

Abstract
Nociceptin/Orphanin FQ (N/OFQ) is a 17 amino acid peptide whose receptor is designated ORL1 or nociceptin receptor (NOP). We utilized a potent, selective, and orally bioavailable antagonist with documented engagement with NOP receptors in vivo to assess antidepressant‐ and anxiolytic‐related pharmacological effects of NOP receptor blockade along with measures of cognitive and motor impingement. LY2940094 ([2‐[4‐[(2‐chloro‐4,4‐difluoro‐spiro[5H‐thieno[2,3‐c]pyran‐7,4′‐piperidine]‐1′‐yl)methyl]‐3‐methyl‐pyrazol‐1‐yl]‐3‐pyridyl]methanol) displayed antidepressant‐like behavioral effects in the forced‐swim test in mice, an effect absent in NOP −/− mice. LY2940094 also augmented the behavioral effect of fluoxetine without changing target occupancies (NOP and serotonin reuptake transporter [SERT]). LY2940094 did not have effects under a differential‐reinforcement of low rate schedule. Although anxiolytic‐like effects were not observed in some animal models (conditioned suppression, 4‐plate test, novelty‐suppressed feeding), LY2940094 had effects like that of anxiolytic drugs in three assays: fear‐conditioned freezing in mice, stress‐induced increases in cerebellar cGMP in mice, and stress‐induced hyperthermia in rats. These are the first reports of anxiolytic‐like activity with a systemically viable NOP receptor antagonist. LY2940094 did not disrupt performance in either a 5‐choice serial reaction time or delayed matching‐to‐position assay. LY2940094 was also not an activator or suppressor of locomotion in rodents nor did it induce failures of rotarod performance. These data suggest that LY2940094 has unique antidepressant‐ and anxiolytic‐related pharmacological effects in rodents. Clinical proof of concept data on this molecule in depressed patients have been reported elsewhere.
Keywords: Antidepressant, anxiolytic, LY2940094, nociceptin
Abbreviation
- 5‐CSRTT
choice serial reaction time test
- CER
conditioned emotional response
- cGMP
cyclic guanosine monophosphate
- DMTP
delayed‐matching to position
- DRL72s
differential reinforcement of low rate 72s schedule
- FR
fixed ratio
- HAMA
hamilton anxiety rating scale
- ITI
intertrial interval
- LY2940094
[2‐[4‐[(2‐chloro‐4,4‐difluoro‐spiro[5H‐thieno[2,3‐c]pyran‐7,4′‐piperidine]‐1′‐yl)methyl]‐3‐methyl‐pyrazol‐1‐yl]‐3‐pyridyl]methanol
- MDD
major depressive disorder
- MPEP
2‐Methyl‐6‐(phenylethynyl)pyridine hydrochloride
- N/OFQ
Nociceptin/Orphanin
- NOP
nociceptin receptor
- SB612111
[(‐)‐cis‐1‐methyl‐7‐[[4‐(2,6‐dichlorophenyl)piperidin‐1‐yl]methyl]‐6,7,8,9‐tetrahydro‐5H‐benzocyclohepten‐5‐ol]
- SERT
serotonin reuptake transporter
- SSRI
selective serotonin uptake inhibitor
Introduction
The biogenic amine hypothesis of depression continues to be the prevailing heuristic model for major depressive disorder (MDD). Although multiple pieces of clinical and preclinical data contribute to the veracity of this mechanistic model, the principle data come from the efficacy of antidepressant drugs in patients that increase the central bioavailability of monoamine neurotransmitters (Iversen 2005). Despite efficacy of multiple mechanistically and structurally distinct monoamine facilitators including tricyclic antidepressants, monoamine uptake inhibitors, selective serotonin reuptake inhibitors (SSRIs), and dual and triple uptake inhibitors (serotonin, norepinephrine, and dopamine), these drugs do not produce response in all patients, do not produce remission in all patients for which response has been established, and have a diversity of side‐effects that impede either safety or compliance (c.f., Rush et al. 2006).
Although multiple new avenues for the discovery and development of improved antidepressants continue to be interrogated, novel, non‐monoamine‐based antidepressants with improvements in efficacy and tolerability have yet to be put into widespread clinical practice. Multiple reasons likely exist for this paucity of alternative medicines for a devastating and life‐endangering disease. One reason is overarching; the neurobiology and pathophysiology of depression is not well understood. This issue results in an inability to provide homology and disease‐based models in the drug discovery laboratory. It has also been argued that current preclinical models do not sufficiently predict efficacy in MDD patients (Belzung 2014). The present experiments were undertaken to provide the preclinical basis for predicting translational efficacy of a compound with a novel mechanism of action. These data provided the foundation for which the first nociceptin receptor (NOP) antagonist, LY2940094, was placed into clinical investigation to test the hypothesis that blockade of this receptor would be effective and tolerable in MDD patients (Post et al., 2016). In this proof of concept study, clinical support for the N/OFQ hypothesis of depression was obtained under double‐blind, placebo‐controlled conditions in a small patient population (Post et al. 2016).
Nociceptin/Orphanin FQ (N/OFQ) is a 17 amino acid peptide that was identified to bind the NOP receptor (Meunier et al. 1995; Reinscheid et al. 1995). Multiple biological tools have been generated to enable inquiries into the physiological and pathophysiological roles of N/OFQ including specific agonists and antagonists as well as receptor‐deficient mice and rats. Results of such investigations have led to increased understanding of the potential utility of employing small molecule modulators of NOP receptors to help treat a number of disease states (Lambert 2008; Witkin et al. 2014a). In addition to pain, obesity, and drug dependence, the psychiatric areas of mood and anxiety have received a great deal of experimental scrutiny.
The hypothesis predicting the therapeutic potential of NOP antagonists in depression has some support. N/OFQ is upregulated by stressors and the enhanced nociception levels are reduced by antagonists (see Witkin et al. 2014a); N/OFQ is increased in plasma of patients with bipolar depression, postpartum depression, and MDD (Gu et al. 2003; Wang et al. 2009; Zhang et al. 2009). Furthermore, NOP receptor antagonists engender antidepressant‐like effects in rodents (see full summaries on these data in Witkin et al. 2014a).
In this study, we describe results of effects of LY2940094 in rodent assays that detect antidepressant drugs, and in an array of rodent assays that detect anxiolytic drugs since most conventional antidepressants are also anxiolytic in patients. The findings from these studies document the unique antidepressant and anxiolytic preclinical profile of LY2940094. These data were used to support the clinical investigation that led to the first proof of concept study showing antidepressant efficacy of this mechanism (Post et al., 2016). The NOP receptor antagonist LY2940094 was an ideal tool to test the translational hypothesis that NOP receptor blockade would be antidepressant in MDD patients. The molecule demonstrates high potency and selectivity for NOP receptors along with good oral bioavailability (Toledo et al. 2014; compound 36; Statnick et al. 2016), which translates to the ability to sufficiently occupy NOP receptors in the brain as measured with PET imaging (Raddad et al., 2016). LY2940094 has nM potency for binding and functional activity at human and rat NOP receptors with over a 4000× separation in affinity for mu, kappa, or delta opioid receptors (Toledo et al. 2014; Statnick et al. 2016).
Materials and Methods
Animal care and welfare
All studies were conducted under the guidance of US NIH guidelines and under approved and monitored animal care and use protocols within our local institutional research facilities.
Mouse forced‐swim test
This assay utilized both male, NIH‐Swiss mice (20–25 g, Harlan Sprague‐Dawley, Indianapolis, IN) and male, NOP receptor‐deficient mice. The NOP−/− mice and the wild‐type littermate controls (NOP+/+) were created on a 129S6 background by heterozygous breeding (Kest et al. 2001) and were ~12 weeks old at the time of testing. NIH‐Swiss mice were placed in clear plastic cylinders (diameter 10 cm; height: 25 cm) filled to 6 cm with 22–25°C water for 6 min. These parameters were minor modifications of the method utilizing 6 cm water depth that has been used on multiple occasions by our lab (e.g., Li et al. 2003; Li et al. 2006b) and others (Porsolt et al. 1977; Trullas and Skolnick 1990; Popik et al. 2008) to detect antidepressant agents of diverse structure and mechanism including tricyclics, monoamine oxidase inhibitors, atypical agents, electroconvusive shock, PDE4 inhibitors, and NMDA receptor antagonists. For the NOP mice, ~12‐week‐old 129S6 male mice (wild type or NOP KO) were tested. Mice were tested for 6 min in room temperature water. Immobility time was measured from time +2 min till the end of the trial. The container for the test was a 4 L beaker. The beaker was filled ~1/2 full with room temperature water. The water was deep enough so that the mouse would not be able to stand but not so high as to allow the mouse to jump out of the beaker. The duration of immobility was recorded during the last 4 min of a 6‐min trial. A mouse was regarded as immobile when floating motionless or making only those movements necessary to keep its head above the water. Means ± S.E.M. were subjected to ANOVA followed by Dunnett's test with P < 0.05 set as the error rate for statistical significance. However, in the case of the NOP KO mice, two‐way ANOVA was utilized with genotye and treatment as factors. For the drug combination studies with LY2940094 and fluoxetine, two‐way ANOVA was utilized with dose and drug treatment as factors.
Mouse locomotor activity
Male CD1 mice (Charles River) were used. Activity was measured in a 20‐station Photobeam Activity System (San Diego Instruments, San Diego, CA) with seven photocells per station. Animals were weighed, dosed p.o, and immediately placed individually in a polypropylene cage (40.6 × 20.36 × 15.2 cm). Data were collected for 2 h expressed as total ambulations, where ambulation was defined as the sequential breaking of adjacent photobeams. Data were analyzed by two‐way ANOVA comparing vehicle, 10 and 30 mg/kg‐treated groups of mice over time.
Mouse fear‐conditioning
Mice (C57B1/6) received a single training session of five trials, separated by 30 sec intertrial intervals, in which a neutral tone conditioned stimulus (CS: 5 kHz, 100 dB, 30 sec duration) was paired with a scrambled foot‐shock unconditioned stimulus (0.57 mA, 2 sec duration, coterminating with tone offset). The next day, mice were dosed with vehicle, LY2940094 (3, 10, or 30 mg/kg, p.o.) or paroxetine (1, 3, 10 mg/kg, i.p.) or the mGluR5 negative allosteric modulator MPEP (30 mg/kg, i.p.). We used MPEP as a quality control for this assay since C57Bl/6 mice are very sensitive to sedating effects of benzodiazepine anxiolytics under these conditions in our hands (data not shown). Negative allosteric modulation is an acknowledged potential anxiolytic mechanism (Spooren and Gasparini 2004) that is further supported by the clinical use of fenobam, an mGlu5 receptor antagonist, for the treatment of anxiety (Porter et al. 2005).
