

Abstract
Lymphocytes respond to a variety of stimuli by activating intracellular signaling pathways, which in turn leads to rapid cellular proliferation, migration and differentiation, and cytokine production. All of these events are tightly linked to the energy status of the cell, and therefore studying the energy-producing pathways may give clues about the overall functionality of these cells. The extracellular flux analyzer is a commonly used device for evaluating the performance of glycolysis and mitochondrial respiration in many cell types. This system has been used to study immune cells in a few published reports, yet a comprehensive protocol optimized particularly for lymphocytes is lacking. Lymphocytes are fragile cells that survive poorly in ex vivo conditions. Oftentimes lymphocyte subsets are rare, and working with low cell numbers is inevitable. Thus, an experimental strategy that addresses these difficulties is required. Here, we provide a protocol that allows for rapid isolation of viable lymphocytes from lymphoid tissues, and for the analysis of their metabolic states in the extracellular flux analyzer. Furthermore, we provide results of experiments in which the metabolic activities of several lymphocyte subtypes at different cell densities were compared. These observations suggest that our protocol can be used to achieve consistent, well-standardized results even at low cell concentrations, and thus it may have broad applications in future studies focusing on the characterization of metabolic events in immune cells.
Keywords: Immunology, Issue 117, extracellular flux, mouse, lymphocytes, T cells, B cells, metabolism, glycolysis, respiration, mitochondria, magnetic separation
Introduction
The immune response against antigens is a tightly regulated balance between immune activation and immune suppression. Immune activation drives rapid cell proliferation and migration, as well as cytokine production, antibody secretion and increased phagocytosis in response to the stimulant, whereas immune suppression simply inhibits these events and therefore is important in preventing unnecessary immune responses1-6. Recent studies have shown that a direct link exists between the activation status of immune cells and the activity of various metabolic pathways7. Immune cells can shift between resting and activated states by switching energy producing pathways on and off. Furthermore, it has been observed that different immune cell types may use different metabolic strategies to fuel their increased energy needs during activation. For example, while activation of T lymphocytes directs cells into an almost completely glycolytic state8, activated B lymphocytes use a balance of glycolysis and oxidative phosphorylation9,10. These studies point out the importance of investigating the effects of immune cell activation on cellular metabolism.
Real time, simultaneous measurements of oxygen consumption rate (OCR) and extracellular acidification rate (ECAR), as indicators of oxidative phosphorylation and glycolysis, is a common strategy to address the states of energy producing pathways11-13. In order to achieve this, an extracellular flux analyzer, such as the Seahorse XF 96, is routinely used. Such an instrument can rapidly compare changes in OCR and ECAR across cell types or upon different stimulation conditions. So far, various cell types, including immune cells, have been studied using these devices. However, an optimized protocol specifically designed for immune cells is not available.
Immune cells, particularly lymphocytes, differ from other cell types in several critical ways. Lymphocytes are fragile cells that do not survive for long durations in ex vivo conditions14-16. This is an even greater issue when they are cultured in suboptimal growth media lacking essential nutrients, such as those used in extracellular flux analysis. Unlike macrophages and many cell lines, lymphocytes do not adhere to plastic surfaces; therefore it is critical to attach them to the analysis plate without generating stress. Finally, some lymphocyte subpopulations may be extremely rare and harvesting them at the required, optimal amounts might be challenging17-19.
Here, we provide an optimized protocol that is specifically developed for lymphocytes. Using splenocytes, B lymphocytes and naïve CD4+ T lymphocytes isolated from mouse spleen and lymph nodes20, we show the characteristics of their resting state glycolysis and oxidative phosphorylation at different cell densities. Data were normalized to account for the differences in the initial cell numbers for each well by measuring the end assay cell lysate protein concentrations, which were directly proportional to the cell numbers. Our protocol not only provides guidelines for the rapid isolation of viable lymphocytes for extracellular flux assays, but it also allows for working at suboptimal cell concentrations without compromising the data quality.
Protocol
All animal experiments were carried out in accordance with the LIG-4 animal protocol, which was approved by the NIAID Animal Care and Use Committee.
1. Preparation of Reagents
Prepare magnetic separation (MS) buffer: PBS (pH 7.2) supplemented with 0.5% BSA and 2 mM EDTA or automatic separation solution supplemented with 0.5% BSA. Sterile filter (0.22 µm) and store at 2-8 °C.
Prepare RF10 medium: RPMI 1640 medium supplemented with 10% heat-inactivated fetal bovine serum, 50 U/ml penicillin, 50 µM streptomycin, 1 mM sodium pyruvate, 2 mM L-glutamine, 0.1 mM non-essential amino acids, 50 µM 2-mercaptoethanol, 10 mM HEPES. Use sterile reagents. Sterile filter and store at 2-8 °C.
Prepare mitochondrial stress test medium: XF base medium supplemented with 1 mM sodium pyruvate, 2 mM L-glutamine and 25 mM glucose.
Prepare glycolysis stress test medium: XF base medium supplemented with 2 mM L-glutamine.
Adjust the pH of both the mitochondrial and glycolysis stress test media to 7.4, sterile filter and store at 2-8 °C. Warm media to 37 °C and re-adjust the pH before use (carry out pH measurements at 37 °C). NOTE: Using XF medium, which lacks bicarbonate, is critical. Since the atmosphere in the extracellular flux analyzer contains only ambient CO2, any bicarbonate in the medium, upon binding to protons released as a byproduct of glycolysis, will dissociate to form CO2 and water. CO2 will degas during the experiment, causing drifts in pH that would obscure the glycolytic acidification signal.
Prepare oligomycin: Prepare 10 mM stock solution in DMSO; store at -20 °C.
Prepare 2,4-DNP: Prepare 1 M stock solution in DMSO; store at -20 °C.
Prepare antimycin A: Prepare 10 mM stock solution in DMSO; store at -20 °C.
Prepare rotenone: Prepare 10 mM stock solution in DMSO; store at -20 °C.
Prepare glucose: Dissolve glucose in glycolysis stress test medium to form a 250 mM solution; prepare fresh before each experiment and do not freeze.
Prepare 2-DG: Dissolve solid 2-DG in glycolysis stress test medium to form a 500 mM solution; prepare fresh before each experiment and do not freeze.
