

Abstract
Background
Metformin is a widely used biguanide drug for the treatment of type 2 diabetes. It has been revaluated as a potential anti-cancer drug with promising activity in various tumors. However, the precise mechanisms underlying the suppression of cancer cells by metformin remain not well understood.
Material/Methods
In this study, human renal cell carcinoma cell line ACHN was used to investigate the anti-proliferation effect of metformin. A cell counting kit-8 assay was used to detect the cell viability. The cell cycle distribution and apoptosis were analyzed by flow cytometry. The expression of cyclin D1 and p27KIP1 was detected by Western blot. The underlying mechanism involving miRNA34a was further investigated by quantitative RT-PCR and transfection with miRNA inhibitor specific for miRNA34a in ACHN, 769-P, and A498 cells.
Results
Metformin could significantly inhibit the proliferation of ACHN cells in a dose- and time-dependent manner. In addition, the results showed that metformin induced G0/G1 phase arrest and delayed entry into S phase in ACHN cells. It was shown that metformin downregulates the expression of cyclin D1 and increases the p27KIP1 level. Furthermore, metformin increased ACHN cell death. Lastly, miRNA34a was found to be upregulated by metformin in ACHN, 769-P, and A498 cells. Subsequently, it was demonstrated that inhibition of miRNA34a could partially attenuate the suppressive effect of metformin on renal cancer cell proliferation.
Conclusions
The study data revealed that metformin induced cell growth inhibition and cell cycle arrest partially by upregulating miRNA34a in renal cancer cells.
MeSH Keywords: Cell Cycle, Cell Proliferation, Kidney Neoplasms, Metformin, MicroRNAs
Background
Renal cell carcinoma (RCC) is the most common malignant cancer in kidney, accounting for about 90% of adult kidney cancers. Clear cell renal cell carcinoma (ccRCC) is the most common histological subtype of RCC, which represents approximately 80% of RCC [1]. Surgical intervention is the primary treatment for RCC. However, approximately 30–40% of patients develop metastases after surgery [2]. Furthermore, ccRCC exhibits resistance to chemotherapy and radiation. Therefore, novel therapeutic strategies are urgently required.
Metformin (1,1-dimethylbiguanide hydrochloride), which belongs to the family of biguanides, is a widely used anti-diabetic drug for the treatment of type 2 diabetes. It can reduce hepatic glucose production, improve insulin sensitivity, and modify the serum lipid profile. It has been demonstrated to be a well-tolerated drug with limited and transient side effects. In the past few years, multiple epidemiologic studies have shown that diabetic patients treated with metformin had a decreased incidence of different tumors [3]. Due to its potential as a promising candidate for cancer therapy, metformin has received more attention, and increasing evidence has indicated that it can inhibit cell proliferation in various cancer cells and animal models, including prostate, lung, and liver cancers, as well as RCC [4–7]. However, the precise mechanism underlying the suppression of RCC by metformin remains unclear.
The best known effect of metformin is the activation of AMPK, a key regulator of multiple metabolic pathways. However, in several previous studies, it has been shown that the inactivation of AMPK by siRNA or the pharmacological inhibitor did not totally abolish the anti-cancer effects of metformin in prostate and hepatic cancer cells [8,9], suggesting that the inhibitory effect of metformin in cancers can be independent of AMPK activation. Moreover, the alteration of the microRNA (miRNA) profiles in vivo and in vitro has been shown to be associated with the anti-tumor effect of metformin [10,11].
The miRNAs are a family of conserved non-coding small RNAs, which negatively regulate the coding mRNAs at the post-transcriptional level and further play important roles in many biological processes. A number of studies have revealed that miRNAs have a significant impact in the pathogenesis of RCC [12]. Several miRNAs, such as miRNA148b, act as oncogenes in RCC [13], while some other miRNAs were identified as tumor suppressors, including miRNA-451 and 34a [14,15]. miRNA34a was found to be downregulated in RCC and inhibited cell proliferation and metastasis by affecting its downstream target genes [15–17], which suggested that miRNA34a might be a potential novel target in RCC therapy. In the present study, we used human RCC cell line ACHN, 769-P, and A498 cells to investigate the effect of metformin on the cell growth and the mechanisms involving miRNA34a.