Following a 60‐min pretreatment period, mice were placed in a novel environment and allowed to explore for 2 min, during which time no stimuli were presented. Following the 2 min pre‐CS period, the tone CS was presented continuously for 4 min, in the absence of further shock presentation. Data were analyzed by ANOVA. Fear conditioning experiments were conducted in standard Plexiglas operant chambers (Med‐Associates, Burlington, VT) equipped with shock grid floors and white side panels, housed within sound‐attenuating chambers containing a speaker and ceiling‐mounted videocamera connected to a 4‐port multiplexer to facilitate videotracking. All behavior was monitored, recorded, and analyzed using the TopScan software (CleverSys Inc., Reston, VA). Tones and shocks were controlled by a PC using MedPC IV (Med‐Associates, Fairfax, VT) software. Cued fear responses were tested in a novel environment consisting of large black Plexiglas open‐field chambers (40 × 40 × 50 cm) with white Plexiglas floors below a ceiling‐mounted videocamera. Data were analyzed by ANOVA, followed by Dunnett's posthoc tests.
Rat DRL 72s schedule
Adult, male Sprague–Dawley rats weighing between 300 and 350 g at the beginning of the behavioral experiments (Holtzman, Madison, WI) were housed in pairs.
Sixteen operant‐conditioning chambers (30.5 × 24.1 × 29.2 cm; MED Associates, St. Albans, VT) were enclosed in sound‐attenuating cubicles with white masking noise. The interior of each chamber consisted of three levers mounted on one wall with a house light mounted on the opposite wall. The house light was turned on at initiation of each test session and was turned off at the termination of each session. A water access port was situated next to the lever in the middle of the wall, wherein a reinforced response caused a clicker apparatus to sound paired with a dipper (0.02‐mL cup) to be lifted from a water trough to an opening in the floor of the access port for 4 sec.
Rats were water deprived for ~22.5 h before each session with daily 20‐min water access occurring following each operant session. Rats were initially trained under a fixed ratio (FR)‐1 water reinforcement schedule with a fixed 1‐min time schedule for automatic reinforcement. Thus, each response was reinforced with water and water was also provided every minute in the absence of a response. Rats that did not acquire lever‐pressing behavior following three daily 1 h sessions under this schedule were trained using the method of successive approximation. Following acquisition of lever‐pressing behavior, rats were trained daily on DRL 18‐s schedule for ~2 weeks, following which they were advanced to DRL 72‐s sessions. Responding on these sessions became stable after ~8 weeks. Under the DRL schedule, water reinforcement was delivered after each lever press that followed the last lever press by at least the 18 or 72s required by the schedule. Experimental test sessions lasted for 1 h and were conducted 5 days/week during light hours.
Once behavior stabilized, drugs were administered to the animals once or twice weekly with at least 1 week between subsequent drugs to minimize possible carry‐over effects. Drug treatments were administered on Tuesdays and Fridays. No treatments were administered on other test days. Wednesdays were control days. “Control” behavior was calculated as the pooled mean lever presses or reinforcers received during Wednesday sessions across all Wednesdays for the duration of each study. Performance was normalized to percent of control by dividing total responses (or reinforcers) for each drug or vehicle treatment by the mean number of responses (reinforcers) in the respective pooled control condition, multiplied by 100.
The data analyses were conducted on the raw response and reinforcement data. All behavioral data are expressed as the mean ± S.E.M. normalized to the control condition. The effects of LY2940094 (30 mg/kg) and imipramine (10 mg/kg) were analyzed using ANOVA on responses and reinforcements, followed by Dunnett's post hoc tests (α = 0.05).
Rat conditioned emotional response assay
The conditioned emotional response (CER) assay has been well‐validated as an acute assay for detecting anxiolytic‐like activity of clinically validated agents as well as potential novel mechanisms of action (George et al. 2009; Rorick‐Kehn et al. 2006). Subjects were pair‐housed male, Sprague Dawley rats (Harlan, Indianapolis, IN), maintained under a food‐restricted schedule consisting of 20–30 g of food per pair per day (immediately post‐session, Monday–Friday) with ad libitum water in the homecage. Rats were first trained to lever‐press for a sweet pellet reward (45 mg; Bioserv) and were subsequently progressed to the operant schedule comprising the conditioned suppression paradigm in standard operant boxes (Med Associates, Inc.) until conditioned response suppression was achieved.
Following training to lever‐press on continuous reinforcement (FR‐1 schedule) training, rats were progressed to a Variable Interval (VI)‐30 sec schedule for a daily (Monday–Friday) 32 min session. Upon acquisition of robust responding on this schedule, suppression training began where superimposed on the VI‐30 sec schedule was a second schedule in which a houselight CS was twice presented in the session for 2 min and terminated with a footshock (0.5 sec, 0.5 mA). The first and second CS presentations (termed the “cued” period) began 10 and 22 min, respectively, after the onset of the session. Responding during the non‐cued period reflects baseline operant behavior and detects potential non‐selective effects upon motor or food motivation, whereas increases in responding during the suppressed cued periods reflect anxiolytic‐like activity (Davis 1990). The number of responses occurring in the 2 min prior to each CS presentation (No CS responses) and those occurring in the 2‐min CS presentation (CS responses) were used to calculate the suppression ratio. The suppression ratio (# of CS responses/[# of CS responses + # of No CS responses]) ranges from 0 to 0.5, where 0 indicates a maximal conditioned emotional response (complete suppression characterized by zero CS responses) and 0.5 indicates no conditioned emotional response (no suppression characterized by equivalent CS and No CS responses).
Following adequate suppression training (demonstration of reliable suppression of responding during the CS presentation), 16 rats (approximately 400 g, 1.54 years old) were administered vehicle, LY2940094 (10 or 30 mg/kg) or the mGluR5‐negative allosteric modulator 3‐((2‐Methyl‐1,3‐thiazol‐4‐yl)ethynyl)pyridine hydrochloride (MTEP) (10 mg/kg) orally 1 h prior to behavioral testing in a within‐subjects design, where drug‐testing occurred twice per week (Tuesday, Friday) with the vehicle training otherwise occurring (Monday, Wednesday, Thursday). Drug assignments within and across days were determined using a Latin square. We used the mGlu5 receptor antagonist as a positive control in this study based upon literature findings in this assay (Rorick‐Kehn et al. 2006) and the anxiolytic properties of this mechanism (Spooren and Gasparini 2004; Porter et al. 2005). Data were analyzed by ANOVA followed by post hoc Dunnett's test.
The three dependent measures, suppression ratio, No CS responses, and CS responses, were each analyzed using a 3‐way ANOVA (Animal, Dose‐Order, Dose), where the main effect of dose was reported. Contrasts between Vehicle and LY2940094 were performed using the Bonferroni correction and the contrast between Vehicle and Positive Control, MTEP, was performed using Student's t‐test (α < 0.05). Statistical analyses were performed in JMP 5.1.1 statistical software.
Mouse four plate test
Male, CF1 mice were used. The apparatus (Panlabs, Barcelona, Spain) consisted of a cage (25 cm, 18 cm, 16 cm) [Model LE830] floored by four identical rectangular plates (11 cm, 8 cm) separated from one another by a gap of 4 mm. The plates were connected to a device that was capable of generating electric foot shocks (0.5 mA; 0.5 sec) [Model LE10026]. The top of the cage was covered by a transparent Perspex lid that prevented escape from the arena. Following an 18 sec habituation period, mice (n = 10) were subjected to an electric shock when crossing (transition) from one plate to another that is two legs on one plate and two legs on another. The number of punished crossings was calculated during a test period of 120 sec.
Statistical analysis was performed using JMP software (SAS Institute, Cary, NC). Dunnett's test following ANOVA was used (α < 0.05).
Rat novelty‐suppressed feeding
Male, Sprague‐Dawley rats (Harlan SD), 200–250 g were housed 4/cage. Standard Lab Chow and water were freely available with the exception of the food deprivation period which was in effect for 4 days; during this time, the animals were fed with standard lab chow placed on the floor of the homecage. Testing occurred on day 5 in experimentally naive rats.
The novel environment consisted of a Plexiglas cage of the same dimensions as the rat homecage (36 × 28 × 21 cm). Unlike the homecage, the novel cage was brightly lit and had a stainless steel grid rack, rather than a sawdust bedding floor. The light (67‐W, 110‐V) was placed on the top of the cage lid (clear Plexiglas), directly above the food dish. The food dish was a 12–cm‐diameter Petri dish lined with filter paper. One piece of standard lab chow was placed in the center of the dish. Each animal was placed into the novel cage to initiate an experimental session. The duration of the observation period and hence the maximum latency was 300 sec. The latency to begin eating was defined as chewing of food and constituted the dependent measure.
Rat stress‐induced hyperthermia
This assay can detect acute activity of clinically established anxiolytics as well as putative anxiolytic‐like activity, such as MTEP (Spooren and Gasparini 2004; Porter et al. 2005) which was used as a quality control compound as used previously (Fell et al. 2011). Male, Fischer F‐344 (300–400 g; Harlan Laboratories, Indianapolis, IN) were individually housed with food and water available ad libitum. Animals were fasted ~12–18 h before the experiment. Rats were dosed orally with 3, 10, or 30 mg/kg LY2940094 and MTEP (10 mg/kg, water vehicle) was used as a quality control. Following a 60‐min pretreatment period, core baseline body temperature was measured (T1, °C; Physitemp BAT‐12 Microprobe Thermometer, RET‐2 rat rectal probe), and then 10 min later, a second body temperature measurement was recorded (T2). The change in body temperature (T2 minus T1) was defined as the stress‐induced hyperthermic response. Data were analyzed using ANOVA, followed by Dunnett's post hoc tests (α < 0.05).
