

Abstract
High-resolution episcopic microscopic (HREM) technology enables rapid acquisition of high-resolution digital volumetric and three-dimensional (3D) morphometric data. Here, we describe the detailed protocol to image the entire mouse embryo. The protocol consists of four major sections: sample preparation, embedding, image acquisition and finally, 3D visualization. The technology requires specimens to be stained with a fluorescent dye, which can be problematic for large or dense specimens. To overcome this limitation, we have improved the existing protocol to enhance tissue penetration of the dye by pretreating the specimen with a solution containing urea and sodium dodecyl sulfate. The protocol uses only routine laboratory equipment and reagents for easy adaptation in standard laboratory settings. We show that the resulting high-resolution 3D images faithfully recapitulate the detailed morphologic features of the internal organs of mouse embryos, thereby permitting morphometric analyses. Together, we present a detailed and improved protocol using standard laboratory equipment to acquire high-resolution 3D images of small and large sized specimens.
Keywords: Developmental Biology, Issue 117, High-resolution episcopic microscopy (HREM), Episcopic fluorescence image capture (EFIC), 3D reconstruction, Amira, mouse, morphogenesis, urinary tract
Introduction
The advent of 3D imaging technologies has opened the possibility of systemic analysis of detailed morphological features during fetal development. A variety of 3D imaging modalities is now available include micro-magnetic resonance imaging (micro-MRI), micro computed tomography, high frequency ultrasound, optical coherence tomography, optical project tomography and episcopic microscopy1-4. Each modality has its own unique features in resolution, contrast, speed, cost, in utero capability and availability.
Episcopic fluorescence image capture (EFIC) and high-resolution episcopic microscopy (HREM) are episcopic microscopy imaging methods4. Here, high-resolution serial images are captured continuously from the block face instead of tissue sections. The resulting images faithfully reflect detailed morphological features with minimal or no tissue distortion. The acquired volumetric data is readily converted to high-resolution 3-D images suitable for accurate morphometric analysis. Because of relative low cost and high resolution, HREM and EPIC have become the superior alternatives for systemic assessment of mouse models of human birth defects2,4-8.
Both EFIC and HREM methods require specimens to be embedded in the suitable medium. EFIC detects autofluorescence emitted from the embedded tissue. Because of this, it is limited to specimens emitting high levels of autofluorescence but not those with relatively weak autofluorescence such as early stage embryos9. To overcome the dependency of autofluorescence, specimens are stained with a fluorescent dye such as eosin for HREM imaging. The choices of dyes and staining methods also make HREM more compatible to detect molecular signals in the context of tissue architecture and morphology4,10.
In this article, we present an improved HREM protocol suitable for whole body assessment of the late stage mouse embryos. To facilitate staining, we include a pretreatment step to increase tissue penetration, thereby enabling HREM imaging of older mouse embryos.
Protocol
All animal uses are approved by the Institutional Animal Care and Use Committee at Boston Children's Hospital.
1. Sample Preparation
- Timed mating
- Place a C57Bl6 wild type male and a female together in the same cage.
- Check for the formation of a mating plug early next morning. The mating plug (aka, vaginal or copulation plug) is a hardened gelatinous deposition that blocks the vagina.
- Set the plug date as embryonic day 0.5 (e0.5).
- Isolation of mouse embryos
- Euthanize pregnant females by CO2 asphyxiation.
- Put mice supine on an absorbent pad and spray abdominal region with 70% ethanol.
- Lift the skin and make an initial 5 mm incision at the caudal abdominal region using surgical scissors. Cut and remove the abdominal skin to fully expose the internal organs
- Cut near vagina to remove the entire uterus.
- Cut between implantation sites along the uterine horn. Each segment or conceptus should have only one embryo inside.
- Keep the uterine segments in 1x ice-cold phosphate buffered saline (PBS).
- Embryo dissection
- Place each conceptus in a separate dish with ice-cold PBS under a stereoscope.