Material and Methods
Cell culture and reagents
ACHN, 769-P, and A498 cells were obtained from the American type culture collection (ATCC). ACHN and A498 cells were maintained in minimum essential medium (MEM, Hyclone) supplemented with 10% (vol/vol) fetal bovine serum. 769-P cells were maintained in RPMI-1640 medium (Hyclone) supplemented with 10% (vol/vol) fetal bovine serum. Metformin (1,1-dimethylbiguanide hydrochloride) was purchased from Beyotime.
Cell counting kit-8 (CCK-8) assay
Cells were seeded on 96-well plates at an initial density of 2×103 cells per well and allowed to attach for 12 h. Then a series of concentrations of metformin (0.2, 1, and 5 mM) were added to each well, and cells were further cultured for 24, 48, 72, and 96 h. At each time point, cells were stained with a CCK-8 kit (Dojindo) for 1 h at 37°C. The absorbance was measured using a microplate reader at a wavelength of 450 nm. All experiments were performed in triplicate.
Cell cycle analysis
Cell cycle analysis was performed as previously described [18]. Briefly, cells were fixed in 70% ethanol at 4°C overnight. After incubation with RNase (0.1 mg/mL) for 30 min at 37°C, the cells were stained with propidium iodide (PI) 50 μg/mL. Then the cell samples were analyzed with a MoFlo XDP flow cytometer (Beckman). The cell cycle distribution was calculated using ModFit LT software.
Cell apoptosis analysis
After cells were exposed to the indicated concentration of metformin for 48 h, the apoptotic cell death was quantified with an FITC-Annexin V/PI apoptosis detection assay according to the manufacturer’s protocol (BD Biosciences).
Analysis of miRNA expression using quantitative RT-PCR
Expression of mature miRNAs was analyzed with Bulge-Loop™ miRNA quantitative RT-PCR primer kit (RIBOBIO, Guangzhou, China). Total cellular RNA was extracted with RNAiso Plus reagent (Takara) and reversely transcribed to cDNA with Bulge-Loop™ RT primers (RIBOBIO). The quantitative PCR was carried out with the ABI Vii7 Real-Time PCR system (Applied Biosystems) using SYBR Premix Ex Taq II (Takara) according to the manufacturer’s instructions. All reactions were carried out in triplicate. Relative gene expression was calculated using the 2−ΔΔCt method. Small nuclear U6 snRNA was used as an internal control. The Bulge-Loop Forward and Reverse Primers for miRNA34a, U6 snRNA, and 5S rRNA were obtained from RIBOBIO.
Western blot analysis
Western blot was performed to detect the specific protein expression levels as previously described [19]. The primary antibodies used in Western blot were as follows: cyclin D1 (#2926) and p27KIP1 (#3686), purchased from Cell Signaling; and GAPDH (60004-1, ProteinTech).
Cell transfection
The micrOFF™ miRNA inhibitor specific for miRNA34a and negative control were obtained from RIBOBIO. Cells were transfected with these miRNA inhibitors using Lipofectamine 2000 (Invitrogen) following the manufacturer’s instructions.
Statistical analysis
Data analysis was performed using SPSS 16.0 (SPSS Inc.). Statistical analysis was performed by Student’s t-test or analysis of variance (ANOVA) followed by the Newman-Keuls test. Differences were considered statistically significant at p<0.05.
Results
Metformin inhibits cell proliferation in ACHN cells
It is well established that metformin could inhibit cell proliferation in several cancer cells in vitro [5–7]. However, the effect and mechanism of metformin on cell proliferation in some certain cancer cells remain poorly understood. To explore the restriction effect of metformin on the proliferation of ACHN cells, we carried out the cell proliferation assay by using the CCK-8 kit to determine the cell viability. ACHN cells were treated with metformin at a series of concentrations (0.2, 1, and 5 mM), and the cell viability was measured at 24, 48, 72, and 96 hours of treatment. Compared with the control, metformin significantly suppressed the proliferation of ACHN cells after 48 h in a dose- and time-dependent manner (Figure 1), which was similar to the results in other cancer cell types [4,6]. These results demonstrated that metformin was capable of inhibiting ACHN cell proliferation.
Figure 1.
