

Abstract
Adoptive transfer of ex vivo expanded autologous tumor-infiltrating lymphocytes (TILs) can mediate durable and complete responses in significant subsets of patients with metastatic melanoma. Major obstacles of this approach are the reduced viability of transferred T cells, caused by telomere shortening, and the limited number of TILs obtained from patients. Less-differentiated T cells with long telomeres would be an ideal T cell subset for adoptive T cell therapy;however, generating large numbers of these less-differentiated T cells is problematic. This limitation of adoptive T cell therapy can be theoretically overcome by using induced pluripotent stem cells (iPSCs) that self-renew, maintain pluripotency, have elongated telomeres, and provide an unlimited source of autologous T cells for immunotherapy. Here, we present a protocol to generate iPSCs using Sendai virus vectors for the transduction of reprogramming factors into TILs. This protocol generates fully reprogrammed, vector-free clones. These TIL-derived iPSCs might be able to generate less-differentiated patient- and tumor-specific T cells for adoptive T cell therapy.
Keywords: Developmental Biology, Issue 117, Induced pluripotent stem cells, Reprogramming, Tumor-Infiltrating Lymphocytes, T cells, Melanoma, Sendai virus vector, Human, CD3, CD28, IL-2
Introduction
Cellular reprogramming technology that allows generation of induced pluripotent stem cells (iPSCs) via overexpression of a defined set of transcription factors holds great promise in the field of cell-based therapies1,2. These iPSCs exhibit transcriptional and epigenetic features and have the capacity for self-renewal and pluripotency, similarly to embryonic stem cells (ESCs)3-5. Remarkable progress made in reprogramming technology over the past decade has allowed us to generate human iPSCs even from terminally differentiated cells, such as T cells6-8. T cell-derived iPSCs (TiPSCs) retain the same rearranged configuration of T cell receptor (TCR) chain genes as the original T cells, which allows regeneration of antigen-specific T cells from TiPSCs9-11.
Nearly 80% of melanoma-infiltrating lymphocytes (TILs) obtained from a patient's tumor specifically recognize tumor-associated antigens and maintain cytotoxicity against the original cancer cells12. Notably, the expression of programmed cell death protein-1 (PD-1) on TILs was found to identify the autologous tumor-reactive repertoire, including mutated neoantigen-specific CD8+ lymphocytes13. Adoptive transfer of ex-vivo expanded autologous TILs in combination with preparative lymphodepleting regimens and systemic administration of Interleukin-2 (IL-2) can cause substantial regression of metastatic melanoma in subsets of patients14. Despite encouraging results in preclinical models and in patients, poor survival of infused T cells and the existence of immune suppressive pathways appear to compromise the full potential of adoptive T cell therapy. Current clinical protocols require extensive ex vivo manipulation of autologous T cells in order to obtain large numbers. This results in the generation of terminally differentiated T cells that have poor survival, reduced proliferative capacity, and high levels of PD-115.
This limitation of adoptive T cell therapy can be theoretically overcome by using iPSCs that can provide an unlimited source of autologous T cells for immunotherapy. We have recently reported the reprogramming of melanoma TILs expressing high levels of PD-1 by Sendai virus (SeV)-mediated transduction of the four transcription factors, OCT3/4, SOX2, KLF4, and c-MYC16. While retrovirus vectors require integration into host chromosomes to express reprogramming genes, SeV vectors are non-integrating and are eventually eliminated from the cytoplasm. Reprogramming efficiency is much higher with a SeV system compared with lentivirus or retrovirus vectors6-8. Furthermore, SeV can specifically reprogram T cells in peripheral blood mononuclear cells (PBMCs), while some iPSC clones generated by lentivirus or retrovirus vectors can be from nonlymphoid lineages6-8. Here, we detail the procedures implemented for the isolation and activation of human melanoma TILs and for the generation of TIL-derived iPSCs using a SeV reprogramming system.