- Remove uterine tissues, placenta and then fetal membranes sequentially using fine dissecting forceps.
- Transfer the dissected embryo to a 50 ml tube with 1x ice cold PBS using a large transfer pipette. Avoid picking up embryo directly with forceps to minimize tissue damage.
- Fixation
- Fix the dissected embryos in 10% neutral buffered Formalin solution at 4 °C for more than 24 hr with at least 10 sample volumes of fixative.
- Pretreatment
- Prepare the Clearing Solution: 4 M urea, 10% glycerol, 4% Sodium Dodecyl Sulfate (SDS) in double distilled water.
- Replace the fixative with the Clearing Solution.
- Gently rotate the specimen on a rocker at room temperature for 1 - 2 weeks. Exchange the Clearing Solution once on the second and the third day. The specimen slowly becomes clear and translucent.
- Replace the Clearing Solution with 10% formalin and leave on a rocker for 2 days.
- Dehydration
- Wash the pretreated samples with 1x PBS 3 times for 5 min each.
- Dehydrate samples in an ascending ethanol series at room temperature on a gentle rocker. For E15.5 embryos, use an ascending ethanol series including 50%, 60%, 70%, 80%, 85%, 90%, 95% and 100% ethanol, 2 hr each step. For older embryos, the incubation time needs to be increased.
- Staining
- Prepare Staining Solution: add 0.1375 grams of Eosin Y Disodium Salt and 0.0275 grams of Acridine Orange hemi (zinc chloride) salt to 50 ml of 100% ethanol. Stir for 2 hr. Filter with a filter paper. Store at room temperature and avoid light by covering with aluminum foil.
- Transfer specimen to the Staining Solution for 1 hr.
- Replace with fresh Staining Solution and stain overnight.
- Infiltration
- Prepare Infiltration Solution: combine 100 ml of Solution A of JB-4 embedding kit, 1.25 g of Catalyst C, 0.275 g of Eosin Y Disodium Salt and 0.055 g of Acridine Orange hemi (zinc chloride) salt. Stir to mix (200 rpm) in an ice bucket for 2 hr, and then filter with the filter paper.
- Store the Infiltration Solution at 4 °C and avoid light by covering with aluminum foil.
- Transfer the embryos to Infiltration Solution:Staining Solution (1:1), and incubate for at least 3 hr on a rocker at 4 °C.
- Transfer the embryos to Infiltration Solution and incubate for 3 hr on a rocker in a cold room.
- Replace Infiltration Solution once every day, and leave in a cold room on a gentle rocker for three more days.
2. Embedding
Keep Solution B and Infiltration Solution on ice.
Make Embedding Solution (1:25 of Solution B and Infiltration Solution) immediately before embedding.
Orient the specimen in a embedding mold, then pour cold Embedding Solution into the embedding mold gently. Re-orient with a pin if needed.
Use a pin to examine whether the Embedding Solution is solidified. Push out the specimen block, and trim it if needed.
Reorient the specimen block using the separate mold to obtain a cross orientation.
Put a block holder on top of the embedding mold. Gently pour Embedding Solution into the mold until the mold and the block holder are submerged by Embedding Solution.
Keep the specimen in a secondary container and leave it on ice for 3 - 4 hr in a fume hood.
Store the solidified specimens in a 4 °C.
Peel away the embedding mold. Put the block under the stereomicroscope with oblique illumination to inspect position of the specimen in the block. On the block face, mark grid lines around the specimen.
Trim the block to remove access amount of embedding material using the marked grid lines as references.
3. Image Acquisition
Clamp the specimen block to the microtome and obtain a fresh cut surface. Set section thickness (e.g., e9.5-10.5, 1.5 µm; e11.5-12.5, 2.0 µm; e13.5-15.5, 2.5 µm; e16.5-older, 3 µm). Keep the specimen/block at the home position of microtome.