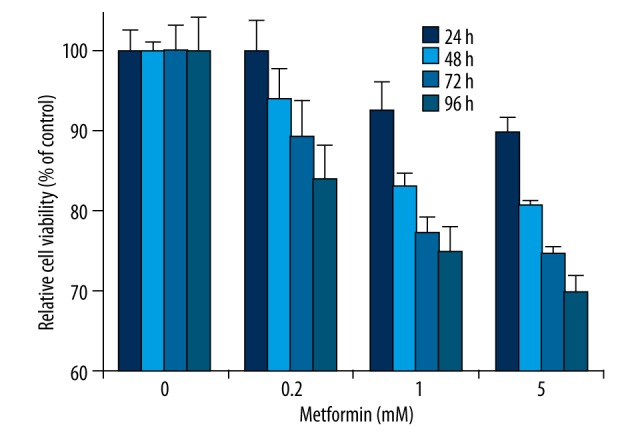
Metformin inhibits cell proliferation in ACHN cells. ACHN cells were treated with metformin at different concentrations (0.2, 1, and 5 mM), while an equal volume of phosphate-buffered saline (PBS) was used as vehicle control. The cell viability was measured at 24, 48, 72, and 96 hours of treatment by CCK-8 assay. The resulting cell viability was set to 100% for PBS-treated control cells at each time point. Data show mean values ±SD from three independent experiments.
Metformin induces cell cycle arrest in ACHN cells
To determine whether the inhibitory effect of metformin on cell growth was mediated through cell cycle arrest, flow cytometry analysis was performed to investigate the effect of metformin on cell cycle distribution. ACHN cells were treated with 5 and 20 mM metformin for 48 h and harvested for analysis by PI staining and flow cytometry. Notably, metformin treatment increased the proportion of cells in G0/G1 phase while decreasing the proportion of cells in S phase (Figure 2A). Specifically, 5 and 20 mM metformin increased the percentage of cells in G0/G1 phase by 4.24% and 12.20%, respectively (Figure 2B). These data suggested that metformin treatment resulted in G0/G1 phase arrest and delayed entry into S phase in ACHN cells.
Figure 2.

Metformin induces cell cycle arrest in ACHN cells. ACHN cells were treated with 5 and 20 mM metformin for 48 hours, while an equal volume of phosphate-buffered saline (PBS) was used as vehicle control. (A) Cells were subjected to analysis of cell cycle distribution through flow cytometry. Representative data from triplicate experiments are shown. (B) Quantification of cell cycle distributions was derived from three independent experiments. Data show mean values ±SD from three independent experiments. (C) Western blot was performed to detect the expression of cyclin D1 and p27KIP1. GAPDH was used as a loading control.
To explore the molecular mechanism responsible for the metformin-induced cell cycle arrest, the expression of several cell cycle-related genes was analyzed. As indicated by Western blot, the expression of cyclin D1 protein was significantly decreased by metformin compared with the controls, while the expression of p27KIP1 was upregulated in the metformin-treated cells (Figure 2C). These data suggested that metformin modulated the expression of cyclin D1 and p27KIP1 to induce the cell cycle arrest at the G0/G1 phase.
Metformin increases the cell death in ACHN cells
We next assessed the effect of metformin on the induction of ACHN cell apoptosis because cell cycle arrest sometimes proceeds to apoptosis or cell death. The Annexin V-FITC/PI staining and flow cytometry were performed to quantify the proportion of apoptotic and dead ACHN cells. Our results showed that metformin treatment significantly induced cell death in ACHN cells (Figure 3A). Compared with the control, 5 and 20 mM metformin increased the percentage of dead cells by 5.85% and 14.32%, respectively, in ACHN cells (Figure 3B). At the same time, 20 mM metformin increased the proportion of late-stage apoptosis slightly (Figure 3A). Therefore, it was suggested that metformin induced the death of ACHN cells.
Figure 3.

Metformin increases cell death in ACHN cells. ACHN cells were treated with 5 and 20 mM metformin for 48 hours, while an equal volume of phosphate-buffered saline (PBS) was used as vehicle control. (A) Cells were subjected to analysis of apoptosis with the FITC-Annexin V/PI apoptosis detection kit. Representative data from triplicate experiments are shown. (B) Quantification of the percentage of dead cells was derived from three independent experiments. Data show mean values ±SD from three independent experiments. * – Indicates p<0.05 compared with the control.
