

Abstract
The evolutionarily conserved extracellular signal transducing RTK-RAS-ERK pathway is an important kinase-signaling cascade that controls multiple cellular and developmental processes principally via activation of ERK, the terminal kinase of the pathway. Tight regulation of ERK activity is essential for normal development and homeostasis; overly active ERK results in excessive cellular proliferation, while underactive ERK causes cell death. C. elegans is a powerful model system that has helped characterize the function and regulation of RTK-RAS-ERK signaling pathway during development. In particular, the RTK-RAS-ERK pathway is essential for C. elegans germline development, which is the focus of this method. Using antibodies specific to the active, diphosphorylated form of ERK (dpERK), the stereotypical localization pattern can be visualized within the germline. Because this pattern is both spatially and temporally controlled, the ability to reproducibly assay dpERK is useful to identify regulators of the pathway that affect dpERK signal duration and amplitude and thus germline development. Here we demonstrate how to successfully dissect, stain, and image dpERK within the C. elegans gonad. This method can be adapted for spatial localization of any signaling or structural protein in the C. elegans gonad, provided an antibody compatible with immunofluorescence is available.
Keywords: Developmental Biology, Issue 117, RAS-ERK, C. elegans, germline, in vivo antibody staining, immunofluorescence, signaling proteins
Introduction
The Receptor Tyrosine Kinase (RTK)-RAt Sarcoma (RAS)-Extracellular signal Regulated Kinase (ERK) pathway relays extracellular signals through a conserved kinase cascade that results in the phosphorylation and activation of ERK1-3. ERK proteins are members of the conserved proline-directed serine/threonine MAP (Mitogen Activated Protein) kinase family, and are directly activated by MEK via dual phosphorylation on the threonine (T) and tyrosine (Y) of the conserved TEY motif. Active ERK (referred to as diphosphorylated ERK, or dpERK) then regulates many cellular and developmental processes through its phosphorylation of a battery of downstream substrates1-3. Thus abnormal ERK activity leads to many cell and developmental defects4-7.
Stringent regulation of ERK activity is critical for normal development: in mammalian systems too much ERK activity leads to excessive cellular proliferation leading to oncogenic growth; too little activity leads to cell death4,6. Additionally, changes in the duration of ERK activity can also lead to distinct outcomes: in PC12 cells, ERK activation for 30 min or less induces cell proliferation, but ERK activation for 60 min or more induces neuronal differentiation8,9. Tight regulation of ERK activity is thus clearly essential for normal development and homeostasis.
C. elegans is a powerful, and genetically malleable model system to dissect the function and regulation of the RTK-RAS-ERK signaling pathway3,10-15. Relative to mammalian systems, which contain multiple genes for RAS and ERK, C. elegans contains one RAS gene (let-60) and one ERK gene (mpk-1), rendering it a genetically more facile system in which to dissect the function of this pathway3,10-14. The C. elegans germline is essentially a tube that consists of mitotic stem cells at its distal end and mature oocytes at its proximal end (Figure 1)16. Germ cells initiate meiosis just proximal to the distal mitotic zone, and progress through an extended meiotic prophase (pachytene), after which they begin to form oocytes in the loop region, finally undergoing oocyte maturation in the proximal region16.
Genetic studies from multiple labs, including our own, have shown that the RTK-RAS-ERK pathway is essential for germline development in C. elegans12,13,15,17-19. Specifically, we found that ERK controls and coordinates at least nine distinct biological processes during germline development, ranging from developmental switches such as germ cell apoptosis to cell biological processes such as oocyte growth11,18. Much like in mammalian systems, too much ERK activity in the C. elegans germline results in production of multiple small oocytes, while too little activity results in one large oocyte11. Thus tight regulation of dpERK is essential for normal germline development. The active form of ERK, as visualized by an antibody specific to dpERK, displays a stereotypical, dynamic, bimodal localization pattern: dpERK is high during mid-pachytene (Zone 1, Figure 1), low in the loop region and high again in mature oocytes (Zone 2, Figure 1). Recently, we found that nutrition acts through the Insulin-like Growth Factor receptor-1 (daf-2) to activate ERK in Zone 112; prior work showed that the sperm signal (via the Ephrin receptor tyrosine kinase) activates ERK in Zone 213.
Given that active ERK functions as a rheostat in the germline to regulate oocyte growth, spatial and temporal localization, as well as amplitude of dpERK, is key to understanding its normal signaling outcome. Using the method described here, changes in the stereotypical localization of dpERK can be easily monitored and predictions derived on the impact of the environmental or genetic perturbations on ERK activity and thus function. Thus, assaying for dpERK enables a comprehensive understanding of its role during germline development.