Protocol
NOTE: Patients should give informed consent to participate in the Institutional Review Board and Human Pluripotent Stem Cell Committee approved study.
1. Isolation and Culture of TILs
Obtain tumor material that is not required for histopathologic diagnosis from the pathology service/tissue procurement core. Place 20-100 g of tumor specimens in a 50-ml tube with 30 ml tumor collecting media (Table 1).
Dissect solid, firm, normal tissue of the tumor specimen from fragile and/or bloody necrotic areas using scissors. After removing the necrotic tissue, use the scissors to mince the specimen as small as possible.
Dissociate the minced specimen into a single-cell suspension using a dissociator and tumor dissociation kit (human) according to the manufacturer's instructions. Filter the suspension with a 70 µm cell strainer that is set on a 50 ml tube and wash the strainer 2 times with 2 ml of RPMI 1640.
Centrifuge at 200 x g at room temperature for 5 min. Discard the supernatant and resuspend the cells in 10 ml of T cell media (Table 1).
Prepare a two-step gradient solution such as Ficoll (gradient solution) in a 50-ml tube, with a lower step of 10 ml of 100% gradient solution and a middle step of 30 ml of 75% gradient solution diluted with Dulbecco's phosphate-buffered saline, no calcium, no magnesium (D-PBS(-)).
Layer the cell suspension (from Step 1.4) on the gradient solution (from Step 1.5). Centrifuge at 400 x g at 20 °C for 45 min.
Collect the layer containing the enriched TILs at the interface between the 100% gradient solution and the 75% gradient solution in a 50 ml tube. Dilute the layer of the solution with the enriched TILs by adding 20-30 ml of D-PBS(-).
Centrifuge at 200 x g at room temperature for 5 min. Discard the supernatant and resuspend the TIL-enriched fraction in 10 ml of T cell media.
Culture the fraction (from Step 1.8) with 2 ml of T cell media and 6,000 IU/ml recombinant human (rh) IL-2 in a 6-well plate at 37 °C, 5% CO2.
Change half the media on day 5 after culture initiation and every 2-3 days thereafter.
Split the cells at a ratio of 1:2 without trypsinization after softly suspending, doubling the number of wells when reaching 80-90% confluency. Add a half volume (1 ml) of fresh T cell media and rhIL-2 at 6,000 IU/ml.
2. Preparation of Mitomycin-C-treated SNL Feeder Cell Plate
Culture SNL feeder cells in 10 ml of SNL feeder cell media (Table 1) on a 0.1% gelatin-coated 10 cm dish until 80-90% confluence (3-4 x 106 cells) is reached.
Add 310 µl of 0.4 mg/ml mitomycin C solution directly into the SNL feeder cell culture dish and incubate at 37 °C, 5% CO2 for 2 hr and 15 min. Aspirate the media and wash the cells twice with 5 ml D-PBS(-).
Add 0.5 ml of 0.25% trypsin/1 mM EDTA and incubate at room temperature for 1 min to dissociate the cells. Add 4.5 ml of SNL feeder cell media to neutralize and re-suspend the cells into a single suspension.
Transfer the cells into a 15 ml tube and centrifuge at 200 x g for 5 min. Aspirate the media and count the cells with a hemocytometer after re-suspending in 10 ml of SNL feeder cell media.
Plate 1.5 x 106 cells per well of the SNL feeder cells on a 10 cm dish and incubate at 37 °C, 5% CO2. Use the SNL feeder cells within 3 days.
3. Generation of iPSCs Using the Sendai Virus (SeV) Vector
Harvest the TILs (from Step 1.11) and activate 5 x 105 cells of TILs with plate-bound mouse anti-human CD3 (1 µg/ml) and soluble mouse anti-human CD28 (5 µg/ml), with rhIL-2 (6,000 IU/ml) in 2 ml of T cell media in a 6-well plate at 37 °C, 5% CO2 for 5 days.
