstract
Cancer cell vascular invasion and extravasation is a hallmark of metastatic progression. Traditional in vitro models of cancer cell invasion of endothelia typically lack the fluid dynamics that invading cells are otherwise exposed to in vivo. However, in vivo systems such as mouse models, though more physiologically relevant, require longer experimental timescales and present unique challenges associated with monitoring and data analysis. Here we describe a zebrafish assay that seeks to bridge this technical gap by allowing for the rapid assessment of cancer cell vascular invasion and extravasation. The approach involves injecting fluorescent cancer cells into the precardiac sinus of transparent 2-day old zebrafish embryos whose vasculature is marked by a contrasting fluorescent reporter. Following injection, the cancer cells must survive in circulation and subsequently extravasate from vessels into tissues in the caudal region of the embryo. Extravasated cancer cells are efficiently identified and scored in live embryos via fluorescence imaging at a fixed timepoint. This technique can be modified to study intravasation and/or competition amongst a heterogeneous mixture of cancer cells by changing the injection site to the yolk sac. Together, these methods can evaluate a hallmark behavior of cancer cells and help uncover mechanisms indicative of malignant progression to the metastatic phenotype.
Keywords: Cancer Research, Issue 117, Cancer, Extravasation, Zebrafish, Embryo, Fluorescence, Vascular Invasion, Metastasis
Introduction
Metastatic disease is a major cause of cancer mortality and many mechanisms that enable cancer cell dissemination remain to be discovered1. In order for a cancer cell to successfully metastasize, it must first invade through the stroma that surrounds a primary tumor, enter (intravasate) into the circulatory system, survive in transit, exit (extravasate) from the circulation, and lastly establish a viable colony at the distant organ site2. Intravasation and extravasation are thus crucial steps in the metastatic cascade, yet every cancer cell is not inherently adept at disrupting and migrating through endothelial junctions3. In fact, there are a series of unique selection pressures that surround cancer cell vascular invasion and the process can be further influenced by a variety of endogenous and exogenous factors4. For these reasons, techniques that probe the aggressive behavior of advanced stage cancer often focus on vascular invasive ability as a means to predict metastatic spread.
Various model systems exist to facilitate the study of cancer cell vascular invasion in vitro. The most used in vitro assays involve either transwell systems to assess cancer cell migration through an endothelial barrier5 or Electric Cell-Substrate Impedance Sensing (ECIS) technology to monitor the real-time disruption of an intact endothelial monolayer by cancer cells6. These assays typically lack the fluid dynamics and stromal factors that would otherwise impact cancer cell attachment to an endothelial wall. This issue is somewhat circumvented by perfusable vascular networks that arise from the 3D culture of endothelia with supporting stromal cells, and these 3D microfluidic systems now represent the forefront of current in vitro options7,8. Still, these approaches omit the robust microenvironment of a functional circulatory system and therefore only in part substitute for in vivo models.
The most widely used in vivo model of vascular invasion is the mouse, in which experimental metastasis assays are commonly performed because they occur on relatively short timescales and are generally indicative of metastatic ability9. These assays involve direct injection of cancer cells into circulation and therefore model the end stages of metastasis, namely extravasation and cancer cell colonization of organs. The experimental metastasis assays differ based on the site of cancer cell injection and the organs ultimately analyzed. In the first assay type, cancer cells are injected into the tail vein of mice and cancer cell seeding in the lungs is monitored10,11. The second assay involves performing intracardiac injections to direct metastatic seeding toward the bone microenvironment12-14, but also the brain15. In the third assay, cancer cells are injected into the spleen in order to permit colonization of the liver16 whereas the fourth delivery route into the carotid artery carries cancer cells to the brain17,18. Irrespective of the cancer cell delivery method, organ colonization is the accepted experimental endpoint and is generally determined via luminescence, histology, or PCR-based techniques. Despite the physiologic advantages of conducting experimental metastasis assays within a murine host, these experiments still require weeks to months to complete and analyze.
The zebrafish (Danio rerio) model has recently emerged as a new system to study cancer progression19,20, and allows for the assessment of cancer cell vascular invasion within a functional circulatory system over a much shorter timescale when compared with mice21-24. The method utilizes a transparent zebrafish strain that has its endothelia tagged with a green reef coral fluorescent protein reporter driven by the kdrl promoter, the zebrafish receptor for vascular endothelial growth factor25. In the assay, cancer cells are labeled with a red fluorescent marker and injected into the precardiac sinus of 2-day old embryos. Anywhere between 48 to 96 hr after the injection, cancer cells that have invaded out of the vasculature and into the caudal region of embryos can be scored efficiently on a fluorescent microscope. Here we apply the technique to a panel of commonly used human breast cancer cell lines to demonstrate stark differences in their vascular invasive ability. Furthermore, we demonstrate that changing the injection site to the embryo yolk sac allows for the study of heterogeneous cell interactions, as cancer cell populations can be differentially labeled with fluorescent dyes and injected into zebrafish embryos lacking fluorescent vasculature. In this latter assay, cancer cells that have invaded the yolk and intravasated into the vasculature are scored in the caudal region 24 to 48 hr after injection. Due to the effectiveness and convenience of this model, zebrafish are increasingly employed to rapidly test the vascular invasive ability of cancer cells under a physiologic setting.
