

Abstract
Proper neuronal development and function is the prerequisite of the developing and the adult brain. However, the mechanisms underlying the highly controlled formation and maintenance of complex neuronal networks are not completely understood thus far. The open questions concerning neurons in health and disease are diverse and reaching from understanding the basic development to investigating human related pathologies, e.g., Alzheimer's disease and Schizophrenia. The most detailed analysis of neurons can be performed in vitro. However, neurons are demanding cells and need the additional support of astrocytes for their long-term survival. This cellular heterogeneity is in conflict with the aim to dissect the analysis of neurons and astrocytes. We present here a cell-culture assay that allows for the long-term cocultivation of pure primary neurons and astrocytes, which share the same chemically defined medium, while being physically separated. In this setup, the cultures survive for up to four weeks and the assay is suitable for a diversity of investigations concerning neuron-glia interaction.
Keywords: Neuroscience, Issue 117, hippocampus, cell culture, indirect coculture, hippocampal neurons, astrocytes, synaptogenesis
Introduction
Throughout the past decades, the general interpretation of neuroglia function has evolved from the attribution of a merely supportive towards an active regulatory role concerning neuronal function1. Because of their prominent impact on brain homeostasis in health and disease2, astrocytes are of special interest for the scientific community. In the past few years, a diversity of studies have focused on neuron-glia interactions in vivo and in vitro3. However, most of the culture systems do not allow for the separate analysis of both cell types and of their respective secretomes.
Several approaches exploit the direct cocultivation of neurons and glia to achieve long lasting survival and physiologically relevant neuronal network development4-6. The present protocol reaches the same goals while keeping both cell types physically separated7. Compared to conditioned medium approaches8,9, our system allows to study the bidirectional communication between neurons and astrocytes. The expression of secreted signaling molecules can be monitored while the cells maturate in the shared medium. This opportunity is especially relevant, as astrocytes release soluble factors, such as cytokines, growth factors and extracellular matrix molecules10,11, thereby regulating neuronal growth and function7,12. Thus, it has been demonstrated that the addition of thrombospondin to retinal ganglion cells in vitro induces the formation of synapses13. However, other yet unknown factors are necessary to render synapses functional13. Furthermore, molecules released by astrocytes have to be identified in order to understand the basis of neuron-glia interactions.
The cultivation of primary neurons and astrocytes from mouse and rat has been described previously14-16. Here we present an elegant and versatile tool to combine both cell types in an indirect coculture approach. Since the two cultures are physically separated yet sharing the same medium, the impact of neurons, astrocytes and soluble molecules, can be separately analyzed, thus creating a powerful tool for neuron-glia interaction studies.
Protocol
The experiments with mice were in accordance with the German Law and the German Society for Neuroscience guidelines of animal husbandry. The animal care and utilization committees of the Ruhr-University Bochum have granted the appropriate permits.
1. Preparation and Cultivation of Cortical Astrocytes
Note: Complete these steps of the protocol at least 7 d before proceeding to the next steps, as the astrocyte cultures should develop into confluent monolayers before the neurons are prepared. Primary astrocytes are derived from mixed glial cultures obtained from mouse pups around postnatal day (P) 0-3. Three brains (6 cortices) per T75 flask are to be used.
Prepare the astrocyte medium by supplementing Dulbecco's Modified Eagle Medium (DMEM) with 10% v/v horse serum and 0.1% v/v gentamycin under sterile conditions. Store the medium at 4 °C for up to 2 weeks.
Coat 3x T75 flasks with 10 µg/mL Poly-D-Lysine (PDL; in cell-culture-grade water) for at least 1 h at 37 °C in the presence of 6% v/v CO2. After coating, wash the flasks twice with Phosphate-Buffered Saline (PBS) to remove unbound cations, and add 9 mL astrocyte medium.
For the preparation of the brains, add 10 mL Hank's Balanced Salt Solution (HBSS) to 1x 10 cm Petri dish and 1 mL HBSS to 1x 15 mL tube (regardless of the number of brains that will be used). Keep a pair of surgical scissors, 2 fine forceps and a binocular within reach for the procedure.
- Decapitate 3 pups quickly with the surgical scissors and collect the heads in an empty 10 cm dish.
- Perform the preparation of the cortices under the binocular. Using two fine forceps, remove the dorsal skin from the skull at the midline, starting from the neck up towards the level of the eyes. Thereafter, the brain with its blood vessels is visible under the skull.