Protocol
The protocol described here is primarily for the invertebrate model system C. elegans, and follows all the ethical guidelines set forth by the institution.
1. Animal Maintenance
Maintain C. elegans working stock cultures on Nematode Growth Medium20,21 (NGM, see Materials Table) agar seeded with E. coli OP50.
Maintain worm stocks at temperatures between 16 °C and 25 °C. NOTE: Culturing worms at higher temperatures results in faster growth rate, often, aberrant germline development and lower brood sizes. Additionally, temperature sensitive genotypes will display different phenotypes at different temperatures.
Transfer worms to new NGM plates every 2 - 3 days depending on their growth rate so as to maintain a constant supply of well-fed worms. NOTE: For any given experiment involving dpERK levels, worms should NOT be obtained from a starved or crowded plate. Crowding or starvation impacts insulin signaling in worms22, and insulin signaling regulates ERK12 activity. Thus worms from starved or overcrowded plates will result in variable and difficult-to-reproduce staining patterns.
2. Dissection of Adult Worms for Obtaining Gonads
Pick 100 - 150 WT (N2) or desired genotype of worms at the desired and specific developmental stage (L1, L2, L3, L4, adult, etc.) in 100 µL of M9 buffer in a 1.5 mL microcentrifuge tube.
Fill the microcentrifuge tube with 900 µL of M9 buffer (see Materials Table). Centrifuge the tubes at 1,000 x g in a microcentrifuge for 1 min at ambient temperature.
Gently remove 900 µL of the M9 buffer and discard. Remove the buffer under a dissecting microscope to ensure that the worms are not accidently removed.
Repeat steps 2.2 - 2.3 two more times with fresh M9 buffer each time. This ensures that any bacteria that were carried over with the worms are effectively washed away.
After the final wash remove 900 µL of M9 buffer from the tube and discard.
Transfer the remaining 100 µL of M9 buffer containing the 100 - 150 worms with a 200 µL-micropipette tip to a flat bottom glass watch dish. Ensure that the micropipette tip is cut at the 10 µL mark before use. Not cutting the tip will result in shearing of the worms.
Add 1 - 3 µL of 0.1 M levamisole to the glass watch dish containing the worms in M9 buffer. Gently swirl the levamisole and the M9 to enable even mixing until the worms stop moving. NOTE: Levamisole stocks can be stored at -20 °C for long-term storage. Here, use the higher concentration of 1 - 3 mM than what has been described in literature23 of 0.2 - 0.25 mM of levamisole in order to obtain rapid loss of mobility in the animals. If the animals flay around for too long in the liquid suspension, the resulting patterns of dpERK in the gonads can be variable (due to stress or lack of nutrition or both). More reproducible staining patterns have been achieved with 1 - 3 mM levamisole for dissection.
Attach two 25 G needles to two 1 mL syringes (the size of the syringe does not matter, the gauge of the needle is critical). Position one syringe in each hand (Figure 2).
- Under a dissecting microscope, position each needle under and over each worm as shown in Figure 2 and place a fine cut on the worm near the second pharyngeal bulb in a scissor-like motion. After one cut in the worm move on to the next animal until all the animals in the dish have been cut at least once. NOTE: Be mindful that steps 2.8 - 2.9 are time sensitive. Dissect all worms in the dish in under five min for a reproducible dpERK pattern. Longer dissection times will result in a turning off of the dpERK signal, especially in Zone 1. Adjust the number of worms per round of dissection (i.e., 50 worms vs. 150) if necessary to stay in the allotted time frame.
- In case an extruded gonad is not visible during the dissection, do not attempt another cut in the same animal. Usually that will only shear the animal and not result in extrusion of the gonad. Instead, move on to the next animal. Worms where the gonads do not extrude will appear as such on the slides that will be made in section 6 and can be ignored during imaging NOTE: Through all of the dissection and processing steps, it is important to bear in mind that the gonad will remain attached to the body/carcass. Do not force the gonad and the body apart with the needle. Often times an aberrant tear in the gonadal tissue in an attempt to sever the gonad from the body can result in spurious antibody signal. The body/carcass can be ignored during the imaging steps (section 6), and only the gonad imaged. Additionally, sometimes the intestinal cells can also serve as internal controls/somatic controls for the antibody being assayed.
3. Gonad Fixation
After the dissection is complete, add 2 mL of 3% paraformaldehyde (PFA) directly to the glass watch dish containing the 100 μL of M9 and the dissected animals and cover with Parafilm. Place the covered watch glass dish in a chemical fume hood. Caution: PFA is a hazardous chemical. Thus, the fixation should be performed in the chemical fume hood.
Incubate at RT for 10 min.