2. Harvesting Spleen and Lymph Nodes from Mice
Euthanize the mouse by CO2 asphyxiation followed by cervical dislocation.
Spray the mouse with 70% ethanol.
For subsequent T cell isolation, harvest the spleen, superficial cervical, superficial/deep axillary, mandibular, inguinal, and mesenteric lymph nodes.
For subsequent B cell isolation, harvest only the spleen. NOTE: Use a biosafety cabinet and keep the harvested organs in PBS or RF10 media on ice until processing. Use sterile labware and sterile technique for all processing and isolation steps. Keep all buffers and media on ice to increase isolation efficiency and to reduce cell death.
3. Tissue Processing
Place a 70 µm cell strainer over a 50 ml conical tube and transfer the organs onto the strainer. For T cell isolation, combine all the organs and for B cell isolation transfer only the spleen.
Using the plunger from a sterile 3 ml syringe, mash the tissue through the strainer. During mashing, make sure that the filter and the organs are moist at all times and wet them as required by adding MS buffer. This will both keep cell viability high and prevent clogging of the filter.
After mashing, apply 5-10 ml MS buffer to the filter to increase cell recovery. Centrifuge the suspension at 400 x g for 5 min at 4 °C. (Carry out all further centrifugations under these conditions, unless specified).
Decant the liquid and resuspend the pellet in 5 ml ACK red blood cell lysis buffer. Incubate for 5 min on ice to lyse red blood cells (red blood cells need to be removed because they interfere with the magnetic separation). Add 10-20 ml MS buffer and centrifuge.
Place a 30 µm cell filter over a 15 ml conical tube and prime it with 1 ml MS buffer (this filtration will remove debris from the erythrocyte lysis). Decant the liquid from the centrifuged tube, resuspend the pellet in 5 ml MS buffer and pass it through the filter. Wash the initial tube with 4 ml MS buffer to increase cell recovery, and use this to rinse the filter.
Using a hemocytometer or an automated cell counter, determine the total number of cells. This cell number will be used to determine the amount of magnetic separation reagents required in Section 4. If splenocytes are to be used in the subsequent extracellular flux assay, set aside the appropriate number of cells at this time.
Centrifuge the suspension and proceed to magnetic separation. Resuspend splenocytes not used for B cell isolation in 5 ml RF10 and keep on ice until the extracellular flux assay.
4. Magnetic Separation of B Cells and Naïve CD4+ T Cells
Determine the necessary volumes of biotin-antibody cocktail, anti-biotin microbeads, and MS buffer. Use the following volumes for 108 cells as a guide and scale up or down as necessary. NOTE: Incubate samples in a refrigerator rather than on ice since incubating on ice may reduce antibody binding efficiency and longer incubation times might be required.
| Reagents for naïve CD4+ T cell separation | Volume for 108 cells |
| (scale appropriately) | |
| Biotin-antibody cocktail | 100 μl |
| MS buffer for first incubation | 400 μl |
| Anti-biotin microbeads | 200 μl |
| CD44 microbeads | 100 μl |
| MS buffer for second incubation | 200 μl |
| Incubate the biotin-antibody cocktail and cells at 4 °C for 5 min. Add anti-biotin microbeads, CD44 microbeads, MS buffer and incubate at 4 °C for 15 min. |
Table 1: T cell isolation volumes and instructions.
| Reagents for B cell separation | Volume for 108 cells |
| (scale appropriately) | |
| Biotin-antibody cocktail | 100 μl |
| MS buffer for first incubation | 400 μl |
| Anti-biotin microbeads | 200 μl |
| MS buffer for second incubation | 300 μl |
| Incubate the biotin-antibody cocktail and cells at 4 °C for 15 min. Add the appropriate volume of biotin-antibody cocktail and MS buffer, and incubate the mixture at 4 °C for 15 min. Add the appropriate volume of anti-biotin microbeads and MS buffer and incubate the mixture at 4 °C for 15 min. After the second incubation, fill the tube with MS buffer and centrifuge. |
Table 2: B cell isolation volumes and instructions.
Resuspend the pellet in MS buffer: 500 µl for manual separation, 3-4 ml for automatic separation.
- Continue with either manual or automated separations as follows:
- Manual Separation:
- Insert an LS column into the separation magnet, place a sterile 15 ml conical tube below to collect the flow through, and prime the column by washing with 3 ml MS buffer. Discard the flow through.
- Transfer the cell suspension to the primed LS column and allow it to pass through; wash the tube used during incubation with 3 ml MS buffer and pass this through the column as well. The eluate will contain the purified cells. For B cell isolation, pass the purified cells through the column a second time into a new 15 ml tube, and repeat the washing step. This will increase purity and yield.
- Automated Separation:
- Switch on the automatic separator and check the levels of running buffer, rinsing solution and 70% Ethanol. Ensure that the waste is empty before starting the separation.
- Select the appropriate chilled rack depending on the tube size of the unpurified sample (e.g., 15 ml). In order to keep the cells viable throughout the separation process, use racks that have been previously chilled at 2-8 °C for 3-4 hr, or until the coolant becomes solid.
- Mount the chilled rack on the sample stage of the automatic separator.
- Place the sample tube in slot A1, a tube for the negative fraction in slot B1, and a tube for the positive fraction in C1. If collecting more than one sample, place additional tubes in the next columns (A2, B2, C2, etc.).
- Select the separation tab on the touchscreen, and indicate the arrangement of tubes on the rack. From the separation menu, select the "Depletes" program and complete the program cycle with the appropriate type of wash (see note below). NOTE: The "Depletes" program carries out depletion using the machine's most sensitive mode, which prioritizes purity. Additionally, there are three different wash options: quick rinse, rinse, and sleep. If the samples are from the same source and are to be combined, select quick rinse. If the samples are to be kept separate, select rinse. Select the sleep option if no further isolations will be carried out that day and if the machine will be shut down following isolation
- Start the isolation by pressing "Run," and confirm the buffer levels when prompted. After the program has ended, the negative fraction will contain purified cells.
- Fill the conical tube up to 15 ml with MS buffer and centrifuge.
- Decant the MS buffer and resuspend cells in 5 ml RF10 media. Keep cells on ice until the extracellular flux assay. NOTE: Ideally, the extracellular flux assay should be carried out immediately after isolation. However, in preliminary experiments no significant differences in assay outcomes were observed in cells used within 3 hr of isolation versus freshly-isolated ones.