Mount the stereo zoom microscope horizontally facing the block surface of specimen.
Turn on the computer and microscope, and start the image acquisition software.
Visualize and focus the optics to the block-face under the "live view" mode of imaging software.
Align microscope and microtome if needed so that the block-face is at the center of viewfinder.
Adjust zoom so that the entire block-face is within the viewfinder.
Select green fluorescent protein (GFP) filter (excitation 470 ± 20 nm, dichroic 495 nm, emission 525 ± 25 nm) and refocus if needed.
Measure and set the exposure time manually (e.g., 80 - 400 mini sec).
Acquire images of the block-face after each fresh cut and save images automatically if possible.
Record important parameters including resolution, section thickness and zoom.
After finishing the entire specimen, export and convert all image files to JPEG format if necessary.
Invert all original images using an imaging processing software, and adjust brightness and contrast.
Save all the processed images in a separate folder.
4. 3D Visualization
Upload all the processed images to a 3D visualization software following the instructions.
Input resolution and section thickness to convert all images to volumetric data (i.e., voxel size) and save the 3D file.
Align all images using the automatic mode. Adjust individual images manually if needed.
Save the aligned image to a new file name.
Analyze morphometric features of the specimen using the 3D visualization software.
Representative Results
We have reported high quality HREM images of mouse embryos between e9.5 and e13.58. Image quality of the late staged embryos, however, is significantly compromised because of the limited tissue penetration of the fluorescent dye eosin. To increase the staining efficiency, we have tested several pretreatment methods that are compatible to fluorescent imaging11. Specifically, the e15.5 mouse embryos were treated with either Sca/e A212 or Whole-Body CUBIC13. We have also tested the simplified formula, the Clearing Solution (4 M urea, 10% glycerol and 4% sodium dodecyl sulfate). All pretreatment methods increased staining efficiency. Embryos became translucent after 1 week of treatment with the Clearing Solution. No obvious tissue damage was detected after the treatment. The pretreated embryos were processed through steps including dehydration, staining and infiltration before being embedded in the plastic embedding media. HREM images were acquired from the embedded specimen as described above in section 3 (Image Acquisition) and displayed using a 3D visualization software. An example of whole mount surface view of e15.5 embryo demonstrated detailed morphological features of the skin including whiskers pads and developing hair follicles (Figure 1). Resolution of the virtual section view was comparable to histological section (Figure 2).
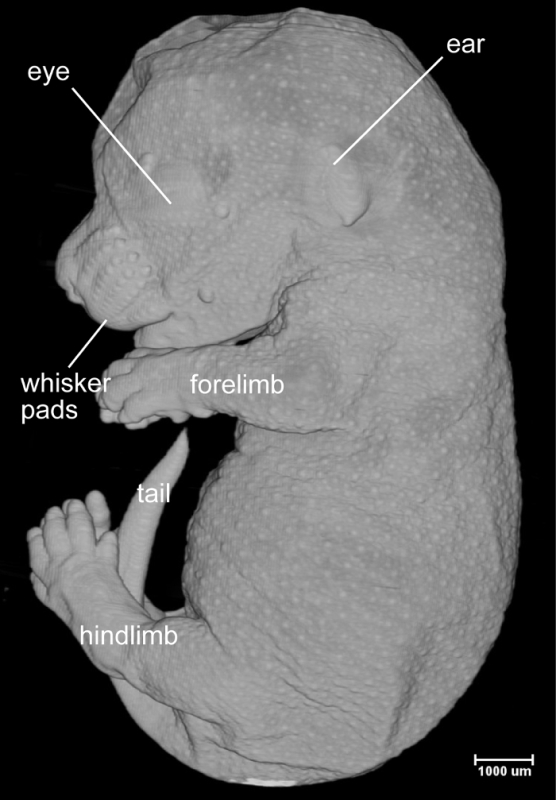 Figure 1:3D Surface View of the e15.5 Embryo. It shows the detailed morphological features of the skin. The developing hair follicles (small white dots) are apparent, demonstrating high-resolution of the image. Please click here to view a larger version of this figure.