Protocol
Ethics Statement: Zebrafish embryos were generated according to an approved IACUC protocol. These experiments were carried out in compliance with recommendations by the Georgetown University Animal Care and Use Committee.
1. Organize Embryos for Injection and Create Stock Solutions
- Generate requisite zebrafish larvae to assess cancer cell vascular invasion.
- Set up pair-wise or group in-cross mating with Tg(kdrl:grcfp)zn1;mitfab692;ednrb1b140 fish. NOTE: We generated Tg(kdrl:grcfp)zn1;mitfab692;ednrb1b140 zebrafish, by crossing Tg(kdrl:grcfp)zn125, which express green reef coral fluorescent protein in endothelial cells, with a line that lacks pigment cells, mitfab692;ednrb1b140, developed at the Zebrafish International Resource Center.
- Collect eggs, clean, and remove unfertilized or deformed embryos the next day22.
- Incubate embryos at 28.5 °C until ready for injection with cancer cells, to occur when the zebrafish embryos are 2-days post-fertilization (2 dpf).
- Make injection plates.
- Melt 25 ml of 1.5% agarose in dH2O for each plate.
- Pour 12 ml of the agarose into a 100 mm x 15 mm petri dish and let it harden.
- Re-melt then pour the remaining agarose in the plate.
- Immediately position a cut glass mold (3 mm x 7.2 cm wide x 7.5 cm long) so that it is at a 30 degree angle to the agarose and positioned in the center of the plate. NOTE: This will create a steep 60° wall and a 30° sloped ramp.
- Tape the glass mold in place and let the agarose harden.
- Gently remove the glass mold and be careful not to tear the agarose. NOTE: Molds can be stored in dH2O at 4 °C.
- Equilibrate injection plate with fish water (0.3 g/L sea salt).
- Rinse plate twice with distilled water.
- Equilibrate plate by adding 10 ml of fish water to the plate and place on shaker for 10 min.
- Equilibrate plate a second time with fish water.
Split 2-day post-fertilization (dpf) embryos into injection groups by transferring them into dishes containing fish water22.
Prepare recovery dishes for each group to utilize after injection. Ensure that the recovery dish contains 10 ml fish water, plus penicillin (25 µg/ml) and streptomycin (50 µg/ml).
Prepare 2x tricaine solution by adding 4 ml of buffered tricaine stock (4 mg/ml,10 mM Tris, pH 7) to 50 ml of fish water plus penicillin and streptomycin.
Dissolve 15 mg low melting-point agarose in 10 ml of 2x tricaine solution to generate a mounting anesthetic medium that will immobilize live embryos for imaging. NOTE: Mounting medium consists of 1.5 % agarose.
- Pull microinjection needles.
- Place glass capillary tubing in a vertical pipette puller. Pull long tapered pipettes using 20 mAmp current and a 2-coil heating element.
2. Labeling Cancer Cells with Lipophilic Fluorescent Dye
Maintain cancer cells in their recommended culture conditions. NOTE: These lines were maintained in DMEM + 10% FBS: BT-474, MCF-7, MDA-MB-231, MDA-MB-468, and SK-BR-3. These lines were maintained in RPMI + 10% FBS: HCC 1806 and T-47D. All cell lines were maintained at 37 oC and 5% CO2 during incubation.
- Generate a single-cell suspension by dissociating an adherent culture of cancer cells.
- Wash cells first with PBS and then treat with 0.05% trypsin-EDTA solution. NOTE: Trypsin exposure time will depend on the cell line.
- Neutralize the trypsin solution with serum-containing cell culture media after the cells detach.
Centrifuge the trypsin-neutralized cell suspension for 5 min at 200 x g, then resuspend the cell pellet in fresh culture media for cell counting.
- Count the cell suspension using an automated counter and prepare 1 million cells in 200 µl of cell culture media.
- Verify cell viability with trypan blue dye exclusion before injection into zebrafish embryos. NOTE: Only viable cell populations should be injected into zebrafish embryos, as injection of dead cells will not reflect true vascular invasion.
Add 2 µl of red lipophilic dye to the cancer cell suspension for a 1:100 dilution, mix well, and then incubate the mixture at 37 °C for 20 min. NOTE: Concentration of the dye and labeling time may need to be optimized for each cell line.
Following the incubation, add 1 ml of fresh media to the tube and then centrifuge for 5 min at 200 x g.
- Wash away residual fluorescent dye from the cancer cells.
- Aspirate the supernatant from the cell pellet, resuspend the pellet in 1 ml of fresh culture media, and centrifuge for 5 min at 200 x g.
- Repeat the washing step a second time: aspirate the supernatant, resuspend the cell pellet in 1 ml of fresh media and then centrifuge again for 5 min at 200 x g.
- Repeat the washing step third time: aspirate the supernatant, resuspend the cell pellet in 1 ml of fresh media and then centrifuge again for 5 min at 200 x g.