Metformin upregulates the level of miRNA34a in ACHN cells
In order to further investigate the possible molecular mechanisms of the anti-tumor effect of metformin involving miRNA34a in ACHN cells, we performed quantitative RT-PCR to determine the effect of metformin on miRNA-34a expression level in ACHN cells. These cells were treated with metformin (5 and 20 mM) for 24 h and harvested for miRNA-34a quantitative analysis. U6 snRNA and 5S rRNA are the most frequently used small RNA reference genes for miRNA normalization in plants, animals, and humans. Firstly, we measured the expression levels of these small RNAs and showed that U6 snRNA was not affected by metformin (Figure 4A). Therefore, U6 snRNA was used for normalization in further quantitative RT-PCR analysis of miRNA34a in our present study.
Figure 4.

Metformin upregulates the level of miRNA34a in ACHN cells. (A) ACHN cells were treated with 5 and 20 mM metformin for 48 hours, while an equal volume of phosphate-buffered saline (PBS) was used as vehicle control. Cell lysates were subjected to quantitative PCR to measure the level of U6 snRNA. 5S rRNA was used as an internal control. (B) Quantitative PCR was performed to quantify the expression level of miRNA34a. U6 snRNA was used as an endogenous control in B and C. (C) ACHN cells were transfected with either the inhibitor-miRNA34a (inh-miRNA34a) or negative control (inh-NC) for 48 h. Quantitative PCR was performed to verify the inhibition efficacy of miRNA34a. (D) Cells were transfected with indicated inhibitors (inh-NC or inh-miRNA34a) for 24 h. After treatment with 5 mM metformin or PBS for another 72 h, the cell viability was measured by CCK-8 assay. The resulting cell viability was set to 100% for PBS-treated control cells. Data show mean values ±SD from three independent experiments. n.s. – Indicates no significance. * – Indicates p<0.05.
As shown in Figure 4B, it was found that miRNA34a was significantly upregulated in metformin-treated cells. In 5 and 20 mM metformin-treated cells, the levels of miRNA34a expression were 1.66- and 2.46-fold greater, respectively, compared with the levels in the control group (Figure 4B). In some previous studies, miRNA34a has been shown to be capable of inhibiting cell proliferation in renal cancer cells [15,16]. These data implied that the upregulation of miRNA34a induced by metformin probably contributed to the inhibitory effect of metformin on cell proliferation in ACHN cells.
To further investigate whether the anti-proliferation activity of metformin in ACHN cells was miRNA34a dependent, we transfected miRNA34a inhibitor into ACHN cells to downregulate the miRNA34a. The inhibition efficacy of miRNA34a was then verified by real-time PCR. As shown in Figure 4C, the miRNA34a expression level was significantly suppressed in the specific inhibitor-transfected cells compared with the negative control. Subsequently, we analyzed the effects of transient transfection with miRNA34a inhibitor and additional metformin treatment on the ACHN cell proliferation. As shown in Figure 4D, treatment with 5 mM metformin could significantly suppress the cell viability, which was consistent with the results in Figure 1. Moreover, it was found that the transfection with miRNA34a inhibitor could partially reverse the inhibition effect of metformin on cell proliferation in ACHN cells (Figure 4D).
Metformin has been demonstrated to inhibit cell proliferation in several types of cancer cells [4–7], but the underlying mechanisms vary in various cells. Therefore, we tested whether metformin inhibited other renal cancer cells also by upregulating microRNA-34a. As shown in Figure 5A and 5C, miRNA34a was significantly upregulated by metformin treatment in 769-P and A498 cells. In addition, treatment with 10 mM metformin could inhibit the cell viability (Figure 5B, 5D), which was consistent with our results in ACHN cells (Figure 1, Figure 4D). Furthermore, both 769-P and A498 cells were transfected with miRNA34a inhibitor (data not shown), and it was found that the inhibitor could partially reverse the suppressive effect of metformin on cell proliferation in these cells (Figure 5B, 5D). All these data suggested that metformin inhibited renal cancer cell proliferation partially by upregulating miRNA34a.
Figure 5.