Using a disposable 9" glass Pasteur pipette add 3 mL of 1x PBS-T (see Materials Table) to the watch glass dish containing the dissected animals. Mix by drawing the PFA solution and the 1x PBS-T with the dissected animals into the Pasteur pipette and releasing gently back into the watch glass dish. Do not vortex the tubes during the washes.
Using a fresh glass Pasteur pipette draw the 5 mL of liquid (containing dissected worms, PFA and 1x PBS-T) from the watch glass dish into the Pasteur pipette and transfer the contents into a fresh 5 mL glass conical tube.
Centrifuge the conical tubes for 30 sec in a clinical centrifuge at 1,000 x g. Use glass Pasteur pipettes and conical tubes as dissected gonads stick to plastic.
Remove and discard the supernatant taking care so as not to disturb the dissected animals. Remove the liquid after each wash from the conical tube under a dissecting microscope so as not to affect the dissected gonads, and minimize their loss.
Using a fresh Pasteur pipette, add 5 mL of 1x PBS-T to the conical tubes. Centrifuge the conical tubes in a clinical centrifuge for 30 s at 1,000 x g. Remove and discard the supernatant taking care so as not to disturb the dissected animals.
Repeat step 3.7 for a total of three times.
Using a fresh Pasteur pipette, add 2 mL of 100% methanol to the tubes, and gently mix the gonads with the methanol by drawing up into the Pasteur pipette. Incubate the tube at -20 °C for a minimum of 1 h. NOTE: Dissected gonads can be stored in methanol for up to 2 days for dpERK staining. Storage for more than 2 days and up to 2 weeks is compatible with other antibody staining applications, such as anti-Lamin antibody, but not with the dpERK antibody.
After the desired incubation time, repeat step 3.7 for a total of three times in order to wash the gonads. Do not vortex the tubes during the washes. After the final wash leave 500 μL of 1x PBS-T in the 5 mL conical tube.
Using a fresh Pasteur pipette transfer the contents of the conical glass tube into a fresh 1 mL glass tube (6 mm x 50 mm). Allow the dissected animals to settle by gravity for 5 - 10 min.
Using a fresh Pasteur pipette and under a dissecting microscope, remove as much of the wash as possible from the 1 mL glass tubes without disturbing the dissected gonads.
4. Blocking and Primary Antibody Treatment
Add 100 µL of 30% Normal Goat Serum (NGS) diluted in 1x PBS-T (see Materials Table) to the dissected animals in the 1 mL glass tubes. 30% NGS serves as the blocking buffer. Cover the tube with Parafilm to avoid evaporation of the liquid. Incubate at RT for 1 h or at 4 °C overnight.
After the desired time of incubation, remove the blocking buffer using a fresh Pasteur pipette, removing as much liquid as possible. Use a dissecting microscope to ensure that gonads are not drawn up in the Pasteur pipette.
Dilute the MAPKYT antibody (see Materials Table, referred to in this method as dpERK) at 1:400 in 30% NGS. Prepare enough antibody dilution to use 100 µL per tube. Unused, diluted antibody can be stored at 4 °C for a week.
Add 100 µL of the diluted anti-dpERK antibody solution to the 1 mL glass tubes containing the dissected animals. Seal the tubes with Parafilm. Incubate the tubes at 4 °C overnight.
After overnight incubation, add 800 µL of 1x PBST to the tubes at RT.
Draw the sample up in a fresh glass Pasteur pipette and release gently to ensure that the gonads are evenly suspended in the 1x PBS-T. Let the gonads settle to the bottom with gravity. Remove the supernatant. This ensures that the dissected gonads are washed but not damaged during the process.
Repeat steps 4.5 - 4.6 for a total of three washes. Do not vortex the tubes during the washes. After the third wash be sure to remove and discard as much supernatant as possible without disturbing the dissected animals. Ensure that a fresh Pasteur pipette is used each time.
5. Secondary Antibody Treatment
During the primary antibody washes prepare the dilution for the secondary antibody. Dilute the anti-mouse secondary antibody at 1:500 in 30% NGS. As with the primary antibody, use 100 µL per tube. Unused antibody can be stored at 4 °C for a week.
Add 100 µL secondary antibody solution to the 1 mL glass tubes containing the dissected animals. Cover each tube with Parafilm, and then wrap the tubes in aluminum foil to keep the contents of the tube in dark. Incubate the tubes at RT for 2 h, or alternatively at 4 °C overnight.
After the desired time of incubation, add 800 µL of 1x PBS-T to the tubes and repeat steps 4.5 - 4.6 for a total of three times. After the final wash, remove and discard as much of the supernatant as possible under a dissecting microscope using a fresh Pasteur pipette.