Aspirate the supernatant and resuspend the cell pellet containing 1 million labeled cancer cells in 500 µl of fresh media. NOTE: 0.5 mM EDTA can be added to the media to prevent cell clumping.
3. Injecting Cancer Cells into the Pre-cardiac Sinus of Zebrafish Embryos
- Attach microinjection dispense system to a pressurized air source and turn on the microinjection dispense system power source.
- Test pressure by depressing the foot pedal. A brief pulse of air should emit from the needle holder.
- Equilibrate the injection plates twice with the 2x tricaine solution.
- For each equilibration step, add 20 ml of 2x tricaine solution to the injection plate and place on shaker for 10 min.
Use plastic pipette to transfer a group of embryos to a small dish containing the 2x tricaine solution.
- Backfill the microinjection injection needle with cancer cells using a gel-loading pipet tip.
- Place the needle in an electrode storage jar with the pointed end facing down so cells settle near the tip.
- Transfer 20 - 30 anesthetized embryos to an injection plate by collecting the embryos with plenty of 2x tricaine in a plastic pipette.
- Allow embryos to settle in the tip of the pipette.
- Gently expel embryos into the trough of the injection plate, spreading the embryos along the length of the trough.
- Align embryos with heads facing up and bellies facing the steep wall of the trough. NOTE: Tricaine solution should cover both the trough length and the flat agarose surface, with embryos only residing in the trough. Embryos are now ready for injection.
- Inject 50 - 100 cancer cells (2 - 5 nl) into the precardiac sinus of the zebrafish embryos using the microinjection dispense system.
- Attach the needle to the needle holder of a micromanipulator.
- Position the injection plate under the stereoscope with the 60 degree wall to the left and focus on the top embryo at 25x magnification.
- Position the micromanipulator so that, when extended, the needle will pierce the embryo.
- Extend the needle by eye until it is nearly touching the embryo.
- Looking under the microscope, align the needle so that it will pierce the embryo upon further extension.
- Pierce the embryo through the yolk sac placing the tip just at, but not in, the pre-cardiac sinus.
- Inject cells by depressing the foot pedal. The force of the injection expels the cells into the cardiac sinus. Retract the needle.
- Using the right hand, extend and retract the injection needle. With the left hand, make fine adjustments to position next embryo.
- Return the needle to the electrode storage jar while setting up to inject another plate.
- Transfer the embryos to the recovery dish once the entire plate is injected.
- Tilt the injection plate to pool the embryos at the bottom, washing any remaining embryos out of the trough, and collecting them with the plastic pipette.
- Allow the embryos to settle in the bottom of the pipette.
- Transfer the embryos to the recovery dish in a minimal volume of tricaine.
- Incubate recovery dish at 28 °C for 1 hr.
- Separate viable zebrafish embryos from dead embryos and other debris.
Incubate dish at 33 °C until ready for scoring, typically 24 - 96 hr. NOTE: This temperature is determined as a compromise between 37 °C, the ideal temperature for cancer cells, and 28.5 °C, the ideal temperature for zebrafish.
4. Scoring Extravasation
Anesthetize the batch of embryos to be scored by placing them in a dish with tricaine solution.
- Place an anesthetized larvae on a depression microscopy slide in a drop of tricaine.
- Orient larvae laterally for optimal imaging of the caudal region.
- Count the number of cancer cells that have successfully invaded out of the vasculature by focusing up and down through the tail region to clearly discern intact cells. NOTE: It is best to have at least two individuals involved in this process, where the individual scoring the fish is blind to the experimental condition being assessed.
- Score larvae on a compound fluorescence microscope with the 10x objective lens. Use the 20x objective for any difficult calls.
5. Mounting Embryos onto Slides and Subsequent Fluorescence Imaging
Melt 1.5% agarose/tricaine solution and bring to 37 °C.
Anesthetize the embryo to be imaged by placing it in tricaine solution.
Transfer the embryo in a drop of tricaine solution to the imaging surface. Optionally use a glass-bottom dish or microscope slide.
Use a glass pipette to remove the excess tricaine solution, retaining the embryo on the imaging surface.
Overlay one drop of melted agarose solution over the embryo.
Quickly, before the agarose polymerizes, use a delicate tool, like an eyelash brush, to orient the embryo laterally for imaging, giving extra care to ensure the embryo is flattened along the imaging surface.
Submerge the now polymerized agarose drop under tricaine solution.
Subject the live zebrafish embryo to microscopic imaging.
6. Modification: Injecting Cancer Cells Into the Yolk Sac of Zebrafish Embryos
Prepare the microinjection dispense system and injection plates as previously described in section (3) of this protocol.
Label two cell populations with contrasting fluorescent dyes as previously described in section (2) of this protocol.
Inject 5 - 10 nl of 2 x 10^7 cells/ml into the yolk sac. Keep the injection volume constant to inject identical cell numbers (100 - 200 cancer cells) from each cell population NOTE: One may use transparent zebrafish embryos lacking fluorescent vasculature for this assay. Passive entry of particles into the vasculature can be controlled for by injecting fluorescent beads (< 10 µm) or, alternatively, a cell line that does not intravsate.