Metformin upregulates the level of miRNA34a in 769-P and A498 cells. (A, C) 769-P and A498 cells were treated with 5 and 20 mM metformin for 48 hours, while an equal volume of phosphate-buffered saline (PBS) was used as vehicle control. Cell lysates were subjected to quantitative PCR to quantify the expression level of miRNA34a. U6 snRNA was used as an endogenous control. (B, D) 769-P and A498 cells were transfected with indicated inhibitors (inh-NC or inh-miRNA34a) for 24 h. After treatment with 10 mM metformin or PBS for another 48 h, the cell viability was measured by CCK-8 assay. The resulting cell viability was set to 100% for PBS-treated control cells. Data show mean values ±SD from three independent experiments. * – Indicates p<0.05.
Discussion
In the present study, we investigated the effect of metformin in suppressing the proliferation of human RCC cells and the molecular events underlying the anti-proliferation activity. Our data showed that treatment with metformin significantly reduced ACHN cell viability in a dose- and time-dependent manner (Figure 1). Furthermore, metformin treatment induced ACHN cell cycle arrest at the G0/G1 phase and cell death (Figures 2, 3). Molecularly, metformin decreased cyclin D1 expression level and enhanced the p27KIP1 expression level (Figure 2C). We further found evidence to suggest that the anti-proliferation effects of metformin in renal cancer cells may be partially through the upregulation of miRNA34a (Figures 4, 5).
As an anti-diabetic biguanide, metformin has been widely used in the treatment of type 2 diabetes around the world for more than 40 years because there are no major contraindications and the cost of the drug is low. In the past decades, various studies have revealed the beneficial roles of this drug in different pathologies, including heart failure, obesity, aging, and virus infections [20–22]. Especially, some recent epidemiologic and clinical studies reported that metformin may reduce overall cancer risk in diabetic patients [3], so metformin was revaluated as a potential anti-cancer drug. However, the contribution of metformin to the anti-cancer effect remains to be ascertained. There is increasing evidence that metformin could directly reduce the cell proliferation in numerous types of cancers such as prostate, lung, and liver [4–6]. Consistent with these previous reports, it was shown that metformin inhibited cell proliferation significantly in human renal cancer cells in our present study (Figures 1, 5). A recent study reported that metformin could also regulate inflammation and might play a role in the tumor microenvironment [23]. Moreover, metformin has been found to improve the sensitivity and efficiency of some established chemotherapeutic agents and small-molecule inhibitors such as gefitinib and erlotinib [24,25]. Therefore, a deep understanding of the functions and mechanisms of metformin in various cancers is necessary to determine its therapeutic effects and its clinical indications.
We next investigated the potential mechanism of metformin in suppressing cell proliferation, and found that metformin treatment caused G0/G1 phase cell cycle arrest in ACHN cells (Figure 2). Cell cycle is a series of events leading to cell division and replication, which is regulated strictly by cyclins, cyclin-dependent kinase (CDKs), and CDK inhibitors (CDKIs). It has been shown that cyclin D1 and CDKs are essential for driving cells to pass the G1/S restriction point. CDKIs, such as p21CIP1 and p27KIP1, can prevent inappropriate cyclin/CDK activity in the G1 phase [26]. To explore the molecular mechanism responsible for the metformin-induced cell cycle arrest in ACHN cells, we detected the expression of some key regulators. As shown in Figure 2C, the cyclin D1 was significantly decreased and the p27KIP1 was upregulated by metformin, effects that were consistent with some previous reports on pancreatic and esophageal squamous cancer cells [27,28].
MicroRNAs have been demonstrated to play important roles in numbers of biological processes, including cell differentiation, proliferation, and apoptosis. In recent years, some researchers have focused on the potential miRNAs involved in anti-tumor effects of metformin in different cancer cells. MiRNA34a has been identified to function as a tumor suppressor in different types of cancers [29] and has gained more attention from researchers investigating metformin. In breast cancer cells, metformin induces miRNA34a to suppress the Sirt1/Pgc-1α/Nrf2 pathway and increases the susceptibility of wild-type p53 cancer cells to oxidative stress and TRAIL-induced apoptosis [30]. Another study reported that metformin could increase miRNA-34a expression and downregulate its direct targets Notch, Slug, and Snail in pancreatic Panc02 cells [31]. We also found that miRNA34a was significantly upregulated in metformin-treated renal cancer cells (Figures 4B, 5A, 5C). Furthermore, we have demonstrated that miRNA34a inhibitor could partially reverse the inhibition effect of metformin on cell proliferation (Figures 4D, 5B, 5D), which suggested that metformin suppressed renal cancer cell proliferation partially by upregulating miRNA34a. However, the mechanism of miRNA34a upregulation with metformin treatment remains unclear. A previous study reported that promoter methylation contributed to miRNA34a loss in ACHN, 786-O, and SN12PM6 renal carcinoma cell lines [15], suggesting that the upregulation of miRNA34a by metformin was probably due to promoter demethylation. Although the precise role of metformin in DNA demethylation needs to be defined, we have provided novel evidence for a mechanism involving miRNA34a that might contribute to the anticancer effect of metformin in renal cancer cells.