Mouse stress‐induced increases in cerebellar cGMP
This assay is a stress‐based assay that can detect anxiolytic effects of acutely administered drugs (c.f., Tang et al. 1997) and was studied here using alprazolam as a positive control as in Fell et al. (2011). Male, CF‐1 mice in the stress group were exposed to inescapable footshock (10 sec., 1.0 mA) and then immediately killed using a focused‐beam microwave (Thermatron, KY) and the cerebellum dissected for cGMP level determination on ice. For footshock, the Habitest Modular Test System (Ryder et al. 2006) was used (Coulbourn Instruments, Allentown, PA).
Tissue dissection and cGMP radioimmunoassay: After microwaving, a small piece (10–20 mg) of cerebellar cortex was quickly removed from the skull, the tissue weight was recorded and homogenized in 2 mL of 1.0% perchloric acid (Sigma, St. Louis, MO). Tissue homogenates were kept on ice for 30 min, then placed in a boiling water bath for 5 min followed by centrifugation at 11,700g for 20 min. A quantity of 1.0 mL of supernatant from each sample was acetylated using 40 μL triethylamine (Sigma) and 20 μL acetic anhydride (Sigma), vortexed and centrifuged at 13,000g for 20 min at 4°C. The acetylated mouse cerebellar supernatant samples were stored at 4°C until cGMP analysis by radioimmunoassay. Cerebellar cGMP content in the acetylated samples was determined using the cGMP [125I] Flash Plate™ radioimmunoassay (Perkin Elmer Sciences, Boston, MA) on duplicate samples from each animal as per manufacturer's instructions.
For each animal, cGMP levels were normalized to wet tissue weight and used to compute group averages and SEM. All statistical analyses were performed using ANOVA followed by the Dunnett's test (α < 0.05).
Rat delayed‐matching‐to‐position
Under this procedure, rats were trained to press the same lever (left or right) as the one presented prior to a delay interval. Male, Lister hooded rats (N = 61) were housed in groups of four in plastic cages containing sawdust. The rats were maintained on a 12‐h light–dark cycle with lights on at 7 am. The experiments were conducted during the same part of the light phase each day (between 0730 and 1330). Starting weight of the animals was 210 ± 1.1 g (mean ± S.E.M.) and at the time of testing they weighed 487 ± 6.2 g (mean ± S.E.M.). They were maintained on a food‐restricted diet, which allowed for growth, with ad libitum access to water.
Sixteen standard operant chambers, housed in sound and light attenuating chambers were used. Each chamber comprised a house light, two retractable levers, each with a stimulus light above it. The levers were located either side of a recessed magazine where food pellets (Noyes, 45 mg, Formula P) were delivered from an automatic pellet dispenser. Start of a session was signalled by onset of the house light, and its permanent offset indicated the end of a session. Experimental sessions were controlled and data recorded using programs written in house using MedPC‐IV software.
Sample phase
A trial began with one of the levers extending into the chamber. The stimulus light above the lever would also be illuminated. If the lever was pressed, it would be retracted and the stimulus light extinguished. This initiated the pseudorandomly selected delay period of 1, 2, 4, 8, or 16 sec during training or 1, 12, or 32 sec during test.
Head entry
After this delay period was completed, animals were required to make a head‐entry into the food hopper within 10 sec; if an appropriate response was made both levers were extended into the chamber. If no response was made during this period, the house‐light would be extinguished and the trial counted as an omission.
Choice phase
If animals made the appropriate head entry response and the levers extended into the operant chamber, they would then have 10 sec within which to make their response. A correct response consisted of an animal pressing the same lever that had been extended into the chamber during the sample phase. This response would result in both levers retracting and a single food pellet being delivered into the food hopper. After a 5 sec intertrial interval, the next trial would begin. Following an incorrect response, (the animal pressing the opposite lever as had been extended during the sample phase), both levers would retract and the house‐light would be extinguished signaling the end of that trial. After a 5 sec time out, the next trial would begin, signalled by onset of the house‐light. If the animal failed to respond within 10 sec during the choice phase, the levers would be retracted, the house‐light extinguished and the trial recorded as an omission.
Each session terminated after 75 trials (15 or 25 pseudorandom presentations of each delay period). Animals were trained to a criterion of greater than 70% accuracy over the entire session with less than 10% omissions of responding. After criterion had been reached, rats were trained two times a week in a baseline condition and once a week in a test condition.
Dependent measures and statistical analyses
%Accuracy = correct/(correct + incorrect)×100; %Omissions = omissions/(correct + incorrect + omissions)×100.
Average latency correct = sum correct latencies/#of correct trials
Animals were assigned to treatment groups counterbalanced for previous treatments and baseline performance. The same statistical tests were used for both matching and test analysis, Treatment (or assigned treatment) groups were analyzed for statistical significance using a two‐way mixed repeated measures ANOVA with within‐subjects factor of Delay and between‐subjects factor of assigned Treatment (4 levels). Analysis of simple effects followed with planned comparisons was used to investigate significant effects on treatment. If significant effects of treatment were present but no significant interaction, the interaction would be further assessed with planned comparisons and analysis of simple effects. Treatment effects on simple measures (e.g., total percent accuracy, latency to make in/correct responses) were analyzed using a one‐way analysis of variance with Treatment (4 levels) as the sole factor. Planned comparisons were used to examine appropriate significant effects.
Rat five choice serial‐reaction time test
Rats were trained to nose poke into one of five holes when illuminated for a brief time. Male, Lister hooded rats (N = 59) were housed in groups of four in plastic cages containing sawdust. The rats were maintained on a 12‐h light–dark cycle with lights on at 7 am. The experiments were conducted during the same part of the light phase each day (between 0800 and 1330). Starting weight of the animals was 200–250 g and at the time of testing they weighed 398 ± 4.1 g (mean ± S.E.M.). They were maintained on a food‐restricted diet, which allowed for growth with ad libitum access to water.
Standard operant chambers (12), housed in sound and light attenuating chambers were used. Each chamber comprised a house light, a recessed magazine where food pellets (Test Diet™, AIN‐76A Rodent Tablet 45 mg) were delivered from an automatic pellet dispenser, opposite to a standard rat 5 hole wall, consisting of five nose pokes each containing a stimulus light at the back. Start of a session was signaled by onset of the house light, and its permanent offset indicated the end of a session. Experimental sessions were controlled and data were recorded using programs written in house using MedPC‐IV software.
A trial began with 0.1 sec illumination of the house light, followed by the illumination of one of the five stimulus lights. The light was illuminated for 0.25 sec. A nose poke in the correct hole, either while the stimulus light was on, or during the 5 sec limited hold following the stimulus light onset, initiated release of a food pellet and the intertrial interval (ITI) of 5 sec. An incorrect response results in the start of ITI. If the animal did not respond within 5 sec following the stimulus light onset, an omission was recorded and the ITI started. A premature response, where the rat responded during the ITI, resulted in a 5 sec punishment period with the house light on before restarting the trial. Each session terminated after 100 trials (20 pseudorandom presentations of each stimulus light).
Rats were assigned to dose groups based on task performance on the day prior to test. Statistical approach was the same for both counterbalancing and test performance. Data were subjected to between‐subjects ANOVA with between‐subjects factor of (assigned) treatment. The dependent variables were % correct, % omissions, average latency to make a correct response, average latency to make an incorrect response, number of premature responses, number of preservative responses, and average latency to retrieve food from the magazine.
If significant main effects were found, then these were further investigated using planned comparisons analysis. In the absence of main effects, a priori, planned comparisons would be carried out based on the frequent likely occurrence of U‐shaped dose response curves.
The following measures were analyzed: % correct responding (i.e., accuracy) = total correct/(total correct + incorrect) × 100; % omissions = total omissions/(total correct + incorrect + omissions) × 100; Average latency correct = latency to correct/no. trials correct; Premature responding = no of responses during ITI preceding trial; Perseverative responding = no of repeated responses at the response aperture; Food magazine latency = latency to retrieve food from magazine.
Mouse rotarod performance
Experimentally naive, male NIH Swiss mice (Harlan Sprague‐Dawley, Indianapolis, IN) were allowed to acclimate to the vivarium for at least 3 days prior to testing. They weighed between 28 and 32 g and were housed in groups of 10–12 in plastic cages (24 × 45 × 15 cm high) with sawdust bedding in a temperature‐controlled vivarium with a 12‐h light–dark cycle (lights on: 0600–1800 h). Experiments were conducted during the light phase of this cycle. After dosing with diazepam (i.p., 30 min prior) or LY2940094 (p.o., 60 min prior) or vehicle, mice were placed on a rotorod (Ugo Basile 7650) operating at a speed of 6 revolutions/min and observed for falling. Mice that fell off the rotarod on two occasions during 2 min were scored as failing. Mice were not pretrained on this task.
Plasma and brain drug levels
Brain samples were weighed and a threefold volume of water/methanol (4:1, v/v) was added prior to homogenization with an ultrasonic tissue disrupter. Control (naive) brain tissue was also homogenized to generate control homogenate for preparation of calibration standards.
Stock solutions containing 1 mg/mL of LY2940094 and fluoxetine HCl were diluted to produce working solutions which were then used to fortify control plasma or control brain homogenate to yield calibration standards with concentrations ranging from 1 to 5000 ng/mL. Aliquots of each study sample, calibration standard, and control samples were then transferred to 96‐well plates, mixed with acetonitrile/methanol (1:1, v/v) containing an internal standard to precipitate sample proteins then centrifuged to pellet insoluble material. The resulting supernatants were subjected to LC‐MS/MS analysis using an Applied Biosystems/MDS Sciex API 4000 (Foster City, CA) equipped with a TurboIonSpray interface, and operated in positive ion mode. The analytes were chromatographically separated with a gradient LC system and detected and quantified with Selected Reaction Monitoring (SRM) (M+H)+ transitions specific to each compound (2940094, m/z ,481.2 > 202.1 and 110140, m/z 309.0 > 44.0). The mass spectrometer quadrapoles were tuned to achieve unit resolution (0.7 DA at 50% FWHM) and data were acquired and processed with Applied Biosystems/MDS Sciex Analyst software (version 1.4.2).