- Fix the head at the rostral end with a pair of forceps and incise the midline of the skull, starting at the posterior end. Subsequently, clip off the right and the left halves of the skull to expose the brain.
- Close the tip of the forceps and position it ventrally under the brain. Lift the brain out of the skull and transfer it into the HBSS-filled 10 cm Petri dish. Proceed in the same manner with the remaining specimen until all brains are collected in the dish.
After collecting the brains, start with the separation of the cortices by pinching off the hindbrain using a pair of forceps like a pair of scissors. Perform a midline incision between the two hemispheres with one pair of forceps. Carefully fix the brain with a forceps and cut off the midbrain and the olfactory bulbs using a second forceps to end up with the cortical halves.
Fix one cortical half with one pair of forceps and peel the meninges from the hemispheres by detaching them from the lateral edge and subsequently pulling them from the cortical surface using a second forceps.
Flip the cortical half with its surface oriented down towards the dish; carefully dissect and remove the crescent-shaped hippocampus using one pair of forceps. Transfer the single cortices into the conical 15 mL tube using one closed forceps to carry an individual cortex.
After collecting all cortices, transit to the sterile laminar flow hood and begin with the preparations for the dissociation of the cortical tissue.
Add 1 mL DMEM to a 2 mL reaction tube and add 0.1% w/v papain (30 units). Incubate the suspension in a water bath at 37 °C until a clarified solution develops (about 3 min). Add 0.24 mg/mL L-cysteine and 20 µg/mL DNAse and shake gently. NOTE: For digestion, approximately 1 mL of enzyme solution is needed.
Filter (0.22 µm pore size) to obtain 1 mL sterile solution before adding it to the cortical tissue. Incubate the tubes for 30 min - 1 h at 37 °C (i.e., in a water bath). NOTE: Steps 1.11 - 1.14 are critical and should be completed within 15 min.
To terminate the digestion reaction, add 1 mL astrocyte medium and carefully triturate the digested tissue using a 1 mL pipette. For trituration, gently move the pipette plunger, thereby aspirating and extruding the cortical tissue suspension.
Once the tissue has been dissociated into a single-cell suspension, add 5 mL astrocyte medium and centrifuge for 5 min at 216 x g.
Aspirate the supernatant carefully from the resulting pellet and resuspend the cells in 1 mL astrocyte medium.
Add the cell suspensions to T75 flasks replenished with 9 mL astrocyte medium (see 1.2).
Incubate the cells at 37 °C in the incubator in the presence of 6% v/v CO2. After 4 d, perform the first complete medium change (10 mL). Thereafter, change the culture medium every 2 - 3 d.
After 7 d, check if the cells have formed a confluent monolayer. If not, wait for another 2 - 3 days until complete confluence of the culture is attained. NOTE: Astrocytes display large flattened cell bodies, localize at the bottom of the flask and do not develop notable cellular processes. They differ from the small phase-contrast bright progenitor cells and microglia residing on top of the monolayer17.
In order to get rid of the progenitor cells and obtain a pure astrocyte culture, place the T75 flasks on an orbital shaker and shake the cultures O/N at 250 rpm. Ensure that the temperature is adjusted to 37 °C and that the filter of the flask is sealed with laboratory film to prevent evaporation of the CO2.
The next day, aspirate the medium and add 10 mL fresh culture medium replenished with 20 µM cytosine-1-β-D-arabinofuranoside (AraC) to eliminate residual dividing cells. NOTE: incubation with AraC for 2 - 3 d in the incubator at 37 °C, 6% CO2 will result in a pure astrocyte culture.
Monitor the purity of the astrocyte cultures by visual inspection under the light microscope. If residual phase-contrast bright progenitor cells and microglia still reside on top of the astrocytic monolayer17 (see step 1.16), repeat steps 1.18 and 1.19. NOTE: The confluent and pure astrocyte monolayers can be maintained for up to 14 d in the incubator (37 °C, 6% v/v CO2) before proceeding to the next step. Change half of the medium every 3 d. Do not collect, freeze and thaw the astrocytes, as these cells will lose their neuronal supportive properties through the storage procedure.
2. Preparing Astrocytes for the Indirect Coculture
Note: 48 - 72 h before preparing the neurons, transfer astrocytes to the cell-culture inserts. Generally, a single T75 flask delivers a sufficient number of cells to support the neuronal cultures obtained with one preparation.