5. Extracellular Flux Assay
NOTE: The protocol outlined here is for the 96-well format of the instrument. Volumes will need to be adjusted if another format is used.
- Hydration of Sensor Cartridge
- Lift up the sensor cartridge, fill each well of the utility plate with 200 µl of calibrant solution and lower the sensor cartridge onto the utility plate, submerging the sensors in the calibrant solution. NOTE: Verify that the calibrant solution level is high enough to keep the sensors submerged. The cartridge harbors fluorophores associated to O2 and H+ for each well; these need to be hydrated in order for them to work properly.
- Place the cartridge in a 37 °C incubator not supplemented with CO2 or oxygen/nitrogen. Carry out all further incubations under these conditions, unless specified. Incubate the cartridge overnight to hydrate.
- Preparation of Adhesive-coated Plates
- Prepare 2.5 ml of a 22.4 µg/ml adhesive solution in 0.1 M sodium bicarbonate, pH 8.0 (bicarbonate provides the optimum pH for adhesive adsorption).
- Apply 25 µl of the solution to each well of the assay plate, and incubate on the bench at room temperature for 20 min.
- Aspirate adhesive and wash each well twice using 200 µl of sterile water. Wait until wells are dry before seeding the cells.
- Seeding Cells in Adhesive-coated Plates
- Centrifuge cells at room temperature at 200 x g for 5 min. Resuspend cells in 5 ml of the appropriate assay medium (see above for formulation of mitochondrial stress and glucose stress media). Centrifuge cells again and resuspend at the desired concentration in assay medium (resuspension volume will vary based on concentration; each well will contain 180 µl of cell suspension). NOTE: As long as the appropriate assay media and compounds are used, the extracellular flux analyzer can simultaneously run the mitochondrial and glycolysis stress tests on the same plate.
- Plate 180 µl of cell suspension in each well. Use wells A1, A12, H1, and H12 for background temperature correction: add 180 µl of assay medium in these wells (no cells).
- Incubate plates for 25 min at 37 °C.
- Centrifuge the plate at 200 x g for 5 min with no brake to ensure that all cells have completely attached. Visually confirm that the cells are stably adhered to the culture surface, forming a monolayer, by viewing under the microscope. Transfer the plates back to the incubator for an additional 30 min.
- Preparation of 10x Concentrations of Compounds, and Loading of Injector Ports
- For the mitochondrial stress test, prepare 2.5 ml each of 10 µM oligomycin, 1 mM DNP, and a mixture of 10 µM rotenone and 10 µM antimycin A, all in mitochondrial stress assay medium. NOTE: The concentration of 2,4-DNP is based on previously-published studies;21 this is a general guideline but the concentration of uncoupler for each cell type used should be determined by the researcher.
- For the glycolysis stress test, prepare 2.5 ml each of 250 mM glucose, 10 µM oligomycin, and 500 mM 2-deoxyglucose (2-DG) in glycolysis stress assay medium.
- Warm 10x solutions to 37 °C, and adjust pH to 7.4.
- Load the compounds into the appropriate injector ports of the cartridge using a multichannel micropipettor and the loading guides, as follows:
Port Mitochondrial Stress Assay Glycolysis Stress Assay A 20 µl oligomycin 20 µl glucose B 22 µl 2,4-DNP 22 µl oligomycin C 25 µl antimycin A/rotenone 25 µl 2-DG - Incubate the cartridge while setting up the program.
- Setting Up Extracellular Flux Assay Protocols
- Set up the following program (Table 4).
Step Loop Calibration –– Equilibration –– Baseline readings 3 times: Mix 3 min, wait 0 min, measure 3 min End loop –– Inject Port A –– Measurements 3 times: Mix 3 min, wait 0 min, measure 3 min End loop –– Inject Port B –– Measurements 3 times: Mix 3 min, wait 0 min, measure 3 min End loop –– Inject Port C –– Measurements 3 times: Mix 3 min, wait 0 min, measure 3 min End loop –– End Program –– - Begin the program. After the calibration step, replace the calibrant plate for the assay plate (when prompted). NOTE: Using the software, it is possible to indicate groups of wells that have similar conditions. Additionally, it is crucial to indicate which wells are the control wells (in this protocol, they are the four corners of the plate) and to indicate which wells are empty. For a more detailed, step-by-step, protocol for setting up the machine and its software, the manufacturer's website should be consulted.
- Measuring Protein Content
- Remove the remaining assay medium from each well without disturbing the cells. NOTE: If it is not convenient to proceed with the protein concentration assay, it is possible to freeze the whole plate at -20 °C until analysis. If frozen, thaw before continuing.
- Prepare a 1x solution of protease inhibitors (100x stock) in RIPA lysis medium (sufficient for 50 µl/well).
- Add 50 µl RIPA lysis medium supplemented with protease inhibitors to each well. Agitate plate on a shaker for 5 min, and then incubate plate on ice for 30 min to fully lyse cells.
- Spin the plate at 200 x g for 5 min at room temperature to bring lipids and other molecules to the bottom of the plate so that they do not interfere with the bicinchoninic acid (BCA) assay.
- Measure protein concentration by BCA assay according to manufacturer's recommendations.
Representative Results
Eukaryotic cells use an integrated network of glycolysis, the tricarboxylic acid (TCA) cycle, and oxidative phosphorylation to meet the majority of their energy demands and to provide intermediates required for cell growth and proliferation. These pathways start with the intracellular trapping of free glucose in the form of glucose-6-phosphate, which subsequently gets processed into pyruvate. Pyruvate is either reduced to lactate or transported into the mitochondria, where it forms acetyl coenzyme A (CoA). Acetyl CoA then enters into the TCA cycle. High-energy intermediates of the TCA cycle propel the movement of electrons in the electron transport chain (ETC), which in turn expels H+ from the mitochondrial matrix to generate a H+ gradient across the inner mitochondrial membrane. Oxygen acts as the final electron acceptor, and H+ return back to the mitochondrial matrix through the FO/F1 complex, where their potential energy is used to generate ATP (Figure 1A).