Harvest the TILs (from Step 3.1) in a 15 ml tube using a pipette and count the cell number with a hemocytometer.
Reactivate 1 x 105 cells of TILs with plate-bound mouse anti-human CD3 (1 µg/ml) and soluble mouse anti-human CD28 (5 µg/ml), with rhIL-2 (60 IU/ml) in 500 µl of reprogramming media (Table 1) per well in a 24-well plate at 37 °C, 5% CO2 for 24 hr. Prepare 3 wells for SeV infection: OCT3/4, SOX2, KLF4, and c-MYC (1 well); SeV infection (GFP: green fluorescent protein) (1 well); and cell count (1 well).
After 24 hr (from Step 3.3), count the cell number with a hemocytometer and determine the volume of each virus for various (3-20) multiplicities of infection (MOI). Remove one set of Sendai virus vector tubes from the -80 °C storage. Thaw the SeV vectors quickly in a water bath (37 °C) and place them on ice. Volume of virus (µl) = MOI (CIU/cell) x number of cells/titer of virus (CIU/ml) × 10-3 (µl/ml)
Add the calculated volumes of each of the four Sendai virus vector tubes that individually carried OCT3/4, SOX2, KLF4, and c-MYC by adding a mixture of SeV vectors at 10-20 MOI into the media, including activated TILs.
To determine the transduction efficiency, add the calculated volumes of SeV-GFP into a well of activated TILs.
Place the dish (from Step 3.5 and Step 3.6) in the incubator at 37 °C, 5% CO2 for 24 hr. After 24 hr, estimate the infection efficiency using TILs infected with SeV-GFP under fluorescence microscopy.
Collect the cells (from Step 3.5) in a 15 ml tube. Centrifuge at 200 x g at room temperature for 5 min.
Discard the supernatant and resuspend the pellet in 0.5 ml of hESC media with Primate ES Cell Medium and basic fibroblast growth factor (bFGF, 4 ng/ml).
Transfer the suspension into a 10 cm dish that contains mitomycin-C-treated SNL feeder cells in 10 ml of hESC media. Change the hESC media every other day until the colonies are grown.
Around day 18 to 21, remove the SNL feeder cells around the colonies under a microscope using a 10-µl tip in the clean bench.
Scrape the colonies using a 10-µl tip and transfer them using a 200-µl tip into 100 µl of iPSCs media per well of a 96-well tissue culture plate. Pipet up and down at 2-3 times to break the colonies into small clumps with an average size of 50-100 µm.
Transfer the small clumps into 6-well tissue culture plates with mitomycin-C-treated SNL feeder cells (from Step 2) in 2 ml per well of iPSCs media with basic fibroblast growth factor (bFGF, 4 ng/ml). Change iPSCs media every day.
Transfer the iPSCs to 6-well tissue culture plates with fresh mitomycin-C-treated SNL feeder cells with 1 mg/ml collagenase IV every 5-6 days. Prepare 2 wells per plates of each clone for immunostaining.
Determine the full reprogramming of each clone by immunohistochemical staining of pluripotency markers (SSEA3, SSEA4, TRA-1-60, TRA-1-81, and OCT3/4)1. NOTE: For further evaluation of pluripotency potential, confirm that fully-reprogrammed iPSC lines can differentiate into three germ layers by an embryoid body formation and differentiation assay or a teratoma formation assay16.
Confirm that fully-reprogrammed iPSC lines demonstrate a normal karyotype by cytogenetic analysis.
For teratoma formation, inject an undifferentiated TIL-derived iPSC (1 x 106) suspension in 100 µl of DMEM containing 10% FCS into the subcutaneous tissue of 6-8 week old female NOD/SCID mice. Stain teratoma sections with hematoxylin and eosin.
4. Immunofluorescence Staining of iPSCs for SSEA3, SSEA4, TRA1-60 and TRA1-81
Wash iPSCs with 1 ml of PBS and fix the iPSCs with 1 ml of 4% paraformaldehyde solution for 10 min. Remove the 4% paraformaldehyde solution and wash the iPSCs with 1 ml of PBS 3 times. NOTE: Be careful when using paraformaldehyde because it is toxic.