Recently, some pharmacokinetics studies of metformin have revealed that the plasma drug concentrations were achieved in the micromolar (μM) range in mouse models and diabetic patients after conventional administration of metformin [32–34]. However, most in vitro studies, as well as our present study (Figure 1), demonstrated that the millimolar (mM) concentrations of metformin are required to inhibit the cell proliferation [4–7]. This discrepancy remains a discussion point in the scientific community and promotes the studies of tumor-related biological factors to evaluate the effects of metformin. The high concentration of metformin required for its antineoplastic activity in vitro might be related to the low expression levels of organic cation transporters (OCT1, OCT2, and OCT3) that controlled the cellular uptake of metformin in immortalized cell lines [35]. Besides, the high levels of glycine and serine, which were present in culture media in vitro, have been shown to attenuate metformin sensitivity [36], probably leading to the high concentration of metformin that was utilized to achieve its inhibitory effect on cell proliferation in vitro. Thus, although the millimolar metformin concentration in vitro may not be relevant to the drug concentration required in vivo and clinically, the in vitro studies are still thought to be important to investigate the potential antineoplastic activity of metformin in various cancers and the underlying molecular mechanisms. We have demonstrated the anti-tumor effects of metformin and its related mechanisms in human RCC cells in vitro in this study. The pharmacokinetics of metformin and potential therapeutic strategies in RCC in vivo are still under further investigation.
Conclusions
Our study indicated that metformin significantly reduced cell viability in renal cancer cells. We found that metformin treatment induced ACHN cell cycle arrest at the G0/G1 phase and cell death. Furthermore, our data revealed that metformin inhibited cell proliferation partially by upregulating miRNA34a in renal cancer cells.
Footnotes
Conflict of interest
Authors claim no conflict of interest.
Source of support: This work was supported by the National Natural Science Foundation of China (81471067, 81501738) and Chongqing Key Programs for Science and Technology Development (cstc2014yykfB10009)
References
- 1.Decastro GJ, McKiernan JM. Epidemiology, clinical staging, and presentation of renal cell carcinoma. Urol Clin North Am. 2008;35:581–92. doi: 10.1016/j.ucl.2008.07.005. [DOI] [PubMed] [Google Scholar]
- 2.Escudier B, Porta C, Schmidinger M, et al. Renal cell carcinoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2014;25(Suppl 3):49–56. doi: 10.1093/annonc/mdw328. [DOI] [PubMed] [Google Scholar]
- 3.Libby G, Donnelly LA, Donnan PT, et al. New users of metformin are at low risk of incident cancer: a cohort study among people with type 2 diabetes. Diabetes Care. 2009;32:1620–25. doi: 10.2337/dc08-2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kato H, Sekine Y, Furuya Y, et al. Metformin inhibits the proliferation of human prostate cancer PC-3 cells via the downregulation of insulin-like growth factor 1 receptor. Biochem Biophys Res Commun. 2015;461:115–21. doi: 10.1016/j.bbrc.2015.03.178. [DOI] [PubMed] [Google Scholar]
- 5.Wang J, Gao Q, Wang D, et al. Metformin inhibits growth of lung adenocarcinoma cells by inducing apoptosis via the mitochondria-mediated pathway. Oncol Lett. 2015;10:1343–49. doi: 10.3892/ol.2015.3450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cai X, Hu X, Cai B, et al. Metformin suppresses hepatocellular carcinoma cell growth through induction of cell cycle G1/G0 phase arrest and p21CIP and p27KIP expression and downregulation of cyclin D1 in vitro and in vivo. Oncol Rep. 2013;30:2449–57. doi: 10.3892/or.2013.2718. [DOI] [PubMed] [Google Scholar]