- Recover the injected embryos as previously described in section (3) of this protocol and then screen for successful injections.
- Use a stereoscope to screen and transfer viable embryos that were successfully injected to a new dish. NOTE: All embryos should have a consistently sized mass of cells located in the yolk. Embryos are discarded if the mass size differs or if any cells are located outside of the yolk.
- Transfer the viable embryos to a new dish if cancer cells are clearly seen in the yolk sac.
Incubate dish at 33 °C until ready for scoring, typically 24 - 48 hr. NOTE: This temperature is determined as a compromise between 37 °C, the ideal temperature for cancer cells, and 28.5 °C, the ideal temperature for zebrafish.
To score intravasation, follow the guidelines described in section (4) of this protocol, but instead count the number of cancer cells that have successfully invaded into the vasculature of the caudal region.
Representative Results
Here we tested the vascular invasive ability of commonly used breast cancer cell lines in a zebrafish embryo model (Figure 1). Rigorous criteria were employed in scoring extravasation for these different cell lines, where positive events were only counted if the cancer cells had clearly extravasated, this being done chiefly to limit any false-positives that could arise from scoring cellular debris.
Our analysis revealed stark differences amongst the lines when invasion was assessed 96 h after injection into the precardiac sinus (Table 1). Of the 7 cell lines tested, zebrafish embryos injected with the MDA-MB-231 cell line exhibited the greatest number of extravasated cancer cells per embryo, a finding that is consistent with the accepted metastatic nature of the line (Figure 2). We also found that BT-474 cells readily invaded into the caudal region of the embryos, while other cell lines, such as MCF-7, SK-BR-3, and T-47D cells, were minimally invasive (Figure 3). A curious discovery was that roughly half of the zebrafish embryos injected with the MDA-MB-468 cells had edema in the cardial region and these were not scored. As a consequence, a number of embryos injected with MDA-MB-468 cells were discarded before we found enough viable specimens that could be quantitated for extravasation. Despite these ostensible differences, it is notable that extravasation occurred with each cancer cell line tested.
As a modification to the precardiac sinus assay, we performed a competition experiment whereby two populations of MDA-MB-231 cells that differ in their vascular invasive ability26 were simultaneously injected into the yolk sac of developing embryos. MDA-MB-231 cells propagated under confluent culture conditions prior to injection exhibited a reduction in intravasation into the circulation and therefore had a reduced presence in the caudal region of the embryos upon scoring (Figure 4).
| Cell Line | Avg. # Extravasated Cells per Embryo | Range |
| MDA-MB-231 (Subconfluent) | 4 | 0-9 |
| BT-474 | 1.5 | 0-4 |
| MDA-MB-468 | 0.7 | 0-6 |
| MCF-7 | 0.6 | 0-2 |
| T-47D | 0.3 | 0-2 |
| HCC1806 | 0.2 | 0-1 |
| SK-BR-3 | 0.1 | 0-1 |
Table 1: Comparison of the Vascular Invasive Ability of Breast Cancer Cell Lines in Zebrafish Commonly used breast cancer cell lines were injected into the precardiac sinus of 2-day old zebrafish embryos and scored 4 days later. Cancer cells invading from the vasculature and into the caudal region were quantitated in 10 embryos per cell line. The average number of extravasated cancer cells is indicated along with the range.
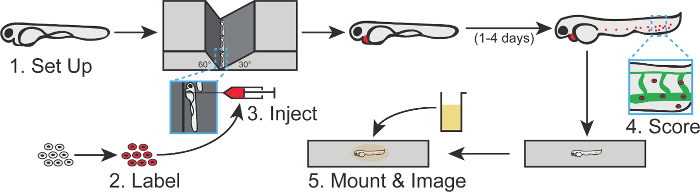 Figure 1. Visual Summary of the Zebrafish Cancer Cell Extravasation Assay. In this assay, cancer cells are labeled with a fluorescent dye and injected into the precardiac sinus of 2-day old zebrafish embryos, whose vasculature is marked by a contrasting fluorescent reporter. Following 2 - 4 additional days, cancer cells that have invaded into the caudal region of the embryo are scored and can be mounted in an anesthetic agarose medium for subsequent imaging. Each step depicted in this schematic corresponds to a section of the protocol. Please click here to view a larger version of this figure.
Figure 1. Visual Summary of the Zebrafish Cancer Cell Extravasation Assay. In this assay, cancer cells are labeled with a fluorescent dye and injected into the precardiac sinus of 2-day old zebrafish embryos, whose vasculature is marked by a contrasting fluorescent reporter. Following 2 - 4 additional days, cancer cells that have invaded into the caudal region of the embryo are scored and can be mounted in an anesthetic agarose medium for subsequent imaging. Each step depicted in this schematic corresponds to a section of the protocol. Please click here to view a larger version of this figure.