Place as many inserts as needed into 24-well-plates. Coat each insert with 10 µg/mL PDL (dissolved in cell-culture-grade water) for about 1 h at 37 °C, 6% v/v CO2. After the incubation, wash the inserts twice with PBS. In the meantime, proceed with the preparation of astrocytes. NOTE: The inserts filled with PBS can be kept until the astrocyte cell suspension is ready for use.
Aspirate the astrocyte medium (with AraC) and wash the culture one time with 10 mL PBS to ensure any residual serum is removed.
Add 3 mL of 0.05% trypsin-EDTA (Ethylenediaminetetraacetic acid) to the flasks and incubate the cells for trypsinization at 37 °C, 6% v/v CO2 for approximately 10 min.
Control the cell detachment by visual inspection using a microscope and facilitate the detachment of the cells by tapping the side of the flasks against the desktop. NOTE: Steps 2.5 - 2.8 are critical and should be finished within 15 - 20 min.
Following the trypsinization, gently re-suspend the cells in 7 mL astrocyte medium and transfer the cell suspension to a 15 mL tube. Centrifuge for 5 min at 216 x g.
Carefully aspirate the supernatant and resuspend the cell pellet in 1 mL astrocyte medium.
Count the cells using a counting chamber.
Fill the bottom of the wells of the 24-well-plate with 500 µL astrocyte medium and transfer 25,000 cells in 500 µL astrocyte medium into each individual insert. Incubate the culture at 37 °C, 6% v/v CO2. NOTE: After 42 - 72 h the cultures will reach approximately 100% confluence. At this stage the purity of the culture can be verified by immunocytochemistry11. The confluent cultures can be maintained for up to 7 d until proceeding to the next step. Change half of the medium every 3 d.
3. Preparation of Primary Hippocampal Neurons
Note: Primary mouse hippocampal neurons should be derived from E15.5 - E16 embryos of timed pregnant mice.
Prepare neuron medium (MEM with 10 mM sodium pyruvate, 0.1% w/v ovalbumin, 2% v/v B27 medium supplement, and 0.1% v/v gentamicin (sterile filtered)), and preparation medium (HBSS with 0.6% w/v glucose and 10mM HEPES (4-(2-hydroxyethyl)-1-piperazine ethanesulfonic acid)). Pre-warm and pre-equilibrate the neuron medium in filter flasks at 37 °C, 6% v/v CO2.
- Place the glass-coverslips (12 mm diameter) into the wells of 24-well-plates (use the special coverslips, which are notched for the use with cell culture inserts).
- Coat for at least 1 h with 15 µg/mL Poly-DL-Ornithine in water (cell-culture-grade) at 37 °C. Use at least 500 µL per well to ensure that the coverslips are completely covered. Wash the wells twice with PBS. Leave the PBS in the wells until plating the cells. Ensure that the wells do not dry out.
To dissect the hippocampi, prepare 3x 10 cm dishes filled with preparation medium and 1x 2 mL tube with 1 mL preparation medium. Prepare scissors and 2 fine forceps.
Sacrifice the pregnant mouse by cervical dislocation, sterilize the abdomen by washing with 70% v/v ethanol, and open the abdomen using sharp scissors. Excise the beaded uterus carefully with scissors and transfer the organ into 1x 10 cm dish filled with preparation medium.
Next, remove the embryos from the uterus. Carefully incise the uterus and remove the amnion without harming the embryos by using 2 forceps. Thereafter, decapitate each embryo with a pair of scissors and transfer the heads into a second 10 cm dish supplied with preparation medium.
- Proceed with the preparation of the heads (similar to the preparation described in 1.4 - 1.7).
- Immobilize the head with a pair of forceps and incise the skull and skin in the neck region close to the hindbrain, perpendicular to the neuraxis. Use a second pair of forceps to lift both skull and covering skin, and bend the soft skull cap towards the rostral end of the head, thereby exposing the brain.
- With the help of 2 forceps, detach the brain from the cranial cavity. To do this, close 1 pair of forceps and separate the brain from the skull starting at the olfactory bulbs and moving towards the hindbrain. Collect the brains in another 10 cm dish filled with preparation medium.
Dissect the hippocampi from the cortex moieties as described above for the astrocyte preparation (step 1.6). Remove the hippocampi from each hemisphere and collect them in the 2 mL tube filled with preparation medium.