Pretreat the iPSCs with blocking buffer (Table 1) for 30 min. Meanwhile, dilute the primary antibodies (rat anti-human SSEA3, mouse anti-human SSEA4, mouse anti-human TRA1-60, and mouse anti-human TRA1-81) 1:50 in 1.5% goat serum solution diluted with PBS and TritonX-100 (total volume: 1 ml).
Remove the blocking buffer (Table 1). Add the diluted primary antibody solution and incubate for 1 hr at room temperature. Meanwhile, dilute the secondary antibody (anti-mouse IgG and IgM or anti-rat IgM) 1:50 in 1.5% goat serum solution.
Remove the diluted primary antibody solution and wash the iPSCs with 1 ml PBS 3 times, each for 5 min. Add the diluted secondary antibody solution and incubate for 45 min at room temperature in a dark room.
Remove the diluted secondary antibody solution and wash the iPSCs with 1 ml of PBS 3 times, each for 5 min. Observe the iPSCs with immunofluorescence microscopy.
5. Immunofluorescence Staining of iPSCs for Oct3/4
Wash iPSCs with 1 ml of PBS and fix the iPSCs with 1 ml of 4% paraformaldehyde solution for 10 min. Remove the 4% paraformaldehyde solution and wash the iPSCs with PBS 3 times.
Add permeabilization buffer (Table 1) and incubate at room temperature for 10 min. Remove the permeabilization buffer and add blocking buffer (Table 1).
Incubate at room temperature for 30 min. Meanwhile, dilute the primary antibody (mouse anti-human Oct3/4) 1:50 in blocking buffer.
Add the diluted primary antibody solution and incubate at 4 °C overnight.
Remove the diluted primary antibody solution and wash the iPSCs with 1ml of PBS 3 times, each for 5 min. Meanwhile, dilute the secondary antibody (goat anti-mouse IgG/IgM) 1:100 in blocking buffer.
Add the diluted secondary antibody solution and incubate at room temperature in a dark room for 45 min.
Remove the diluted secondary solution and wash the iPSCs with 1 ml of PBS 3 times, each for 5 min. Observe the iPSCs under immunofluorescence microscopy.
Representative Results
Figure 1 shows the overview of the procedure that involves the initial expansion of melanoma TILs with rhIL-2, which is followed by activation with anti-CD3/CD28 and gene transfer of OCT3/4, KLF4, SOX2, and c-MYC to TILs for the generation of iPSCs. Usually, TILs on culture with rhIL-2 start to form spheres 21-28 days after initiation of culture. At this point, TILs are ready to be activated with anti-CD3/CD28. Figure 2A shows TILs, on culture with rhIL-2 on day 21, that are ready to be activated with anti-CD3/CD28. Figure 2B shows GFP introduction by Sendai virus (SeV) transfected at 20 MOI. Figure 2C shows a typical pluripotent clone on SNL feeder cells that appears 18-21 days after SeV infection. Figure 2D shows that the generated TIL-derived iPSCs have a normal karyotype. Figure 3 shows immunofluorescent staining to confirm pluripotency marker expression on iPSCs (SSEA3, SSEA4, TRA1-81, TRA1-60, and OCT3/4). Figure 4 shows that iPSCs derived from melanoma TILs are able to form teratoma that contain a variety of cells from three germ layers (neural tissue, respiratory epithelium, and cartilage). Figure 5 shows that generated TIL-derived iPSCs retain TCR rearrangements.