- 7.Liu J, Li M, Song B, et al. Metformin inhibits renal cell carcinoma in vitro and in vivo xenograft. Urol Oncol. 2013;31:264–70. doi: 10.1016/j.urolonc.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 8.Ben SI, Laurent K, Loubat A, et al. The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin D1 level. Oncogene. 2008;27:3576–86. doi: 10.1038/sj.onc.1211024. [DOI] [PubMed] [Google Scholar]
- 9.Do MT, Kim HG, Khanal T, et al. Metformin inhibits heme oxygenase-1 expression in cancer cells through inactivation of Raf-ERK-Nrf2 signaling and AMPK-independent pathways. Toxicol Appl Pharmacol. 2013;271:229–38. doi: 10.1016/j.taap.2013.05.010. [DOI] [PubMed] [Google Scholar]
- 10.Fujimori T, Kato K, Fujihara S, et al. Antitumor effect of metformin on cholangiocarcinoma: In vitro and in vivo studies. Oncol Rep. 2015;34:2987–96. doi: 10.3892/or.2015.4284. [DOI] [PubMed] [Google Scholar]
- 11.Kato K, Iwama H, Yamashita T, et al. The anti-diabetic drug metformin inhibits pancreatic cancer cell proliferation in vitro and in vivo: Study of the microRNAs associated with the antitumor effect of metformin. Oncol Rep. 2016;35:1582–92. doi: 10.3892/or.2015.4496. [DOI] [PubMed] [Google Scholar]
- 12.Li M, Wang Y, Song Y, et al. MicroRNAs in renal cell carcinoma: A systematic review of clinical implications (Review) Oncol Rep. 2015;33:1571–78. doi: 10.3892/or.2015.3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nie F, Liu T, Zhong L, et al. MicroRNA-148b enhances proliferation and apoptosis in human renal cancer cells via directly targeting MAP3K9. Mol Med Rep. 2016;13:83–90. doi: 10.3892/mmr.2015.4555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhu S, Huang Y, Su X. Mir-451 correlates with prognosis of renal cell carcinoma patients and inhibits cellular proliferation of renal cell carcinoma. Med Sci Monit. 2016;22:183–90. doi: 10.12659/MSM.896792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu G, Li H, Wang J, et al. miRNA-34a suppresses cell proliferation and metastasis by targeting CD44 in human renal carcinoma cells. J Urol. 2014;192:1229–37. doi: 10.1016/j.juro.2014.05.094. [DOI] [PubMed] [Google Scholar]
- 16.Zhang C, Mo R, Yin B, et al. Tumor suppressor microRNA-34a inhibits cell proliferation by targeting Notch1 in renal cell carcinoma. Oncol Lett. 2014;7:1689–94. doi: 10.3892/ol.2014.1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yamamura S, Saini S, Majid S, et al. MicroRNA-34a suppresses malignant transformation by targeting c-Myc transcriptional complexes in human renal cell carcinoma. Carcinogenesis. 2012;33:294–300. doi: 10.1093/carcin/bgr286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xie W, Feng Q, Su Y, et al. Transcriptional regulation of PES1 expression by c-Jun in colon cancer. PLoS One. 2012;7:e42253. doi: 10.1371/journal.pone.0042253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xie W, Qu L, Meng L, et al. PES1 regulates sensitivity of colorectal cancer cells to anticancer drugs. Biochem Biophys Res Commun. 2013;431:460–65. doi: 10.1016/j.bbrc.2012.12.145. [DOI] [PubMed] [Google Scholar]
- 20.Mahmood K, Naeem M, Rahimnajjad NA. Metformin: the hidden chronicles of a magic drug. Eur J Intern Med. 2013;24:20–26. doi: 10.1016/j.ejim.2012.10.011. [DOI] [PubMed] [Google Scholar]
- 21.Joven J, Menendez JA, Fernandez-Sender L, et al. Metformin: A cheap and well-tolerated drug that provides benefits for viral infections. HIV Med. 2013;14:233–40. doi: 10.1111/hiv.12000. [DOI] [PubMed] [Google Scholar]