After completing the dissection, transfer the tube to the sterile laminar flow bench and digest the hippocampal tissue with 1 mL digestion solution containing papain. Prepare the digestion solution as described in step 1.9 - 1.10, but use MEM instead DMEM. NOTE: Steps 3.9 - 3.12 are critical and should be completed within 10 - 15 min.
After completion of digestion, carefully withdraw the digestion solution by gentle suction with a pipette.
Wash the hippocampi 3 times with neuron medium by sequentially adding and thereafter carefully aspirating 1 mL fresh culture medium per wash cycle. Do not centrifuge the cell suspension.
After the final washing step, carefully triturate the tissue in 1 mL neuron medium.
Count the cells using a counting chamber and plate 35,000 cells in 500 µL neuron medium per a single well of a 24-well-plate (the plate mentioned in 3.2). NOTE: Optionally, a cell aliquot can be counterstained with trypan blue to assess the vitality of the cell preparation. Control the plating density by visual inspection using the microscope (typically 90 - 110 cells per visual field of a standard 10X objective).
Place neurons in the incubator at 37 °C, 6% v/v CO2 for 1 h.
Post incubation, take the prepared inserts (seeded with confluent astrocyte monolayers) out of the incubator (see step 2.8). Exchange the medium by aspirating the astrocyte medium and replacing it with 500 µl fresh neuron medium. NOTE: The inserts should harbor confluent astrocyte monolayers after 2 - 3 DIV (Days In Vitro).
Place the inserts with astrocytes carefully into the wells that contain the neuron cultures (step 3.13) using sterile forceps.
Place the resulting indirect neuron-astrocyte coculture back into the incubator at 37 °C, 6% v/v CO2. NOTE: Under these conditions, neurons can be cultivated for up to 4 weeks in completely defined medium. No medium change is necessary. For long-time cultures, it is recommended to fill empty wells with sterile water in order to reduce the evaporation from the 24-well-plate.
Perform in vitro cell analyses after desired culture periods (e.g., for immunocytochemistry, PCR, Western blot, electrophysiological recordings (patch clamp or multielectrode array))7,15,18. NOTE: The culture system is also suitable for acute and chronic pharmacological treatment of the cultures11.
Representative Results
The analysis of the neuronal cultures via the indirect coculture system is multifarious and can be performed at different stages of culture maturation. Due to the fact that the cells can be maintained for up to 4 weeks, long-term investigations of the cultures are possible.
The schematic in the middle left panel of Figure 1 demonstrates the cocultivation setup. With the use of this system, live cell imaging of both cell types can be performed, as exemplified by phase contrast pictures of the astrocyte monolayer (top left panel) and the neuronal network (bottom left panel). After fixation, immunocytochemical assessment is possible, as shown in top right and bottom right panels.
Neurons begin to establish synaptic connections starting at approximately 7 DIV in our model (data not shown). By 14 DIV multiple synapses are formed within neuronal networks, as identified by the combined immunocytochemical detection of pre and postsynaptic markers (Figure 1, bottom right panel). Importantly, not only the amount of synaptic marker puncta can be visualized and quantified using this approach, but also the structurally completed synapses, which are most likely to contribute to network connectivity.
The astrocyte cultures not only provide trophic support to neurons11, but also synthesize several secreted factors19,20 involved in neural plasticity. The expression of one of these factors, namely matrix metalloproteinase 2 (MMP2), is demonstrated in the top right panel of Figure 1. Interestingly, 2 distinct MMP2-isoforms were detected in the coculture medium using Western Blot (Figure 1, middle right panel). Potentially astrocytes secrete MMP2 into the shared medium, thus affecting the plasticity of neurons. Also multiple isoforms of Tenascin-C, a known regulator of axon outgrowth, cell migration and differentiation21 were identified.
Taken together, these results demonstrate two examples and thereby provide proof of principle for the variety of investigations of astrocytes, neurons and their reciprocal communication that can be realized using the indirect cocultivation method described herein.