The working principle of the extracellular flux assay depends on interfering with glycolysis and oxidative phosphorylation at specific points, and assessing the resulting effects. For this purpose, we used glucose to propagate glycolysis in starved cells, and 2-deoxyglucose (2-DG), which is converted to 2-deoxyglucose-6-phosphate, a competitive inhibitor of phosphoglucoisomerase23, by hexokinase, in order to block glycolysis. Rotenone (a complex I-specific inhibitor of the ETC), antimycin A (a complex III-specific inhibitor of the ETC), oligomycin (inhibitor of ATP synthase24), and the uncoupling agent 2,4-dinotrophenol (DNP)25 were used to intervene specific events related to electron transport, proton gradient and ATP synthesis (Figure 1A). Instead of 2,4-DNP, carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP) can also be used, but further optimization may be required. In either case, uncouplers should always be titrated for a specific cell type before use to determine the optimal concentration necessary.
The glycolysis stress test (Figure 1B) starts with a baseline measurement of the extracellular acidification rate (ECAR) in the previously starved cells. Since these cells are at their basal minimum, and can therefore be practically considered as non-glycolytic, the ECAR measured at this point is referred to as the non-glycolytic acidification. This acidification likely corresponds to respiratory CO2 generated in the TCA cycle being converted to HCO3- and H+. This is followed by the injection of glucose to activate glycolysis, which presents as an increase in the extracellular acidification due to the formation of lactate. This increase represents the normal rate of glycolysis. The cells are then challenged with the injection of oligomycin, which blocks the generation of ATP through oxidative phosphorylation. Cells respond to this dramatic decrease in ATP production by activating glycolysis to its maximum level, and that results in a secondary increase in the ECAR level (glycolytic reserve). The test is terminated by total inhibition of glycolysis using the glucose analog 2-DG, which returns the ECAR to its non-glycolytic level. Interestingly, it has been proposed that, contrary to the manufacturer's recommendations, maximal glycolysis is not necessarily attained by the injection of oligomycin26. In cells with a high glycolytic capacity, if there is not a significant increase in ATP demand, glycolysis may be perfectly able to cope with the loss of mitochondrial ATP without it needing to be up-regulated. The glycolytic rate with oligomycin could be surpassed by adding respiratory inhibitors, such as rotenone and myxothiazol, which provide a few benefits over oligomycin: 1) They increase the ATP demand, as they cause the reversal of ATP Synthase, with ATP hydrolysis, to pump protons in an attempt to recover the mitochondrial membrane potential26; 2) They prevent respiratory acidification of the medium, which can confound the ECAR results (see below). Other ways to increase ATP demand include the addition of compounds that stimulate the hydrolysis of ATP by the plasma membrane ATPases26. All this should be considered carefully by researchers when planning a glycolysis stress test.
The mitochondrial stress test (Figure 1C) starts with a baseline measurement of the oxygen consumption rate (OCR) in non-starved cells. This is followed by the injection of oligomycin, which inhibits the return of protons through the FO/F1 complex and thus rapidly hyperpolarizes the mitochondrial membrane. Hyperpolarization prevents further proton pumping through respiratory complexes, and the respiratory rate decreases. The remaining respiration is called proton leak, which represents the flow of protons through lipids or other channels. This hyperpolarized state is rapidly reversed by the addition of the uncoupling agent 2,4-DNP, which acts as a proton ionophore. In response, cells try to recover the membrane potential in a futile attempt by increasing the electron transport rate to its maximum, and this in turn increases the OCR. Finally, with the addition of two ETC inhibitors (antimycin A and rotenone), mitochondrial respiration completely stops and OCR decreases to its lowest level. At this level, oxygen consumption is not due to mitochondrial activity (non-mitochondrial). The difference in the OCR generated by these inhibitors is called the maximum mitochondrial respiration, which is the sum of the baseline respiration and the spare capacity.
B and T cell isolations yield highly viable pure lymphocyte populations.
B cells and naïve CD4+ T cells were isolated as outlined in Section 4 of the protocol, and splenocytes were simply obtained by lysing the red blood cells as described in Section 3.3.
The sensitivity of cell type-specific metabolic assays depends on the viability and the purity of the starting cell population. Therefore, in order to verify viability of the isolated mouse T cells, splenocytes, and B cells, and the purity of T and B cells, small aliquots of cells were stained for flow cytometric analysis (Figure 2). In the forward scatter-area vs. side scatter-area (FSC-A vs. SSC-A) plot, lymphocytes were gated, and within this gate, the population along the diagonal in the forward scatter-height (FSC-H) vs. FSC-A plot were determined as the singlets. Within the singlet population, viability was measured by gating the cells that stained negative for the live/dead marker (Figure 2A); T cell viability was 97.9%, splenocyte viability was 92%, and B cell viability was 94%. B cell purity, as measured by the B220+ CD19+ population26, was 99%, while CD4+ T cell purity, as measured by the CD44- CD4+ population27, was 98.3% (Figure 2B).
Protein concentration of the cell lysate can be used as a direct indicator of plated cell number.
Isolated lymphocytes and splenocytes were plated in an adhesive-coated 96-well assay plate at 5 x 105 cells/well, 2.5 x 105 cells/well, 1.25 x 105 cells/well, and 0.625 x 105 cells/well. The confluence at each plating density of the three cell types was visualized under the light microscope (Figure 3A). As expected, confluence correlated with the initial plating densities. Upon completion of the extracellular flux assay, plated cells were lysed and their protein concentrations were quantified using the BCA assay. For all cell types, lysate protein concentrations were shown to be linearly correlated with the initial plating densities (Figure 3B), which confirms that lysate protein concentrations can be used as an accurate measure for the normalization of cell numbers when interpreting the extracellular flux data. The plating densities used in this experiment have been optimized for naïve, unstimulated lymphocytes. If stimulated or previously cultured cells are to be used, further optimization may be required.
Mitochondrial and glycolytic stress assays are dependent on plated cell number.