Mouse NOP receptor occupancy
Simultaneous measurement of NOP receptor (Pedregal et al. 2012) and serotonin reuptake transporter (SERT) protein occupancy (Dreyfus et al. 2013) were assessed in drug naive male, NIH Swiss mice (same as used in the forced swim studies). Fluoxetine HCl (i.p.) was dosed 30 min prior and LY2940094 (p.o.) was dosed 60 min prior to killing (dosed as per the forced‐swim experiments). Tracers for NOP and SERT were dosed (i.v. lateral tail vein) and the mice were killed by cervical dislocation after 60 min of their injection. The whole brain is rapidly removed, and lightly rinsed with sterile water. Frontal cortex and hypothalamus brain tissue are dissected, weighed, stored in 1.5 mL eppendorf tubes, and placed on dry ice until tissue extraction (see below). Using a drug naive rat, six cortical brain tissues samples are collected for use in generating blank and standard curve samples.
Tissue samples are thawed on wet ice. Acetonitrile containing 0.1% formic acid is added to each sample at a volume of four times (eight times for hypothalamus) the weight of the tissue sample. For standard curve (0.3–30 ng/g) samples, a calculated volume of standard reduces the volume of acetonitrile. The sample is homogenized (7–8 watts power using sonic probe dismembrator; Fisher Scientific) and centrifuged for 16–min at 14,000 rpm. The (50 μL) supernatant solution is diluted by 100 μL of sterile water (pH 6.5) for NOP. The (50 μL) supernatant solution is diluted by 150 μL of sterile water (pH 6.5) for SERT. This solution is then mixed thoroughly and analyzed via LC/MS/MS for tracer compound. The tissue levels of the tracer are analyzed by LC/MS/MS.
The molecules used in this assay were handled as follows:
DASB, (3‐amino‐4‐[2‐[(di(methyl)amino)methyl]phenyl]sulfanyl‐benzonitrile) used as the SERT tracer, was dissolved into sterile water (concentration 1000 μg/mL). The 2 μg/mL solution is prepared by diluting the 100 μg/mL stock with saline. Frozen aliquots of the tracer stock were stored in a −80 freezer for future use. Tracer dose = 10 μg/kg, IV + 40‐min pretreatment. Dose volume = 5 mL/kg.
Paroxetine, used as the SERT‐positive control, was dissolved in 8% hydroxyl‐propyl‐β‐cyclodextrin + 0.05% acetic acid. Positive control dose = 12 mg/kg, IP + 1‐h pretreatment. Dose volume = 10 mL/kg.
(2S)‐2‐[(2‐fluorophenyl)methyl]‐3‐(2‐fluorospiro[4,5‐dihydrothieno[2,3‐c]pyran‐7,4′‐piperidine]‐1′‐yl)‐N,N‐dimethyl‐propanamide (L)‐tartaric acid, the NOP receptor tracer (Pedregal et al. 2012, cpd. S‐(27)), was dissolved into 25% HP‐BCD (1 mg/mL). The 0.6 μg/mL solution was prepared by diluting the 1 mg/mL stock with 25% HP‐BCD. Frozen aliquots of the tracer stock were stored in a −80 freezer for future use. Tracer dose = 3 μg/kg, IV + 40‐min pretreatment. Dose volume = 5 mL/kg.
SB612111, used as the NOP receptor‐positive control, was dissolved in 25% HP‐BCD. The quality control dose = 3 mg/kg, PO + 60‐min pretreatment. The positive control dose = 30 mg/kg, IV + 60‐min pretreatment. Dose volume = 10 mL/kg.
Compounds for in vivo studies
LY2940094 was synthesized as described by Eli Lilly and Company (Toledo et al. 2014). The compound was dissolved in 20% captisol®, a cyclodextrin derivative (Cydex, Inc., Lenexa, KS) in 25 mmol/L phosphate buffer (pH2) and dosed orally 60 min prior to testing. Imipramine HCl, amitriptyline HCl, and chlordiazepoxide HCl were obtained from Sigma‐Aldrich. They were dissolved in 0.9% NaCl and injected i.p., 30 min prior to testing. Paroxetine HCl and fluoxetine HCl were synthesized at Eli Lilly and Company, dissolved in 0.9% NaCl, and dosed i.p., 30 min prior to testing. MTEP and MPEP (Tocris) were dissolved in water and dosed i.p. Alprazolam (Sigma‐Aldrich) was suspended in 10% hydroxypropyl beta‐cyclodextrin. Compounds were dosed in the forms noted in a volume of 1 or 2 mL/kg for rat studies and 10 mL/kg for mouse studies.
Results
LY2940094 produced anti‐immobility effect in the forced‐swim assay that detects standard of care antidepressants
Oral administration of LY2940094 produced dose‐dependent decreases in immobility in mice (F4,31 = 4.39, P < 0.01) (Fig. 1A). The minimal effective dose under this assay was 30 mg/kg and the maximal effect was comparable to the positive control, imipramine (15 mg/kg, i.p.). The antidepressant‐like effect of LY2940094 did not tolerate after 5 days of consecutive oral dosing (F4,31 = 4.45, P < 0.01) (Fig. 1B). The on‐target activity of LY2940094 to produce these antidepressant‐like effects was demonstrated in nociceptin null mice. In this study, the antidepressant‐like effects of LY2940094 were completely prevented in NOP −/− mice (Fig. 2). The specificity of this effect was shown by the fact that comparable effects of the tricyclic antidepressant, imipramine, were unchanged in the knockout mice (Fig. 2). A two‐way ANOVA confirmed these findings where the effect of treatment (F2,40 = 10.15, P < 0.001), and genotype (F2,40 = 6.42, P < 0.05) were significant, whereas the interaction term was not (F2,40 = 1.76, P = 0.186).
Figure 1.
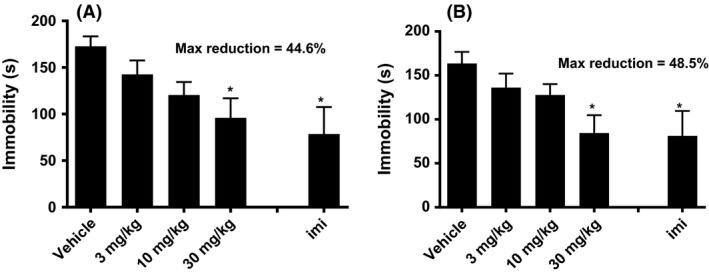
(A) LY2940094 dose‐dependently decreased immobility in the mouse forced‐swim test (n = 8), an effect shared by the tricyclic antidepressant imipramine (imi). Mice were dosed p.o. with LY2940094 and tested 60 min later. IMI: 15 mg/kg, 30 min prior by i.p. (n = 4). *P < 0.05 by Dunnett's test. (B) LY2940094 retained antidepressant‐like efficacy in the mouse forced‐swim test after 5 days of dosing. Mice were dosed p.o. with LY2940094 (n = 8) for 5 days and then tested on day 5, 60 min later. IMI: 15 mg/kg, 30 min prior by i.p. (n = 4). *P < 0.05 by Dunnett's test compared to vehicle.
Figure 2.
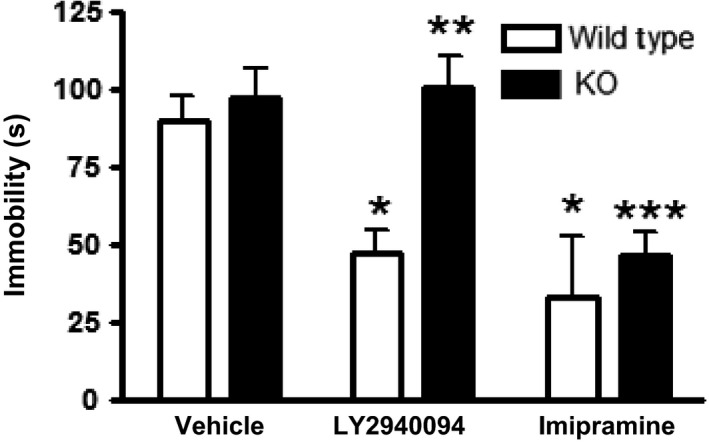
LY2940094 decreased immobility or float time in wild‐type mice (unfilled bars) but was devoid of activity in NOP receptor knockout mice (filled bars). Imipramine was active in the mouse‐forced swim test in both genotypes. Mice were dosed p.o. with LY2940094 (30 mg/kg) then tested 60 min later. IMI: 15 mg/kg, 30 min prior i.p. Wildtype: vehicle and LY2940094 (n = 10); imipramine (n = 3). Knockout: vehicle (n = 11), LY2940094 (n = 9), imipramine (n = 4). *P < 0.05 versus wildtype vehicle; **P < 0.05 versus wildtype LY2940094; ***P < 0.01 versus knockout vehicle. NOP, nociceptin receptor.
Antidepressant‐like efficacy of fluoxetine was enhanced by addition of LY2940094 in mice
Doses of 3 and 10 mg/kg LY2940094 were combined with 10 mg/kg fluoxetine. Although inactive when given alone, the drug combinations demonstrated effects not observed with either drug alone. There was a significant effect of addition of LY2940094 (F1,42 = 8.72, P < 0.01) and a significant effect of dose of LY294004 (F2,42 = 8.09, P < 0.01) but not a significant treatment x dose interaction (F2,42 = 1.83, P = 0.17). Doses of LY2940094 that did not significantly alter immobility time when given alone were able to increase the anti‐immobility effects of fluoxetine. In these studies, a dose of 3 mg/kg (p.o.) LY2940094 when given with an ineffective dose of fluoxetine (3 mg/kg, i.p.) produced effects that were statistically greater than either drug alone (Fig. 3).
Figure 3.