Figure 1:3D Surface View of the e15.5 Embryo. It shows the detailed morphological features of the skin. The developing hair follicles (small white dots) are apparent, demonstrating high-resolution of the image. Please click here to view a larger version of this figure.
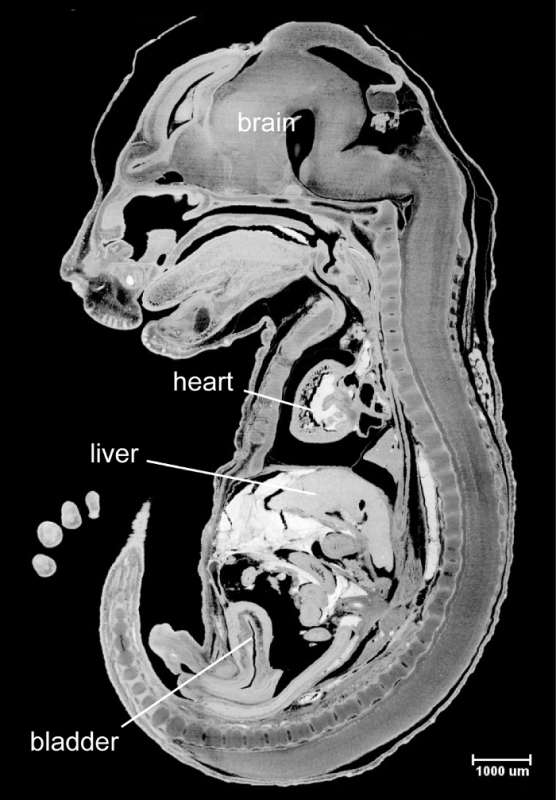 Figure 2:The Virtual Section View of Midline Section of the e15.5 Embryo. The internal structures, including the heart, liver, gut and bladder are clearly visible. Please click here to view a larger version of this figure.
Figure 2:The Virtual Section View of Midline Section of the e15.5 Embryo. The internal structures, including the heart, liver, gut and bladder are clearly visible. Please click here to view a larger version of this figure.
Discussion
Here, we present a modified protocol using routine laboratory equipment to acquire serial HREM images that are compatible for rapid 3D visualization and morphometric analysis of complex structures. Because the high-resolution images are taken directly from the block face instead of individual sections, the fine morphological features are preserved and rapidly reconstructed digitally in a 3D visualization program.
3D imaging offers significant advantages in visualizing and understanding complex morphological features. Most 3D imaging technologies depend on specialized equipment1-4. In contrast, EFIC and HREM imaging only utilize standard laboratory equipment including a stereomicroscope equipped with a digital camera and a microtome14. Functional coupling between microscope and microtome enables automation of image acquisition enables automation of the entire image acquisition process. However, we have shown that functional coupling is not obligatory8. Instead, we simply mount the stereomicroscope horizontally so that it faces directly to the block face of a standard microtome at the resting stop position. The major limitation of this simple setup is that the HREM images have to be taken manually. We have estimated that it takes approximately 5 sec per image on average. Another limitation is that individual image may shift from one position to another but this is not an issue because they are readily realigned during 3D reconstruction.
Using the HREM technology, we have analyzed a series of 3D images of mouse embryos from e9.5 to e13.5, and uncovered a novel mechanism by which the solitary embryonic cloaca transforms into two separate structures, the distal digestive tract and the lower urinary tracts8. Quality of the HREM images of late staged embryos is limited due to inefficient penetration of eosin (data not shown). To increase tissue penetration of e15.5 and e18.5 embryos, we have used two different pretreatment methods that are compatible to fluorescent imaging11 and tested a new formula, the Clearing Solution (4 M urea, 10% glycerol and 4% SDS). All of these methods have shown significant improvement of tissue penetration and eosin staining. An important consideration before and after pretreatment is that the specimen is prone to skin swelling or shrinking due to osmotic pressure. To limit the sudden change of osmotic pressure thereby reduce tissue deformation, we recommend gradual exchange of the solution. In addition, we found that inclusion of ethanol helps significantly (the step 1.5.4). During infiltration and embedding steps, it is important to keep the specimen from light and at 4 ˚C.