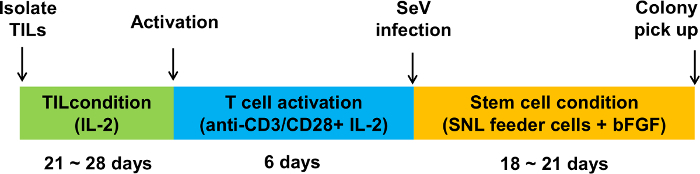 Figure 1:Schematic overview of the generation of iPSCs from melanoma TILs. The protocol involves three stages: isolation and culture of TILs with IL-2, T cell activation with anti-CD3/CD28, and cell reprogramming with the Sendai virus (SeV) vector encoding OCT3/4, SOX2, KLF4, and c-MYC. Please click here to view a larger version of this figure.
Figure 1:Schematic overview of the generation of iPSCs from melanoma TILs. The protocol involves three stages: isolation and culture of TILs with IL-2, T cell activation with anti-CD3/CD28, and cell reprogramming with the Sendai virus (SeV) vector encoding OCT3/4, SOX2, KLF4, and c-MYC. Please click here to view a larger version of this figure.
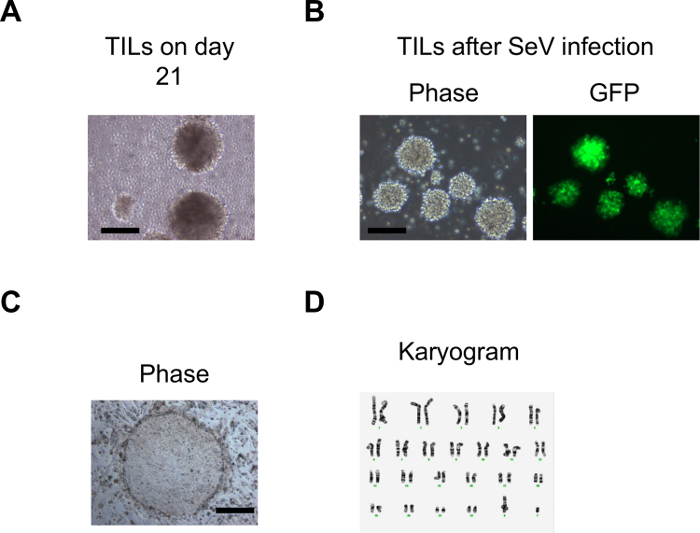 Figure 2: Generation of iPSCs from melanoma TILs. (A) An image representing the morphology of the TILs in rhIL-2 when they started to expand 2-3 weeks after initiation of the culture. (B) Images representing morphology and GFP expression of TILs one day after SeV infection. (C) A typical ESC-like iPSC colony on day 21 after SeV infection. The Scale bars = 200 µm. (D) Cytogenetic analysis on twenty G-banded metaphase cells from one of the iPSCs derived from melanoma TILs. Figure (D) is adapted from Saito et al16. Please click here to view a larger version of this figure.
Figure 2: Generation of iPSCs from melanoma TILs. (A) An image representing the morphology of the TILs in rhIL-2 when they started to expand 2-3 weeks after initiation of the culture. (B) Images representing morphology and GFP expression of TILs one day after SeV infection. (C) A typical ESC-like iPSC colony on day 21 after SeV infection. The Scale bars = 200 µm. (D) Cytogenetic analysis on twenty G-banded metaphase cells from one of the iPSCs derived from melanoma TILs. Figure (D) is adapted from Saito et al16. Please click here to view a larger version of this figure.
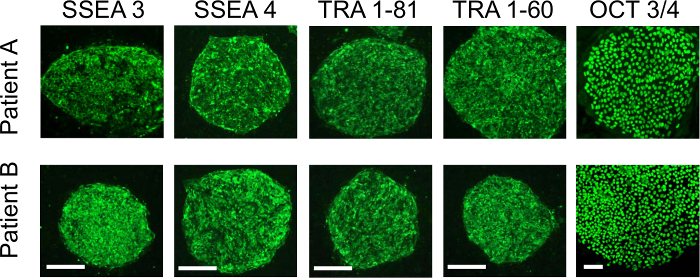 Figure 3: Immunofluorescence staining with stem cell pluripotent markers. The immunofluorescence assay shows that the TIL-derived iPSCs from two patients are positive for SSEA3, SSEA4, TRA-1-81, TRA-1-60, and OCT3/4. The Scale bars = 100 µm.The figure is adapted from Saito et al16. Please click here to view a larger version of this figure.