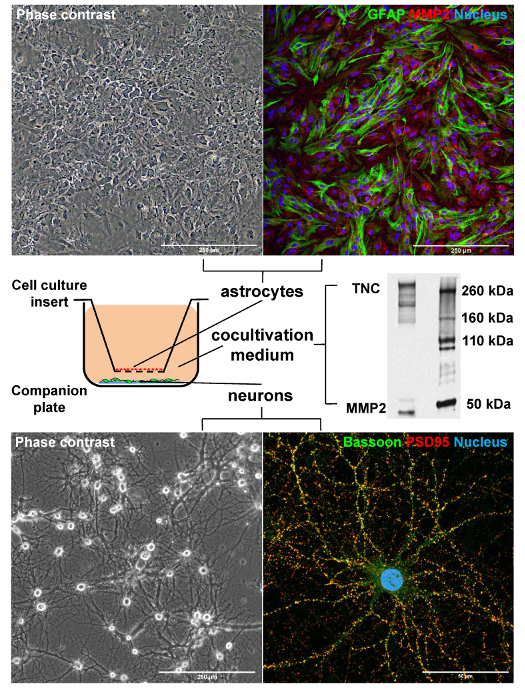 Figure 1: The Indirect Astrocyte-Neuron Coculture System Allows for Separate Analysis of Astrocytes, Neurons and Secreted Molecular Mediators. Top panel, left: the astrocytes form monolayers on the cell culture insert membrane, phase contrast; scale bar: 250 microns. Top panel, right: astrocytes are immunostained for Glial Acidic Fibrillar Protein (GFAP) (mouse clone GA5) and Matrix Metalloprotease 2 (MMP2) (rabbit polyclonal); scale bar: 250 microns. Middle panel, left: the scheme illustrates the indirect coculture setup. Although two cultures are physically separated, they share the same medium. Middle panel, right: two secreted molecular mediators of neuron-glia interactions are revealed in the co-cultivation medium using Western Blot. Multiple isoforms of Tenascin C (TNC), a neurite outgrowth regulator, and MMP2, an extracellular matrix modifier, are documented. Bottom panel, left: primary neurons develop highly interconnected networks by the 14th day of cultivation, phase contrast; scale bar: 250 microns. Bottom panel, right: starting with 14 d in vitro, multiple synaptic connections are established between neurons, as detected by immunocytochemical labeling; scale bar: 50 microns. The co-localization of the presynaptic marker Bassoon (rabbit polyclonal) with the postsynaptic PSD95 scaffolding protein (mouse clone 6G6-1C9) documents the structurally completed synapses formation. Please click here to view a larger version of this figure.
Figure 1: The Indirect Astrocyte-Neuron Coculture System Allows for Separate Analysis of Astrocytes, Neurons and Secreted Molecular Mediators. Top panel, left: the astrocytes form monolayers on the cell culture insert membrane, phase contrast; scale bar: 250 microns. Top panel, right: astrocytes are immunostained for Glial Acidic Fibrillar Protein (GFAP) (mouse clone GA5) and Matrix Metalloprotease 2 (MMP2) (rabbit polyclonal); scale bar: 250 microns. Middle panel, left: the scheme illustrates the indirect coculture setup. Although two cultures are physically separated, they share the same medium. Middle panel, right: two secreted molecular mediators of neuron-glia interactions are revealed in the co-cultivation medium using Western Blot. Multiple isoforms of Tenascin C (TNC), a neurite outgrowth regulator, and MMP2, an extracellular matrix modifier, are documented. Bottom panel, left: primary neurons develop highly interconnected networks by the 14th day of cultivation, phase contrast; scale bar: 250 microns. Bottom panel, right: starting with 14 d in vitro, multiple synaptic connections are established between neurons, as detected by immunocytochemical labeling; scale bar: 50 microns. The co-localization of the presynaptic marker Bassoon (rabbit polyclonal) with the postsynaptic PSD95 scaffolding protein (mouse clone 6G6-1C9) documents the structurally completed synapses formation. Please click here to view a larger version of this figure.
Discussion
The main goal of the current protocol is to completely separate neuronal and astrocytic cultures, while maintaining them in shared medium. For this reason, the purity of the cultures obtained should be verified at the beginning of the procedure. We recommend the use of neuron-specific tubulin, neurofilaments or NeuN protein as neuronal markers, GFAP as astrocytic marker, O4 antigen as oligodendrocyte precursor marker and Iba1 protein to identify microglia.