Prepare the DAPI solution during the washes for the secondary antibody. Dilute 1 µL of DAPI (from a 1,000x stock of 1 mg/mL stored at -20 °C) in- 1 mL of 1x PBST.
Add 800 µL of the diluted DAPI solution to the tubes containing the dissected animals. Cover the mouth of the tube with Parafilm, and wrap each tube in aluminum foil. Incubate at RT for 20 min.
After the incubation with DAPI, remove as much of the DAPI solution as possible with a fresh Pasteur pipette, under a dissecting microscope so as not to disturb the dissected animals.
Add 1 drop of mounting solution to the tubes. Wrap each tube in aluminum foil. NOTE: For visualizing dpERK staining, stained gonads should be mounted for microscopy immediately. For other antibody applications such as phosphorylated serine 10 Histone H3, gonads can be kept in mounting media for up to 2 weeks at 4 °C in the dark.
6. Assembling Slides and Imaging
Place a layer of lab tape lengthwise, on top, of two glass microscope slides (25 mm x 75 mm x 1 mm) as seen in Figure 3. Place a fresh clean glass microscope slide in between the two taped slides. NOTE: The thickness of the tape helps generate an agarose pad on the middle slide. Thickness of the agarose pad is almost equal to that of the tape, as the tape serves as a spacer for creating the agarose pad.
Drop ~ 200 µL of melted 2% agarose (made in distilled water) on the microscope slide in the middle. Quickly place a fresh clean slide, perpendicular to and on top, of the agarose.
Let the agarose solidify (1 - 2 min).
Remove the top microscope slide very gently, such that the agarose pad is not destroyed. The agarose pad can remain attached either to the top or the bottom slide. It does not matter which slide the agarose pad remains attached to, as long as it is a smooth surface of agarose.
Using a Pasteur pipette transfer the dissected animals onto the agarose pad. Remove any excess liquid from the slide using a Pasteur pipette under a dissecting microscope.
Using an eyelash hair or finely drawn capillary, push the gonads and intestines away from one another in order to spread the gonads out on the slide.
Cover the microscope slide with a 24 mm x 50 mm size coverslip. Take care to ensure that the coverslip is lowered gently onto the slide with minimal air bubbles being formed between the coverslip and the slide.
- Store at 4 °C overnight in a slide box (an opaque slide box with the slides lying flat, coverslip up) to allow excess liquid to evaporate and gonads to become somewhat flattened (due to the weight of the coverslip on the gonads). The slight flattening of the gonads allows for better imaging of an otherwise round gonadal tube.
- Once the coverslip is mounted, do not to apply pressure to the top of the coverslip with fingers. Often this results in distortion of gonads and loss of the dpERK signal.
- Flatten the gonads overnight only for more effective imaging. The slides are ready to be viewed as soon as they are assembled. To take images immediately, gently absorb the excess liquid between the coverslip and the slide from the corners using a clean lab tissue.
After the overnight period, seal the slide and coverslip with transparent nail polish topcoat on the four corners of the coverslip and the slide boundary. The nail polish/coverslip seal ensures that the coverslip does not move while imaging and protects against destruction of the samples.
- Image the gonads with a compound microscope at 63X. However the objective lens and microscope can be varied based on the availability of microscopes and user application. Because the size of the entire gonad from the proliferative region to the oocytes is larger than can be captured in one image, the images are taken as a montage with overlapping boundaries as shown in Figure 4.11-14, 18.
- In many cases, edit the carcass after the final imaging and assembly, as long as no major alterations are made to the germline image. Store the raw images along the assembled image to enable comparison of the 'before' assembly/editing and 'after' assembly/ editing of the images. NOTE: The microscopy images must not be altered for signal strength or saturation during the assembly of the montage.
Representative Results
In WT adult hermaphroditic animals, dpERK is typically visualized in mid-pachytene region, Zone 1, and in the most mature oocytes from -1 through -4 or -5 (Zone 2). Perturbations in this activation pattern reflect changes to the signaling pathway. Female germlines do not specify sperm, and thus do not display activation of ERK in Zone 2, with only weak activation in Zone 1. These are represented in Figure 5.
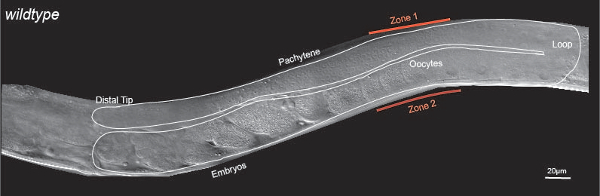 Figure 1: C. elegans germline Morphology. Differential Interference contrast (DIC) image of an adult hermaphrodite with one U-shaped gonad arm outlined with the white line. The adult hermaphroditic gonad contains mitotic cells at the distal tip. The mitotic cells enter meiosis and progress through meiotic prophase (pachytene) developing into mature oocytes in the proximal gonad. Zone 1 and Zone 2 mark stereotypical localization of dpERK in an adult hermaphroditic gonad. Scale: 20 µm. Please click here to view a larger version of this figure.