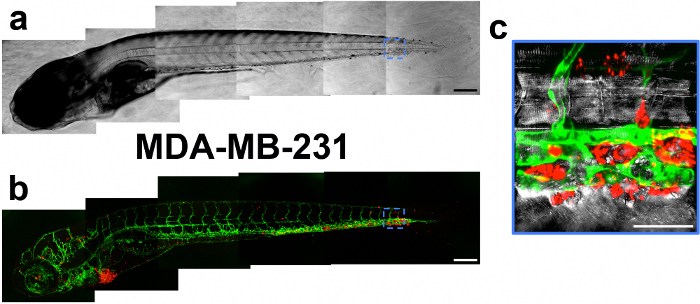 Figure 2. Image Mosaic of a Zebrafish Embryo 4 days after Injection with MDA-MB-231 Cells. Brightfield (A) and fluorescent (B) mosaics shown. Fluorescent images are depicted as the maximum projection of a z-stack, in which a green color denotes the vasculature and a red color indicates the cancer cells. Scale bar, 200 µm. (C) The embryo's caudal region is magnified to show extravasated cancer cells. Punctate fluorescent labeling is seen upon magnification. Scale bar, 50 µm. Please click here to view a larger version of this figure.
Figure 2. Image Mosaic of a Zebrafish Embryo 4 days after Injection with MDA-MB-231 Cells. Brightfield (A) and fluorescent (B) mosaics shown. Fluorescent images are depicted as the maximum projection of a z-stack, in which a green color denotes the vasculature and a red color indicates the cancer cells. Scale bar, 200 µm. (C) The embryo's caudal region is magnified to show extravasated cancer cells. Punctate fluorescent labeling is seen upon magnification. Scale bar, 50 µm. Please click here to view a larger version of this figure.
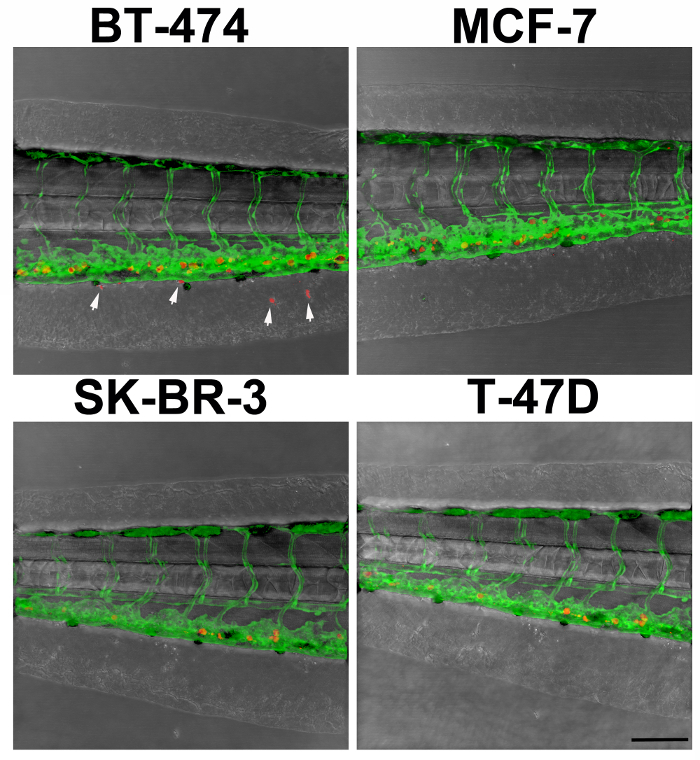 Figure 3. Example of Experimental Outcomes. Here extravasation is demonstrated in an embryo injected with BT-474 cancer cells but not in an embryo injected with MCF-7, SK-BR-3, and T-47D cancer cells. Arrowheads denote extravasated cells. Scale bar, 100 µm. Please click here to view a larger version of this figure.
Figure 3. Example of Experimental Outcomes. Here extravasation is demonstrated in an embryo injected with BT-474 cancer cells but not in an embryo injected with MCF-7, SK-BR-3, and T-47D cancer cells. Arrowheads denote extravasated cells. Scale bar, 100 µm. Please click here to view a larger version of this figure.
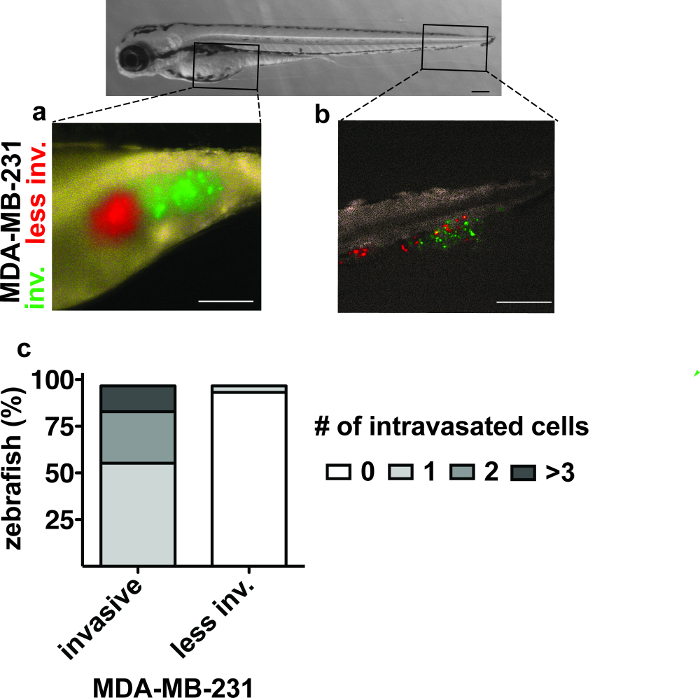 Figure 4.Intravasation of MDA-MB-231 Cell Populations 2 days after Yolk Sac Injection.(A) Yolk sac with green (invasive) and red (less invasive) labeled MDA-MB-231 cells. (B) The caudal region with MDA-MB-231 cells that invaded the vasculature to reach the tail region. (C) The number of zebrafish embryos with intravasated MDA-MB-231 cells in the caudal region 2 days after injection26. Scale bars, 250 µm. Please click here to view a larger version of this figure.