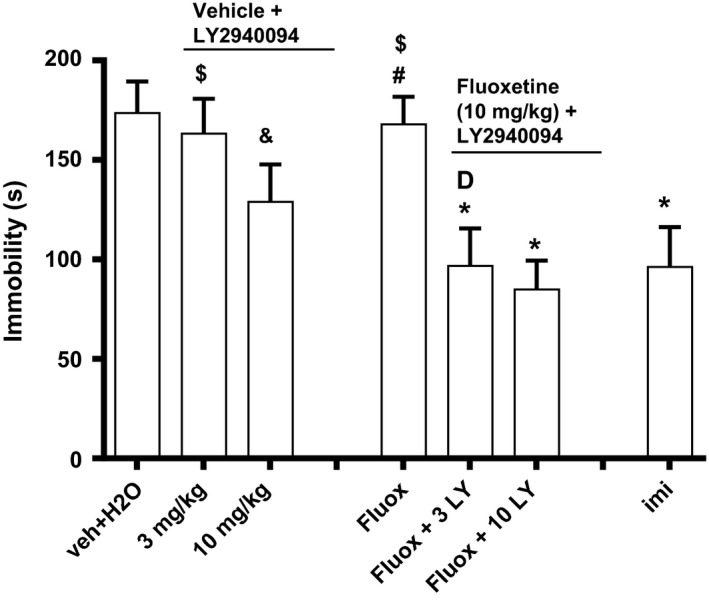
LY2940094 enhanced the antidepressant‐like effect of fluoxetine in the mouse forced‐swim test. LY2940094 was dosed p.o., 1 h prior to the test or 30 min prior to fluoxetine (i.p.). The test was started 30 min after fluoxetine in the combination study. IMI: imipramine at 15 mg/kg, i.p. *P < 0.05 compared to vehicle (Dunnett's test); $ P < 0.05 compared to 3 + flu10 (t‐test); # P < 0.05 compared to 10 + flu10 (t‐test); & P < 0.05 compared to 10 + flu10 (t‐test, one tail); D P < 0.05 versus veh, 3 alone and fluoxetine alone (Dunnett's test). Each bar represents the mean ± S.E.M of 8 mice.
The enhancement of effects of fluoxetine by LY2940094 were not associated with changes in either NOP receptor occupancy by LY2940094 or by changes in serotonin uptake site occupancy by fluoxetine; both receptors were already highly occupied (Table 1). Likewise, the synergy with fluoxetine/LY2940094 combinations were not likely due to pharmacokinetic alterations in either plasma or brain exposures of either drug alone as there were no significant differences observed between drug exposures when the molecules were dosed together (Table 1).
Table 1.
Plasma and whole brain levels of fluoxetine and LY2940094 and occupancy of NOP receptors and the serotonin transporter (SERT) after co‐administration of LY2940094 and fluoxetine in micea
| Compounds | Plasma [Fluoxetine] | Plasma [LY294009] | Brain [Fluoxetine] | Brain [LY2940094] | % NOP RO | % 5HTT RO |
|---|---|---|---|---|---|---|
| Fluoxetime + Vehicle | 450 ± 40 | — | 7349 ± 520 | — | 16.5 ± 4.4 | 95.7 ± 1.3 |
| Vehicle + LY2940094 | — | 140 ± 14 | — | 123 ± 25 | 97.0 ± 0.8 | 6.0 ± 1.3 |
| Fluoxetime + LY2940094 | 400 ± 34 | 150 ± 37 | 6960 ± 781 | 131 ± 30 | 99.9 ± 0.5 | 95.5 ± 1.8 |
Data are from mice from the drug interaction study for which data are shown in Figure 3. Fluoxetine (10 mg/kg, i.p.) was given 30 min prior and LY2940094 (3 mg/kg, p.o.) was given 60 min prior to testing. Data represent means ± S.E.M. of 4 mice/group. Values are ng/mL for plasma concentrations and ng/g for brain. NOP, nociceptin receptor.
Lack of effect of LY2940094 under a DRL‐72s assay
LY2940094 was inactive up to oral doses of 30 mg/kg under these assay conditions. LY2940094 did not significantly affect either the number of responses (F3,28 = 0.13, P = 0.94) or the number of reinforcers (F3,28 = 0.06, P = 0.98) occurring under the DRL schedule. In contrast, imipramine produced an antidepressant‐like signature in this assay (Fig. 4).
Figure 4.

LY2940094 was without significant effect under the DRL72 s assay in rats. In contrast, imipramine significantly enhanced response efficiency, increasing the number of reinforcers delivered. LY2940094 was dosed orally 60 min prior to testing. Each point represents the mean ± S.E.M of data from 8 rats. *P < 0.05 compared to vehicle (Dunnett's test).
LY2940094 had mixed effects in in vivo assays that detect effects of anxiolytic drugs
We studied only a few assays known to detect anxiolytic drugs and we only studied acute dosing under these procedures. In three assays that detect anxiolytic drugs, LY2940084 was not active. Under a conditioned‐suppression paradigm (conditioned emotional response), LY2940094 did not attenuate response suppression (F2,45 = 0.15, P = 0.86) like that of the positive control agent MTEP up to 30 mg/kg that also decreased response rates in the absence of shock‐paired stimuli (Fig. 5A, bottom panel). LY2940094 also did not affect responding in the absence of the shock‐paired stimulus (F2,45 = 1.13, P = 0.33) LY2940094 thus did not alter the suppression ratio as did MTEP under these conditions (F2,45 = 0.55, P = 0.58) (Fig. 5A, top panel). LY2940094 was also not active in mice under either a novelty‐suppressed feeding paradigm (F2,21 = 0.01, P = 0.99) (Fig. 5B) or the 4‐plate test (F3,28 = 0.13, P = 0.94) (Fig. 5C). In contrast to the lack of efficacy of LY2940094 under these procedures, chlordiazepoxide increased responding (Fig. 5B and C).
Figure 5.
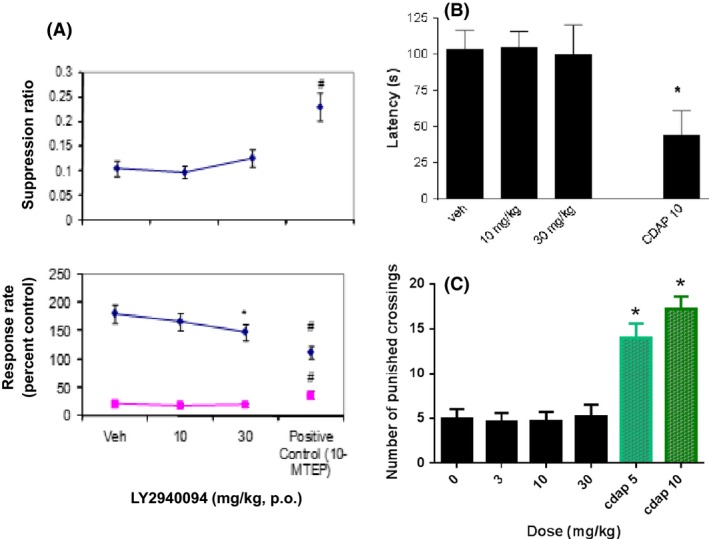
LY2940094 was not active in multiple assays that detect effects of traditional anxiolytics (SSRIs or benzodiazepine anxiolytics). (A) LY2940094 was not active in attenuating conditioned suppression (i.e., ‘cued’ periods) of food‐maintained responses under a VI30 sec schedule in rats, in contrast to the positive effects of MTEP in increasing responding in the presence of the shock‐associated cue. LY2940094 or MTEP were dosed orally and behavior evaluated beginning 60 min post dosing. # P < 0.05 compared to vehicle control (Dunnett's test). (B) LY2940094 was not active in reducing suppression of latencies of rats to eat in a novel environment. In contrast, chlordiazepoxide (10 mg/kg) was effective. LY2940094 was dosed orally and behavior evaluated beginning 60 min post dosing. Chlordiazepoxide was given i.p., and behavioral testing began 30 min post dosing. *P < 0.05 compared to vehicle control by Dunnett's test. n = 8/group. (C) LY2940094 was not active in reducing suppression of behavior in a four‐plate test in mice. In contrast, chlordiazepoxide markedly increased responses. LY2940094 was dosed orally and behavior evaluated beginning 60 min post dosing. Chlordiazepoxide was given i.p., and behavioral testing began 30 min post dosing. n = 8/group *P < 0.05 compared to vehicle control by Dunnett's test.
In contrast, anxiolytic‐like activity was detected in some assay systems with LY2940094. In mice, LY2940094 significantly attenuated fear‐conditioned immobility with a minimal effective dose of 30 mg/kg (F4,43 = 4.1, P < 0.01) (Fig. 6A), an effect comparable to that observed with the SSRI paroxetine where doses of 3 and 10 mg/kg were significantly different than vehicle control values as was the mGlu5 receptor antagonist MTEP (Fig. 6B). Stress‐induced hyperthermia in rats was likewise attenuated by LY2940094 (F3,39 = 5.87, P < 0.01) as with the positive control MTEP (Fig. 6C). LY2940094 also dose‐dependently suppressed stress‐induced increases in cerebellar cGMP as did the anxiolytic agent alprazolam (F3,28 = 12.1, P < 0.0001) (Fig. 6D). Although statistically significant in all of these assays, the positive control drugs appeared more efficacious than LY2940094 at the doses tested.
Figure 6.
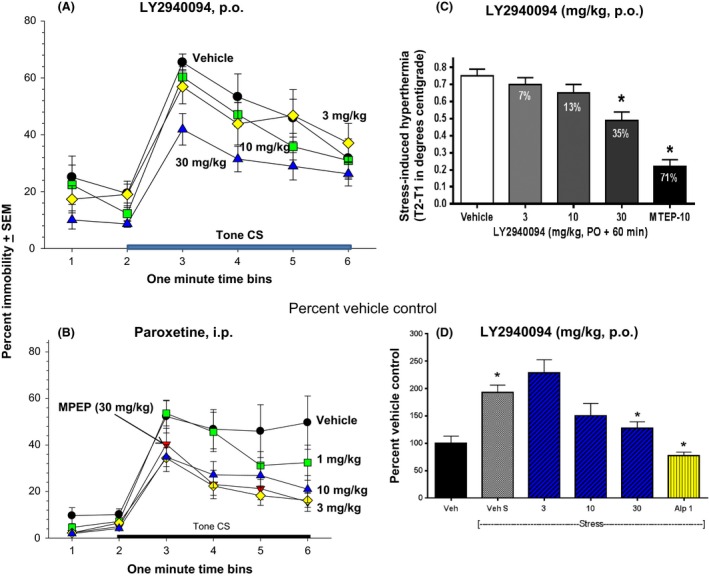
LY2940094 was efficacious in several rodent models in which conventional anxiolytic drugs are active. (A) LY2940094 significantly attenuated immobility in the mouse fear‐conditioned freezing assay at 30 mg/kg. LY2940094 was dosed orally 60 min prior to testing. (B) Paroxetine was studied as a positive comparator in the fear‐conditioned freezing assay with effects at 3 and 10 mg/kg being significantly different than vehicle (Dunnett's test). MPEP was also used as a positive control in this study. Each point represents the mean ± S.E.M effect in 10–11 mice. (C) Dose‐dependent inhibition of stress‐induced hyperthermia in Fischer F‐344 rats after oral administration of LY2940094:. Data represent means (±S.E.M) for n = 10 rats/group. MTEP was used as a positive control at 10 mg/kg. Neither LY2940094 nor MTEP affected baseline core body temperature (data not shown). *P < 0.05 compared to vehicle (Dunnett's test). n = 8/group. (D) Decreases in stress‐induced elevations in cerebellar cGMP in CF‐1 mice by LY2940094 or alprazolam. *P < 0.05 compared to vehicle by Dunnett's test (n = 8/group).