Figure 1: C. elegans germline Morphology. Differential Interference contrast (DIC) image of an adult hermaphrodite with one U-shaped gonad arm outlined with the white line. The adult hermaphroditic gonad contains mitotic cells at the distal tip. The mitotic cells enter meiosis and progress through meiotic prophase (pachytene) developing into mature oocytes in the proximal gonad. Zone 1 and Zone 2 mark stereotypical localization of dpERK in an adult hermaphroditic gonad. Scale: 20 µm. Please click here to view a larger version of this figure.
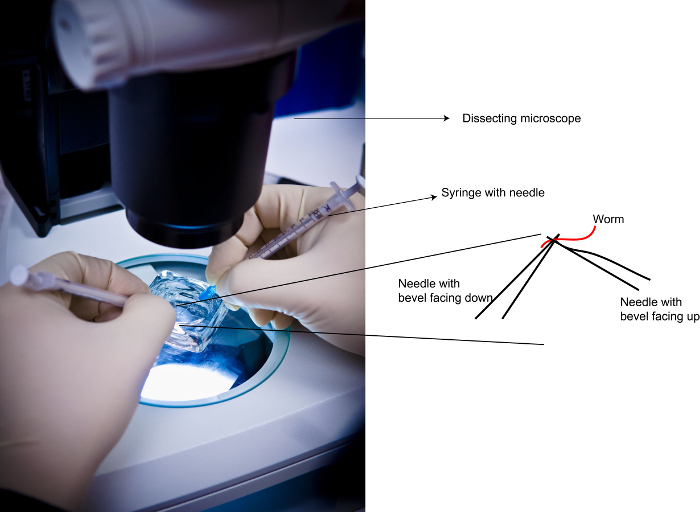 Figure 2: Demonstration of Needle Positions during dissections. Left: Photograph of a dissection in process. Right: Needle positions with reference to the worm. Please click here to view a larger version of this figure.
Figure 2: Demonstration of Needle Positions during dissections. Left: Photograph of a dissection in process. Right: Needle positions with reference to the worm. Please click here to view a larger version of this figure.
 Figure 3: Demonstration of Agarose Slide Preparation. (A) Place a tape lengthwise across 2 clean microscope slides. Place a fresh clean microscope slide between the two slides with the tape as shown. The three slides are next to each other lengthwise. (B) Add melted agarose to the slide in the center. (C) Place a fresh microscope slide perpendicular to, and on top of, the middle slide, carrying the drop of agarose. (D) Allow the agarose to solidify. (E) Remove the top slide leaving behind the solidified agarose pad. Use the bottom slide with the agarose pad for mounting dissected and stained germlines. Please click here to view a larger version of this figure.
Figure 3: Demonstration of Agarose Slide Preparation. (A) Place a tape lengthwise across 2 clean microscope slides. Place a fresh clean microscope slide between the two slides with the tape as shown. The three slides are next to each other lengthwise. (B) Add melted agarose to the slide in the center. (C) Place a fresh microscope slide perpendicular to, and on top of, the middle slide, carrying the drop of agarose. (D) Allow the agarose to solidify. (E) Remove the top slide leaving behind the solidified agarose pad. Use the bottom slide with the agarose pad for mounting dissected and stained germlines. Please click here to view a larger version of this figure.
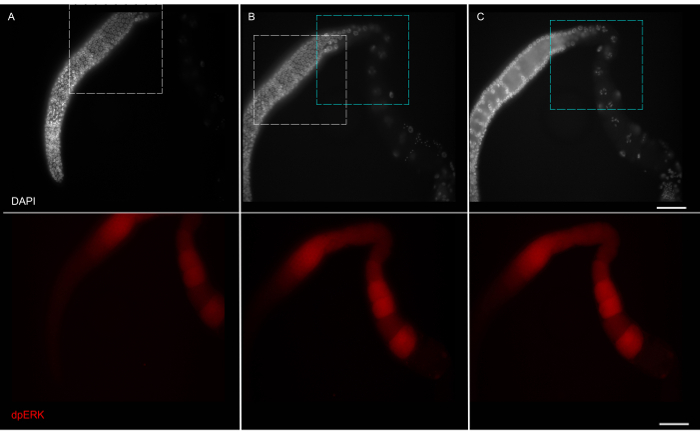 Figure 4: Imaging the Slides as a Montage. Montage of a wild type germline with DAPI (top) and dpERK (bottom) channel. Images taken at 63X with overlapping boundaries, white boxes for panel A and B and green boxes for panel B and C. The DAPI and dpERK images for each panel were taken simultaneously in two different channels. The assembled image is shown in Figure 5A. Scale bar: 20 μm. Please click here to view a larger version of this figure.