The OCR was measured for each cell type and plating density in the extracellular flux analyzer. As expected, higher cell numbers have a higher measured OCR, as well as more dramatic responses to oligomycin, 2,4-DNP, and antimycin A/rotenone (Figure 4A). Standardizing OCR measurements to each sample's protein concentration reveals that in general, larger numbers of cells lead to more accurate OCR measurements. Plating at 5 and 2.5 x 105 cells/well resulted in similar normalized OCR measurements in the T cells and splenocytes, and 5 and 1.25 x 105 cells/well resulted similar normalized OCR measurements in B cells (Figure 4B). The slight differences in the normalized B cell OCR measurements might be an indirect indication that B cells perform better at 2.5 x 105 cells/well compared to other cell densities. By all measures, 0.625 x 105 cells/well gave suboptimal results, demonstrating that this plating density is insufficient. Baseline respiration, proton leak, maximum mitochondrial respiration, non-mitochondrial respiration, and ATP production linearly correlated with plating densities for all cell types (Figure 4C). Additionally, most of the baseline respiration is used towards synthesizing ATP, as indicated by the low proton leak in the three cell types. While the primary focus of the mitochondrial stress experiment is to measure changes in OCR, the ECAR is still useful to record to ensure that the assay was successfully carried out. Similar to the OCR, the two higher plating densities resulted in higher ECAR and more dramatic responses to oligomycin, 2,4-DNP, and antimycin A/rotenone (Figure 4D). The dramatic changes in ECAR upon addition of 2,4-DNP, and antimycin A/rotenone may be due to changes in respiration in addition to changes in glycolysis, since CO2 generated in the TCA cycle is converted to HCO3 - and H+. This issue has been recently addressed, and there exists a simple method for correcting the total extracellular acidification signal using oxygen consumption data, to obtain the real glycolytic rate12,29.
The glycolysis stress assay was most successful at the highest plating density (Figure 5A, B). In all cell types, the 5 x 105 cells/well samples had the greatest changes in ECAR after the addition of glucose, oligomycin, and 2-DG (Figure 5B). Additionally, normalizing to protein concentration demonstrated that for all cell types, the 5 x 105 cells/well samples yielded optimal results, while lower concentrations-especially 0.625 x 105 cells/well-did not display a robust glycolytic reserve capacity. Non-glycolytic acidification, glycolysis, and apparent glycolytic capacity linearly correlated with plating densities for all cell types (Figure 5C). For the glycolysis stress experiment, the OCR graph shows that glucose slightly stimulates mitochondrial respiration, which is then inhibited by oligomycin treatment; the addition of 2-DG did not affect the OCR (Figure 5D). The OCR graph can be used as a further indicator that the glycolysis stress experiment was successfully carried out. Also, the OCR graph can be used to correct the ECAR graph, if required, as explained above in the case of the mitochondrial stress test12,29.
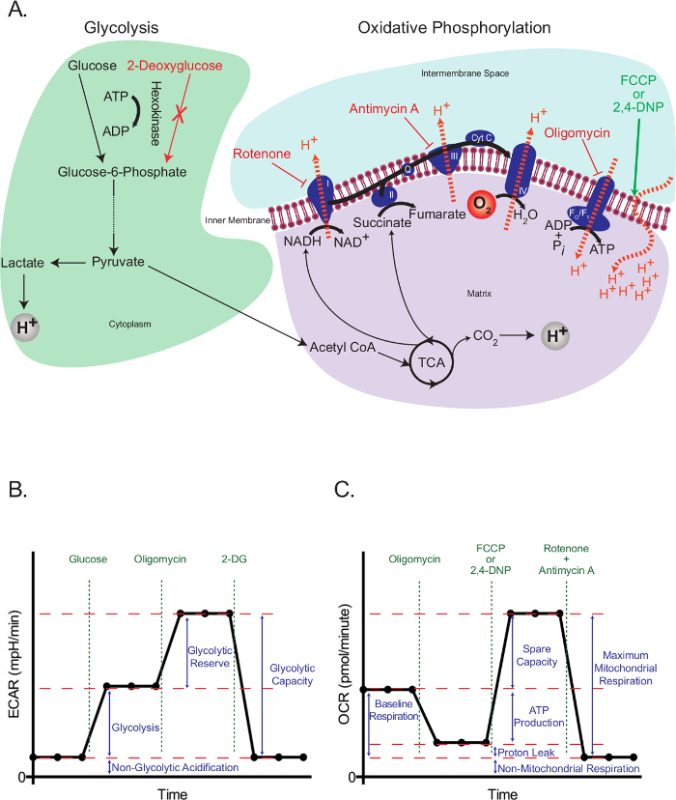 Figure 1:Outline of extracellular flux assays. (A) Illustration of glycolysis (left) and oxidative phosphorylation (right) showing the action of the metabolic drugs used in the extracellular flux assays. (B) Schematic of the extracellular acidification rate (ECAR) graph; schematic of the oxygen consumption rate (OCR) graph (C). Please click here to view a larger version of this figure.
Figure 1:Outline of extracellular flux assays. (A) Illustration of glycolysis (left) and oxidative phosphorylation (right) showing the action of the metabolic drugs used in the extracellular flux assays. (B) Schematic of the extracellular acidification rate (ECAR) graph; schematic of the oxygen consumption rate (OCR) graph (C). Please click here to view a larger version of this figure.
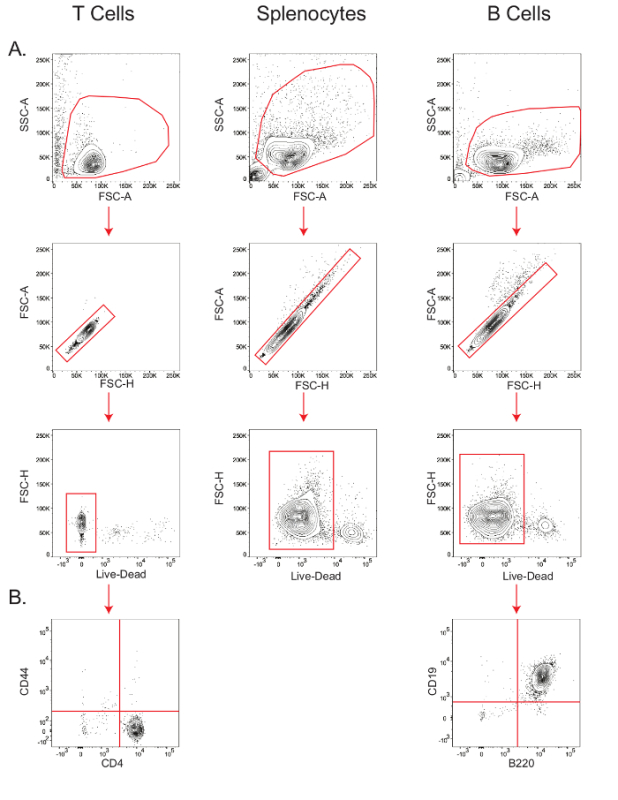 Figure 2:Stepwise gating of T cells, B cells, and splenocytes to determine viability and purity. Flow cytometry plots showing the viability of T cells, splenocytes, and B cells (A); and purity of T and B cells (B). Results are representative of at least three independent experiments. Please click here to view a larger version of this figure.