Figure 3: Immunofluorescence staining with stem cell pluripotent markers. The immunofluorescence assay shows that the TIL-derived iPSCs from two patients are positive for SSEA3, SSEA4, TRA-1-81, TRA-1-60, and OCT3/4. The Scale bars = 100 µm.The figure is adapted from Saito et al16. Please click here to view a larger version of this figure.
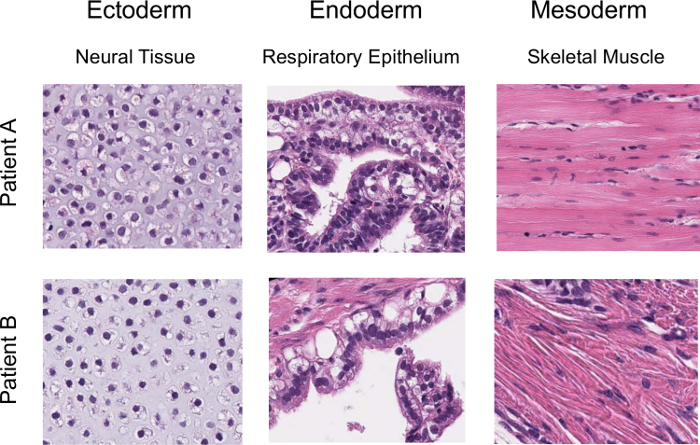 Figure 4:Confirmation of pluripotency for iPSCs with teratoma formation. Hematoxylin- and eosin-stained representative teratoma sections derived from the TIL-derived iPSCs clone (6 weeks post-injection into NOD/SCID mice) are shown. Figure adapted from Saito et al16. Please click here to view a larger version of this figure.
Figure 4:Confirmation of pluripotency for iPSCs with teratoma formation. Hematoxylin- and eosin-stained representative teratoma sections derived from the TIL-derived iPSCs clone (6 weeks post-injection into NOD/SCID mice) are shown. Figure adapted from Saito et al16. Please click here to view a larger version of this figure.
 Figure 5: A wide variety of TCR-β gene arrangement patterns in TIL-iPSCs. TCR-β gene rearrangements in TIL-iPSCs are identified by capillary electrophoresis. The green line is derived from the band for the Jβ1 gene, and the blue line is derived from the band for the Jβ2 gene. The figure is adapted from Saito et al16. Please click here to view a larger version of this figure.
Figure 5: A wide variety of TCR-β gene arrangement patterns in TIL-iPSCs. TCR-β gene rearrangements in TIL-iPSCs are identified by capillary electrophoresis. The green line is derived from the band for the Jβ1 gene, and the blue line is derived from the band for the Jβ2 gene. The figure is adapted from Saito et al16. Please click here to view a larger version of this figure.
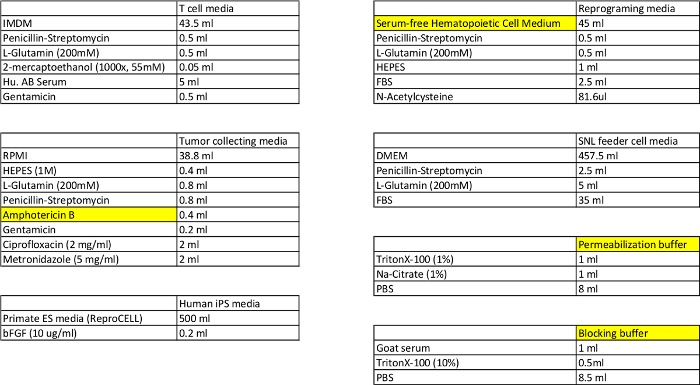 Table 1: Composition for the T cell, reprogramming, tumor collecting, SNL feeder cell, and iPSC media, and the permeabilization and blocking buffers.