Pay special attention when performing a critical step of the protocol, as specified by a 'NOTE' before the step description. Take into consideration that both primary cultures of neurons and astrocytes are rather sensitive. Therefore, several problems may arise at the beginning of the procedure. If the astrocytes do not reach a confluent monolayer in T75 flasks after 10 DIV (protocol section 1), check the components of the astrocyte medium (for expiration date). In addition, increase the amount of cortical tissue prepared to initiate the culture (up to 10 cortices per flask). If the astrocytes do not reach a confluent monolayer on cell culture inserts after 72 h (protocol section 2), check the components of the astrocyte medium and PDL (for expiration date).Try to plate out freshly prepared cultures, determine the proportion of viable cells prior to plating and adjust cell numbers accordingly (e.g., use trypan blue counterstaining). If low survival of neurons is detected after 24 h, hippocampus tissue was not triturated gently enough (protocol step 3.11).Pay attention to avoid the formation of bubbles when dissociating the tissue. If survival of neurons is significantly lowered after 7 - 14 DIV and if neurons form aggregates of 3 - 5 cells after 7 DIV, use freshly prepared inserts with astrocytes, check for astrocyte purity by immunocytochemistry, and check the neuron medium components. If big aggregates of neurons (more than 10 cells) are visible, it is possible that the hippocampus tissue was not sufficiently triturated, resulting in incomplete dissociation (protocol step 3.11). Pay attention to suspension homogeneity. If neuronal culture is contaminated with non-neuronal cells (microglia, astrocytes, oligodendrocyte precursor cells, etc.), completely remove the neighboring cortical tissue adjacent to the hippocampus (step 3.7) and also check the neuron medium components.
The proposed indirect coculture of primary neurons and astrocytes provides a versatile tool for the long-term investigation of neuron-glia interactions. Importantly, this protocol is optimized for mouse cells, which opens perspective for the implementation of mouse genetic models. Because of the complete physical separation of the 2 cell types, neurons and astrocytes of distinct genotypes can be combined in the desired way.
Among a variety of applications, this approach can be used to investigate neural networks development, synaptogenesis, network activity and astrocyte-neuron signaling. Although the indirect cocultivation of neurons and astrocytes opens vast opportunities for their segregated analysis and long-range signaling, the lack of direct contact may compromise the subtle regulation of synaptic plasticity22. Thus, the lack of interaction between astrocytic protrusions and neuronal synapses has to be treated carefully when using this model.
The assay is based on two previous protocols for astrocyte and neuronal preparations14,17 and has been improved in the course of the past years. The approach originally has been established for mouse7 and rat11 cells. Previous publications of the laboratory provide a detailed characterization of the assays concerning cell survival, synapse formation and electrophysiological characterization of the resulting cells7,11,15,18. Furthermore, the assay can be adapted to other settings, such as the micro-electrode-arrays (MEAs)7. For future applications, the reader should consider other protocols for the detailed investigation of synapse formation23, as well as for MEA analysis in vitro. In conclusion, the assay provides a versatile tool for laboratories focusing on various aspects of neuron-glia interactions.
Disclosures
The authors have nothing to disclose.
Acknowledgments
The present work was supported by the German research foundation (Deutsche Forschungsgemeinschaft DFG: GRK 736, Fa 159/22-1; the research school of the Ruhr University Bochum (GSC98/1) and the priority program SSP 1172 "Glia and Synapse", Fa 159/11-1,2,3).
References
- Volterra A, Meldolesi J. Astrocytes, from brain glue to communication elements: the revolution continues. Nat Rev Neurosci. 2005;6:626–640. doi: 10.1038/nrn1722. [DOI] [PubMed] [Google Scholar]
- Barreto GE, Gonzalez J, Torres Y, Morales L. Astrocytic-neuronal crosstalk: implications for neuroprotection from brain injury. Neurosci Res. 2011;71:107–113. doi: 10.1016/j.neures.2011.06.004. [DOI] [PubMed] [Google Scholar]
- Araque A, Carmignoto G, Haydon PG. Dynamic signaling between astrocytes and neurons. Annu Rev Physiol. 2001;63:795–813. doi: 10.1146/annurev.physiol.63.1.795. [DOI] [PubMed] [Google Scholar]