Figure 4: Imaging the Slides as a Montage. Montage of a wild type germline with DAPI (top) and dpERK (bottom) channel. Images taken at 63X with overlapping boundaries, white boxes for panel A and B and green boxes for panel B and C. The DAPI and dpERK images for each panel were taken simultaneously in two different channels. The assembled image is shown in Figure 5A. Scale bar: 20 μm. Please click here to view a larger version of this figure.
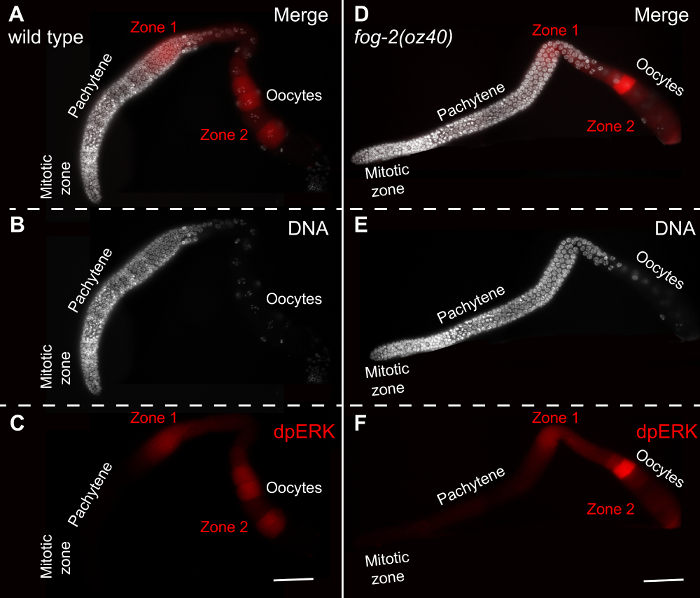 Figure 5: Dynamic Temporal/Spatial Activation of ERK. (A-C) WT (N2) adult (24 hours past mid-L4 stage of development) hermaphrodite germlines stained for DNA (B, DAPI, white) and dpERK (C, red). The dpERK signal in Zone 1, the pachytene region, and Zone 2, the mature oocytes is highlighted. (D-F) fog-2(oz40) female germlines (at 8 h past mid-L4 stage of development) stained for DNA (E, DAPI) and dpERK (F). Young female germlines display weak dpERK in Zone 1, and in a single oocyte in a sperm independent manner. Zone 2 is sperm dependent, and thus absent in the female germline. Scale bar: 20 μm. Please click here to view a larger version of this figure.
Figure 5: Dynamic Temporal/Spatial Activation of ERK. (A-C) WT (N2) adult (24 hours past mid-L4 stage of development) hermaphrodite germlines stained for DNA (B, DAPI, white) and dpERK (C, red). The dpERK signal in Zone 1, the pachytene region, and Zone 2, the mature oocytes is highlighted. (D-F) fog-2(oz40) female germlines (at 8 h past mid-L4 stage of development) stained for DNA (E, DAPI) and dpERK (F). Young female germlines display weak dpERK in Zone 1, and in a single oocyte in a sperm independent manner. Zone 2 is sperm dependent, and thus absent in the female germline. Scale bar: 20 μm. Please click here to view a larger version of this figure.
Discussion
Active ERK (dpERK) follows a stereotypical spatial and dynamic localization pattern in the C. elegans germline. This stereotyped dpERK spatial pattern (Figure 5), and amplitude in an adult C. elegans gonad, can be effectively correlated with the many biological processes that ERK regulates. For example, an inability to activate ERK in Zone 2 results in an arrest in oocytes in prophase of meiosis, a phenotype observed in female germlines that do not specify sperm, and thus do not activate ERK in mature oocytes (Figure 5). Mating with males' results in effective accumulation of dpERK in Zone 2 in the female germlines11,13, coupled with onset of oocyte maturation and ovulation. Thus analysis of spatial patterns and temporal activation of dpERK upon certain genetic perturbations (such as RNA interference mediated gene inactivation) or chemical treatments allows for identification of factors that regulate ERK signaling and thus ERK-dependent biological processes. Such analyses also enable discovery of novel genetic interactions between signaling pathways.