Figure 2:Stepwise gating of T cells, B cells, and splenocytes to determine viability and purity. Flow cytometry plots showing the viability of T cells, splenocytes, and B cells (A); and purity of T and B cells (B). Results are representative of at least three independent experiments. Please click here to view a larger version of this figure.
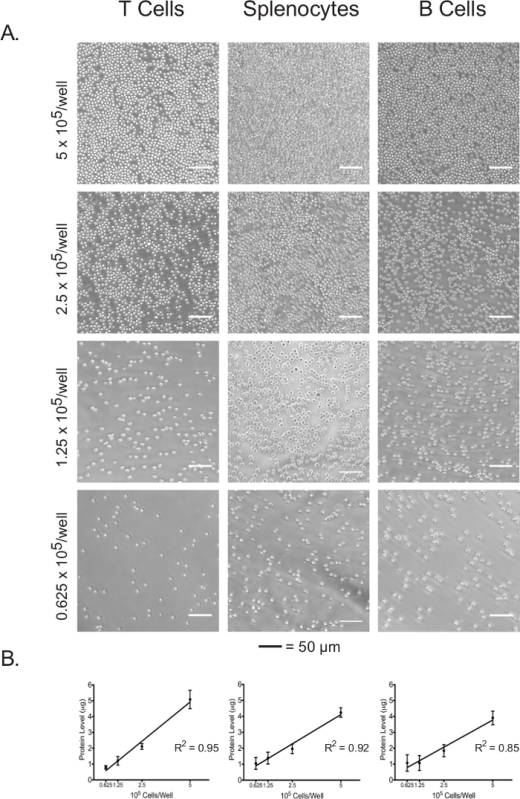 Figure 3:Cell confluence correlates with the lysate protein concentrations of each cell type at different plating densities. (A) Light micrographs of cells in assay plate wells at plating densities ranging from 5 x 105 cells/well to 0.625 x 105 cells/well. Scale bars denote 50 µm. (B) Lysate protein concentrations at different plating densities, as measured by the BCA assay. Results are representative of at least three independent experiments. Please click here to view a larger version of this figure.
Figure 3:Cell confluence correlates with the lysate protein concentrations of each cell type at different plating densities. (A) Light micrographs of cells in assay plate wells at plating densities ranging from 5 x 105 cells/well to 0.625 x 105 cells/well. Scale bars denote 50 µm. (B) Lysate protein concentrations at different plating densities, as measured by the BCA assay. Results are representative of at least three independent experiments. Please click here to view a larger version of this figure.
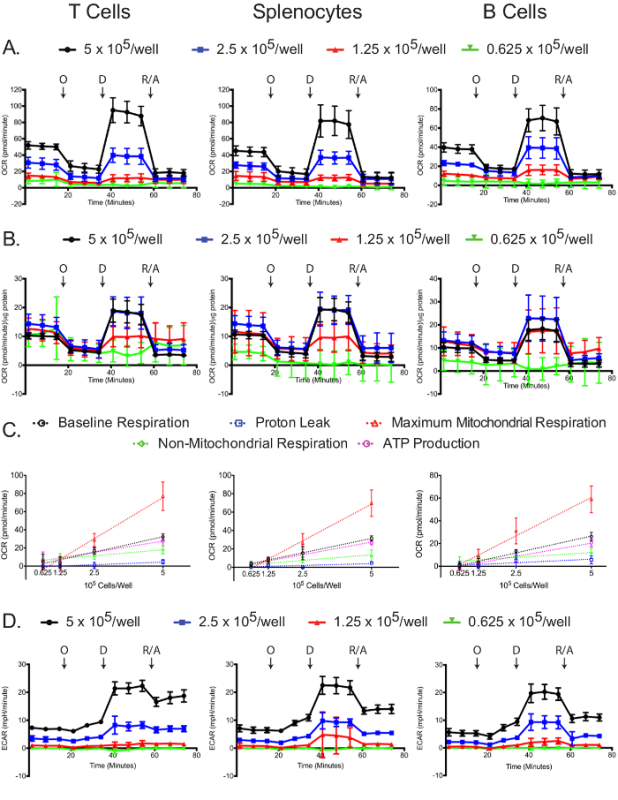 Figure 4:Mitochondrial stress assay. Raw (A) and standardized (B) OCR for each cell type and plating density are shown. (C) Cell number-dependent changes in the levels of baseline, maximum mitochondrial and non-mitochondrial respiration, as well as the oxygen consumptions linked to proton leak or ATP production, are shown for each cell types. (D) Raw ECAR values obtained from the mitochondrial stress tests are shown. Each data point represents the mean of 7-8 wells with standard deviation. Labeled arrows denote injections of oligomycin (O), 2,4-DNP (D), and rotenone/antimycin A (R/A). Results are representative of at least three independent experiments. Please click here to view a larger version of this figure.
Figure 4:Mitochondrial stress assay. Raw (A) and standardized (B) OCR for each cell type and plating density are shown. (C) Cell number-dependent changes in the levels of baseline, maximum mitochondrial and non-mitochondrial respiration, as well as the oxygen consumptions linked to proton leak or ATP production, are shown for each cell types. (D) Raw ECAR values obtained from the mitochondrial stress tests are shown. Each data point represents the mean of 7-8 wells with standard deviation. Labeled arrows denote injections of oligomycin (O), 2,4-DNP (D), and rotenone/antimycin A (R/A). Results are representative of at least three independent experiments. Please click here to view a larger version of this figure.