LY2940094 did not impact cognitive performances of rats
In two assays that can detect drug‐induced impairment of cognitive performance, LY2940094 was inactive under both the 5CSRTT (F4,52 = 0.89, P > 0.1) and delayed‐matching to position (DMTP) (F4,57 = 2.2, P > 0.05), whereas the antidepressant amitriptyline was active (Fig. 7).
Figure 7.

LY2940094 did not disrupt cognitive performances of rats. Amitriptyline but not LY2940094 decreased the accuracy of responding and increased the number of response omissions of rats under a 5‐choice serial reaction time task (5‐CSRTT). ***P < 0.001. Amitripyline but not LY2940094 decreased the accuracy of responding and increased the number of response omissions of rats under a delayed matching to position (DMTP) task. ***P < 0.001. MTEP, 3‐((2‐Methyl‐1,3‐thiazol‐4‐yl)ethynyl)pyridine hydrochloride.
LY2940094 did not impair other ongoing behaviors
The specificity of the behavioral effects of LY2940094 for the behavioral changes reported directly above can be assessed by the negative effects observed with this NOP receptor antagonist. In a test of motor impairment, chlordiazepoxide but not LY2940094 impaired performance of mice on a rotarod performance task (Fig. 8A). Likewise, LY2940094 did not significantly affect locomotor performance of mice (Fig. 8B) (Time: F11,180 = 40.8, P < 0.05; Dose: F2,180 = 3.34; P < 0.05 with vehicle vs. dose being nonsignificant).
Figure 8.
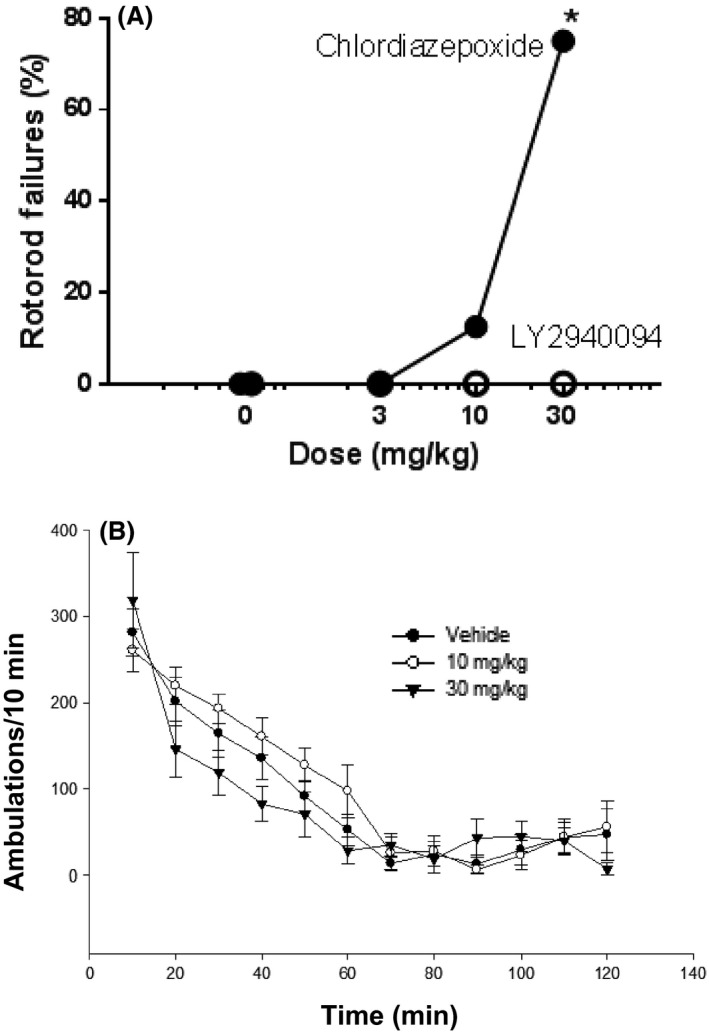
There was no significant impact of LY2940094 on motor coordination or movement. (A) Chlordiazepoxide but not LY2940094 increased the percentage of rotarod failures of mice. *P < 0.05 compared to vehicle by Dunnett's test. (B) LY2940094 did not significantly alter the locomotor activity of mice compared to vehicle.
Discussion
The present series of experiments was conducted in order ascertain the potential of LY2940094 to engender antidepressant and anxiolytic‐like effects in rodents as well as to evualte some potential side‐effects of this molecule. Unlike conventional antidepressants that robustly enhance cortical monoamine levels (c.f., Jordan et al. 1994), LY2940094 only marginally enhanced synaptic concentrations of serotonin, without significantly altering norepinephrine or dopamine in rat medial prefrontal cortex (Post et al. 2016). Despite this non‐monoaminergic signature, LY2940094 generated an antidepressant‐like phenotype in mice in the forced‐swim assay comparable in magnitude to that of the monoamine‐based antidepressant imipramine (Fig. 1A) in an assay that detects standard antidepressant drugs (Porsolt et al. 1977; Cryan et al. 2002). However, LY2940094 was not active under a DRL 72‐sec assay. The antidepressant signature of LY2940094 was due to blockade of NOP receptors since the antidepressant signature was absent in mice lacking the NOP receptor (Fig. 3). The findings with the NOP receptor knockout mice (NOP −/−) are consistent with the research literature under these experimental conditions with NOP receptor antagonists other than LY2940094 (see Witkin et al. 2014a for summary). Notable here too is the identification of LY2940094 as a potential antidepressant (Post et al. 2016) using the forced‐swim test (Porsolt et al. 1977). This and related behavioral models have come under scrutiny for their potential lack of translational power into MDD patients (c.f., Belzung 2014). Nonetheless, data have continued to confirm the predictive value of this assay. For example, other nondirect monoaminergic agents including NMDA receptor antagonists such as ketamine (Abdallah et al. 2015), the antimuscarinic scopolamine (Witkin et al. 2014b), and the PDE4 inhibitor rolipram (Li et al. 2003; Zhang et al. 2009) have shown efficacy in MDD patients and activity in the forced‐swim assay.
Further, LY2940094 was able to enhance the antidepressant‐like effects of fluoxetine in mice without altering the plasma or brain exposures of either drug alone, and without altering the occupancy of either drug at NOP receptors or serotonin transporters (Table 1). These are thus the first data to document a pharmacodynamic drug interaction between NOP receptor blockade and monoamine mechanisms. The mechanism of this drug interaction was not explored here nor were drugs other than fluoxetine studied. The possible augmentation of serotonergic neurotransmission by LY2940094 is one possible mechanism (see Le Maître et al. 2005; Gavioli and Calo 2013). In rats, LY2940094 also produced activity in the forced‐swim test with a minimal effective dose of 30 mg/kg, p.o. (Post et al. 2016). In contrast, under a DRL 72s assay that detected effects of the antidepressant imipramine, LY2940094 was not active suggesting the possibility that LY2940094 might not control impulsivity (c.f., Marek et al. 2016). These findings indicate that NOP receptor antagonism with LY2940094 overlaps biologically with some, but not all, of the substrates sub‐serving the antidepressant‐like effects of biogenic amine‐based antidepressants.
In assays that detect effects of anxiolytic drugs, LY2940094 was inactive under multiple conditions for which anxiolytic drugs were active (Fig. 5). Thus, LY2940094 was not active in counteracting conditioned suppression, novelty‐suppressed feeding, or reducing suppression produced in the four plate test in mice. LY2940094 was also not active when evaluated under the Vogel conflict test in rats or marble‐burying in mice (Post et al. 2016). Unfortunately, there are no studies in the literature with other NOP receptor antagonists studied under these conditions against which to make comparisons with LY2940094 (see Witkin et al. 2014a for summary). It is also important to reiterate the fact that multiple strains of rodents were used in the present set of studies and therefore, direct comparisons across one strain is not possible. It is important for the reader to be aware of the potential for strain‐dependent drug effects.
In contrast, LY2940094 attenuated fear‐conditioned freezing behavior in mice, similar to the SSRI paroxetine (Fig. 6), reduced stress‐induced hyperthermia in rats like the positive control MTEP, an mGlu5 receptor antagonist (Fig. 6), and reduced stress‐induced increases in cerebellar cGMP as did alprazolam (Fig. 6). Thus, as with the antidepressant‐detecting assays, LY2940094 produced effects in anxiolytic‐detecting assays in vivo that only partially overlapped with conventional anxiolytic agents and SSRI anxiolytics. These findings with mixed results in multiple assays in mice and rats make prediction of efficacy against anxiety symptoms difficult. Compounding this state of affairs is a consistent scientific literature demonstrating anxiolytic‐like effects of NOP receptor agonists (c.f. Witkin et al. 2014a for a review). Further, it is recognized that in many assays, antidepressants require subchronic treatment to demonstrate anxiolytic efficacy and only acute dosing with LY2940094 was studied here as in other studies with NOP receptor antagonists (see Witkin et al. 2014a for summary). In a mouse marble‐burying assay which detects the effects of SSRI antidepressants after acute dosing (Li et al. 2006a), LY2940094 was also inactive. The possibility of creating broader increases in anxiolytic‐like efficacy with subchronic dosing of NOP receptor antagonists remains an empirical question.
We could only find one report of an antagonist (icv dosing of the peptide UFP‐101) producing anxiolytic‐like efficacy (elevated plus‐maze) (Duzzioni et al. 2011). In contrast, there are at least 15 reports in the experimental literature showing efficacy of N/OFQ and other agonists in in vivo assays that detect anxiolytic agents such as diazepam (see Witkin et al. 2014a). Although the mechanisms by which antagonists engender such anxiolytic signatures in rodents is not understood, examples of antagonists producing effects previously thought to be agonist‐driven have been observed in alcohol studies that have led to speculations on mechanism relating to receptor adaptations (Rorick‐Kehn et al. 2016). In clinical investigation in MDD patients, LY2940094 separated from placebo in reducing HAMD scores but did not separate from placebo on either the Hamilton anxiety rating scale (HAMA) total score at Week 4 or the patient‐rated HADS anxiety subscale (Post et al. 2016). The MDD patients in the Post et al. (2016) study, however, were not enriched in anxiety symptoms or ratings. Future clinical data will thus help to better define the therapeutic potential in this domain.