Figure 4.Intravasation of MDA-MB-231 Cell Populations 2 days after Yolk Sac Injection.(A) Yolk sac with green (invasive) and red (less invasive) labeled MDA-MB-231 cells. (B) The caudal region with MDA-MB-231 cells that invaded the vasculature to reach the tail region. (C) The number of zebrafish embryos with intravasated MDA-MB-231 cells in the caudal region 2 days after injection26. Scale bars, 250 µm. Please click here to view a larger version of this figure.
Discussion
This technique utilizes the zebrafish model to efficiently test the vascular invasive ability of cancer cells (see Figure 1). Here we applied the technique to a panel of breast cancer cell lines in order to provide a baseline onto which other investigators can then build their own studies (see Table 1; Figures 2 - 3). The observation that MDA-MB-231 cells readily invaded into the caudal region of zebrafish embryos would make this cell line ideal for testing agents that might inhibit vascular invasion. Comparatively less invasive cell lines, like HCC1806 or MDA-MB-468 cells for example, could be employed to study factors that would otherwise potentiate vascular invasion. If these types of experiments require the inclusion of drugs, then one has two distinct routes of delivery, depending on whether the treatment effect is expected to be durable. Cancer cells can be pretreated with the drug prior to injection into the embryos or the drug can be directly added to the water housing the embryos where it will impact the cancer cells by diffusion.
In our comparison, we analyzed cancer cell extravasation in the zebrafish embryo tail 96 h after injection into the precardiac sinus. For the MDA-MB-231 cell competition assay, intravasation into circulation was assessed 48 hr after the yolk sac injection. The timepoints chosen for analysis typically require optimization for each cell line where, for some experiments, peak extravasation can occur just 24 hr following the injection. It is worth noting that the zebrafish embryos have a functional innate immune system when they are injected with cancer cells27 and, consequently, fluorescent debris from the cancer cells may be engulfed by immune cells like macrophages or neutrophils. Activity of the innate immune system can therefore potentially create false-positive fluorescent labeling when attempting to score extravasation; care must be given to only score cells that robustly fluoresce in the appropriate channel. Since current cancer research is implicating the immune system in enhancing cancer metastasis28, the zebrafish embryo model may provide unique insights into this area by allowing for examination of crosstalk occurring between cancer cells and the innate immune system. This idea is exemplified by a couple of recent studies. The first of these studies demonstrated that VEGFR inhibition within zebrafish enhanced the ability of neutrophils to elicit metastatic behavior from cancer cells29. A second study showed that the CXCR4-CXL12 immune chemokine signaling axis is intact within zebrafish, as the receptor on human cancer cells was capable of sensing and responding to the zebrafish ligand30, and the expression of CXCR4 in cancer cells correlated with their invasive ability within the animal model. Furthermore, it was demonstrated that zebrafish macrophages exhibited a chemotactic response towards human CXCL12. A zebrafish embryo strain with fluorescently labeled neutrophils31, for example, could be utilized to further explore cancer cell interactions with the immune system in this model.
A few steps in the protocol require particular attention. First, it is essential that cancer cells be dissociated into a single-cell suspension in order to prevent clogging the needle during injection into the embryos. Cellular aggregates can also block the flow of blood and interfere with the ability of the cancer cells to travel into the caudal region, thus skewing the analysis. Cellular aggregates may also induce blood vessels to rupture and, if this occurs, scoring the invasive ability of live cancer cells in these regions is neither reliable nor recommended. Another potential problem area pertains to labeling the cancer cells with the fluorescent dye. In experiments run by our lab, we have found that lipophilic dyes, like DiI for example, tend to produce the best labeling, yet its fluorescence level can vary between cell lines. For this reason, the fluorescent labeling of cancer cells should be optimized prior to injection into the embryos in order to prevent over-labeling. It is critical that the excess dye is washed away from the cancer cells after they have been labeled, and this is achieved by the various centrifugation steps indicated in the labeling section of the protocol; these steps may seem superfluous, but they are absolutely necessary. Too much fluorescent dye can be toxic to cells or leak into the bloodstream, producing an artifactual fluorescent background. Many of these issues can be circumvented via the use of cancer cells expressing fluorescent proteins, and it is recommended that these systems be employed in lieu of fluorescent dye when possible. Finally, when scoring the embryos for cancer extravasation, it is imperative to apply consistent criteria to all conditions. We find that involving at least two individuals in the scoring step helps maintain scientific rigor: one individual is tasked with preparing the slides and recording the results, while the other individual renders the extravasation verdict and is kept blind to the condition being evaluated. When scoring, one can either quantitate extravasated cancer cells in the tail region or create binary criteria for ease of comparison. Fluorescently dyeing the cancer cells produces punctate labeling that will be visible at very high magnification (Figure 2c), though whole cells are easier to discern at lower magnification (Figure 3), the latter of which is recommended for scoring. Judging more ambiguous cases of extravasation may be aided by the acquisition of optical slices on a confocal microscope, where quantitating extravasation as a percent of all cells in the tail region may control for unequal loading of cancer cells into the embryo.