Please click here to view a larger version of this table.
Table 1: Composition for the T cell, reprogramming, tumor collecting, SNL feeder cell, and iPSC media, and the permeabilization and blocking buffers.
Please click here to view a larger version of this table.
Discussion
Here, we demonstrated a protocol for reprogramming melanoma TILs to iPSCs by SeV-mediated transduction of the four transcription factors OCT3/4, SOX2, KLF4, and c-MYC. This approach, using a SeV system to reprogram T cells, offers the advantage of a non-integrating method7.
A previous study showed that a SeV reprogramming system was highly efficient and reliable to reprogram not only fibroblasts but also peripheral blood T cells7,17. In addition, we have recently shown that melanoma TILs that are more differentiated and express higher levels of inhibitory receptors such as PD-1 than peripheral blood T cells can also be reprogrammed using SeV vectors16. Timing of stimulation with anti-CD3/CD28 and infection with SeV vector was critical in reprogramming TILs. Peripheral blood T cells can be stimulated with anti-CD3 and IL-2 for reprogramming immediately after harvesting from the subject7, while TILs need to be cultured for 3-4 weeks with IL-2 before stimulation.
Although more than 99% of TILs were CD8 T cells after 3-4 weeks of culture with IL-2 and activation with anti-CD3/CD2816, we recommend analysis of TCR rearrangement in iPSCs to confirm that generated iPSCs are from TILs (Figure 5). Of note, previous studies including ours indicated that every iPSCs generated using SeV from peripheral blood mononuclear cells or TILs had TCR rearrangement7,16.
Although generation of iPSCs from melanoma TILs was feasible, we found that the reprogramming efficiency of melanoma TILs was lower (0.01-0.05 %)16 than that of peripheral blood T cells (0.1%)7. The reason for this remains unclear, but it might be associated with the higher expression of inhibitory receptors or the greater number of differentiated T-cell subsets in TILs13,16. Recent significant progress for reprogramming technology may improve the reprogramming efficiency for generation of TIL-iPSCs. The second generation SeV vector, TS12KOS, was found to have higher efficiency of iPSC generation than the conventional SeV vector used in the previous study18,19.
Although the derivation of human iPSCs with a SeV reprogramming system is feasible, there are several limitations to be considered in this protocol. We use fetal bovine serum during SeV infection in this protocol. We found that the reprogramming efficiency of melanoma TILs was significantly lower if using media with human serum during SeV infection compared to one with fetal bovine serum, despite similar proliferation of TILs. We also use a mouse feeder layer for iPSC generation and maintenance, but a defined feeder-free and xeno-free system may be alternative methods in future studies.
It takes around two months from the time of tumor harvest to generate iPSCs from melanoma TILs. In addition, it would take an additional 1-2 months to obtain transgene-free iPSCs. This might be shortened by the use of the new type of SeV vector, TS12KOS, which has shown a more efficient virus elimination rate19.
In conclusion, the generation of human iPSCs from melanoma TILs is feasible. The current protocol might be an important step for generating an infinite number of tumor-specific T cells for adoptive T cell therapy.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
We thank Ms. Deborah Postiff and Ms. Jackline Barikdar in the Tissue Procurement Core and Dr. Cindy DeLong in the Pluripotent Stem Cell Core Laboratory at the University of Michigan for her technical assistance. This study was supported by University of Michigan startup funding and grants from the Central Surgical Association, American College of Surgeons, Melanoma Research Alliance, and NIH/NCI (1K08CA197966-01) to F. Ito.