In evaluation of potential side‐effect liabilities of NOP receptor antagonism, LY2940094 did not engender cognitive impairment, whereas impaired performance on two cognitive tasks was observed with amitriptyline (Fig. 7), an antidepressant with high affinity for muscarinic receptors (Rathbun and Slater 1963) that engenders cognitive impairment in humans (Kerr et al. 1996). In contrast to anxiolytic doses of chlordiazepoxide (Fig. 8), motor impairment was not observed with LY2940094. Based upon these findings, it was predicted that LY2940094 would be tolerable at predicted human efficacious exposures. Indeed, LY2940094 engendered a relatively inert adverse event profile in patients (Post et al. 2016).
It would have been possible to reject the hypothesis that LY2940094 might be antidepressant in patients based upon the lack of fully congruent antidepressant‐ and anxiolytic‐like efficacy in the preclinical models discussed. However, given that preclinical models are not disease models, the overall basis for a bet of translational success into humans must be built upon sound biological grounds that utilize multiple convergent data on a molecule and mechanism. It follows logically from the lack of disease homology models that increasing the number of tests in which activity is detected does not necessarily prescribe an increased probability of success in clinical translation.
The preclinical findings reported here provided the opportunity to determine the necessary and sufficient biological outputs in rodents that predicted the translation to efficacy in MDD patients. In the case of LY2940094, high NOP receptor occupancy in both rodent (Toledo et al. 2014) and human brain (Raddad et al. 2016) appear to be required for efficacy in preclinical models (current report) and in patients (Post et al. 2016). The high levels of monoamines detected in rodent prefrontal cortex in response to conventional antidepressants (e.g., Jordan et al. 1994) are clearly not required for predicting patient efficacy (Post et al. 2016). Although not a model of depression, the forced‐swim assay has provided valuable predictive guidance as to potential efficacy in MDD patients (Porsolt et al. 1977; Cryan et al. 2002). LY2940094 was active in both rats (Post et al. 2016) and in mice under the forced‐swim test, an effect shown to require NOP receptors (present data). In contrast, although the DRL 72 s assay detects multiple antidepressant mechanisms (Marek et al. 2005; Ardayfio et al. 2008; Marek et al. 2016), it did not detect effects of LY2940094 at a dose active in the rat forced‐swim test (Post et al. 2016). Another predictive model, marble‐burying in mice, detects acutely administered conventional antidepressants but did not detect effects of LY2940094 (Post et al. 2016). An initial Phase 2 proof‐of‐concept trial with LY2940094 in MDD patients (Post et al. 2016) appeared to be best predicted from the biology of the nociceptin system in regulating stress biology (c.f., Witkin et al. 2014a), efficacy in a model detecting conventional antidepressants (forced‐swim) at ≥80% NOP receptor occupancy levels, and the fact that this behavioral output in mice was dependent upon the presence of NOP receptors.
The preclinical data presented here were used to predict the therapeutic success of LY2940094 in moderating or alleviating symptoms of depression. An 8‐week, double‐blind, placebo‐controlled, proof‐of‐concept study was completed that evaluated LY2940094 as a novel oral medication for the treatment of patients with MDD in which 136 patients were randomized to placebo or LY2940094. This study provided statistically significant evidence for an antidepressant effect of 40‐mg once‐daily dosing of LY2940094 versus placebo based on the change from baseline to Week 8 of the GRID‐HAMD‐17 total score although the effect was not large. The dose used was based upon human NOP receptor occupancy of ~80% (Post et al. 2016). This finding was supported by the CGI‐I endpoint score. Moreover, there was a positive effect on the mood item #1. Emotional processing assessment at Week 1 revealed a positive outcome on facial recognition tasks as well as on other parameters in favor of LY2940094. LY2940094 was safe and well‐tolerated in this first Phase 2 study. The data from Post et al. (2016) represent the first human data providing evidence that blockade of NOP receptor signaling could represent a promising strategy for the treatment of patients with MDD. Furthermore, augmentation of the antidepressant signature of fluoxetine by LY2940094 suggests a potential use of this novel mechanism as an add‐on therapy for MDD patients that do not achieve an adequate clinical response to an SSRI.
The overlapping, along with the unique, pharmacological actions of LY2940094 relative to conventional antidepressants and anxioltyics point to the possibility that LY2940094 will ultimately demonstrate both overlapping and distinct clinical pharmacological effects in MDD patients. The potential of NOP receptor blockade for body weight and binge‐eating control (Statnick et al. 2016; Witkin et al. 2014a) and for reduction in ethanol intake (Rorick‐Kehn et al. 2016) suggest additional potential differentiating features of LY2940094 from conventional antidepressant drugs that would be valuable in an antidepressant. LY2940094 was also active in some assays that detect antioltyic drug effects after acute dosing (e.g., stress‐induced cGMP production, and stress‐induced freezing), findings not typically observed with acutely administered antidepressants. Therefore, LY2940094 presents a unique biological signature that might result in novel and beneficial therapeutic outcomes.
Author Contributions
Participated in research design: J. M. Witkin, J. E. Pintar, M. J. Benvenga, D. L. Mckinzie, K. A. Svensson, M. A. Statnick, L. Rorick‐Kehn, J. Smith, V. N. Barth. Conducted experiments: J. M. Witkin, M. A. Ansonoff, B. L. Adams, S. D. Gleason, K. M. Knitowski, X. Li, S. Chaney, J. F. Falcone, J. Foss, K. Lloyd, J. Catlow. Contributed new reagents or analytic tools: M. A. Toledo, N. Diaz, C. Lafuente, A. Jiménez, A. Benito, C. Pedregal, M. A. Martínez‐Grau. Performed data analysis: J. M. Witkin, M. J. Benvenga, L. M. Rorick‐Kehn, X. Li, S. D. Gleason, M. A. Ansonoff. Wrote or contributed to the writing of the manuscript: J. M. Witkin, L. M. Rorick‐Kehn, B. L. Adams, S. D. Gleason, J. Smith, J. T. Catlow, J. E. Pintar, M. J. Benvenga, V. N. Barth, D. L. McKinzie, K. M. Knitowski, M. A. Toledo, N. Diaz, C. Lafuente, A. Jiménez, J. F. Falcone, A. Benito, M. A. Martínez‐Grau, A. Post, and M. A. Statnick.
Disclosure
None declared.
Acknowledgements
We are grateful for the experimental support of Patrick Love from Covance Laboratories, Inc. for his expert attention to the work on receptor occupancy in the drug combination experiments.
Witkin J. M., Rorick‐Kehn L. M., Benvenga M. J., Adams B. L., Gleason S. D., Knitowski K. M., Li X., Chaney S., Falcone J. F., Smith J. W., Foss J., Lloyd K., Catlow J. T., McKinzie D. L., Svensson K. A., Barth V. N., Toledo M. A., Diaz N., Lafuente C., Jiménez A., Benito A., Pedregal C., Martínez‐Grau M. A., Post A., Ansonoff M. A., Pintar J. E., Statnick M. A.. Preclinical findings predicting efficacy and side‐effect profile of LY2940094, an antagonist of nociceptin receptors, Pharma Res Per, 4(6), 2016, e00275, doi: 10.1002/prp2.275
A brief report of some of these data were presented in poster form at the Experimental Biology meeting in San Diego, CA, April 2014.