A critical bottleneck for any imaging based analysis is the availability of functional reagents such as antibodies that are specific to the application. If such a reagent exists then methods such as the one described above can be easily adapted for the analysis of localization pattern for any protein of interest. The application is thus limited by the availability of a functioning antibody. Additionally, because the method describes dissection and staining at a given developmental time, it only enables the capturing of information in one time frame, and does not shed light on the dynamics or protein regulation in real time or rate of protein turnover.
The method described above is adapted from Francis et al.23, which is distinct from the Seydoux and Dunn method24 for gonad extrusion and antibody staining. The method based on Francis et al. will be called the "suspension method" and the Seydoux and Dunn method called the "freeze cracking method" for comparison. The suspension method has been very effective in our hands for obtaining reproducible localization patterns of signaling molecules such as dpERK, that are inherently more sensitive to harsh handling conditions. In the case of dpERK localization, in our experience, the signal in Zone 1 is virtually undetectable with the freeze cracking method. Additionally, sample drying, which can often occur with the freeze cracking method, results in spurious antibody signals. The suspension method minimizes sample drying, since the gonads are suspended in liquid throughout the process. We also find that the suspension method is useful for imaging multiple different genotypes on a single slide. For example, if two genotypes, such as hermaphrodites vs females are to be directly compared for dpERK localization, the two genotypes can be mixed together and processed. This is possible because the phenotypes are distinct and can be distinguished via DAPI morphologies. Staining in the same tube followed by imaging on the same slide allows for direct comparison between the gonads from different genotypes. Alternatively, if the DAPI morphologies are not distinguishable, then a two-step antibody staining can be adopted. First each individual genotype is treated with two distinct antibodies, Antibody A for WT and Antibody B for mutant (both antibodies need to be raised in a host distinct from the dpERK antibody). Once the two genotypes have been stained with the primary antibody, and washed, they can be mixed together in one tube and now stained with the dpERK antibody, followed by a secondary antibody treatment to visualize all the antibodies in the tube. An ability to compare different genotypes on a single slide, especially when deductions of activation patterns are being made ensures accurate interpretations not affected by distinct staining conditions.
While advantageous, there are multiple steps throughout the suspension method that require careful manipulation. It is critical that the dissections occur in a time efficient manner; importantly, the dissections should not take over 5 min. Longer dissection times result in variable dpERK signal, which can often be misinterpreted as regulated changes in ERK activation, rather than as technical failures. It is crucial to start with over 100 - 150 animals for each dissection because throughout the various wash steps the gonads can easily be lost from the tubes due to pipetting errors. Loss of gonads can be reduced either by dissecting in multiple small batches (such as three batches of 50 animals each) that can be combined, or by dissecting one large batch of 150. Another challenge is getting the gonads to lie in a well-spaced formation on the slide so that the entire distal gonad can be imaged. To enable this, use an eyelash on a matchstick or a pulled pipette to space out the gonads. However, care should be taken so as not to damage the gonads during this process. Often with these techniques it takes multiple attempts to master the dissections as well as arrangements of the gonads onto the slides.
Once this technique has been mastered, it serves as a powerful method for varied biological analyses, and can be used to study more than just dpERK levels. For example this method can be used to visualize active p38 levels, or active CDK levels, or any protein for which an antibody reagent exists. Additionally, the method can be coupled with a chemical inhibition screen in whole animals to assay for novel inhibitors or activators of the pathway, via assaying for dpERK levels. This will allow for an in vivo readout of both reaction and sensitivity to any inhibitors that may interact with the RTK-RAS-ERK signaling pathway. Moreover, this method can be used in antibody combinations with multiple signaling outputs that can all be used and visualized at one time. Using multiple antibodies allows correlation between multiple pathways, their activation status, and outcomes all within the same tissue, enabling better understanding of these interactions. These are just a few examples of further applications of this method, but this system can be modified to study multiple research interests.
Disclosures
The authors declared that no competing interests exist.
Acknowledgments
Work in the Arur Lab is supported by grants from the National Institutes of Health, NIHGM98200; Cancer Prevention Research Institute of Texas, CPRIT RP160023; the American Cancer Society Research Scholar Award (ACS RSG014-044-DDC); and by the Anna Fuller Funds.