References
- Takahashi K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Wernig M, et al. In vitro reprogramming of fibroblasts into a pluripotent ES-cell-like state. Nature. 2007;448:318–324. doi: 10.1038/nature05944. [DOI] [PubMed] [Google Scholar]
- Maherali N, et al. Directly reprogrammed fibroblasts show global epigenetic remodeling and widespread tissue contribution. Cell Stem Cell. 2007;1:55–70. doi: 10.1016/j.stem.2007.05.014. [DOI] [PubMed] [Google Scholar]
- Okita K, Ichisaka T, Yamanaka S. Generation of germline-competent induced pluripotent stem cells. Nature. 2007;448:313–317. doi: 10.1038/nature05934. [DOI] [PubMed] [Google Scholar]
- Loh YH, et al. Reprogramming of T cells from human peripheral blood. Cell Stem Cell. 2010;7:15–19. doi: 10.1016/j.stem.2010.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki T, et al. Generation of induced pluripotent stem cells from human terminally differentiated circulating T cells. Cell Stem Cell. 2010;7:11–14. doi: 10.1016/j.stem.2010.06.003. [DOI] [PubMed] [Google Scholar]
- Staerk J, et al. Reprogramming of human peripheral blood cells to induced pluripotent stem cells. Cell Stem Cell. 2010;7:20–24. doi: 10.1016/j.stem.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura T, et al. Generation of rejuvenated antigen-specific T cells by reprogramming to pluripotency and redifferentiation. Cell Stem Cell. 2013;12:114–126. doi: 10.1016/j.stem.2012.11.002. [DOI] [PubMed] [Google Scholar]
- Vizcardo R, et al. Regeneration of Human Tumor Antigen-Specific T Cells from iPSCs Derived from Mature CD8(+) T Cells. Cell Stem Cell. 2013;12:31–36. doi: 10.1016/j.stem.2012.12.006. [DOI] [PubMed] [Google Scholar]
- Wakao H, et al. Expansion of functional human mucosal-associated invariant T cells via reprogramming to pluripotency and redifferentiation. Cell Stem Cell. 2013;12:546–558. doi: 10.1016/j.stem.2013.03.001. [DOI] [PubMed] [Google Scholar]
- Dudley ME, Wunderlich JR, Shelton TE, Even J, Rosenberg SA. Generation of tumor-infiltrating lymphocyte cultures for use in adoptive transfer therapy for melanoma patients. J Immunother. 2003;26:332–342. doi: 10.1097/00002371-200307000-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gros A, et al. PD-1 identifies the patient-specific CD8(+) tumor-reactive repertoire infiltrating human tumors. J Clin Invest. 2014;124:2246–2259. doi: 10.1172/JCI73639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg SA, et al. Durable Complete Responses in Heavily Pretreated Patients with Metastatic Melanoma Using T-Cell Transfer Immunotherapy. Clinical Cancer Research. 2011;17:4550–4557. doi: 10.1158/1078-0432.CCR-11-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Restifo NP, Dudley ME, Rosenberg SA. Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol. 2012;12:269–281. doi: 10.1038/nri3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito H, et al. Reprogramming of Melanoma Tumor-Infiltrating Lymphocytes to Induced Pluripotent Stem Cells. Stem Cells International. 2016;2016:11. doi: 10.1155/2016/8394960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusaki N, Ban H, Nishiyama A, Saeki K, Hasegawa M. Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc Jpn Acad Ser B Phys Biol Sci. 2009;85:348–362. doi: 10.2183/pjab.85.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ban H, et al. Efficient generation of transgene-free human induced pluripotent stem cells (iPSCs) by temperature-sensitive Sendai virus vectors. Proc Natl Acad Sci U S A. 2011;108:14234–14239. doi: 10.1073/pnas.1103509108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujie Y, et al. New Type of Sendai Virus Vector Provides Transgene-Free iPS Cells Derived from Chimpanzee Blood. PLoS One. 2014;9:e113052. doi: 10.1371/journal.pone.0113052. [DOI] [PMC free article] [PubMed] [Google Scholar]