The yolk sac injection route is different from the precardiac sinus as it allows for a higher number of cells and therefore better accommodates the heterogeneity within a cell population. As shown in Figure 4, several differentially labeled cell populations can be simultaneously injected into the yolk sac to compare their invasive ability and pattern. The angiogenic ability of injected cells and enhanced recruitment of blood vessels may facilitate cancer cell entry into the tail region but this aspect has not yet been analyzed. Technically, yolk sac injections are easier to perform than a precardiac sinus injection though, on the other hand, the yolk sac is a nutrient-rich environment that may hinder the exit of cancer cells. It is also possible that injection near blood vessels in the yolk may prematurely introduce cancer cells into the circulation and, therefore, one must carefully screen for botched injections and omit these embryos from scoring. Lastly, it is important to note that the yolk microenvironment is somewhat artificial, as it lacks an analogous primary site in mice or humans.
The various model systems that seek to predict cancer metastasis are not without their advantages and disadvantages, and the zebrafish extravasation assay is no exception. As a major advantage, this assay allows for the assessment of vascular invasive ability within a functional circulatory system and experimental questions can be answered after just a few days. Given the rapid turnaround time, such a system could be utilized to probe the aggressive behavior of not only established cell lines, but also freshly isolated samples from human cancer patients. The major disadvantage is that this model omits the earliest and latest events of the metastatic cascade by focusing solely on vascular invasion. Moreover, the human cancer cells injected into these zebrafish embryos are obviously not operating in a syngeneic milieu and some interplay with the stroma may consequently be lost. Despite these limitations, the zebrafish embryo model has a deserved place in both basic and translational cancer research, as we have employed it show a role for the Hippo signaling pathway26 and keratin-associated protein 5-532 in the vascular invasive ability of cancer cells. Hence, this model has the potential to aid the investigation of a key metastatic hallmark of advanced stage cancer.
Disclosures
The authors have no conflicts of interest to disclose.
Acknowledgments
We thank Peter Johnson of the Georgetown University Microscopy Core for assistance with imaging the zebrafish embryos. The Microscopy & Imaging Shared Resource and the Zebrafish Shared Resource are partially supported by NIH/NCI grant P30-CA051008. This work was also supported by NIH/NCI CA71508 (AW) and CA177466 (AW).
References
- Cummings MC, et al. Metastatic progression of breast cancer: insights from 50 years of autopsies. Am J Path. 2014;232:23–31. doi: 10.1002/path.4288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen DX, Bos PD, Massagué J. Metastasis: from dissemination to organ-specific colonization. Nat Rev Cancer. 2009;9(4):274–284. doi: 10.1038/nrc2622. [DOI] [PubMed] [Google Scholar]
- Reymond N, d'Água BB, Ridley AJ. Crossing the endothelial barrier during metastasis. Nat Rev Cancer. 2013;13(12):858–870. doi: 10.1038/nrc3628. [DOI] [PubMed] [Google Scholar]
- Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19(11):1423–1437. doi: 10.1038/nm.3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper S, Marshall JF, Sahai E. Tumor cell migration in three dimensions. Method Enzymol. 2006;409:625–642. doi: 10.1016/S0076-6879(06)06049-6. [DOI] [PubMed] [Google Scholar]
- Rahim S, Üren A. A real-time electrical impedance based technique to measure invasion of endothelial cell monolayer by cancer cells. J Vis Exp. 2011. [DOI] [PMC free article] [PubMed]
- Jeon JS, et al. Human 3D vascularized organotypic microfluidic assays to study breast cancer cell extravasation. Proc Natl Acad Sci USA. 2015;112(1):214–219. doi: 10.1073/pnas.1417115112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin Y, et al. Microfluidic assay for simultaneous culture of multiple cell types on surfaces or within hydrogels. Nat Protoc. 2012;7(7):1247–1259. doi: 10.1038/nprot.2012.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargo-Gogola T, Rosen JM. Modelling breast cancer: one size does not fit all. Nat Rev Cancer. 2007;7(9):659–672. doi: 10.1038/nrc2193. [DOI] [PubMed] [Google Scholar]