References
- Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410:37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- McKay MM, Morrison DK. Integrating signals from RTKs to ERK/MAPK. Oncogene. 2007;26:3113–3121. doi: 10.1038/sj.onc.1210394. [DOI] [PubMed] [Google Scholar]
- Sundaram MV. Canonical RTK-Ras-ERK signaling and related alternative pathways. WormBook. 2013. pp. 1–38. [DOI] [PMC free article] [PubMed]
- Fabregat I, Roncero C, Fernandez M. Survival and apoptosis: a dysregulated balance in liver cancer. Liver Int. 2007;27:155–162. doi: 10.1111/j.1478-3231.2006.01409.x. [DOI] [PubMed] [Google Scholar]
- Hackett A, Rowe L. FGFR1 Pfeiffer syndrome without craniosynostosis: an additional case report. Clin Dysmorphol. 2006;15:207–210. doi: 10.1097/01.mcd.0000220608.40155.d4. [DOI] [PubMed] [Google Scholar]
- Murphy LO, Blenis J. MAPK signal specificity: the right place at the right time. Trends Biochem Sci. 2006;31:268–275. doi: 10.1016/j.tibs.2006.03.009. [DOI] [PubMed] [Google Scholar]
- Vogels A, Fryns JP. Pfeiffer syndrome. Orphanet J Rare Dis. 2006;1:19. doi: 10.1186/1750-1172-1-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klesse LJ, Meyers KA, Marshall CJ, Parada LF. Nerve growth factor induces survival and differentiation through two distinct signaling cascades in PC12 cells. Oncogene. 1999;18:2055–2068. doi: 10.1038/sj.onc.1202524. [DOI] [PubMed] [Google Scholar]
- Marshall CJ. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell. 1995;80:179–185. doi: 10.1016/0092-8674(95)90401-8. [DOI] [PubMed] [Google Scholar]
- Chang C, Sternberg PW. C. elegans vulval development as a model system to study the cancer biology of EGFR signaling. Cancer Metastasis Rev. 1999;18:203–213. doi: 10.1023/a:1006317206443. [DOI] [PubMed] [Google Scholar]
- Lee MH, et al. Multiple functions and dynamic activation of MPK-1 extracellular signal-regulated kinase signaling in Caenorhabditis elegans germline development. Genetics. 2007;177:2039–2062. doi: 10.1534/genetics.107.081356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez AL, et al. DAF-2 and ERK couple nutrient availability to meiotic progression during Caenorhabditis elegans oogenesis. Dev Cell. 2013;27:227–240. doi: 10.1016/j.devcel.2013.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MA, et al. A sperm cytoskeletal protein that signals oocyte meiotic maturation and ovulation. Science. 2001;291:2144–2147. doi: 10.1126/science.1057586. [DOI] [PubMed] [Google Scholar]
- Ohmachi M, et al. C. elegans ksr-1 and ksr-2 have both unique and redundant functions and are required for MPK-1 ERK phosphorylation. Curr Biol. 2002;12:427–433. doi: 10.1016/s0960-9822(02)00690-5. [DOI] [PubMed] [Google Scholar]
- Church DL, Guan KL, Lambie EJ. Three genes of the MAP kinase cascade, mek-2, mpk-1/sur-1 and let-60 ras, are required for meiotic cell cycle progression in Caenorhabditis elegans. Development. 1995;121:2525–2535. doi: 10.1242/dev.121.8.2525. [DOI] [PubMed] [Google Scholar]
- Hubbard EJ, Greenstein D. The Caenorhabditis elegans gonad: a test tube for cell and developmental biology. Dev Dyn. 2000;218:2–22. doi: 10.1002/(SICI)1097-0177(200005)218:1<2::AID-DVDY2>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Arur S, et al. MPK-1 ERK controls membrane organization in C. elegans oogenesis via a sex-determination module. Dev Cell. 2011;20:677–688. doi: 10.1016/j.devcel.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arur S, et al. Multiple ERK substrates execute single biological processes in Caenorhabditis elegans germ-line development. Proc Natl Acad Sci U S A. 2009;106:4776–4781. doi: 10.1073/pnas.0812285106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattingly HH, Chen JJ, Arur S, Shvartsman SY. A Transport Model for Estimating the Time Course of ERK Activation in the C. elegans Germline. Biophys J. 2015;109:2436–2445. doi: 10.1016/j.bpj.2015.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiernagle T. Maintenance of C. elegans. WormBook. 2006. pp. 1–11. [DOI] [PMC free article] [PubMed]
- Dorman JB, Albinder B, Shroyer T, Kenyon C. The age-1 and daf-2 genes function in a common pathway to control the lifespan of Caenorhabditis elegans. Genetics. 1995;141:1399–1406. doi: 10.1093/genetics/141.4.1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis R, Barton MK, Kimble J, Schedl T. gld-1, a tumor suppressor gene required for oocyte development in Caenorhabditis elegans. Genetics. 1995;139:579–606. doi: 10.1093/genetics/139.2.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seydoux G, Dunn MA. Transcriptionally repressed germ cells lack a subpopulation of phosphorylated RNA polymerase II in early embryos of Caenorhabditis elegans and Drosophila melanogaster. Development. 1997;124:2191–2201. doi: 10.1242/dev.124.11.2191. [DOI] [PubMed] [Google Scholar]