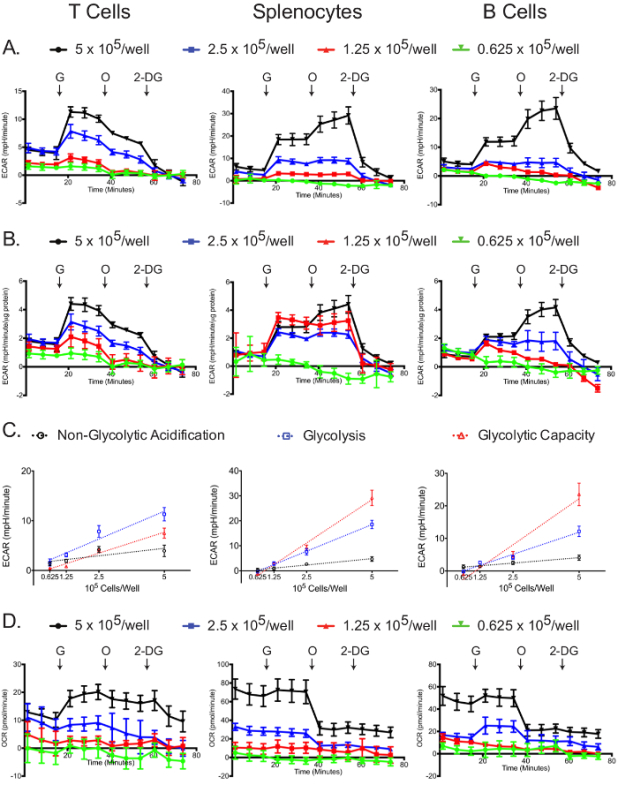 Figure 5:Glycolytic stress assay. Raw (A) and standardized (B) ECAR for each cell type and plating density are shown. (C) Cell number-dependent changes in ECAR for non-glycolytic acidification, glucose-induced glycolysis and total glycolytic capacity are shown. (D) Raw OCR values obtained from the glycolytic stress tests are shown. Each data point represents the mean of 7-8 wells with standard deviation. Labeled arrows denote injections of glucose (G), oligomycin (O), and 2-deoxyglucose (2-DG). Results are representative of at least three independent experiments. Please click here to view a larger version of this figure.
Figure 5:Glycolytic stress assay. Raw (A) and standardized (B) ECAR for each cell type and plating density are shown. (C) Cell number-dependent changes in ECAR for non-glycolytic acidification, glucose-induced glycolysis and total glycolytic capacity are shown. (D) Raw OCR values obtained from the glycolytic stress tests are shown. Each data point represents the mean of 7-8 wells with standard deviation. Labeled arrows denote injections of glucose (G), oligomycin (O), and 2-deoxyglucose (2-DG). Results are representative of at least three independent experiments. Please click here to view a larger version of this figure.
Discussion
The protocol we developed allowed for efficient isolation of pure and viable lymphocyte subsets, which were subsequently used in extracellular flux analysis at different concentrations to evaluate the differences in the glycolysis and mitochondrial respiration performance. This protocol is designed specifically for lymphocytes and addresses the special considerations related to these cell types, such as low basal metabolic activity, fragility, low frequency, and their inability to adhere to assay plates. Therefore, compared to previously published protocols11,12, our method offers a more convenient and better-optimized guide for researchers working in the field of immunology. There are several critical steps in this protocol, including getting pure and viable cell populations, plating the cells at optimal confluence, and standardizing the extracellular flux assay measurements to the protein concentration in each well.
The initial number of cells plated could both be visualized by light microscopy and also be quantified using protein concentration measurements. The linear correlation between cell number and protein concentration confirmed that the protein concentration can indeed be used as a way to standardize the effects of changes in cell numbers between different wells.
By standardizing the ECAR and OCR results to the protein concentrations, we showed that, despite lower cell confluence, as few as 2.5 x 105 cells/well could be used in most assays without compromising the quality of the data. However, the lower limit of cells that can be used varied between cell types and assays. For example, while as few as 1.25 x 105 cells could be used for B cell OCR measurements, even 2.5 x 105 cells/well were not enough to assess the glycolytic performance of the same cell type. Therefore, as long as cell number is not a limiting factor, plating at over 90% confluence, which approximately corresponds to 5 x 105 lymphocytes/well, is preferable. Further optimization might be required when cells of different sizes-such as previously activated and partially differentiated lymphocytes-are used. Additionally, when using previously cultured primary lymphocytes, there might be a variation in cell viability between different treatment conditions, which would decrease the reliability of protein concentration as a measure of the cell number since dead or dying cells can also contribute to the measured protein levels. In such instances, it might be helpful to sort live cells by flow cytometry before carrying out the extracellular flux assays.
In our assays using freshly isolated and highly viable lymphocyte populations, we obtained reliable functional data for both OCR and ECAR measurements. While all cell types behaved similarly in the mitochondrial stress test, striking differences between cell types were observed in the glycolysis stress test. For instance, the glycolytic performance of naïve T cells was low compared to splenocytes or B cells, and it did not change with the addition of oligomycin. This observation is in line with previously published studies7,30, confirming the validity of our protocol.
In conclusion, our method offers an efficient and convenient way of testing the metabolic activity of lymphocytes using an extracellular flux analyzer, and it can be useful in a wide range of immunological studies exploring the metabolic changes in immune cells upon activation, cell differentiation or due to disease phenotypes such as infection, autoimmunity and hematologic malignancies.
Disclosures
The authors declare that they have no competing financial interests. Open Access fees for this article were provided by Agilent Technologies.
Acknowledgments
The authors thank Dr. Michael N. Sack (National Heart, Lung, and Blood Institute) for support and discussion, Ms. Ann Kim for optimizing the B cell isolation protocol, Dr. Joseph Brzostowski for his help with microscopy, and Ms. Mirna Peña for maintaining the animals used. This study was supported by the Intramural Research Programs of the National Institutes of Health, National Institute of Allergy and Infectious Diseases, and National Heart, Lung, and Blood Institute.