- 22.Xie W, Wang L, Dai Q, et al. Activation of AMPK restricts coxsackievirus B3 replication by inhibiting lipid accumulation. J Mol Cell Cardiol. 2015;85:155–67. doi: 10.1016/j.yjmcc.2015.05.021. [DOI] [PubMed] [Google Scholar]
- 23.Incio J, Suboj P, Chin SM, et al. Metformin reduces desmoplasia in pancreatic cancer by reprogramming stellate cells and tumor-associated macrophages. PLoS One. 2015;10:e141392. doi: 10.1371/journal.pone.0141392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Soo JS, Ng CH, Tan SH, et al. Metformin synergizes 5-fluorouracil, epirubicin, and cyclophosphamide (FEC) combination therapy through impairing intracellular ATP production and DNA repair in breast cancer stem cells. Apoptosis. 2015;20:1373–87. doi: 10.1007/s10495-015-1158-5. [DOI] [PubMed] [Google Scholar]
- 25.Li L, Han R, Xiao H, et al. Metformin sensitizes EGFR-TKI-resistant human lung cancer cells in vitro and in vivo through inhibition of IL-6 signaling and EMT reversal. Clin Cancer Res. 2014;20:2714–26. doi: 10.1158/1078-0432.CCR-13-2613. [DOI] [PubMed] [Google Scholar]
- 26.Pestell RG. New roles of cyclin D1. Am J Pathol. 2013;183:3–9. doi: 10.1016/j.ajpath.2013.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tanaka R, Tomosugi M, Horinaka M, et al. Metformin causes G1-phase arrest via down-regulation of MiR-221 and enhances TRAIL sensitivity through DR5 up-regulation in pancreatic cancer cells. PLoS One. 2015;10:e125779. doi: 10.1371/journal.pone.0125779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cai X, Hu X, Tan X, et al. Metformin induced AMPK activation, G0/G1 phase cell cycle arrest and the inhibition of growth of esophageal squamous cell carcinomas in vitro and in vivo. PLoS One. 2015;10:e133349. doi: 10.1371/journal.pone.0133349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Adams BD, Parsons C, Slack FJ. The tumor-suppressive and potential therapeutic functions of miR-34a in epithelial carcinomas. Expert Opin Ther Targets. 2016;20(6):737–53. doi: 10.1517/14728222.2016.1114102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Do MT, Kim HG, Choi JH, Jeong HG. Metformin induces microRNA-34a to downregulate the Sirt1/Pgc-1alpha/Nrf2 pathway, leading to increased susceptibility of wild-type p53 cancer cells to oxidative stress and therapeutic agents. Free Radic Biol Med. 2014;74:21–34. doi: 10.1016/j.freeradbiomed.2014.06.010. [DOI] [PubMed] [Google Scholar]
- 31.Cifarelli V, Lashinger LM, Devlin KL, et al. Metformin and rapamycin reduce pancreatic cancer growth in obese prediabetic mice by distinct MicroRNA-regulated mechanisms. Diabetes. 2015;64:1632–42. doi: 10.2337/db14-1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dowling RJ, Lam S, Bassi C, et al. Metformin pharmacokinetics in mouse tumors: Implications for human therapy. Cell Metab. 2016;23:567–68. doi: 10.1016/j.cmet.2016.03.006. [DOI] [PubMed] [Google Scholar]
- 33.Chandel NS, Avizonis D, Reczek CR, et al. Are metformin doses used in murine cancer models clinically relevant? Cell Metab. 2016;23:569–70. doi: 10.1016/j.cmet.2016.03.010. [DOI] [PubMed] [Google Scholar]
- 34.Foretz M, Guigas B, Bertrand L, et al. Metformin: From mechanisms of action to therapies. Cell Metab. 2014;20:953–66. doi: 10.1016/j.cmet.2014.09.018. [DOI] [PubMed] [Google Scholar]
- 35.Viollet B, Guigas B, Sanz GN, et al. Cellular and molecular mechanisms of metformin: an overview. Clin Sci (Lond) 2012;122:253–70. doi: 10.1042/CS20110386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gravel SP, Hulea L, Toban N, et al. Serine deprivation enhances antineoplastic activity of biguanides. Cancer Res. 2014;74:7521–33. doi: 10.1158/0008-5472.CAN-14-2643-T. [DOI] [PubMed] [Google Scholar]