Collectively, this protocol uses routine laboratory equipment to acquire HREM images, which is compatible for high-resolution 3D visualization and morphometric analysis. The protocol should be readily adapted in any standard laboratory setting.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We thank Drs. Yichen Huang, Chunming Guo and Zhenfang Zhou for their technical support. This research was funded by NIH/NIDDK (1R01DK091645-01A1, XL) and American Heart Association (AHA, 13GRNT16950006, XL).
References
- Gregg CL, Butcher JT. Quantitative in vivo imaging of embryonic development: opportunities and challenges. Differentiation. 2012;84:149–162. doi: 10.1016/j.diff.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Tobita K, Francis RJ, Lo CW. Imaging techniques for visualizing and phenotyping congenital heart defects in murine models. Birth defects Res C. 2013;99:93–105. doi: 10.1002/bdrc.21037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris FC, et al. A coming of age: advanced imaging technologies for characterising the developing mouse. Trends Genet. 2013;29:700–711. doi: 10.1016/j.tig.2013.08.004. [DOI] [PubMed] [Google Scholar]
- Weninger WJ, et al. Phenotyping structural abnormalities in mouse embryos using high-resolution episcopic microscopy. Dis model mech. 2014;7:1143–1152. doi: 10.1242/dmm.016337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohun TJ, Weninger WJ. Episcopic three-dimensional imaging of embryos. Cold Spring Harb protoc. 2012. pp. 641–646. [DOI] [PubMed]
- Sizarov A, et al. Three-dimensional and molecular analysis of the arterial pole of the developing human heart. J Anat. 2012;220:336–349. doi: 10.1111/j.1469-7580.2012.01474.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson RH, et al. Normal and abnormal development of the intrapericardial arterial trunks in humans and mice. Cardiovasc Res. 2012;95:108–115. doi: 10.1093/cvr/cvs147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YC, Chen F, Li X. Clarification of mammalian cloacal morphogenesis using high-resolution episcopic microscopy. Dev Biol. 2016;409:106–113. doi: 10.1016/j.ydbio.2015.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohun TJ, Weninger WJ. Embedding embryos for episcopic fluorescence image capturing (EFIC) Cold Spring Harb protoc. 2012. pp. 675–677. [DOI] [PubMed]
- Weninger WJ, Mohun TJ. Three-dimensional analysis of molecular signals with episcopic imaging techniques. Methods mol biol. 2007;411:35–46. doi: 10.1007/978-1-59745-549-7_4. [DOI] [PubMed] [Google Scholar]
- Richardson DS, Lichtman JW. Clarifying Tissue Clearing. Cell. 2015;162:246–257. doi: 10.1016/j.cell.2015.06.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hama H, et al. Scale: a chemical approach for fluorescence imaging and reconstruction of transparent mouse brain. Nat Neurosci. 2011;14:1481–1488. doi: 10.1038/nn.2928. [DOI] [PubMed] [Google Scholar]
- Susaki EA, et al. Whole-brain imaging with single-cell resolution using chemical cocktails and computational analysis. Cell. 2014;157:726–739. doi: 10.1016/j.cell.2014.03.042. [DOI] [PubMed] [Google Scholar]
- Mohun TJ, Weninger WJ. Generation of volume data by episcopic three-dimensional imaging of embryos. Cold Spring Harb protoc. 2012. pp. 681–682. [DOI] [PubMed]