- Mohanty S, Xu L. Experimental metastasis assay. J Vis Exp. 2010. [DOI] [PMC free article] [PubMed]
- Minn AJ, et al. Genes that mediate breast cancer metastasis to lung. Nature. 2005;436(7050):518–524. doi: 10.1038/nature03799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell JP, Merkel AR, Masood-Campbell SK, Elefteriou F, Sterling JA. Models of bone metastasis. J Vis Exp. 2012. p. e4260. [DOI] [PMC free article] [PubMed]
- Arguello F, Baggs RB, Frantz CN. A murine model of experimental metastasis to bone and bone marrow. Cancer Res. 1988;48(23):6876–6881. [PubMed] [Google Scholar]
- Minn AJ, et al. Distinct organ-specific metastatic potential of individual breast cancer cells and primary tumors. J Clin Invest. 2005;115(1):44–55. doi: 10.1172/JCI22320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bos PD, et al. Genes that mediate breast cancer metastasis to the brain. Nature. 2009;459(7249):1005–1009. doi: 10.1038/nature08021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares KC, et al. A preclinical murine model of hepatic metastases. J Vis Exp. 2014. p. e51677. [DOI] [PMC free article] [PubMed]
- Kienast Y, et al. Real-time imaging reveals the single steps of brain metastasis formation. Nature Med. 2010;16(1):116–122. doi: 10.1038/nm.2072. [DOI] [PubMed] [Google Scholar]
- Hasegawa H, Ushio Y, Hayakawa T, Yamada K, Mogami H. Changes of the blood-brain barrier in experimental metastatic brain tumors. J Neurosurg. 1983;59(2):304–310. doi: 10.3171/jns.1983.59.2.0304. [DOI] [PubMed] [Google Scholar]
- Amatruda JF, Shepard JL, Stern HM, Zon LI. Zebrafish as a cancer model system. Cancer Cell. 2002;1(3):229–231. doi: 10.1016/s1535-6108(02)00052-1. [DOI] [PubMed] [Google Scholar]
- Feitsma H, Cuppen E. Zebrafish as a cancer model. Molecular Cancer Res. 2008;6(5):685–694. doi: 10.1158/1541-7786.MCR-07-2167. [DOI] [PubMed] [Google Scholar]
- Stoletov K, Klemke R. Catch of the day: zebrafish as a human cancer model. Oncogene. 2008;27(33):4509–4520. doi: 10.1038/onc.2008.95. [DOI] [PubMed] [Google Scholar]
- Stoletov K, et al. Visualizing extravasation dynamics of metastatic tumor cells. J Cell Sci. 2010;123(13):2332–2341. doi: 10.1242/jcs.069443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanada M, Zhang J, Yan L, Sakurai T, Terakawa S. Endothelial cell-initiated extravasation of cancer cells visualized in zebrafish. PeerJ. 2014. [DOI] [PMC free article] [PubMed]
- Teng Y, Xie X, Walker S, White DT, Mumm JS, Cowell JK. Evaluating human cancer cell metastasis in zebrafish. BMC Cancer. 2013;13(453) doi: 10.1186/1471-2407-13-453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross LM, Cook MA, Lin S, Chen J-N, Rubinstein AL. Rapid analysis of angiogenesis drugs in a live fluorescent zebrafish assay. Arterioscl Throm Vas. 2003;23(5):911–912. doi: 10.1161/01.ATV.0000068685.72914.7E. [DOI] [PubMed] [Google Scholar]
- Sharif GM, et al. Cell growth density modulates cancer cell vascular invasion via Hippo pathway activity and CXCR2 signaling. Oncogene. 2015;34:5879–5889. doi: 10.1038/onc.2015.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novoa B, Figueras A. Zebrafish: model for the study of inflammation and the innate immune response to infectious diseases. Adv Exp Med Biol. 2012;946:253–275. doi: 10.1007/978-1-4614-0106-3_15. [DOI] [PubMed] [Google Scholar]
- Kitamura T, Qian B-Z, Pollard JW. Immune cell promotion of metastasis. Nature Rev Immunol. 2015;15(2):73–86. doi: 10.1038/nri3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S, et al. Neutrophil-mediated experimental metastasis is enhanced by VEGFR inhibition in a zebrafish xenograft model. J Pathol. 2012;227(4):431–445. doi: 10.1002/path.4013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tulotta C, et al. Inhibition of signaling between human CXCR4 and zebrafish ligands by the small molecule IT1t impairs the formation of triple-negative breast cancer early metastases in a zebrafish xenograft model. Dis Model Mech. 2016;9(2):141–153. doi: 10.1242/dmm.023275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renshaw SA, Loynes CA, Trushell DMI, Elworthy S, Ingham PW, Whyte MKB. A transgenic zebrafish model of neutrophilic inflammation. Blood. 2006;108(13):3976–3978. doi: 10.1182/blood-2006-05-024075. [DOI] [PubMed] [Google Scholar]
- Berens EB, et al. Keratin-associated protein 5-5 controls cytoskeletal function and cancer cell vascular invasion. Oncogene. 2016. Jul 4, Epub ahead of print. [DOI] [PMC free article] [PubMed]