- Dityatev A, et al. Activity-dependent formation and functions of chondroitin sulfate-rich extracellular matrix of perineuronal nets. Dev Neurobiol. 2007;67:570–588. doi: 10.1002/dneu.20361. [DOI] [PubMed] [Google Scholar]
- Robinette BL, Harrill JA, Mundy WR, Shafer TJ. In vitro assessment of developmental neurotoxicity: use of microelectrode arrays to measure functional changes in neuronal network ontogeny. Front Neuroeng. 2011;4:1. doi: 10.3389/fneng.2011.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voigt T, Opitz T, de Lima AD. Synchronous Oscillatory Activity in Immature Cortical Network Is Driven by GABAergic Preplate Neurons. J Neurosci. 2001;21(22):8895–8905. doi: 10.1523/JNEUROSCI.21-22-08895.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissler M, Faissner A. A new indirect co-culture set up of mouse hippocampal neurons and cortical astrocytes on microelectrode arrays. J Neurosci Methods. 2012;204:262–272. doi: 10.1016/j.jneumeth.2011.11.030. [DOI] [PubMed] [Google Scholar]
- Yu CY, et al. Neuronal and astroglial TGFbeta-Smad3 signaling pathways differentially regulate dendrite growth and synaptogenesis. Neuromolecular Med. 2014;16:457–472. doi: 10.1007/s12017-014-8293-y. [DOI] [PubMed] [Google Scholar]
- Yu P, Wang H, Katagiri Y, Geller HM. An in vitro model of reactive astrogliosis and its effect on neuronal growth. Methods Mol Biol. 2012;814:327–340. doi: 10.1007/978-1-61779-452-0_21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucukdereli H, et al. Control of excitatory CNS synaptogenesis by astrocyte-secreted proteins Hevin and SPARC. PNAS. 2011;108:E440–E449. doi: 10.1073/pnas.1104977108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyka M, Busse C, Seidenbecher C, Gundelfinger ED, Faissner A. Astrocytes are crucial for survival and maturation of embryonic hippocampal neurons in a neuron-glia cell-insert coculture assay. Synapse. 2011;65:41–53. doi: 10.1002/syn.20816. [DOI] [PubMed] [Google Scholar]
- Navarrete M, Araque A. Basal synaptic transmission: astrocytes rule! Cell. 2011;146:675–677. doi: 10.1016/j.cell.2011.08.006. [DOI] [PubMed] [Google Scholar]
- Christopherson KS, et al. Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell. 2005;120:421–433. doi: 10.1016/j.cell.2004.12.020. [DOI] [PubMed] [Google Scholar]
- Kaech S, Banker G. Culturing hippocampal neurons. Nat Protoc. 2006;1:2406–2415. doi: 10.1038/nprot.2006.356. [DOI] [PubMed] [Google Scholar]
- Geissler M, et al. Primary hippocampal neurons, which lack four crucial extracellular matrix molecules, display abnormalities of synaptic structure and function and severe deficits in perineuronal net formation. J Neurosci. 2013;33:7742–7755. doi: 10.1523/JNEUROSCI.3275-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzyubenko E, Gottschling C, Faissner A. Neuron-Glia Interactions in Neural Plasticity: Contributions of Neural Extracellular Matrix and Perineuronal Nets. Neural Plast. 2016;2016:5214961. doi: 10.1155/2016/5214961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy KD, de Vellis J. Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J Cell Biol. 1980;85:890–902. doi: 10.1083/jcb.85.3.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyka M, et al. Chondroitin sulfate proteoglycans regulate astrocyte-dependent synaptogenesis and modulate synaptic activity in primary embryonic hippocampal neurons. Eur J Neurosci. 2011;33:2187–2202. doi: 10.1111/j.1460-9568.2011.07690.x. [DOI] [PubMed] [Google Scholar]
- Eroglu C. The role of astrocyte-secreted matricellular proteins in central nervous system development and function. J Cell Commun Signal. 2009;3:167–176. doi: 10.1007/s12079-009-0078-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ethell IM, Ethell DW. Matrix metalloproteinases in brain development and remodeling: synaptic functions and targets. J Neurosci Res. 2007;85:2813–2823. doi: 10.1002/jnr.21273. [DOI] [PubMed] [Google Scholar]
- Theocharidis U, Long K, ffrench-Constant C, Faissner A. In: Prog Brain Res. Dityatev Alexander, Wehrle-Haller Bernhard, Asla Pitkänen., editors. Vol. 214. Elsevier; 2014. pp. 3–28. [DOI] [PubMed] [Google Scholar]
- Dityatev A, Rusakov DA. Molecular signals of plasticity at the tetrapartite synapse. Curr Opin Neurobiol. 2011;21:353–359. doi: 10.1016/j.conb.2010.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ippolito DM, Eroglu C. Quantifying synapses: an immunocytochemistry-based assay to quantify synapse. JoVe. 2010. [DOI] [PMC free article] [PubMed]