References
- Abdallah CG, Sanacora G, Duman RS, Krystal JH (2015). Ketamine and rapid‐acting antidepressants: a window into a new neurobiology for mood disorder therapeutics. Annu Rev Med 66: 509–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ardayfio PA, Benvenga MJ, Chaney SF, Love PL, Catlow J, Swanson SP, et al. (2008). The 5‐hydroxytryptamine2A receptor antagonist R‐(+)‐alpha‐(2,3‐dimethoxyphenyl)‐1‐[2‐(4‐fluorophenyl)ethyl‐4‐piperidinemethanol (M100907) attenuates impulsivity after both drug‐induced disruption (dizocilpine) and enhancement (antidepressant drugs) of differential‐reinforcement‐of‐low‐rate 72‐s behavior in the rat. J Pharmacol Exp Ther 327: 891–897. [DOI] [PubMed] [Google Scholar]
- Belzung C (2014). Innovative drugs to treat depression: did animal models fail to be predictive or did clinical trials fail to detect effects? Neuropsychopharmacology 39: 1041–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cryan JF, Markou A, Lucki I (2002). Assessing antidepressant activity in rodents: recent developments and future needs. Trends Pharmacol Sci 23: 238–245. [DOI] [PubMed] [Google Scholar]
- Davis M (1990). Animal models of anxiety based on classical conditioning: the conditioned emotional response (CER) and the fear‐potentiated startle effect. Pharmacol Ther 47: 147–165. [DOI] [PubMed] [Google Scholar]
- Dreyfus N, Myers JK, Badescu VO, de Frutos O, de la Puente ML, Ding C, et al. (2013). Discovery of a potent, dual serotonin and norepinephrine reuptake inhibitor. ACS Med Chem Lett 4: 560–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duzzioni M, Duarte FS, Leme LR, Gavioli EC, De Lima TC (2011). Anxiolytic‐like effect of central administration of NOP receptor antagonist UFP‐101 in rats submitted to the elevated T‐maze. Behav Brain Res 222: 206–211. [DOI] [PubMed] [Google Scholar]
- Fell MJ, Witkin JM, Falcone JF, Katner JF, Perry KW, Hart J, et al. (2011). N‐(4‐((2‐(trifluoromethyl)‐3‐hydroxy‐4‐(isobutyryl)phenoxy)methyl)benzyl)‐1‐methyl‐1H‐imidazole‐4‐carboxamide (THIIC), a novel mGlu2 potentiator with potential anxiolytic/antidepressant properties: In vivo profiling suggests a link between behavioral and CNS neurochemical changes. J Pharmacol Exp Ther 336: 165–177. [DOI] [PubMed] [Google Scholar]
- Gavioli EC, Calo G (2013). Nociceptin/orphanin FQ receptor antagonists as innovative antidepressant drugs. Pharmacol Ther 140: 10–25. [DOI] [PubMed] [Google Scholar]
- George SA, Hutson PH, Stephens DN (2009). Differential effects of MPEP and diazepam in tests of conditioned emotional response and Pavlovian‐to‐instrumental transfer suggests ‘anxiolytic’ effects are mediated by different mechanisms. Psychopharmacology 204: 499–509. [DOI] [PubMed] [Google Scholar]
- Gu H, Hu D, Hong XR, Mao J, Cui Y, Hui N, et al. (2003). Changes and significance of orphanin and serotonin in patients with postpartum depression. Zhonghua Fu Chan Ke Za Zhi 38: 727–728. [PubMed] [Google Scholar]
- Iversen L. (2005). The monoamine hypothesis of depression Pp. 71–86 in: Licinio J. and Wong M. L., eds. Biology of Depression.Wiley‐VCH, Weinheim. [Google Scholar]
- Jordan S, Kramer GL, Zukas PK, Moeller M, Petty F (1994). In vivo biogenic amine efflux in medial prefrontal cortex with imipramine, fluoxetine, and fluvoxamine. Synapse 18: 294–297. [DOI] [PubMed] [Google Scholar]
- Kerr JS, Powell J, Hindmarch I (1996). The effects of reboxetine and amitriptyline, with and without alcohol on cognitive function and psychomotor performance. Br J Clin Pharmacol 42: 239–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kest B, Hopkins E, Palmese CA, Chen ZP, Mogil JS, Pintar JE (2001). Morphine tolerance and dependence in nociceptin/orphanin FQ transgenic knock‐out mice. Neuroscience 104: 217–222. [DOI] [PubMed] [Google Scholar]
- Lambert DG (2008). The nociceptin/orphanin FQ receptor: a target with broad therapeutic potential. Nat Rev Drug Discov 7: 694–710. [DOI] [PubMed] [Google Scholar]
- Le Maître E, Vilpoux C, Costentin J, Leroux‐Nicollet I (2005). Opioid receptor‐like 1 (NOP) receptors in the rat dorsal raphe nucleus: evidence for localization on serotoninergic neurons and functional adaptation after 5,7‐ dihydroxytryptamine lesion. J Neurosci Res 81: 488–496. [DOI] [PubMed] [Google Scholar]
- Li X, Witkin JM, Nead AB, Skolnick P (2003). Enhancement of antidepressant potency by a potentiator of AMPA receptors. Cell Mol Neurobiol 23: 419–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Morrow D, Witkin JM (2006a). Decreases in nestlet shredding of mice by serotonin uptake inhibitors: comparison with marble burying. Life Sci 78: 1933–1939. [DOI] [PubMed] [Google Scholar]
- Li X, Need AB, Baez M, Witkin JM (2006b). mGlu5 receptor antagonism is associated with antidepressant‐like effects in mice. J Pharmacol Exp Ther 319: 254–259. [DOI] [PubMed] [Google Scholar]
- Marek GJ, Martin‐Ruiz R, Abo A, Artigas F (2005). The selective 5‐HT2A receptor antagonist M100907 enhances antidepressant‐like behavioral effects of the SSRI fluoxetine. Neuropsychopharmacology 30: 2205–2215. [DOI] [PubMed] [Google Scholar]
- Marek GJ, Day M, Hudzik TJ (2016). The utility of impulsive bias and altered decision making as predictors of drug efficacy and target selection: rethinking behavioral screening for antidepressant drugs. J Pharmacol Exp Ther 356: 534–548. [DOI] [PubMed] [Google Scholar]
- Meunier JC, Mollereau C, Toll L, Suaudeau C, Moisand C, Alvinerie P, et al. (1995). Isolation and structure of the endogenous agonist of opioid receptor‐like ORL1 receptor. Nature 377: 532–535. [DOI] [PubMed] [Google Scholar]
- Pedregal C, Joshi EM, Toledo MA, Lafuente C, Diaz N, Martinez‐Grau MA, et al. (2012). Development of LC‐MS/MS‐based receptor occupancy tracers and positron emission tomography radioligands for the nociceptin/orphanin FQ (NOP) receptor. J Med Chem 55: 4955–4967. [DOI] [PubMed] [Google Scholar]
- Popik P, Kos T, Sowa‐Kućma M, Nowak G (2008). Lack of persistent effects of ketamine in rodent models of depression. Psychopharmacology 198: 421–430. [DOI] [PubMed] [Google Scholar]
- Porsolt RD, Bertin A, Jalfre M (1977). Behavioral despair in mice: a primary screening test for antidepressants. Arch Int Pharmacodyn Ther 229: 327–336. [PubMed] [Google Scholar]
- Porter RH, Jaeschke G, Spooren W, Ballard TM, Büttelmann B, Kolczewski S, et al. (2005). Fenobam: a clinically validated nonbenzodiazepine anxiolytic is a potent, selective, and noncompetitive mGlu5 receptor antagonist with inverse agonist activity. J Pharmacol Exp Ther 315: 711–721. [DOI] [PubMed] [Google Scholar]
- Post A, Smart TS, Krikke‐Workel J, Dawson GR, Harmer CJ, Browning M, et al. (2016). A selective nociceptin receptor antagonist to treat depression: evidence from preclinical and clinical studies. Neuropsychopharmacology 41: 1803–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raddad E, Chappell A, Meyer J, Wilson A, Tauscher J, Statnick MA, et al. (2016). Occupancy of nociceptin/orphanin fq peptide receptors by the antagonist LY2940094 in rats and healthy human subjects. Drug Metab Disp 44: 1536–1542. [DOI] [PubMed] [Google Scholar]
- Rathbun RC, Slater IH (1963). Amitriptyline and nortriptyline as antagonists of central and peripheral cholinergic activation. Psychopharmacology 4: 114–125. [DOI] [PubMed] [Google Scholar]
- Reinscheid RK, Nothacker HP, Bourson A, Ardati A, Henningsen RA, Bunzow JR, et al. (1995). Orphanin FQ: a neuropeptide that activates an opioidlike G protein‐coupled receptor. Science 270: 792–794. [DOI] [PubMed] [Google Scholar]
- Rorick‐Kehn LM, Ciccocioppo R, Wong CJ, Witkin JM, Martinez‐Grau MA, Adams BL, et al. (2016). A novel, orally‐bioavailable nociceptin receptor antagonist, LY2940094, reduces ethanol self‐administration and ethanol‐seeking in animal models. Alcohol Clin Exp Res 40: 945–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rush AJ, Trivedi MH, Wisniewski SR, Nierenberg AA, Stewart JW, Warden D, et al. (2006). Acute and longer‐term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Psychiatry 163: 1905–1917. [DOI] [PubMed] [Google Scholar]
- Rorick‐Kehn LM, Perkins EJ, Knitowski KM, Hart JC, Johnson BG, Schoepp DD, McKinzie DL (2006). Improved bioavailability of the mGlu2/3 receptor agonist LY354740 using a prodrug strategy: in vivo pharmacology of LY544344. J Pharmacol Exp Ther. 316(2): 905–913. [DOI] [PubMed] [Google Scholar]
- Ryder JW, Falcone JF, Manro JR, Svensson KA, Merchant KM (2006). Pharmacological characterization of cGMP regulation by the biarylpropylsulfonamide class of positive, allosteric modulators of alpha‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid receptors. J Pharmacol Exp Ther 319: 293–298. [DOI] [PubMed] [Google Scholar]
- Spooren W, Gasparini F (2004). mGlu5 receptor antagonists: a novel class of anxiolytics? Drug News Perspect 17: 251–257. [DOI] [PubMed] [Google Scholar]
- Statnick MA, Chen Y, Ansonoff MA, Witkin JM, Rorick‐Kehn L, Suter TM, et al. (2016). A novel nociceptin receptor antagonist LY2940094 inhibits excessive feeding behavior in rodents: a possible mechanism for the treatment of binge eating disorder. J Pharmacol Exp Ther 356: 493–502. [DOI] [PubMed] [Google Scholar]
- Tang AH, Franklin SR, Carter DB, Sethy VH, Needham LM, Jacobsen EJ, et al. (1997). Psychopharmacology 131: 255–263. [DOI] [PubMed] [Google Scholar]
- Toledo MA, Pedregal C, Lafuente C, Diaz N, Martinez‐Grau MA, Jiménez A, et al. (2014). Discovery of a novel series of orally active nociceptin/orphanin FQ (NOP) receptor antagonists based on a dihydrospiro(piperidine‐4,7′‐thieno[2,3‐c]pyran) Scaffold. J Med Chem 57: 3418–3429. [DOI] [PubMed] [Google Scholar]
- Trullas R, Skolnick P (1990). Functional antagonists at the NMDA receptor complex exhibit antidepressant actions. Eur J Pharmacol 185: 1–10. [DOI] [PubMed] [Google Scholar]
- Wang LN, Liu LF, Zhang JX, Zhao GF (2009). Plasma levels of nociceptin/orphanin FQ in patients with bipolar disorders and health adults. Zhonghua Yi Xue Za Zhi 89: 916–918. [PubMed] [Google Scholar]
- Witkin JM, Statnick MA, Rorick‐Kehn LM, Pintar JE, Ansonoff MA, Chen Y, et al. (2014a). The biology of Nociceptin/Orphanin FQ (N/OFQ) related to obesity, stress, anxiety, mood, and drug dependence. Pharmacol Ther 141: 283–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witkin JM, Overshiner C, Li X, Catlow JT, Schober DA, Heinz BA, et al. (2014b). The M1 and M2 muscarinic receptor subtypes regulate antidepressant‐like effects of the rapidly‐acting antidepressant scopolamine. J Pharmacol Exp Ther 351: 448–456. [DOI] [PubMed] [Google Scholar]
- Zhang HT (2009). Cyclic AMP specific phosphodiesterase‐4 as a target for the development of antidepressant drugs. Curr Pharm Des 15: 1688–1698. [DOI] [PubMed] [Google Scholar]
- Zhang LL, Zheng HP, Ma C, He ZG, Zheng CD (2009). The plasma orphanin FQ in patients with depression before and after treatment. Chin J Psychiatry 42: 138–144. [Google Scholar]