References
- Motz GT, Coukos G. Deciphering and reversing tumor immune suppression. Immunity. 2013;39(1):61–73. doi: 10.1016/j.immuni.2013.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akkaya M, Barclay AN. How do pathogens drive the evolution of paired receptors? Eur J Immunol. 2013;43(2):303–313. doi: 10.1002/eji.201242896. [DOI] [PubMed] [Google Scholar]
- Smith-Garvin JE, Koretzky GA, Jordan MS. T cell activation. Annu Rev Immunol. 2009;27:591–619. doi: 10.1146/annurev.immunol.021908.132706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinarello CA. Proinflammatory cytokines. Chest. 2000;118(2):503–508. doi: 10.1378/chest.118.2.503. [DOI] [PubMed] [Google Scholar]
- Kurosaki T, Kometani K, Ise W. Memory B cells. Nat Rev Immunol. 2015;15(3):149–159. doi: 10.1038/nri3802. [DOI] [PubMed] [Google Scholar]
- Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8(12):958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce EL, Poffenberger MC, Chang CH, Jones RG. Fueling immunity: insights into metabolism and lymphocyte function. Science. 2013;342(6155):1242454. doi: 10.1126/science.1242454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerriets VA, Rathmell JC. Metabolic pathways in T cell fate and function. Trends Immunol. 2012;33(4):168–173. doi: 10.1016/j.it.2012.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doughty CA, et al. Antigen receptor-mediated changes in glucose metabolism in B lymphocytes: role of phosphatidylinositol 3-kinase signaling in the glycolytic control of growth. Blood. 2006;107(11):4458–4465. doi: 10.1182/blood-2005-12-4788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caro-Maldonado A, et al. Metabolic reprogramming is required for antibody production that is suppressed in anergic but exaggerated in chronically BAFF-exposed B cells. J Immunol. 2014;192(8):3626–3636. doi: 10.4049/jimmunol.1302062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelletier M, Billingham LK, Ramaswamy M, Siegel RM. Extracellular flux analysis to monitor glycolytic rates and mitochondrial oxygen consumption. Methods Enzymol. 2014;542:125–149. doi: 10.1016/B978-0-12-416618-9.00007-8. [DOI] [PubMed] [Google Scholar]
- Mookerjee SA, Brand MD. Measurement and Analysis of Extracellular Acid Production to Determine Glycolytic Rate. J Vis Exp. 2015. p. e53464. [DOI] [PMC free article] [PubMed]
- Chacko BK, et al. Methods for defining distinct bioenergetic profiles in platelets, lymphocytes, monocytes, and neutrophils, and the oxidative burst from human blood. Lab Invest. 2013;93(6):690–700. doi: 10.1038/labinvest.2013.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein AB, et al. Impact of different cell isolation techniques on lymphocyte viability and function. J Immunoassay Immunochem. 2006;27(1):61–76. doi: 10.1080/15321810500403755. [DOI] [PubMed] [Google Scholar]
- Hsueh RC, et al. Purification and Characterization of Mouse Splenic B Lymphocytes. AfCS Research Reports. 2012;1(1):1–11. [Google Scholar]
- Zambrano K, et al. Prolonged ex vivo expansion and differentiation of naive murine CD43 B splenocytes. Biotechnol Prog. 2016. [DOI] [PubMed]
- Costa GL, et al. Targeting rare populations of murine antigen-specific T lymphocytes by retroviral transduction for potential application in gene therapy for autoimmune disease. J Immunol. 2000;164(7):3581–3590. doi: 10.4049/jimmunol.164.7.3581. [DOI] [PubMed] [Google Scholar]
- Franz B, May KF, Dranoff G, Wucherpfennig K. Ex vivo characterization and isolation of rare memory B cells with antigen tetramers. Blood. 2011;118(2):348–357. doi: 10.1182/blood-2011-03-341917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider DF, Glenn CH, Faunce DE. Innate lymphocyte subsets and their immunoregulatory roles in burn injury and sepsis. J Burn Care Res. 2007;28(3):365–379. doi: 10.1097/BCR.0B013E318053D40B. [DOI] [PubMed] [Google Scholar]
- Reeves JP, Reeves PA. Removal of lymphoid organs. Curr Protoc Immunol. 2001;Chapter 1:Unit 1 9. doi: 10.1002/0471142735.im0109s01. [DOI] [PubMed] [Google Scholar]
- Akkaya B, et al. A Simple, Versatile Antibody-Based Barcoding Method for Flow Cytometry. J Immunol. 2016;197(5):2027–2038. doi: 10.4049/jimmunol.1600727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traba J, et al. Fasting and refeeding differentially regulate NLRP3 inflammasome activation in human subjects. J. Clin. Invest. 2015;125(12):4592–4600. doi: 10.1172/JCI83260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wick AN, et al. Localization of the Primary Metabolic Block Produced by 2-Deoxyglucose. J. Biol. Chem. 1957;224(2):963–969. [PubMed] [Google Scholar]
- Linnett PE, Beechey RB. Inhibitors of the ATP synthetase system. Methods Enzymol. 1979;55:472–518. doi: 10.1016/0076-6879(79)55061-7. [DOI] [PubMed] [Google Scholar]
- Terada H. Uncouplers of oxidative phosphorylation. Environmental Health Perspectives. 1990;87:213–218. doi: 10.1289/ehp.9087213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mookerjee SA, Nicholls DG, Brand MD. Determining Maximum Glycolytic Capacity Using Extracellular Flux Measurements. PLOS ONE. 2016;11(3):e0152016. doi: 10.1371/journal.pone.0152016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai L, Alaverdi N, Maltais L, Morse HC., 3rd Mouse cell surface antigens: nomenclature and immunophenotyping. J Immunol. 1998;160(8):3861–3868. [PubMed] [Google Scholar]
- Gerberick GF, Cruse LW, Miller CM, Sikorski EE, Ridder GM. Selective modulation of T cell memory markers CD62L and CD44 on murine draining lymph node cells following allergen and irritant treatment. Toxicol Appl Pharmacol. 1997;146(1):1–10. doi: 10.1006/taap.1997.8218. [DOI] [PubMed] [Google Scholar]
- Mookerjee SA, Goncalves RLS, Gerencser AA, Nicholls DG, Brand MD. The contributions of respiration and glycolysis to extracellular acid production. Biochim. Biophys. Acta - Bioenergetics. 2015;1847(2):171–181. doi: 10.1016/j.bbabio.2014.10.005. [DOI] [PubMed] [Google Scholar]
- Buck MD, O'Sullivan D, Pearce EL. T cell metabolism drives immunity. J Exp Med. 2015;212(9):1345–1360. doi: 10.1084/jem.20151159. [DOI] [PMC free article] [PubMed] [Google Scholar]